

Summary
Upon encounter with their cognate antigen, naive CD4 T cells become activated and are induced to differentiate into several possible T helper (Th) cell subsets. This differentiation depends on a number of factors including antigen‐presenting cells, cytokines and co‐stimulatory molecules. The strength of the T‐cell receptor (TCR) signal, related to the affinity of TCR for antigen and antigen dose, has emerged as a dominant factor in determining Th cell fate. Recent studies have revealed that TCR signals of high or low strength do not simply induce quantitatively different signals in the T cells, but rather qualitatively distinct pathways can be induced based on TCR signal strength. This review examines the recent literature in this area and highlights important new developments in our understanding of Th cell differentiation and TCR signal strength.
Keywords: CD4 cell, regulatory T cells, signal transduction, T‐cell receptors
Introduction
Upon encounter with their cognate antigen, naive CD4+ T cells become activated, proliferate and are induced to differentiate into specific T helper (Th) cell subsets, and there are at least six major subsets identified to date. These include Th1, Th2, Th17, Th9, T follicular helper (Tfh) and regulatory T (Treg) cells.1 These Th cell subsets are distinguished by the cytokines that they produce, the expression of specific transcription factors (TF) and distinct migration patterns. For example, Th1 cells produce interferon‐γ, express T‐bet and are important in the defence against intracellular bacteria and viruses; Th2 cells produce interleukin‐5 (IL‐5), IL‐4 and IL‐13, express GATA‐3 and are important in worm infections and contribute to allergic immunopathology.2, 3, 4 The differentiation of Th subsets is determined by multiple factors5 including cytokines produced by innate immune cells, antigen dose6, 7, 8, 9, 10 and specific co‐stimulatory molecules.11, 12 Early studies revealed the importance of T‐cell receptor (TCR) signal strength6, 7 in Th differentiation and subsequent studies have provided more insight into the mechanisms by which TCR signal strength influences Th cell differentiation. These have included the identification of specific signalling pathways downstream of the TCR13 leading to the induction of unique genetic programmes.14 In this review, we will examine the recent advances in this area and discuss how this knowledge may lead to the development of novel therapies aimed at inducing specific Th cell responses.
TCR signalling pathways and Th cell differentiation
Shortly after the first discovery of Th1 and Th2 cells it was observed that peptide affinity and dose played an important role in determining the differentiation of naive CD4 T cells.6, 7 Initial studies aimed to identify the mechanisms by which altered peptide ligands could induce changes in signalling pathways responsible for Th1 versus Th2 differentiation. Two early studies identified early signalling components downstream of the TCR as having a role in determining Th1 or Th2 differentiation.15, 16 Both of these studies showed that Th1 differentiation required strong TCR signals, as shown by a robust calcium flux whereas cells induced to differentiate into Th2 cells, with altered peptide ligands, exhibited weaker early signals and less calcium response.15, 16 Interestingly, even though the calcium signal was weak it was necessary for robust Th2 differentiation as treatment of cells with cyclosporine abrogated the Th2 response.15 Hence, it appeared that weaker TCR signals favoured Th2 differentiation whereas strong signals were needed for Th1 differentiation.13 This finding has been supported by additional studies showing that signalling down the extracellular signal‐regulated kinase (ERK) pathway was important for Th1 differentiation.17 Furthermore, it was shown that stimulation of T cells with low antigen doses allows the rapid up‐regulation of GATA‐3, which is dependent on IL‐2 signalling.18 Stimulation of cells with higher dose antigen induces a high level of ERK activation, which reduces the ability of cells to respond to IL‐2 and so GATA‐3 is not induced.18 Furthermore, inhibition of the ERK pathway during high‐dose stimulation restored the ability of cells to respond to IL‐2 and GATA‐3 and IL‐4 expression was restored. Inhibition of the ERK pathway in T cells stimulated with low‐dose antigen prevented the early induction of GATA‐3 and IL‐4 and this was reversed by the addition of exogenous IL‐2.18 These results suggest that Th2 differentiation requires that activation of the ERK pathway downstream of the TCR be transient and weak to allow for the early induction of GATA‐3 and IL‐2, which are responsible for IL‐4 production.13
In vivo studies described the role of antigen dose in determining Th1 versus Th2 differentiation.9, 10 Van Panhuys et al. examined the relative role of skewing cytokines and antigen dose on Th cell differentiation,10 using dendritic cells (DC) pulsed with various doses of antigen as immunogens. The DC were either treated with lipopolysaccharide or papain in order to skew Th1 and Th2 differentiation, respectively. T cells interacting with lipopolysaccharide‐treated DC experienced longer contact times with DC, which translated into increased calcium fluxes and Th1 differentiation, whereas T cells had shorter interaction times and reduced calcium with peptide‐pulsed papain‐treated DC.10 The dose of antigen was found to dominate over the DC pretreatment in terms of Th cell differentiation. Hence, papain‐treated DC loaded with high‐dose antigen induced Th1 cells whereas lipopolysaccharide‐treated DC loaded with low‐dose antigen induced Th2 cells. The interactions leading to Th1 cells were associated with increased interaction times and more robust calcium fluxes.10 The studies further showed that high‐dose stimulation was influenced by the level of CD80 expression on DC and this led to increased expression of the IL‐12 receptor β 2 chain, necessary for Th1 development.10 Similar results were obtained by Tubo et al.,9 when they examined the differentiation requirements of Th1 and Tfh cells. In these studies, mice were immunized with Listeria monocytogenes bacteria engineered to express specific peptide antigens and the fate of individual naive T cells was examined. They observed that, under identical immunization conditions, individual T cells could give rise to Th1, Tfh or germinal centre Tfh cells.9 This differentiation was influenced by antigen dose and was associated with changes in TCR dwell time. In the systems examined here, Th1 cells were induced by low and intermediate antigen dose and fewer Th1 cells were observed at the highest immunization doses. In contrast, Tfh and germinal centre Tfh numbers increased as the immunization dose was increased.9 Although these studies did not examine signalling in the differentiating T cells, they were able to show, using well‐defined systems, that Th differentiation was influenced by peptide(p):MHC density and TCR–p:MHC dwell time.9, 19 A recent study analysed the fate of single T cells following in vivo immunization and, using a computational model, showed that individual T cells make lineage decisions based on the quality of the TCR signal.20
T‐cell receptor signal strength has also been shown to play an important role in Th17 versus Treg cell differentiation.13 Th17 cells are important in the control of extracellular and fungal pathogens and also contribute to immunopathology in certain autoimmune diseases. Th17 cells secrete the inflammatory cytokines IL‐17 and IL‐22, and their differentiation requires the presence of transforming growth factor (TGF)‐β, IL‐6, IL‐21 and IL‐23,13, 21, 22, 23 whereas IL‐2 inhibits Th17 differentiation.24 Treg cell differentiation in the periphery requires the presence of TGF‐β, retinoic acid and IL‐2,25, 26, 27 and involves the induction of the TF Foxp3. Low TCR signal strength favours Treg cells, even in the presence of Th17‐inducing factors, whereas Th17 differentiation requires high TCR signals.28, 29, 30, 31, 32, 33
Several studies have examined signalling molecules that play a role in determining Treg versus Th17 differentiation. The Tec family kinase, Itk, has been shown to play an important role in Th2, Treg and Th17 differentiation.34, 35, 36 Itk is activated following TCR engagement and directly phosphorylates phospholipase C‐γ, which leads to induction of calcium flux and activation of mitogen‐activated protein kinase and protein kinase C.37 T cells lacking Itk exhibit altered activation and differentiation patterns. Early studies demonstrated that Itk was necessary for Th2 differentiation in T cells stimulated with low‐dose antigen,36 and this was associated with increased expression of Tbet and Th1 differentiation under these conditions. Reduced Th17 differentiation was observed in CD4 T cells lacking Itk,38 and further studies showed that, in the presence of Th17‐driving conditions, Itk–/– T cells increased expression of Foxp3.35 In addition, Itk–/– CD4 T cells gave rise to increased numbers of Treg cells when cultured in inducible Treg conditions.35 This was associated with decreases in activation of the Akt/mammalian target of rapamycin (mTOR) pathway and increased responsiveness to IL‐2.35 The decrease in Akt/mTOR activation was correlated with increased expression of the lipid phosphatase, phosphatase and tensin homologue (PTEN), that is known to inhibit phosphoinositide 3‐kinase activity.35 Given that mice with a genetic defect in Itk expression also exhibit developmental defects, a recent study34 examined the role of Itk in Th cell subset differentiation using a specific inhibitor that blocks the kinase function of Itk and Rlk.39 Treatment of CD4 T cells with this inhibitor reduced the differentiation of Th1, Th2 and Th17 cells, with a more potent effect on Th1 differentiation.34 Reduced numbers of Treg cells were also observed in vivo and in vitro following treatment with the inhibitor, and this was associated with reduced IL‐2 production.34 The difference between these two studies34, 35 could be explained by the fact that Rlk was also blocked by the inhibitor and also that some kinase‐independent function of Itk may also be important in Th cell differentiation.
The serine/threonine kinase casein kinase (CK)2 has also been implicated in Th cell subset differentiation.40, 41, 42 CK2 is ubiquitously expressed and phosphorylates over 500 substrates, and has been shown to promote the activity of the Akt/mTOR, nuclear factor‐κB and Janus kinase–signal transducer and activator of transcription (STAT) pathways in immune cells.43 One important CK2 target is PTEN,44 an important negative regulator of the Akt/mTOR pathway. Treatment of CD4 T cells with inhibitors of CK2 leads to a decrease in Th17 production and increased Treg cell induction.40, 42 This inhibition was associated with decreased STAT3 activation,40, 42 which resulted in reduced IL‐23 receptor expression,42 necessary for optimal Th17 differentiation and maintenance.22 In addition, in vivo treatment of mice with CK2 inhibitor reduced the incidence and severity of experimental autoimmune encephalomyelitis (EAE).40, 42 Genetic ablation of CK2 in Treg cells also resulted in exacerbation of Th2‐mediated lymphoproliferative condition in the lungs.41 This was due to an increase in the number of immunoglobulin‐like transcript 3 (ILT3)‐positive Treg cells,41 which promote programmed cell death 1 ligand 2‐positive/interferon regulatory factor 4 (IRF4) ‐positive DC,41 important for Th2 differentiation.45, 46 These studies suggested that CK2 in Treg cells functions to regulate ILT3 expression, which is important for the control of Th2 responses. A recent study, showed that engagement of programmed cell death 1 on CD4 T cells led to inhibition of CK2 activity, resulting in increased PTEN stability and reduced Akt/mTOR activity.47 These studies therefore identify CK2 as an important regulator of Th cell differentiation, and also highlight additional roles for this kinase in the function of Treg cells. There is much more to be learned about the role of this ubiquitous and highly active kinase in the control of immune responses.
Several other signalling molecules have been implicated in Th cell subset differentiation, including Nck,48 MALT1,49 Pak2,50 Notch13 and other early components of the TCR signalling pathway.13 Another study identified a role for T‐cell activation RhoGTPase‐activating protein (TAGAP) in Th17 differentiation.51 TAGAP inhibits the binding of Zap70 to the adapter RhoH, thereby reducing the strength of the TCR signal.51 Reduced Th17 differentiation was observed in TAGAP–/– T cells and this was associated with a milder experimental autoimmune encephalomyelitis (EAE) phenotype.51 These studies did not address differentiation of other Th cell subsets, or examine IL‐2 production, so it is not clear how this study fits into the body of work suggesting that high TCR signals are required for Th17 differentiation. It is possible that the absence of TAGAP influences IL‐2 production, such that high TCR signals fail to inhibit IL‐2 production. Nck‐deficient T cells showed defects in the differentiation of Tfh cells48 and this was associated with decreased signalling down the Akt/mTOR pathway. Pak2 was shown to be important for thymic Treg cell development as Pak2–/– T cells had reduced numbers of Treg cells and mice with this deletion developed spontaneous colitis.50 Pak2–/– thymocytes exhibited reduced TCR signalling, as determined by Nur77 expression, suggesting that these cells did not receive adequate signals for Treg cell development in the thymus.50 Overall, these studies point to the effect of various signalling molecules on modulating the strength of the TCR signal. The question remains as to which component of the TCR signalling machinery is the most critical in the determination of T‐cell fate. As discussed in more detail below the Akt/mTOR pathway appears to play a pivotal role in these cell fate decisions.
Pathogens have also evolved ways to influence Th differentiation by altering TCR signal strength. This can occur by direct or indirect mechanisms. It is well known that the Schistosoma mansoni egg antigen drives Th2 responses in mouse and human.52 Several studies identified the glycoprotein omega‐1 as one S. mansoni egg antigen component responsible for Th2 differentiation.53, 54 It was shown that omega‐1 acts on DCs to reduce co‐stimulatory molecule expression and IL‐12 production.53 In addition, it was shown that DC treated with omega‐1 had a changed morphology, which resulted in less efficient conjugate formation with T cells.54 It is speculated that this would reduce TCR signal strength, thereby favouring Th2 differentiation.54
An interesting recent paper55 demonstrates how pathogens may directly influence Th cell differentiation by influencing TCR signal strength. This study examined infection with the enteropathogen Yersinia pseudotuberculosis and showed enhanced Th17 and reduced Treg differentiation in the lymph nodes draining the gut.55 This was caused by translocation of the Y. pseudotuberculosis Yop proteins into T cells using a type III secretion system,56 which resulted in reduced calcium and ERK signalling.55 There were also significant changes in the DC populations following Y. pseudotuberculosis infection, including a marked reduction in the tolerogenic CD103+ DC. The studies also demonstrated a direct effect on in vitro Th17 and Treg but not Th1 differentiation following exposure of CD4 T cells with Y. pseudotuberculosis, suggesting that this pathogen creates an immune environment that is favourable to its survival and dissemination.55 Although this study focused on T cells, it was recently shown that Y. pseudotuberculosis is responsible for changing the transcriptional profile of many immune cells through the Yop proteins and type III secretion system.57 A previous study examined the role of TCR strength in determining T effector (Teff) cell function following influenza infection.58 In this study, immunization with high‐affinity peptides led to robust Th1 differentiation, but these cells accumulated and proliferated poorly upon challenge with an influenza virus expressing the epitope. In contrast, immunization with lower‐affinity epitopes led to increased Th17 responses and the cells expanded rapidly upon influenza challenge. This appeared to be, in part, related to the development of a terminally differentiated phenotype in cells primed with high‐affinity peptides.58 Hence, TCR signal strength not only influences Th cell differentiation but also plays a role in the generation of effective memory cells.
The Akt/mTOR pathway and Th cell differentiation
As discussed above many studies looking at the role of TCR signal strength in Th cell differentiation have suggested a critical role for the Akt/mTOR pathway. Akt is a serine threonine kinase that is activated downstream of the TCR by phosphorylation on two critical sites: Threonine (T) 308 by phosphoinositide‐dependent kinase 1 (PDK1) and Serine (S) 473 by mTORC2. Full Akt activation leads to the phosphorylation of multiple targets that influence Th cell function, survival and differentiation. Important targets include the nuclear factor‐κB pathway through activation of the Carma1/Bcl10/Malt1 complex;59 transcription factors such as Foxo1; and pathways important for cell growth and survival such as mTOR, GSK‐3 and CREB.60
We, and others, have shown that strong activation of the Akt/mTOR pathway, secondary to high TCR signal strength, is negatively correlated with Treg cell differentiation.33, 61, 62 Treg cells induced by low‐dose antigen are able to potently suppress immune responses in vitro 33 and in vivo.28, 63 Furthermore, Treg cells exhibit a reduced capacity to activate Akt and only the T308 site is phosphorylated in activated Treg cells,64 and restoration of S473 phosphorylation leads to a loss of suppressive activity of Treg cells.64 Recently, similar results were obtained in human T cells65 in which the Akt/mTOR pathway was shown to be important for the generation of Th1‐like Treg cells. Activation of Akt is regulated by the lipid phosphatase PTEN, which acts to inhibit the action of phosphoinositide 3‐kinase by converting the active phospholipid PIP3 to the inactive PIP2. PIP3 is required to activate PDK1.66 We developed a mathematical model to better understand the mechanism by which the Akt/mTOR pathway might regulate Treg cell differentiation,67 and this model made the prediction that PTEN was differentially regulated following activation with high or low TCR signal strength. The model predicted that when T cells were destined to become Teff cells PTEN levels would be reduced and would stay down, whereas Treg cells would only show transient down‐regulation of PTEN. We were able to validate this prediction and went on to show that PTEN levels are tightly controlled in T cells.68 Interestingly, we observed that the TF Foxo1 was involved in the transcription of PTEN,68 through binding to an upstream regulatory site. Activation of T cells with high‐dose antigen led to full Akt activation and the phosphorylation of Foxo1.68 This had the consequence of causing Foxo1 to leave the nucleus and so PTEN RNA levels were drastically reduced (Fig. 1). This, coupled with other mechanisms of protein degradation, led to the rapid and lasting loss of PTEN protein in developing Teff cells.68 Other studies have shown the importance of PTEN not only in the generation of Treg cells but also in their maintenance.69, 70 These two studies created mice with PTEN deletions restricted to Foxp3+ Treg cells and in both cases the mice developed lymphoproliferative diseases that were associated with poorly functioning Treg cells. In addition, increased activation of Akt and mTOR were observed in Treg cells lacking PTEN.69, 70
Figure 1.
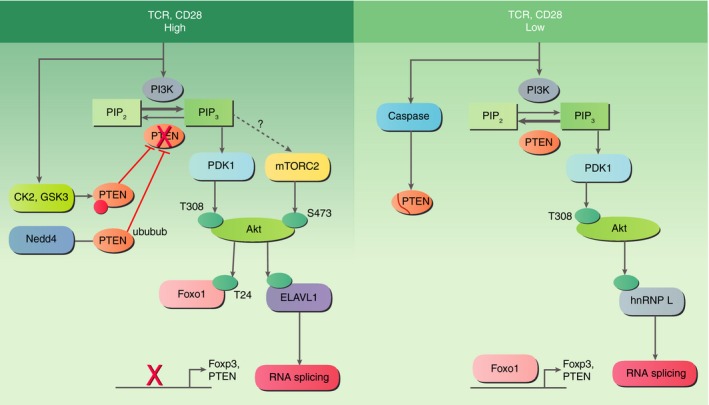
The activity of phosphatase and tensin homologue (PTEN) and Akt and Akt substrate specificity are modulated by T‐cell receptor (TCR) signal strength. High TCR signalling results in activation of CK2 kinase, which phosphorylates and inactivates PTEN. The degradation of PTEN protein via ubiquitination occurs through Nedd4. When PTEN is inactive, phosphorylation of Akt at both S473 and T308 sites occurs. T cells exposed to low TCR strength activate caspases that cleave PTEN to cause transient down‐regulation of PTEN but PTEN activity is maintained and Akt is only phosphorylated at the T308 site. The differential phosphorylation patterns result in different Akt substrates being targeted. For example, Foxo1 is only phosphorylated at T24 following high‐dose stimulation resulting in exclusion from the nucleus, which prevents Foxo1 from inducing expression of Foxp3 and PTEN. Different RNA processing factors are phosphorylated depending on dose and this results in different splicing patterns.
Several studies have taken genetic approaches to altering the signalling of the Akt/mTOR pathway and these include deletion of the tuberous sclerosis, TSC1, gene71 and the various components of the mTORC1 and mTORC2 complexes.72, 73 T cells lacking mTOR, which lack both mTORC1 and mTORC2 complexes, were unable to differentiate into Th1, Th17 or Th2 cells,72 but these cells exhibited enhanced Treg cell differentiation.72 To determine which complex was responsible for these results mice lacking either mTORC1 or mTORC2 in their CD4 T cells were analysed.73 T cells lacking Rheb, which have defects in mTORC1 activation, were inhibited in Th1 and Th17 differentiation.73 CD4 T cells lacking Rictor, the unique component of the mTORC2 complex, failed to undergo Th2 differentiation, whereas Th1 and Th17 differentiation was normal in these mice.73 Interestingly, inhibition of both mTORC1 and mTORC2 was required to induce an increase in Treg cell development,73 suggesting redundancy in these two complexes. TSC1 inhibits mTORC1 activity and deletion of TSC1 leads to constitutive activation of mTORC1. Mice lacking TSC1 in CD4 T cells were found to have increased Th1 and Th17 development71 and the mice developed intestinal inflammation. In addition, Treg from TSC1‐deficient mice were unable to suppress intestinal inflammation in an adoptive transfer model.71 When the TSC1 deficiency was restricted to Foxp3+ Treg cells the TSC1‐deficient Treg cells produced IL‐17, lost Foxp3 expression, and become effector‐like in their function.71 Another study examined differentiation of Tfh and Th1 cells and found that increased signalling via Akt/mTOR, in part mediated by IL‐2, was necessary for Th1 differentiation,74 whereas Tfh cells required low signals via Akt/mTOR. Genetic analyses are useful tools in identifying the important players in a pathway but it is not always possible to identify the mechanisms underlying the precise differentiation pathway. This is because the complete lack of a particular component, such as Rictor, does not provide insight into more subtle variations in function that may occur when T cells are activated in different scenarios. Complexes such as a mTORC2 and mTORC1 have multiple functions in the cell and it is necessary to investigate how each function may impact Th cell differentiation. Our studies of Treg cell differentiation in response to low TCR signal strength indicated that Treg cell induction was negatively correlated with mTORC1 activation.33 Akt plays a pivotal role in the activation of the mTORC1 complex and full Akt activation requires an active mTORC2 complex. Activation of Akt is controlled by TCR signal strength, which results in changes in the phosphorylation state of Akt. We observed that, in T cells stimulated with low TCR signals, only the T308 site on Akt was phosphorylated and that high TCR signals were required for phosphorylation of both T308 and S473.75 In fact, there was a sharp dose–response curve for S473 phosphorylation that was almost switch‐like in behaviour.75 Phosphorylation of the T308 site is mediated by PDK1 whereas mTORC2 phosphorylates S473. We also showed that mTORC2 was only active when cells were stimulated by high‐dose antigen, as demonstrated by reduced phosphorylation of a negative regulatory site on Rictor, the unique mTORC2 component.75 The mechanisms for mTORC2 activation in T cells are not well understood, although in cell lines it has been shown that PIP3 plays a role in mTORC2 activation.76 We speculate, therefore, that under high‐dose conditions when PTEN levels are reduced, sufficient PIP3 is generated to induce both PDK1 and mTORC2 activation (Fig. 1a), whereas under low‐dose conditions only PDK1 is activated (Fig. 1b).
As Akt is a kinase whose activity is controlled by phosphorylation, we asked whether different substrates were phosphorylated by Akt under these two conditions. We took advantage of an antibody that recognizes phosphorylated Akt substrates and, using mass spectrometry analysis of immunoprecipitated substrates, we found that very different sets of substrates were phosphorylated by Akt under conditions of low or high TCR strength.75 The identified Akt targets function in regulating the cytoskeleton, metabolism, nuclear import, RNA processing and transcription (Fig. 1). One area of particular interest to us was the observed differences in RNA processing. Alternative splicing has been described following T‐cell activation77, 78 and several molecules involved in alternative splicing were differentially phosphorylated in cells stimulated with low or high doses. We went on to show that two of the RNA processing factors, hnRNP L and hnRNP A1, played an important role in Treg cell differentiation.75 Further studies are aimed at determining how phosphorylation by Akt alters the function of these RNA processing factors. These studies suggest that different levels of TCR stimulation initiate qualitatively distinct differentiation programmes and that differential regulation of alternative splicing by Akt is one of the key elements determining Th cell fate decisions.
The Akt/mTOR pathway plays an important role in controlling metabolism in cells and recent work has highlighted the importance of metabolic changes in T‐cell function and differentiation.79, 80 In particular, mTOR induces glycolysis and inhibits oxidative phosphorylation, and when T cells are activated they switch their metabolic profile from oxidative phosphorylation to aerobic glycolysis. In addition, different Th cell subsets have been shown to use different metabolic pathways and sources of fuel,79, 80 such as Treg cells, which use fatty acid oxidation.81, 82 Although mTOR complexes are activated through the TCR, they are important amino sensors and the presence of amino acids, such as glutamine, plays an important role in T‐cell activation and function.83 A recent study found that the glutamine transporter, ASCT2, was essential for the development of Th1 and Th17 from naive T cells but was dispensable for the generation of Treg cells.84 ASCT2‐deficient T cells showed a defect in glutamine uptake, mTORC1 activation, and this also involved the Carma1/Bcl10/Malt1 complex.84 Examination of Th1 and Tfh cell differentiation74 revealed that Tfh cells have a reduced ability to undergo aerobic glycolysis due to reduced mTOR activity. Th1 differentiation required strong activation of the mTOR pathway, partly due to signalling via CD25 and IL‐2, for the induction of Tbet and Blimp‐1.74 Tfh cells were also shown to preferentially use fatty acid oxidation, similar to Treg cells. These metabolic profiles are necessary to maintain Th cell function, as shown in a recent study of Treg activation.85 Treg cells express several Toll‐like receptors (TLR) and this study examined the role of TLR signals in modulating Treg cell metabolism, proliferation and suppressive function.85 The addition of TLR1 and TLR2 ligands to activated Treg cells resulted in increases in glycolysis, proliferation and Glut1 expression that were associated with increased mTOR activity but reduced suppressive function.85 These studies suggest that metabolism in Treg cells is highly dynamic and that, depending on the environment, Treg cells can alter their metabolic profile to favour proliferation or suppression.85 Similar results have been observed in human Treg cells.86
Hence, the Akt/mTOR pathway plays a critical role in driving Th cell differentiation and this leads to changes in multiple cell functions including alternative splicing, metabolic reprogramming and cytoskeletal changes, and it is likely that many more functions will be identified in the future.
Genetic programmes regulated by TCR signal strength
All of these signalling pathways culminate in the induction of specific genetic programmes characterized by defined TF and specific epigenetic features.14 Individual Th subsets require specific TF to drive their differentiation and maintenance and these have been well‐described.13, 14 The TF IRF4 plays a critical role in the activation and differentiation of B and T cells and appears to act as a sensor for the strength of the antigen receptor signal. IRF4 is induced rapidly following immune cell activation and the level of expression correlates with the strength of the activating signal.87, 88 In addition, IRF4 influences the differentiation of multiple Th cell subsets, including Th2, Th17 and Tfh cells.89 Two recent studies have examined the role of IRF4 in Th cell differentiation with a particular focus on the role of TCR signal strength and IRF4 function.90, 91 Iwata et al.90 examined the role of IRF4 and basic leucine zipper transcription factor, ATF‐like (BATF) in Th2 differentiation, to address previous controversy in the literature concerning the role of different BATF isoforms in Th2 cell differentiation. IRF4 forms heterodimers with BATF and Jun and both BATF1 and BATF3 can associate with IRF4. This study demonstrated that increasing TCR signal strength resulted in increased expression of both IRF4 and BATF in a coordinated manner. This graded increase resulted in the activation of a hierarchy of genes, with some being sensitive to lower levels of IRF4 and BATF, whereas others required higher expression levels.90 This hierarchy was also observed at the level of binding sites for IRF4, as determined by ChIP‐Seq. Krishnamoorthy et al.91 examined the role of IRF4 in the decision between Tfh and Teff cells that is associated with the induction of the TF Bcl6 and Blimp‐1, respectively. Increasing the strength of the TCR resulted in increased IRF4 levels and enhanced Blimp‐1+ Teff cell induction. Further, increasing the levels of IRF4 in cells stimulated with a fixed TCR signal resulted in an increase in Teff cell differentiation.91 Interestingly, the action of IRF4 was correlated with its ability to bind to sequences of different affinities.91 IRF4 is downstream of mTOR activation,92 so provides a mechanism by which signalling via Akt/mTOR leads to changes in the genetic programming of T cells.
Other TF downstream of mTOR that play important roles in Th cell differentiation include cMyc93 and Foxo1.68, 94 Myc is important in the metabolic reprogramming of T cells following activation.95 Foxo1 is important in the transcription of Foxp3 and PTEN68 and activation of Akt by high TCR signal strength leads to Foxo1 phosphorylation and exit from the nucleus.68 Foxo1 is also a negative regulator of Th17 differentiation by actively binding to RORγt and inhibiting its activity.94 Recently, the RNA‐binding protein roquin‐1 has been shown to inhibit Th17 differentiation96, 97 by repressing RNAs that drive Th17 differentiation such as IL‐6, ICOS, IRF4 and others. Activation of roquin‐1 requires cleavage by Malt1, downstream of TCR signalling, suggesting that this activity is also controlled by TCR signal strength.97
Conclusions
T helper cell activation and differentiation have been topics of fascination for many years and multiple studies have examined the factors required for the generation of the various flavours of Th cell subsets. Early on, it was appreciated that antigen dose, and hence TCR signal strength, played a critical role in determining Th cell fate. Over the years, we have developed a better understanding of how TCR signal strength is translated into divergent cell fates. As this review highlights, there appears to be a coordinated pattern of signalling and genetic programmes initiated following T‐cell activation and individual pathways are dictated by TCR signal strength. The Akt/mTOR pathway plays a pivotal role in regulating Th cell differentiation through its influence on metabolism, alternative splicing and specific TF programmes. The ability to harness these pathways and generate specific Th cell subsets will be of great therapeutic benefit in the context of autoimmunity and cancer immunotherapy.
Disclosures
The author declares no conflicts of interest.
Acknowledgement
This work was supported by ARO grant # W911NF‐17‐1‐0082.
References
- 1. DuPage M, Bluestone JA. Harnessing the plasticity of CD4+ T cells to treat immune‐mediated disease. Nat Rev Immunol 2016; 16:149. [DOI] [PubMed] [Google Scholar]
- 2. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clones. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol 1986; 136:2348–57. [PubMed] [Google Scholar]
- 3. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T‐bet, directs Th1 lineage commitment. Cell 2000; 100:655–69. [DOI] [PubMed] [Google Scholar]
- 4. Zhang DH, Cohn L, Ray P, Bottomly K, Ray A. Transcription factor GATA‐3 is differentially expressed in murine Th1 and Th2 cells and controls Th2‐specific expression of the interleukin‐5 gene. J Biol Chem 1997; 272:21597–603. [DOI] [PubMed] [Google Scholar]
- 5. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol 2010; 28:445–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Constant S, Pfeiffer C, Woodard A, Pasqualini T, Bottomly K. Extent of T cell receptor ligation can determine the functional differentiation of naive CD4+ T cells. J Exp Med 1995; 182:1591–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hosken NA, Shibuya K, Heath AW, Murphy KM, O'Garra A. The effect of antigen dose on CD4+ T helper cell phenotype development in a T cell receptor‐αβ‐transgenic model. J Exp Med 1995; 182:1579–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boonstra A, Asselin‐Paturel C, Gilliet M, Crain C, Trinchieri G, Liu YJ et al Flexibility of mouse classical and plasmacytoid‐derived dendritic cells in directing T helper type 1 and 2 cell development: dependency on antigen dose and differential toll‐like receptor ligation. J Exp Med 2003; 197:101–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tubo NJ, Pagan AJ, Taylor JJ, Nelson RW, Linehan JL, Ertelt JM et al Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell 2013; 153:785–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van Panhuys N, Klauschen F, Germain Ronald N. T‐cell‐receptor‐dependent signal intensity dominantly controls CD4+ T cell polarization in vivo . Immunity 2014; 41:63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. De Becker G, Moulin V, Tielemans F, De Mattia F, Urbain J, Leo O et al Regulation of T helper cell differentiation in vivo by soluble and membrane proteins provided by antigen‐presenting cells. Eur J Immunol 1998; 28:3161–71. [DOI] [PubMed] [Google Scholar]
- 12. Rogers PR, Croft M. CD28, Ox‐40, LFA‐1, and CD4 modulation of Th1/Th2 differentiation is directly dependent on the dose of antigen. J Immunol 2000; 164:2955–63. [DOI] [PubMed] [Google Scholar]
- 13. Yamane H, Paul WE. Early signaling events that underlie fate decisions of naive CD4+ T cells toward distinct T‐helper cell subsets. Immunol Rev 2013; 252:12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kanno Y, Vahedi G, Hirahara K, Singleton K, O'Shea JJ. Transcriptional and epigenetic control of T helper cell specification: molecular mechanisms underlying commitment and plasticity. Annu Rev Immunol 2012; 30:707–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boutin Y, Leitenberg D, Tao X, Bottomly K. Distinct biochemical signals characterize agonist‐ and altered peptide ligand‐induced differentiation of naive CD4+ T cells into Th1 and Th2 subsets. J Immunol 1997; 159:5802–9. [PubMed] [Google Scholar]
- 16. Sloan‐Lancaster J, Steinberg TH, Allen PM. Selective loss of the calcium ion signaling pathway in T cells maturing toward a T helper 2 phenotype. J Immunol 1997; 159:1160–8. [PubMed] [Google Scholar]
- 17. Jorritsma PJ, Brogdon JL, Bottomly K. Role of TCR‐induced extracellular signal‐regulated kinase activation in the regulation of early IL‐4 expression in naive CD4+ T cells. J Immunol 2003; 170:2427–34. [DOI] [PubMed] [Google Scholar]
- 18. Yamane H, Zhu J, Paul WE. Independent roles for IL‐2 and GATA‐3 in stimulating naive CD4+ T cells to generate a Th2‐inducing cytokine environment. J Exp Med 2005; 202:793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tubo NJ, Jenkins MK. TCR signal quantity and quality in CD4+ T cell differentiation. Trends Immunol 2014; 35:591–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cho YL, Flossdorf M, Kretschmer L, Hofer T, Busch DH, Buchholz VR. TCR signal quality modulates fate decisions of single CD4+ T cells in a probabilistic manner. Cell Rep 2017; 20:806–18. [DOI] [PubMed] [Google Scholar]
- 21. McGeachy MJ, Bak‐Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T et al TGF‐β and IL‐6 drive the production of IL‐17 and IL‐10 by T cells and restrain TH‐17 cell‐mediated pathology. Nat Immunol 2007; 8:1390. [DOI] [PubMed] [Google Scholar]
- 22. McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce‐Shaikh B, Blumenschein WM et al The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17‐producing effector T helper cells in vivo . Nat Immunol 2009; 10:314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Laurence A, O'Shea JJ. TH‐17 differentiation: of mice and men. Nat Immunol 2007; 8:903–5. [DOI] [PubMed] [Google Scholar]
- 24. Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z et al Interleukin‐2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 2007; 26:371–81. [DOI] [PubMed] [Google Scholar]
- 25. Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. Cutting edge: IL‐2 is essential for TGF‐β‐mediated induction of Foxp3+ T regulatory cells. J Immunol 2007; 178:4022–6. [DOI] [PubMed] [Google Scholar]
- 26. Elias KM, Laurence A, Davidson TS, Stephens G, Kanno Y, Shevach EM et al Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat‐3/Stat‐5 independent signaling pathway. Blood 2008; 111:1013–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fu S, Zhang N, Yopp AC, Chen D, Mao M, Chen D et al TGF‐β induces Foxp3+ T‐regulatory cells from CD4+ CD25– precursors. Am J Transplant 2004; 4:1614–27. [DOI] [PubMed] [Google Scholar]
- 28. Daniel C, Weigmann B, Bronson R, von Boehmer H. Prevention of type 1 diabetes in mice by tolerogenic vaccination with a strong agonist insulin mimetope. J Exp Med 2011; 208:1501–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gottschalk RA, Corse E, Allison JP. TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo . J Exp Med 2010; 207:1701–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gottschalk RA, Hathorn MM, Beuneu H, Corse E, Dustin ML, Altan‐Bonnet G et al Distinct influences of peptide‐MHC quality and quantity on in vivo T‐cell responses. Proc Natl Acad Sci U S A 2012; 109:881–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Iezzi G, Sonderegger I, Ampenberger F, Schmitz N, Marsland BJ, Kopf M. CD40‐CD40L cross‐talk integrates strong antigenic signals and microbial stimuli to induce development of IL‐17‐producing CD4+ T cells. Proc Natl Acad Sci U S A 2009; 106:876–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Molinero LL, Miller ML, Evaristo C, Alegre ML. High TCR stimuli prevent induced regulatory T cell differentiation in a NF‐κB‐dependent manner. J Immunol 2011; 186:4609–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Turner MS, Kane LP, Morel PA. Dominant role of antigen dose in CD4+ Foxp3+ regulatory T cell induction and expansion. J Immunol 2009; 183:4895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cho H‐S, Shin HM, Haberstock‐Debic H, Xing Y, Owens TD, Funk JO et al A small molecule inhibitor of ITK and RLK impairs Th1 differentiation and prevents colitis disease progression. J Immunol 2015; 195:4822–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gomez‐Rodriguez J, Wohlfert EA, Handon R, Meylan F, Wu JZ, Anderson SM et al Itk‐mediated integration of T cell receptor and cytokine signaling regulates the balance between Th17 and regulatory T cells. J Exp Med 2014; 211:529–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miller AT, Wilcox HM, Lai Z, Berg LJ. Signaling through Itk promotes T helper 2 differentiation via negative regulation of T‐bet. Immunity 2004; 21:67–80. [DOI] [PubMed] [Google Scholar]
- 37. Berg LJ, Finkelstein LD, Lucas JA, Schwartzberg PL. Tec family kinases in T lymphocyte development and function. Annu Rev Immunol 2005; 23:549–600. [DOI] [PubMed] [Google Scholar]
- 38. Gomez‐Rodriguez J, Sahu N, Handon R, Davidson TS, Anderson SM, Kirby MR et al Differential expression of interleukin‐17A and ‐17F is coupled to T cell receptor signaling via inducible T cell kinase. Immunity 2009; 31:587–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhong Y, Dong S, Strattan E, Ren L, Butchar JP, Thornton K et al Targeting interleukin‐2‐inducible T‐cell kinase (ITK) and resting lymphocyte kinase (RLK) using a novel covalent inhibitor PRN694. J Biol Chem 2015; 290:5960–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gibson SA, Yang W, Yan Z, Liu Y, Rowse AL, Weinmann AS et al Protein kinase CK2 controls the fate between Th17 cell and regulatory T cell differentiation. J Immunol 2017; 198:4244–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ulges A, Klein M, Reuter S, Gerlitzki B, Hoffmann M, Grebe N et al Protein kinase CK2 enables regulatory T cells to suppress excessive TH2 responses in vivo . Nat Immunol 2015; 16:267–75. [DOI] [PubMed] [Google Scholar]
- 42. Ulges A, Witsch EJ, Pramanik G, Klein M, Birkner K, Buhler U et al Protein kinase CK2 governs the molecular decision between encephalitogenic TH17 cell and Treg cell development. Proc Natl Acad Sci U S A 2016; 113:10145–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gibson SA, Benveniste EN. Protein kinase CK2: an emerging regulator of immunity. Trends Immunol 2018; 39:82–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome‐mediated degradation. J Biol Chem 2001; 276:993–8. [DOI] [PubMed] [Google Scholar]
- 45. Gao Y, Nish SA, Jiang R, Hou L, Licona‐Limon P, Weinstein JS et al Control of T helper 2 responses by transcription factor IRF4‐dependent dendritic cells. Immunity 2013; 39:722–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Williams JW, Tjota MY, Clay BS, Vander Lugt B, Bandukwala HS, Hrusch CL et al Transcription factor IRF4 drives dendritic cells to promote Th2 differentiation. Nat Commun 2013; 4:2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Patsoukis N, Li L, Sari D, Petkova V, Boussiotis VA. PD‐1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol Cell Biol 2013; 33:3091–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lu KH, Keppler S, Leithauser F, Mattfeldt T, Castello A, Kostezka U et al Nck adaptor proteins modulate differentiation and effector function of T cells. J Leukoc Biol 2015; 98:301–11. [DOI] [PubMed] [Google Scholar]
- 49. Meininger I, Griesbach RA, Hu D, Gehring T, Seeholzer T, Bertossi A et al Alternative splicing of MALT1 controls signalling and activation of CD4+ T cells. Nat Comm 2016; 7:11292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. O'Hagan KL, Choi J, Pryshchep O, Chernoff J, Phee H. Pak2 links TCR signaling strength to the development of regulatory T cells and maintains peripheral tolerance. J Immunol 2015; 195:1564–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tamehiro N, Nishida K, Yanobu‐Takanashi R, Goto M, Okamura T, Suzuki H. T‐cell activation RhoGTPase‐activating protein plays an important role in TH17‐cell differentiation. Immunol Cell Biol 2017; 95:729. [DOI] [PubMed] [Google Scholar]
- 52. Pearce EJ, Kane CM, Sun J, Taylor JJ, McKee AS, Cervi L. Th2 response polarization during infection with the helminth parasite Schistosoma mansoni . Immunol Rev 2004; 201:117–26. [DOI] [PubMed] [Google Scholar]
- 53. Everts B, Perona‐Wright G, Smits HH, Hokke CH, van der Ham AJ, Fitzsimmons CM et al Omega‐1, a glycoprotein secreted by Schistosoma mansoni eggs, drives Th2 responses. J Exp Med 2009; 206:1673–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Steinfelder S, Andersen JF, Cannons JL, Feng CG, Joshi M, Dwyer D et al The major component in schistosome eggs responsible for conditioning dendritic cells for Th2 polarization is a T2 ribonuclease (omega‐1). J Exp Med 2009; 206:1681–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pasztoi M, Bonifacius A, Pezoldt J, Kulkarni D, Niemz J, Yang J et al Yersinia pseudotuberculosis supports Th17 differentiation and limits de novo regulatory T cell induction by directly interfering with T cell receptor signaling. Cell Mol Life Sci 2017; 74:2839–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Viboud GI, Bliska JB. Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu Rev Microbiol 2005; 59:69–89. [DOI] [PubMed] [Google Scholar]
- 57. Nuss AM, Beckstette M, Pimenova M, Schmühl C, Opitz W, Pisano F et al Tissue dual RNA‐seq allows fast discovery of infection‐specific functions and riboregulators shaping host–pathogen transcriptomes. Proc Natl Acad Sci USA 2017; 114:E791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nagaoka M, Hatta Y, Kawaoka Y, Malherbe LP. Antigen signal strength during priming determines effector CD4 T cell function and antigen sensitivity during influenza virus challenge. J Immunol 2014; 193:2812–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cheng J, Hamilton KS, Kane LP. Phosphorylation of Carma1, but not Bcl10, by Akt regulates TCR/CD28‐mediated NF‐κB induction and cytokine production. Mol Immunol 2014; 59:110–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kane LP, Weiss A. The PI‐3 kinase/Akt pathway and T cell activation: pleiotropic pathways downstream of PIP3. Immunol Rev 2003; 192:7–20. [DOI] [PubMed] [Google Scholar]
- 61. Haxhinasto S, Mathis D, Benoist C. The AKT–mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med 2008; 205:565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M et al T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci USA 2008; 105:7797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Turner MS, Isse K, Fischer DK, Turnquist HR, Morel PA. Low TCR signal strength induces combined expansion of Th2 and regulatory T cell populations that protect mice from the development of type 1 diabetes. Diabetologia 2014; 57:1428–36. [DOI] [PubMed] [Google Scholar]
- 64. Crellin NK, Garcia RV, Levings MK. Altered activation of AKT is required for the suppressive function of human CD4+ CD25+ T regulatory cells. Blood 2007; 109:2014–22. [DOI] [PubMed] [Google Scholar]
- 65. Kitz A, de Marcken M, Gautron AS, Mitrovic M, Hafler DA, Dominguez‐Villar M. AKT isoforms modulate Th1‐like Treg generation and function in human autoimmune disease. EMBO Rep 2016; 17:1169–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Waugh C, Sinclair L, Finlay D, Bayascas JR, Cantrell D. Phosphoinositide (3,4,5)‐triphosphate binding to phosphoinositide‐dependent kinase 1 regulates a protein kinase B/Akt signaling threshold that dictates T‐cell migration, not proliferation. Mol Cell Biol 2009; 29:5952–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Miskov‐Zivanov N, Turner MS, Kane LP, Morel PA, Faeder JR. The duration of T cell stimulation is a critical determinant of cell fate and plasticity. Sci Signal 2013; 6:ra97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hawse WF, Sheehan RP, Miskov‐Zivanov N, Menk AV, Kane LP, Faeder JR et al Cutting Edge: differential regulation of PTEN by TCR, Akt, and FoxO1 controls CD4+ T cell fate decisions. J Immunol 2015; 194:4615–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Huynh A, DuPage M, Priyadharshini B, Sage PT, Quiros J, Borges CM et al Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol 2015; 16:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shrestha S, Yang K, Guy C, Vogel P, Neale G, Chi H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol 2015; 16:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Park Y, Jin HS, Lopez J, Elly C, Kim G, Murai M et al TSC1 regulates the balance between effector and regulatory T cells. J Clin Invest 2013; 123:5165–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B et al The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 2009; 30:832–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR et al The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol 2011; 12:295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ray JP, Staron MM, Shyer JA, Ho PC, Marshall HD, Gray SM et al The interleukin‐2‐mTORc1 kinase axis defines the signaling, differentiation, and metabolism of T helper 1 and follicular B helper T cells. Immunity 2015; 43:690–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hawse WF, Boggess WC, Morel PA. TCR signal strength regulates Akt substrate specificity to induce alternate murine Th and T regulatory cell differentiation programs. J Immunol 2017; 199:589–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gan X, Wang J, Su B, Wu D. Evidence for direct activation of mTORC2 kinase activity by phosphatidylinositol 3,4,5‐trisphosphate. J Biol Chem 2011; 286:10998–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ip JY, Tong A, Pan Q, Topp JD, Blencowe BJ, Lynch KW. Global analysis of alternative splicing during T‐cell activation. RNA 2007; 13:563–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Martinez NM, Lynch KW. Control of alternative splicing in immune responses: many regulators, many predictions, much still to learn. Immunol Rev 2013; 253:216–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. van der Windt GJ, Pearce EL. Metabolic switching and fuel choice during T‐cell differentiation and memory development. Immunol Rev 2012; 249:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T‐cell differentiation and function. Immunol Rev 2012; 249:43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O et al Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest 2015; 125:194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF et al Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 2011; 186:3299–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A et al Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol 2010; 185:1037–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X et al Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 2014; 40:692–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG et al Foxp3 and Toll‐like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat Immunol 2016; 17:1459–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C et al Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol 2015; 16:1174–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Man K, Miasari M, Shi W, Xin A, Henstridge DC, Preston S et al The transcription factor IRF4 is essential for TCR affinity‐mediated metabolic programming and clonal expansion of T cells. Nat Immunol 2013; 14:1155. [DOI] [PubMed] [Google Scholar]
- 88. Nayar R, Schutten E, Bautista B, Daniels K, Prince AL, Enos M et al Graded levels of IRF4 regulate CD8+ T cell differentiation and expansion, but not attrition, in response to acute virus infection. J Immunol 2014; 192:5881–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Huber M, Lohoff M. IRF4 at the crossroads of effector T‐cell fate decision. Eur J Immunol 2014; 44:1886–95. [DOI] [PubMed] [Google Scholar]
- 90. Iwata A, Durai V, Tussiwand R, Briseno CG, Wu X, Grajales‐Reyes GE et al Quality of TCR signaling determined by differential affinities of enhancers for the composite BATF‐IRF4 transcription factor complex. Nat Immunol 2017; 18:563–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Krishnamoorthy V, Kannanganat S, Maienschein‐Cline M, Cook SL, Chen J, Bahroos N et al The IRF4 gene regulatory module functions as a read‐write integrator to dynamically coordinate T helper cell fate. Immunity 2017; 47(481–497):e487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zeng H, Chi H. mTOR signaling and transcriptional regulation in T lymphocytes. Transcription 2014; 5:e28263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Preston GC, Sinclair LV, Kaskar A, Hukelmann JL, Navarro MN, Ferrero I et al Single cell tuning of Myc expression by antigen receptor signal strength and interleukin‐2 in T lymphocytes. EMBO J 2015; 34:2008–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Laine A, Martin B, Luka M, Mir L, Auffray C, Lucas B et al Foxo1 is a T cell‐intrinsic inhibitor of the RORγt‐Th17 program. J Immunol 2015; 195:1791–803. [DOI] [PubMed] [Google Scholar]
- 95. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D et al The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011; 35:871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Jeltsch KM, Heissmeyer V. Regulation of T cell signaling and autoimmunity by RNA‐binding proteins. Curr Opin Immunol 2016; 39:127–35. [DOI] [PubMed] [Google Scholar]
- 97. Jeltsch KM, Hu D, Brenner S, Zoller J, Heinz GA, Nagel D et al Cleavage of roquin and regnase‐1 by the paracaspase MALT1 releases their cooperatively repressed targets to promote TH17 differentiation. Nat Immunol 2014; 15:1079–89. [DOI] [PubMed] [Google Scholar]