

Abstract
Coronary artery disease is the most common cause of death globally and is linked to a number of risk factors including serum low density lipoprotein, high density lipoprotein, triglycerides and lipoprotein(a). Recently two proteins, angiopoietin-like protein 3 and 4, have emerged from genetic studies as being factors that significantly modulate plasma triglyceride levels and coronary artery disease. The exact function and mechanism of action of both proteins remains to be elucidated, however, mutations in these proteins results in up to 34% reduction in coronary artery disease and inhibition of function results in reduced plasma triglyceride levels. Here we report the crystal structures of the fibrinogen-like domains of both proteins. These structures offer new insights into the reported loss of function mutations, the mechanisms of action of the proteins and open up the possibility for the rational design of low molecular weight inhibitors for intervention in coronary artery disease.
Introduction
Coronary artery disease (CAD) is the most common form of heart disease and is the most common cause of death globally, with CAD affecting 110 million people in 20151. CAD is linked to a number of risk factors, including smoking2, diabetes3, family history4, stress5 and environmental factors6. Blood fats, including serum low density lipoprotein, high density lipoprotein, triglycerides and lipoprotein(a) are all strongly linked with CAD7–14.
Recently proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors have been developed to inhibit LDL uptake14,15 and to reduce the incidence of CAD16. Parallel to this two other proteins have emerged – primarily from genetic studies – as being factors that significantly modulate plasma triglyceride levels and CAD. These proteins, human angiopoietin-like protein 3 (Angptl3; also known as Ang5) and angiopoietin-like protein 4 (Angptl4) are, as the names suggest, related structurally to angiopoietins which are involved in angiogenesis via their interaction with the Tie2 (tyrosine kinase with immunoglobulin and endothelial growth factor homology domain-2) receptor17. Both angiopoietins and angiopoietin-like proteins (except Angptl8) contain an N-terminal region, predicted to be intrinsically disordered, followed by a coiled-coil oligomerization domain and a C-terminal fibrinogen-like domain. While angiopoietins primarily retain all of these domains, Angptl4 and Angptl3 undergo proteolysis to release the functional fibrinogen-like domain18–20. The molecular mechanisms of action of these proteins are not as well defined, but functionally they appear to play a wider role than that of the angiopoietins. Angptl3 is primarily studied for its role in lipid and glucose metabolism21,22 and is involved in familial hypobetalipoproteinemia 2 (FHBL2), while Angptl4 has been reported to have wider but overlapping functions23 and has been implicated in wound repair24,25.
Recently there has been significant focus on both Angptl3 and Angptl4 being targets for CAD intervention, with Angptl3 being predicted to be the next PCSK926. For Angptl3 loss of function mutations result in significantly lower levels of triglycerides, high density lipoprotein (HDL) cholesterol and low density lipoprotein (LDL) cholesterol27–31, with carrier status associated with a 34% reduction in CAD29. Inhibition of Angptl3 function in humans with the antibody evinacumab reduced triglycerides and LDL cholesterol levels in humans and decreased atherosclerotic lesion area and necrotic content in dyslipidemic mice30. The use of antisense oligos to Angptl3 in human trials significantly decreased triglycerides, LDL cholesterol, very low density lipoprotein (VLDL) cholesterol, non-high density lipoprotein cholesterol, apolipoprotein B and apolipoprotein CIII32. For Angptl4 loss of function mutations are reported to result in significantly lower triglyceride levels and higher HDL levels than among non-carriers and carriers were less likely to have CAD27,33–36.
While there is very significant overlap between the phenotypes associated with loss of function of Angptl3 and Angptl4 there are differences, for example Angptl3 loss of function decreases HDL cholesterol levels while loss of function of Angptl4 increases HDL cholesterol levels. The exact function and mechanisms of action of both proteins remain to be elucidated, but both have been reported to interact with lipoprotein lipase37,38.
Here we determined the structures of the fibrinogen-like domains of Angptl3 and Angptl4, structures which offer new insights into reported loss of function mutations, the mechanisms of action of the proteins and open up the possibility of rational design of low molecular weight inhibitors.
Results and Discussion
Protein production
Angiopoietins and Angiopoietin-like proteins (except Angptl8) have a similar domain architecture. After the signal peptide they have a region predicted to be intrinsically disordered followed by a coiled-coil domain and a C-terminal fibrinogen-like domain (Fig. 1A). Given their importance in CAD and emerging potential to serve as therapeutic drug targets modulating triglyceride levels in human we focused on angiopoietin-like proteins 3 and 4. Initial production trials for Angptl3 and Angptl4 were based on the co-expression of the folding factors Erv1p and PDI and the mature protein (Ser17-Glu460 and Gly26-Ser406 respectively) or for Angptl3, a construct which lacks part of the predicted N-terminal intrinsically disordered region (Phe37-Glu460). These constructs, as well as constructs that lacked the fibrinogen-like domain (Ser17-Gly241, Ser17-Thr225, Phe37-Gly241 and Phe27-Thr225 for Angptl3 and Gly26-Arg183 for Angptl4), were produced as inclusion bodies or in low amounts and/or were prone to proteolysis. Given that the fibrinogen-like domain in angiopoietins is the region which interacts with receptors and is reported to be released from Angptl3 and Angptl4 by proteolysis in vivo, focus switched to the functional C-terminal fibrinogen-like domains (Ile242-Glu460 and Leu184-Ser406 respectively). Both fibrinogen-like domains contain 4 cysteine residues, which by conservation with angiopoietins were thought to form two sequential disulfide bonds. Expression of the Angptl3 and Angptl4 fibrinogen-like domains with an N-terminal His-tag in the cytoplasm of E.coli, along with the folding factors Erv1p, PDI and cypB expressed from a second plasmid, resulted in the production of 80 mg/L of Angptl3 fibrinogen-like domain and 190 mg/L of Angptl4 fibrinogen-like domain.
Figure 1.

Crystal structure of the fibrinogen-like domains of Angptl3 and Angptl4. (A) Schematic of Angiopoietin-like protein domain architecture. Each angiopoietin and angiopoietin-like protein (except Angptl8) contains an N-terminal region predicted to be intrinsically disordered, a coiled-coil region and a C-terminal fibrinogen-like domain. (B) Structure of Angptl3 (left) and Angptl4 (right) fibrinogen-like domains. Each can be split into three subdomains A, B and P. Each structure is colored according to secondary structure elements with α-helices in gold (Angptl3) and violet (Angptl4), β-strands in cyan (Angptl4) and green (Angptl4) and loops in grey. Missing sections in the loops are indicated by dashed lines. Secondary structure elements are labeled and the N- and C-termini are indicated. (C) Electrostatic surface potential of Angptl3, the orientation is equivalent to that in panel B and rotated 90° around horizontal axis to see the underside of the P subdomain. Negative charge is shown in red and positive in blue. (D) As panel C but Angptl4.
Electrospray mass spectrometry of both proteins + /− treatment with N-ethylmaleimide indicated that both fibrinogen-like domains were of the expected mass and both contained two disulfide bonds.
Structure determination of the fibrinogen-like domain of human Angptl3 and Angptl4
Crystals of the fibrinogen-like domains of Angptl3 and Angptl4 diffracted to 2.1 Å and 2.3 Å, respectively. The crystal structures were solved by molecular replacement and the models were refined to R/Rfree values of 19.4/25.3 and 20/25.4 for Angptl3 and Angptl4, respectively. All data collection and refinement statistics are summarized in Table 1.
Table 1.
Data collection and refinement statistics.
| Angptl3 | Angptl4 | |
|---|---|---|
| PDB code | 6EUA | 6EUB |
| Data collection | ||
| Wavelength (Å) | 0.966 | 0.9999 |
| Resolution range (Å) | 43.68–2.1 (2.17–2.1) | 47.16–2.3 (2.382–2.3) |
| Space group | P 21 21 21 | C 2 2 21 |
| Cell dimensions | ||
| a, b, c (Å) | 60.05 63.65 169.43 | 133.02 133.77 39.63 |
| α, β, γ, (°) | 90 90 90 | 90 90 90 |
| Total reflections | 148702 | 102775 |
| Unique reflections | 38793 | 16058 |
| Multiplicity | 3.8 (3.8) | 6.4 (6.3) |
| Completeness (%) | 99.30 (97.05) | 98.88 (95.31) |
| Mean I/ σ Ι | 7.14 (1.02) | 8.45 (1.25) |
| R merge | 0.1317 (1.206) | 0.1492 (1.121) |
| R pim | 0.07577 (0.6875) | 0.06355 (0.4708) |
| CC1/2 | 0.993 (0.398) | 0.995 (0.651) |
| Refinement | ||
| Rwork (%) | 19.4 (27.2) | 20 (31.6) |
| Rfree (%) | 25.3 (31.6) | 25.4 (32) |
| Number of non-hydrogen atoms | 5139 | 1790 |
| macromolecules | 4978 | 1731 |
| ligands | 16 | |
| solvent | 161 | 43 |
| Protein residues | 601 | 216 |
| RMS bonds (Å) | 0.007 | 0.010 |
| RMS angles (°) | 0.79 | 1.06 |
| Average B factor (Å2) | 46.78 | 57.78 |
| macromolecules | 46.99 | 57.76 |
| ligands | 66.35 | |
| solvent | 40.47 | 55.42 |
Statistics for the highest-resolution shell are shown in parentheses.
There are 3 monomers of Angptl3 in the asymmetric unit. The N-terminal His-tag as well as the C-terminal 6 amino acids are not visible. In addition, there are two regions within the fibrinogen-like domain (Lys389 - His391 and Pro420 - Arg429 in the A-chain) for which the electron density is either missing or not of sufficient quality to allow tracing of the chain, suggesting the presence of flexible loops.
The Angptl4 crystal contains 1 molecule per asymmetric unit and the visible polypeptide chain begins with Leu184 and ends with Met400, hence 6 amino acids of the C-terminus are not visible. There is one break in the mainchain of Angptl4 at Gln370, suggesting the loop that it is in is flexible.
Structures of the fibrinogen-like domain of Angptl3 and Angptl4
The fibrinogen-like domains of Angptl3 and Angptl4 form compact structures and, according to fibrinogen nomenclature, can be divided to 3 subdomains A, B and P (Fig. 1B).
The N-terminal domain (subdomain A) consists of an α-helix (α1) and two β-strands (β1, β2) forming a small antiparallel sheet which is stabilized by a disulfide bond formed by Cys246 and Cys274 in Angptl3 and Cys188 and Cys216 in Angptl4. The architecture of subdomain A is the most conserved among the fibrinogen-like domain containing homologs and superimposes well in both Angptl3 and Angptl4.
Subdomain B is the largest subdomain in both molecules. In Angptl3, it is composed of five β-strands (β3, β4, β5, β6 and β10) forming a large twisted antiparallel β-sheet, and three α-helices (α2, α3 and α4). The β-sheet extends from one side of Angptl3 to another and forms a shallow water exposed crevice. The two helices (α2 and α3) are inserted between β3 and β4 on the opposite side of β-sheet and are connected by an extended loop. The short α-helix, α4, connects β-strands β5 and β6. β10 is a part of the C-terminus and packs between strands β3 and β4. The B subdomain of Angptl4 is similar to that of Angptl3; however, the extended β-sheet is formed by four antiparallel β-strands and instead of the long β-strand β6 in Angptl3, it contains 2 short β-strands (β6 and β7), which form an antiparallel β-hairpin and are connected via α-helix α5 and an extended solvent exposed loop.
The third subdomain (P) functions as a site for ligand binding. Consistent with this, the architecture and the sequence of subdomain (P) vary the most among fibrinogen-like domain containing proteins. In Angptl3, the P subdomain contains little regular secondary structure - three short β-strands (β7, β8 and β9) and a short α-helix (α5). β7 and β8 form a small antiparallel β-hairpin, which is braced together with α5 and stabilized by a disulfide bond between Cys394 and Cys408. The short β9 strand is followed by an extended solvent exposed loop and packs against the beginning of the β-strand β10. In contrast to Angptl3, the regular secondary structure of the P subdomain of Angptl4 is comprised of only three short α-helices (α6, α7 and α8). Similarly to Angptl3 it contains one disulfide bond (Cys341 and Cys354).
Comparison to structures of fibrinogen-like domain containing homologs
The fibrinogen-like domains of human Angptl3 and Angptl4 share significant sequence and structural homology with other fibrinogen-like domains. Angptl3 can be superimposed on its closest structural homologues, identified by Dali server39, Angiopoietins 1 and 2 with a rms deviation of 2.3 Å (192 equivalent Cα positions; 4JYO40) or 2.4 Å (192 equivalent Cα positions; 1Z3S41) respectively. Similarly angiopoietins 1 and 2 are the closest structural homologs of Angptl4, with rms deviation of 2.7 Å (200 equivalent Cα positions) and 2.6 Å (200 equivalent Cα positions) respectively. Despite the poor sequence identity for subdomain A (Fig. 2A), for both proteins subdomains A and B superimpose well. In contrast, despite significant sequence identity, there are major structural differences among these proteins within the P subdomain, the subdomain involved in functional interactions and in the segments of the B subdomain immediately preceding P (Fig. 2). For example, the P subdomain of Angptl3 contains only 1 α-helical segment and 3 β-strands and does not superimpose well with structural homologs. In contrast, the P subdomain of Angptl4 is more comparable to those of structural homologs. However, the β-sheet of the B subdomain is narrower in Angptl4 than that of homologous structures and β-strand β6 is much shorter and turns away from the β-sheet towards the solvent and together with β7, α5 and an extended loop creates a separate solvent exposed element in the structure. DALI searches using just the P subdomain of Angptl3 or Angptl4 revealed no close structural homologs.
Figure 2.

Comparison of the Angptl3 and Angptl4 fibrinogen-like domains with angiopoietins. (A) Overlay of the Cα-trace of Angptl3 (blue, left) and Angptl4 (green, right) with those of Ang1 (yellow) and Ang2 (violet). (B) Comparison with Ca2+ binding site of Ang1 (light yellow) and Ang2 (light violet) of the equivalent region in Angptl3 (blue, top) and Angptl4 (green, bottom). Residues involved in Ca2+ binding are shown in stick representation and colored similarly to the protein they belong. Ca2+ ions are shown in ball representation. (C) Alignment of the proteins shown in panel A. Cysteines are highlighted in yellow and conserved amino acids in blue. The P subdomain is boxed and the Asp involved in calcium binding in Ang1 and Ang2 are indicated with arrowheads.
In contrast to Angiopoietin family members and other identified structural homologs, including M- and N-ficolins (PDB codes 2JHI and 2J2P;42,43) and tachylectin 5A (1JC9;44), which contain 3 conserved disulfide bonds, Angptls 3 and 4 are stabilized by only 2 disulfide bonds. The third pair of cysteines in Angiopoietins 1 and 2 (C435-C437 and C433-C435, respectively) forms a part of Ca2+ binding site along with two conserved aspartates (Fig. 2B). Calcium binding is required for fibrinogen polymerization45 and for agglutinating activity of tachylectin 5A46, however, Ca2+ binding site is not directly involved in Angiopoietin/Tie2 interactions but suggested to play a structural role by stabilizing coil elements involved in the interactions41. The two aspartates are conserved in Angptl4, but not in Angptl3 (Fig. 2C). No electron density, which would indicate the presence of Ca2+ or any other ion, was observed in either the Angptl3 or Angptl4 structures.
Electrostatic surface potential
Analysis of the electrostatic surface potential of the fibrinogen-like domain of Angptl3 and Angptl4 revealed that the electrostatic charge is relatively evenly distributed over the majority of the surface of both molecules, except for the presence of an extended positively charged patch at the bottom of the P subdomain of Angptl4 (Fig. 1D), the surface that is most likely involved in the functional interactions. In contrast, no distinct electrostatic properties were identified on the surface of the P subdomain of Angptl3. This, when combined with other structural differences in the P domain, strongly suggests that Angptl3 and Angptl4 have different ligands and hence that they modulate triglyceride levels and CAD risk by distinct mechanisms. This is consistent with the observed difference that Angptl3 loss of function slightly decreases HDL cholesterol levels30 while loss of function of Angptl4 increases HDL cholesterol levels33.
Loss of function mutations
A number of loss of function or reduction of function mutations have been reported for Angptl3 and Angptl427–36. Many of these are frameshifts or premature stop codons, but others result from single amino acid substitutions in the fibrinogen-like domain. Using the determined crystal structures it is possible to elucidate why these mutations cause loss of function.
For Angptl3 there are twelve reported point mutations in the fibrinogen-like domain which are associated with decreased triglyceride levels - M259T, R288Q, S292P, E375K and Y417C27, Y344S31 and, in decreasing order of severity, G253C, I333S, D290H, C408R, Y250C and T383S (;29 S292P also reported). These mutations are found in all three subdomains of Angptl3 (Fig. 3A). Four of the mutations, S292P, R288Q, E375K and Y417C abolish or severely reduce secretion in cellulo27 implying that they significantly affect folding and/or stability of Angptl3. The effects of all four can be explained from the structure by the disruption of structure stabilizing hydrogen bonding (Fig. 3B).
Figure 3.

Structural analysis of mutations in Angptl3 which cause loss of function. (A) Angptl3 fibrinogen-like domain crystal structure with the sites of loss of function mutations depicted in ball and stick representation and highlighted in red. (B) Close up views of the environment of the mutated amino acids in panel A. Mutated residues are shown in ball and stick representation and neighboring or interacting residues are in stick representation. Residues are colored according to their location in the secondary structure elements with nitrogen atoms in blue, oxygen in red and sulfur in yellow. Potential hydrogen bonds are depicted as solid black lines.
Potential structural defects associated with seven of the other eight mutations can similarly be elucidated using the solved crystal structure, either from potential effects on intra- or inter-molecular disulfide bond formation (G253C, Y250C, C408R) and/or disruption of structure stabilizing sidechain or backbone hydrogen bonds (Y250C, D290H, I333S, T383S) and/or disruption of regular secondary structural elements (I333S, Y344S, T383S) (Fig. 3B).
The effect of one mutation cannot easily be explained from the structure obtained here. Met259 is located on β1 in subdomain A with its side chain facing the solvent (Fig. 3A). It is possible that either the M259T changes interactions with the N-terminal region of Angptl3 not included in this structure or another undescribed interaction or influences cleavage to release the fibrinogen-like domain.
For Angptl4 there are six point mutations in the fibrinogen-like domain which are linked to decreased triglyceride levels - G223R, T266M, R336C, W349C, G361S and R384W (Fig. 4A27,33,36). Of these five (G223R, R336C, W349C, G361S and R384W) have been reported to abolish secretion in cellulo27 implying they effect the folding and/or stability of Angptl4. From the crystal structure all six might be predicted to either result in misfolding of the protein or to significantly destabilize the native state (Fig. 4B), by altering intra- or inter-molecular disulfide bonds (R336C, W349C) and/or disrupting structure stabilizing hydrogen bonds (T266M, R336C, W349C, R384W) and/or introducing steric clashes (G233R, T266M, G361S) and/or disruption of regular secondary structural elements (G361S).
Figure 4.

Structural analysis of mutations in Angptl4 which cause loss of function. (A) Angptl4 fibrinogen-like domain crystal structure with the sites of mutations resulting in lower triglyceride levels are indicated in red, mutations leading to the increase of triglyceride levels are shown in blue. (B) Close up views of the environment of the mutated amino acids in panel A. Residues are depicted and colored as in Fig. 3.
For Angptl4 there are two reported mutations (P251T and R371Q) which are linked to increased triglyceride levels33. How these mutations increase triglyceride levels cannot easily be deduced from the structure. Pro251 is located in the middle of a solvent exposed loop between α2 and α3. The P251T mutation is unlikely to directly cause large conformational changes in this region of the protein. Thr has a higher propensity to be in regular secondary structure, especially β-sheet, than Pro and it is possible that the P251T mutation influences local structure and this is transmitted to subdomain P or that it increases the stability of the protein. Arg371 is located in the flexible loop after helix α7. This residue is not clearly visible in the electron density map and hence it is difficult to unambiguously suggest how the R371Q mutation increases triglyceride levels. Given the location of Gln370, Arg371 probably forms part of the positively charged patch of the surface at the bottom of the subdomain P which – by analogy with the angiopoietins – is probably involved in protein-protein interactions. Hence this mutation most likely directly modulates function rather than structure.
To further examine the potential effects on folding and/or structure of the mutations linked to a decrease in triglyceride levels, twelve mutations were made in Angptl3 and six in Angptl4 and the proteins expressed in the E.coli expression system alongside the wild-type proteins. Of the twelve mutations in Angptl3, eight (Y250C, R288Q, S292P, I333S, E375K, T383S, Y417C and C408R) resulted in no soluble protein being produced, while three (G253C, D290H and Y344S) resulted in greatly reduced yields compared with the wild-type protein (Fig. 5A). The one mutation which did not reduce the yield of protein made, M259T, was the one mutation we had been unable to assign a loss of function mutation based on the structure. To further analyze the effects of the mutation we examined thermal stability of the proteins. Wild-type and M259T mutant showed a single unfolding transition with a mid-point of 56.5 + /− 0.5 °C for wild-type and 57.7 + /− 1.1 °C for the M259T (Fig. 5B). Since the mutant again behaved like wild-type we can only assume that the M259T mutation changes interactions either with the N-terminal region of Angptl3 not included in this structure or another undescribed interaction.
Figure 5.
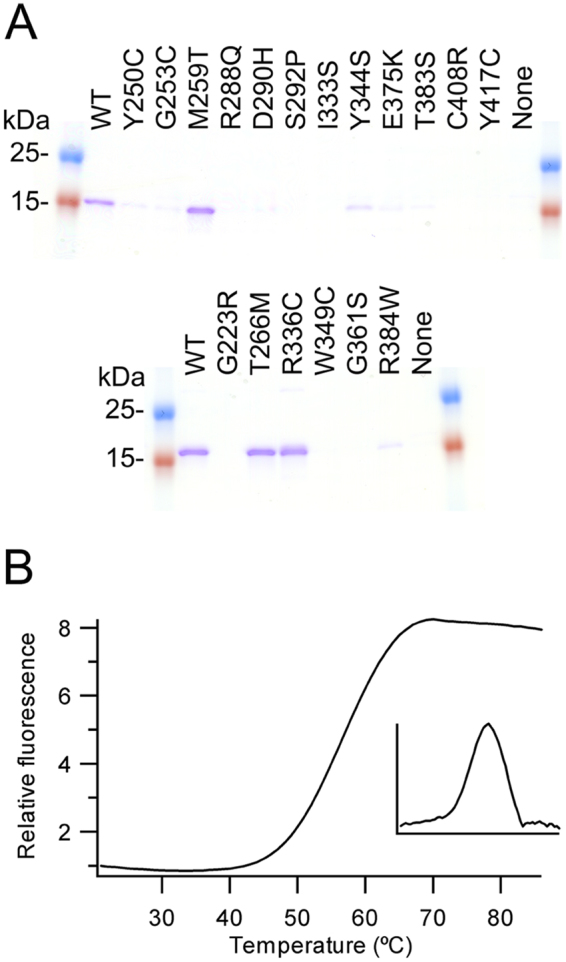
Analysis of loss of function mutations (A) Coomassie stained SDS-PAGE gel of IMAC purified proteins. Upper gel Angptl3, lower gel Angptl4 (Supplementary Fig. 1 shows the full gels) (B) Thermal stability of purified wild-type Angptl3 determined by thermofluor. The insert panel shows the derivative of the change in fluorescence signal. Angptl3 shows cooperative unfolding.
Of the six mutants made in Angptl4, three gave no soluble protein (G223R, W349C and G361S) while one resulted in greatly reduced protein yields (R384W) (Fig. 5A). The remaining two (T266M and R336C) gave yields around those of the wild-type protein. The effects of the R336C mutation were unexpected as this mutation has previously been reported to abolish secretion in cellulo27. This difference may arise due to R336 being surface exposed and hence a cysteine at this position may result in the endoplasmic reticulum exit site machinery perceiving it as being non-native and hence retained. To further analyse the effects of the mutations we examined thermal stability. Wild-type and both mutants showed a single unfolding transition with the mid-point for R336C (62.0 + /− 0.0 °C), being higher than either the wild-type or T266M mutant (both 58.0 + /− 1.0 °C). As per the M259T mutation in Angptl3, the effects of the T266M mutation in Angptl4 on modulating triglyceride levels cannot be explained by structural effects. It is noteworthy that both lie in the A subdomain and hence the T266M mutation may also change interactions either with the N-terminal region of Angptl4 not included in this structure or with another as yet undescribed protein.
Conclusions
The structures of the fibrinogen-like domains of Angptl3 and Angptl4 give new insights into these important proteins strongly linked to CAD, as well as offering the opportunity for the rational design of low molecular weight inhibitors of their function. In addition, the striking differences in the structures of subdomain P of the two proteins strongly suggests that while loss of function mutations of both lower plasma triglyceride levels, they act by different pathways.
Materials and Methods
Strains and expression constructs
All molecular biology in this study was performed using the E.coli strain XL1 Blue (recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac F´[proAB lacI lacZ∆M15 Tn10 Tetr]; Agilent Technologies), while protein production was performed in the strain MDS42 (multiple deletant strain;47).
Expression vectors (Table 2) were made by standard molecular biology techniques. Proteins of interest were cloned into a pET-derivative48 which contains an N-terminal hexahistidine tag (MHHHHHM-) for the protein of interest when cloned in using an in-frame 5′ NdeI restriction site and where the T7 promoter has been replaced with Ptac. E.coli codon optimized genes for mature human Angptl3 and Angptl4 were obtained from Genscript and these were used as templates for all domain constructs. The gene for mature human CypB was amplified from IMAGE clone 3502055 and the internal XbaI restriction site removed by mutagenesis (QuikChange; Agilent). CypB was then cloned into an existing CyDisCo variant pMJS20548 by SpeI/XhoI digestion of the vector and XbaI/XhoI digest of the insert. The resulting ligation gave a tricistronic vector (pME27) encoding Erv1p, PDI and CypB from a Ptac promoter. Plasmid purification was performed using the QIAprep spin miniprepkit (Qiagen) and purification from agarose gels was performed using the gel extraction kit (Qiagen), both according to the manufacturer’s instructions. The plasmids generated were sequenced (Biocenter Oulu core facility) to ensure there were no errors in the cloned gene.
Table 2.
Plasmids used in this study.
| Construct | Plasmid | Selection |
|---|---|---|
| Folding factors: Erv1p, PDI and CypB | pME27 | CmR |
| Angptl3 MH6M-S17-E460 | pME19 | AmpR |
| Angptl3 MH6M-F37-E460 | pME20 | AmpR |
| Angptl3 MH6M-S17-G241 | pME21 | AmpR |
| Angptl3 MH6M-S17-T225 | pME16 | AmpR |
| Angptl3 MH6M-F37-G241 | pME22 | AmpR |
| Angptl3 MH6M-F37-T225 | pME17 | AmpR |
| Angptl3 MH6M-I242-E460 | pME23 | AmpR |
| Angptl4 MH6M-G26-S406 | pME24 | AmpR |
| Angptl4 MH6M-G26-R183 | pME25 | AmpR |
| Angptl4 MH6M-L184-S406 | pME26 | AmpR |
| Angptl3 MH6M-I242-E460, Y250C | pMJS362 | AmpR |
| Angptl3 MH6M-I242-E460, G253C | pMJS363 | AmpR |
| Angptl3 MH6M-I242-E460, M259T | pMJS364 | AmpR |
| Angptl3 MH6M-I242-E460, R288Q | pMJS365 | AmpR |
| Angptl3 MH6M-I242-E460, D290H | pMJS366 | AmpR |
| Angptl3 MH6M-I242-E460, S292P | pMJS367 | AmpR |
| Angptl3 MH6M-I242-E460, I333S | pMJS368 | AmpR |
| Angptl3 MH6M-I242-E460, Y344S | pMJS369 | AmpR |
| Angptl3 MH6M-I242-E460, E375K | pMJS370 | AmpR |
| Angptl3 MH6M-I242-E460, T383S | pMJS371 | AmpR |
| Angptl3 MH6M-I242-E460, C408R | pMJS372 | AmpR |
| Angptl3 MH6M-I242-E460, Y417C | pMJS373 | AmpR |
| Angptl4 MH6M-L184-S406, G223R | pMJS374 | AmpR |
| Angptl4 MH6M-L184-S406, T266M | pMJS375 | AmpR |
| Angptl4 MH6M-L184-S406, R336C | pMJS376 | AmpR |
| Angptl4 MH6M-L184-S406, W349C | pMJS377 | AmpR |
| Angptl4 MH6M-L184-S406, G361S | pMJS378 | AmpR |
| Angptl4 MH6M-L184-S406, R384W | pMJS379 | AmpR |
Selection is indicated by CmR for chloramphenicol resistance and AmpR for ampicillin resistance.
Protein expression and purification
E. coli MDS42 strain transformed with a vector for a Angptl3/4 expression along with the pME27 vector to catalyse disulfide bond formation were streaked onto LB-agar plates containing 100 µg/mL of ampicillin and 35 µg/mL of chloramphenicol and incubated overnight at 37 °C for expression in EnPresso B media (Biosilta Ltd). A single colony was then inoculated into 2 mL of LB media plus 2% glucose containing appropriate antibiotics at 30 °C and 250 rpm for 6 hours. The glucose represses expression from the Ptac promoter used for both CyDisCo components and the proteins of interest. This pre-culture was used to inoculate at a 1:100 ratio the desired culture volume (from 25 mL to 250 mL) of EnPresso B media. When using Ultra Yield (BioSilta) baffled flasks (Thomson Instrument Company), 5 µL Antifoam 204 (Sigma) per 50 mL of the culture was added. The flask was sealed with an oxygen permeable membrane (Thomson Instrument Company). Cultures were grown for 16–18 hours at 30 °C and 250 rpm and then protein expression was induced by isopropyl β-D-thiogalactoside (IPTG) to a final concentration of 0.5 mM. Culturing was continued for 24 hours after induction. Cells were collected by centrifugation at 3220 × g for 20 minutes at 4 °C. The pellet was re-suspended in 50 mM sodium phosphate buffer pH 7.4 supplemented with protease inhibitor cocktail (Roche) in a volume equivalent to half the culture volume.
Protein purification
All constructs expressed had a hexa-histidine tag at their N-terminus to allow purification with standard immobilized metal affinity chromatography (IMAC) using HisPur Cobalt Superflow Agarose resin (Thermofisher). For large scale purifications the following protocol was followed: Lysis was performed by sonication 6 times for 5 seconds at 30 second intervals on ice. The lysate was clarified by 30 minutes centrifugation at 23500 × g at 4 °C and then filtered through a 0.45 µm filter before being applied to a 5 mL IMAC column previously equilibrated with 50 mM sodium phosphate pH 7.5. The resin matrix and lysate were mixed by inversion for 1 hour to allow batch binding and then washed with 4 column volumes of 50 mM sodium phosphate pH 7.5 and 20 column volumes of 50 mM sodium phosphate, 300 mM NaCl, pH 7.5. A manual imidazole step gradient was applied using at least 8 column volumes of 5 then 10 then 20 mM imidazole in 50 mM sodium phosphate pH 7.5 to wash out nonspecifically bound proteins. Angptl3 fibrinogen-like domain was eluted from the column by 3 column volumes of 150 mM imidazole, 50 mM sodium phosphate pH 7.5 followed by 3 column volumes of 200 mM imidazole, 50 mM sodium phosphate pH 7.5. Angptl4 fibrinogen-like domain was eluted applying a manual gradient of imidazole of 3 column volumes of 150, 200, 250 and 500 mM imidazole, 50 mM sodium phosphate, pH 7.4. For both proteins fractions with faint bands of impurities or no visible impurities on 12.5% SDS-PAGE were pooled and taken forward for subsequent purification steps. Pooled fractions from the IMAC were buffer exchanged and concentrated using Amicon® Ultra centrifugal concentrators (Millipore). Size exclusion chromatography was performed using HiLoad™Superdex 75 pg 16/600 column (GE) and 50 mM Tris, 150 mM NaCl, pH 7.5 buffer for Angptl3 fibrinogen-like domain and 50 mM Tris, 200 mM NaCl, pH 7.4 buffer for Angptl4 fibrinogen-like domain. Fractions were analyzed on 12.5% SDS-PAGE and appropriate fractions combined and concentrated using Millipore Amicon® Stirred cell with 10 kDa cut-off membrane.
The small scale purification of mutant constructs of Angptl3 and Angptl4 was performed as described earlier49, except after the elution with 50 mM EDTA, 50 mM sodium phosphate buffer, pH 7.4, the proteins were buffer exchanged and concentrated into 50 mM Tris pH 7.5, 150 mM NaCl buffer using Amicon® Ultra -4 centrifugal concentrators with MWCO 10 000.
Biophysical analysis
Solutions containing 10–40 μM protein and NEM-treated samples supplemented with 1:5 ratio of protein: 0.1% trifluoroacetic acid (TFA) were analyzed by Electrospray Ionization Mass Spectrometry (ESI-MS) (Synapt G2, Waters, Milford, MA, USA) coupled to Acquity Ultra Performance Liquid Chromatography (UPLC) machinery (Waters) with BEH300 C4 1.7 μm 2.1 × 10 mm UPLC column for desalting and protein separation prior to mass spectrometric analysis (Biocenter Oulu core facility). UPLC was performed using 0.4 ml/min flow with 0.1% formic acid and 3% to 70% acetonitrile gradient over 15 minutes.
Thermal shift stability
The thermal shift stability of proteins was measured in 25 μl volume using 5 μg of protein and 5 × SYPRO Orange (Sigma-Aldrich) with 50 mM Tris pH 7.5, 150 mM NaCl. All measurements were performed in triplicates using Applied Biosystems 7500 Real Time PCR instrument, during which the protein samples were heated from 21 to 90 °C with 1 °C increment and the fluorescence was recorded. The melting temperature (Tm) was calculated by examining of the derivative of rate of change versus temperature using IGOR (WaveMetrics Inc.).
Protein crystallization
Crystallization screening was performed in the Biocenter Oulu core facility by the sitting-drop vapour-diffusion method in 96-well plates using TTP Labtech’s Mosquito LCP nanodispenser and commercially available sparse-matrix crystal screens, JCSG-plus™ and PACT premier™ (Molecular Dimensions). Multiple crystal hits were obtained for Angptl3 in JCSG-plus™ crystal screen. The best looking single crystals appeared in the condition containing 0.2 M Magnesium chloride hexahydrate, 0.1 M BIS-Tris pH 5.5, 25% (w/v) PEG 3350. Prior to data collection crystals, crystals were transferred into cryosolution containing 0.2 M Magnesium chloride hexahydrate, 0.1 M BIS-Tris pH 5.5, 35% (w/v) PEG 3350, briefly incubated and flash-frozen in liquid nitrogen. A single crystal of Angptl4 appeared in condition G12 of PACT premier™ HT-96 crystal screen (0.1 M Bis-Tris Propane pH 7.5, 0.2 M Sodium malonate, 20% (w/v) PEG 3350) in 2 weeks and was immediately transferred into cryosolution containing 0.1 M Bis-Tris Propane pH 7.5, 0.2 M Sodium malonate, 35% (w/v) PEG 3350, briefly incubated and flash-cooled in liquid nitrogen.
Data collection
Crystal screening and X-ray diffraction data collection were performed at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. High-resolution data for Angptl3 collected using MASSIF-150, an automated high-throughput facility on beamline ID30a-1, at a wavelength of 0.966 Å and equipped with PILATUS3 2 M detector (DECTRIS). 105° of data were collected using 0.05° oscillation angle. High-resolution data for Angptl4 were collected from a single cryocooled crystal to a resolution of 2.3 Å on beamline ID29–1 equipped with Pilatus_6M detector (DECTRIS). 180° of data were collected at a wavelength of 0.9998 Å using a 0.15° oscillation angle. All data were processed using XDS51. Data collection statistics are presented in Table 1.
Structure determination of the fibrinogen-like domains of Angptl3 and Angptl4
The crystal structures of the fibrinogen-like domain of Angptl3 and Angptl4 were determined by molecular replacement using Phaser-MR52 in the PHENIX software suite53 with previously determined structure of Angiopoietin 2 receptor binding domain (PDB code 1Z3S41) as the search model. Iterative rounds of model building and refinement were performed in Coot54 and phenix.refine53, respectively. In the earlier stages of refinement, 3-fold non-crystallographic symmetry restraints were utilized for Angptl3. In the later stages of refinement, atomic displacement parameters were also refined for both Angptl3 and Angptl4 in phenix.refine by TLS method54, and non-crystallographic symmetry restraints were released for Angptl3. Model geometry was monitored using MolProbity55. Figures were produced using UCSF Chimera56.
Data availability
The structures generated during the current study are available in PDB under accession numbers 6EUA and 6EUB.
Electronic supplementary material
Acknowledgements
This work was supported by the Academy of Finland (grant numbers 266457 and 272573), Sigrid Juselius Foundation and Biocenter Oulu. We also would like to acknowledge the European Synchrotron Radiation Facility (ESRF) in Grenoble, France, for beamtime allocation and assistance during the X-ray data collection. The use of the facilities and expertise of the Biocenter Oulu core facilities, a member of Biocenter Finland, is gratefully acknowledged.
Author Contributions
E.B. performed and analyzed x-ray crystallography data. M.E. and M.S. performed molecular biology, protein expression and purification. M.E. set up crystallization trials for Angptl4. H.I.A. prepared Angptl3 protein. M.S. and L.W.R. designed and coordinated the work. E.B., M.E., M.S. and L.W.R. wrote the manuscript. E.B. prepared the figures.
Competing Interests
A patent for the production system used to make the protein for structural studies using sulfhydryl oxidases in the cytoplasm of E.coli is held by the University of Oulu: Method for producing natively folded proteins in a prokaryotic host (Patent number 9238817; date of patent January 19th 2016). Inventor: Lloyd Ruddock
Footnotes
Ekaterina Biterova and Mariam Esmaeeli contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-25237-7.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Wang H, et al. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388:1775–1812. doi: 10.1016/S0140-6736(16)31470-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakanishi R, et al. Current but not past smoking increases the risk of cardiac events: insights from coronary computed tomographic angiography. Eur Heart J. 2015;36:1031–1040. doi: 10.1093/eurheartj/ehv013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lehrke M, Marx N. Diabetes mellitus and heart failure. Am J Med. 2017;130:S40–S50. doi: 10.1016/j.amjmed.2017.04.010. [DOI] [PubMed] [Google Scholar]
- 4.Khera AV, Kathiresan S. Genetics of coronary artery disease: discovery, biology and clinical translation. Nat Rev Genet. 2017;18:331–344. doi: 10.1038/nrg.2016.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kivimäki M, et al. Job strain as a risk factor for coronary heart disease: a collaborative meta-analysis of individual participant data. Lancet. 2012;380:1491–1497. doi: 10.1016/S0140-6736(12)60994-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cosselman KE, Navas-Acien A, Kaufman JD. Environmental factors in cardiovascular disease. Nat Rev Cardiol. 2015;12:627–642. doi: 10.1038/nrcardio.2015.152. [DOI] [PubMed] [Google Scholar]
- 7.Cohen JC, Boerwinkle E, Mosley TH, Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–1272. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 8.Miller M, et al. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123:2292–2333. doi: 10.1161/CIR.0b013e3182160726. [DOI] [PubMed] [Google Scholar]
- 9.Nikpay M, et al. A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015;47:1121–1130. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nordestgaard BD, Varbo A. Triglycerides and cardiovascular disease. Lancet. 2014;384:626–635. doi: 10.1016/S0140-6736(14)61177-6. [DOI] [PubMed] [Google Scholar]
- 11.TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung and Blood Institute. Loss-of-function mutations in APOC3, triglycerides and coronary disease. N Engl J Med371, 22-31 (2014). [DOI] [PMC free article] [PubMed]
- 12.Do R, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518:102–106. doi: 10.1038/nature13917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenson RS, et al. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat Rev Cardiol. 2016;13:48–60. doi: 10.1038/nrcardio.2015.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reiner Ž. Hypertriglycerideaemia and risk of coronary artery disease. Nat Rev Cardiol. 2017;14:401–411. doi: 10.1038/nrcardio.2017.31. [DOI] [PubMed] [Google Scholar]
- 15.Natarajan P, Kathiresan S. PCSK9 inhibitors. Cell. 2016;165:1037. doi: 10.1016/j.cell.2016.05.016. [DOI] [PubMed] [Google Scholar]
- 16.Sabatine MS, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–1722. doi: 10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- 17.Thomas M, Augustin HG. The role of the angiopoietins in vascular morphogenesis. Angiogenesis. 2009;12:125–137. doi: 10.1007/s10456-009-9147-3. [DOI] [PubMed] [Google Scholar]
- 18.Ono M, et al. Protein region important for regulation of lipid metabolism in Angiopoietin-like 3 (ANGPTL3): ANGPTL3 is cleaved and activated in vivo. J Biol Chem. 2003;278:41804–41809. doi: 10.1074/jbc.M302861200. [DOI] [PubMed] [Google Scholar]
- 19.Quagliarini F, et al. Atypical angiopoietin-like protein that regulates ANGPTL3. Proc Natl Acad Sci USA. 2012;109:19751–19756. doi: 10.1073/pnas.1217552109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chomel C, et al. Interaction of the coiled-coil domain with glycosaminoglycans protects angiopoietin-like 4 from proteolysis and regulates its antiangiogenic activity. FASEB J. 2009;23:940–949. doi: 10.1096/fj.08-115170. [DOI] [PubMed] [Google Scholar]
- 21.Kersten, S. Angiopoietin-like 3 in lipoprotein metabolism Nat Rev Endocrinol, 10.1038/nrendo.2017.119 (2017). [DOI] [PubMed]
- 22.Arca M, Minicocci I, Maranghi M. The angiopoietin-like protein 3: a hepatokine with expanding role in metabolism. Curr Opin Lipidol. 2013;24:313–320. doi: 10.1097/MOL.0b013e3283630cf0. [DOI] [PubMed] [Google Scholar]
- 23.Grootaert C, Van de Wiele T, Verstraete W, Bracke M, Vanhoecke B. Angiopoietin-like protein 4: health effects, modulating agents and structure-function relationships. Expert Rev Proteomics. 2012;9:181–199. doi: 10.1586/epr.12.12. [DOI] [PubMed] [Google Scholar]
- 24.Goh YY, et al. Angiopoietin-like 4 interacts with matrix proteins to modulate wound healing. J Biol Chem. 2010;285:32999–33009. doi: 10.1074/jbc.M110.108175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chong HC, et al. Angiopoietin-like 4 stimulates STAT3-mediated iNOS expression and enhances angiogenesis to accelerate would healing in diabetic mice. Mol Ther. 2014;22:1593–1604. doi: 10.1038/mt.2014.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Musunuru K, Kathiresan S. Is ANGPLT3 the next PCSK9? Nat Rev Endocrinol. 2017;13:503–504. doi: 10.1038/nrendo.2017.88. [DOI] [PubMed] [Google Scholar]
- 27.Romeo S, et al. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J Clin Invest. 2009;119:70–79. doi: 10.1172/JCI37118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pisciotta L, et al. Characterization of three kindreds with familial combined hypolipidemia caused by loss-of-function mutations in ANGPTL3. Circ Cardiovasc Genet. 2012;5:42–50. doi: 10.1161/CIRCGENETICS.111.960674. [DOI] [PubMed] [Google Scholar]
- 29.Stitziel NO, et al. ANGPTL3 deficiency and protection against coronary disease. J Am Coll Cardiol. 2017;69:2054–2063. doi: 10.1016/j.jacc.2017.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dewey, F. E. et al. Genetic and pharmacologic inactivation of ANGPTL3 and cardiovascular disease. N Engl J Med377, 211-221. [DOI] [PMC free article] [PubMed]
- 31.Wang X, Wang D, Shan Z. Clinical and genetic analysis of a family diagnosed with familial hypobetalipoproteinemia in which the proband was diagnosed with diabetes mellitus. Atherosclerosis. 2017;239:552–556. doi: 10.1016/j.atherosclerosis.2015.02.031. [DOI] [PubMed] [Google Scholar]
- 32.Graham MJ, et al. Cardiovascular and metabolic effects of ANGPTL3 antisense oligonucleotides. N Engl J Med. 2017;377:222–232. doi: 10.1056/NEJMoa1701329. [DOI] [PubMed] [Google Scholar]
- 33.Romeo S, et al. Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat Genet. 2007;39:513–516. doi: 10.1038/ng1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dewey FE, et al. Inactivating variants in ANGPTL4 and risk of coronary artery disease. N Engl J Med. 2016;374:1123–33. doi: 10.1056/NEJMoa1510926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Myocardial Infarction Genetics and CARDioGRAM Exome Consortia Investigators. Coding variation in ANGPTL4, LPL, and SVEP1 and the risk of coronary disease. N Engl J Med374, 1134-44 (2016). [DOI] [PMC free article] [PubMed]
- 36.Smart-Halajko MC, et al. ANGPTL4 variants E40K and T266M are associated with lower fasting triglyceride levels in non-Hispanic white Americans from the Look AHEAD clinical trial. BMC Med Genet. 2011;12:89. doi: 10.1186/1471-2350-12-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shimizugawa T, et al. ANGPTL3 decrease very low density lipoprotein triglyceride clearance by inhibition of lipoprotein lipase. J Biol Chem. 2002;277:33742–33748. doi: 10.1074/jbc.M203215200. [DOI] [PubMed] [Google Scholar]
- 38.Yoshida K, Shimizugawa T, Ono M, Furukawa H. Angiopoietin-like protein 4 is a potent hyperlipidemia-inducing factor in mice and inhibitor of lipoprotein lipase. J Lipid Res. 2002;43:1770–1772. doi: 10.1194/jlr.C200010-JLR200. [DOI] [PubMed] [Google Scholar]
- 39.Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucl Acids Res. 2010;38:W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu X, et al. Structural basis for angiopoietin-1 mediated signaling initiation. Proc Natl Acad Sci USA. 2013;110:7205–7210. doi: 10.1073/pnas.1216890110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barton WA, Tzvetkova D, Nikolov DB. Structure of the angiopoietin-2 receptor binding domain and identification of surfaces involved in Tie2 recognition. Structure. 2005;13:825–832. doi: 10.1016/j.str.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 42.Garlatti V, et al. Structural Basis for innate immune sensing by M-Ficolin and its control by a pH-dependent conformational switch. J Biol Chem. 2007;282:35814–35820. doi: 10.1074/jbc.M705741200. [DOI] [PubMed] [Google Scholar]
- 43.Garlatti V, et al. Structural insights into the innate immune recognition specificities of L- and H-Ficolins. EMBO J. 2007;26:623–633. doi: 10.1038/sj.emboj.7601500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kairies N, et al. The 2.0-A crystal structure of tachylectin 5A provides evidence for the common origin of the innate immunity and the blood coagulation systems. Proc Natl Acad Sci USA. 2001;98:13519–13524. doi: 10.1073/pnas.201523798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koopman J, Haverkate F, Briet E, Lord ST. A congenitally abnormal fibrinogen (Vlissingen) with 6-base deletion of γ-chain gene, causing defective calcium binding and impaired fibrin polymerization. J Biol Chem. 1991;266:13456–13461. [PubMed] [Google Scholar]
- 46.Kawabata S, Tsuda R. Molecular basis of non-self recognition by horseshoe crab tachylectins. Biochim Biophys Acta. 2002;1572:414–421. doi: 10.1016/S0304-4165(02)00322-7. [DOI] [PubMed] [Google Scholar]
- 47.Pósfai G, et al. Emergent properties of reduced-genome Escherichia coli. Science. 2006;312:1044–1046. doi: 10.1126/science.1126439. [DOI] [PubMed] [Google Scholar]
- 48.Gaciarz A, et al. Systematic screening of soluble expression of antibody fragments in the cytoplasm of E. coli. Micro Cell Fact. 2016;15:22. doi: 10.1186/s12934-016-0419-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gaciarz A. & Ruddock, L.W. Complementary determining regions and frameworks contribute to the disulfide bond independent folding of intrinsically stable scFv. PLoS One. 2017;12:e0189964. doi: 10.1371/journal.pone.0189964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Svensson O, Monaco S, Popov AN, Nurizzo D, Bowler MW. Fully automatic characterization and data collection from crystals of biological macromolecules. Acta Cryst. 2015;D71:1757–1767. doi: 10.1107/S1399004715011918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kabsch W. XDS. Acta Cryst. 2010;D66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Afonine PV, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2012;68:352–67. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Emsley P, Lohkamp B, Scott W, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Cryst. 2010;D66:16–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pettersen EF, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The structures generated during the current study are available in PDB under accession numbers 6EUA and 6EUB.