

Abstract
Background:
Animal studies suggest that exposure to either of the two widely used drugs of abuse, heroin or cocaine, causes depletion of the antioxidant, reduced glutathione, a hallmark of oxidative stress, in the brain. However, the relevance of the animal findings to the human is uncertain and clinical trials with the antioxidant GSH precursor n-acetylcysteine have produced mixed results in cocaine dependence.
Methods:
Our major objective was to compare glutathione levels, determined by an HPLC-coulometric procedure, in autopsied brain of chronic heroin (n=11) and cocaine users (n=9), who were positive for the drugs in the brain, to those of matched controls (n=16). Six brain regions were examined, including caudate, hippocampus, thalamus and frontal, temporal and insular cortices.
Results:
In contrast to experimental animal findings, we found no statistically significant difference between mean levels of reduced or oxidized glutathione in the drug user vs. control groups. Moreover, no correlation was found between levels of drugs in the brain and those of glutathione.
Conclusions:
Acknowledging the many generic limitations of an autopsied human brain study and the preliminary nature of the findings, our data nevertheless suggest that any oxidative stress caused by heroin or cocaine in chronic users of the drugs might not be sufficient to cause substantial loss of stores of glutathione in the human brain, at least during early withdrawal. These findings, requiring replication, might also have some relevance to future clinical trials employing glutathione supplement therapy as an anti-oxidative strategy in chronic users of the two abused drugs.
Keywords: Glutathione, Oxidative stress, Heroin, Cocaine, Human brain, Postmortem
1. Introduction
It is generally assumed that chronic exposure of some recreational drugs of abuse (e.g., stimulants, heroin) likely “injuries” the human brain to some extent and that neurotoxic injury might be caused at least in part by oxidative stress (Sharma et al., 2007; Cunha-Oliveira et al., 2008; Yamamoto et al., 2010; Sajja et al., 2016). To date, evidence supporting this possibility is largely driven by results of experimental animal investigations. For example, animal (rodent) studies show that high doses of the dopaminergic stimulant methamphetamine can produce increased brain levels of malondialdehyde or malondialdehyde-like lipoperoxidation substances, cause structural damage to (at least) brain dopamine nerve endings, and with reduction of the dopamine neuronal markers lessened by antioxidant treatment (for review see Kish, 2014).
The relevance of animal model findings to the human condition is always uncertain. Further, a practical consideration is that, at present, few tools can or have been used to assess oxidative stress or damage in the human brain directly. This is particularly relevant because of increasing interests in employing antioxidants such as n-acetylcysteine (Baker et al., 2003a,b; Ng et al., 2008; Zhou and Kalivas, 2008; Moussawi et al., 2009; Berk et al., 2013; McClure et al., 2014; Deepmala et al., 2015; Trivedi and Deth, 2015; Duailibi et al., 2017; Nocito Echevarria et al., 2017; Schulte et al., 2017) as a treatment strategy for drug dependence partly based on the assumption that there exists oxidative stress in brain of drug users. One indirect approach has been a measurement in the brain of the tripeptide glutathione (γ-L-glutamyl-L-cysteinylglycine; GSH, the reduced form), a major antioxidant defense. GSH is converted to oxidized glutathione (GSSG) as a consequence of oxidation catalyzed by glutathione peroxidase and can be recycled from the oxidized form to the reduced form by glutathione reductase (Dringen, 2000). One of the most consistent consequences of severe oxidative stress observed in a variety of experimental conditions affecting different organ systems is depletion of GSH (Sen and Packer, 2000; Gu et al., 2015; Won et al., 2015). In this regard, finding of a below normal concentration of GSH can be suggestive of the presence of oxidative stress (Di Monte et al., 1992; Jenner and Olanow, 1996; Won et al., 2015; Ren et al., 2017). Thus, our previous finding, consistent with animal data (Moszczynska et al., 1998), of a trend for GSH reduction in postmortem brain of a subgroup of human methamphetamine users (Mirecki et al., 2004) who had a marked dopamine loss suggests (but does not prove) that some oxidative stress might have occurred in methamphetamine-exposed human brain.
Emerging data, although not always consistent, shows that GSH concentration can be below normal in brain of experimental animals exposed to two other drugs of abuse, namely the heroin metabolite morphine (Goudas et al., 1997; Qiusheng et al., 2005; Guzman et al., 2006, 2009a,b; Ozmen et al., 2007; Abdel-Zaher et al., 2010, 2013a,b; Sumathi et al., 2011; Deng et al., 2012; Hu et al., 2012; Joshi et al., 2014; Motaghinejad et al., 2015a,b; Singh et al., 2015; Yun et al., 2015; Famitafreshi and Karimian, 2017) and the dopaminergic stimulant cocaine (Muriach et al., 2010; Uys et al., 2011; Lopez-Pedrajas et al., 2015; Vitcheva et al., 2015; Hu et al., 2016; Zhang et al., 2016; but see Wiener and Reith, 1990) (see Table 1 for a review). Overall, the findings show that chronic systemic and acute intra-cerebral-spinal morphine exposure consistently deplete brain GSH, with the exception of one study in rat pups showing increased GSH after repeated morphine injection (Traudt et al., 2012). However, results of acute effects of systemic morphine on brain GSH are mixed (increase, Guzman et al., 2009b; Joshi et al., 2014; decrease, Guzman et al., 2006, 2009a,b; and no change, Bien et al., 1992; Goudas et al., 1997). For cocaine, most studies (Muriach et al., 2010; Uys et al., 2011; Lopez-Pedrajas et al., 2015; Vitcheva et al., 2015; Hu et al., 2016; Zhang et al., 2016) except one (no change, Wiener and Reith, 1990; in mice) of chronic cocaine exposure in adults show decreased levels of brain GSH or ratio of GSH/GSSG although the effects of acute cocaine treatment can also be mixed (Wiener and Reith, 1990; Macedo et al., 2010; Uys et al., 2011). Discrepancies in the literature might be explained by the species employed, age at drug exposure (e.g., adolescent cocaine exposure did not result in GSH abnormality measured in adults; Zhu et al., 2016, 2017), brain regions examined, dose regimen and the GSH assay used (Table 1; see also Discussion).
Table 1.
Review of animal literature on morphine- or cocaine-induced changes in brain levels of glutathione (GSH)
| Reference | Methodsa | Speciesb | Treatmentc | Main findings of GSH changes and commentsd | |
|---|---|---|---|---|---|
| Chronic ity |
Dose regimen | ||||
| Morphine: | |||||
| Bien et al. 1992 | DTNB | Rats ♂, Wistar |
Acute | 100 mg/kg IP, i3 h | GSH ↔ WB ([GSH]C ≈ 1.5 mM) |
| Goudas et al. 1997 | HPLC-ECD | Rats ♂, SD |
Acute | 80 mg/kg SC i1–4 h 100 µg ICV i1–5 h |
GSH ↔ cn/ctx/cereb/pons; GSH ↓30% cn/ctx at 3 h; ↔ cereb/pons ([GSH]C = 1.1 – 2.7 mM) |
| Qiusheng et al. 2005 | OPT | Mice♀♂, Kunming |
Chronic | 5–35 mg/kg x 40d IP, escalating dose |
GSH/GSSG ↓40% WB (the ratio decreased along with treatment duration; GSH levels not reported; the OPT fluorescence method might over-estimate GSSG levels (Hissin and Hilf, 1976)) |
|
Guzman et al. 2006, 2009a,b |
HPLC or OPT |
Rats ♀♂, SD, Wistar |
Acute | 3–12 mg/kg, IP i1 h | GSH ↓31–36% WB in adult rats ([GSH]C = 4.7 mM or 0.27 mM in 48h fasted rats); ↓42–88% WB in weaned Wistar rats ([GSH]C = 2.0 mM); ↓24% WB at 3 mg/kg but ↑10% at 6- 12 mg/kg in young malnourished rats at P60 ([GSH]C = 0.41 mM) |
| Ozman et al. 2007 | DTNB | Rabbits ♂ | Acute | 200 µg IT i8 d | GSH ↓48% ctx ([GSH]C = 1.9 mM) |
| Abdel-Zaher et al. 2010, 2013a,b |
DTNB | Mice ♂, Swiss- Webster |
Chronic | 2×5 mg/kg x 1–7d SC, i2 h ± NAL 5 mg/kg |
GSH ↓20–40% WB with 3–7 d morphine treatment; GSH ↓45% WB in naloxone (NAL) challenged ([GSH]C = 2.8 mM) |
| Sumathi et al. 2011 | DTNB | Rats♂, Wistar |
Chronic | 10–160 mg/kg x 21d IP |
GSH ↓46% WB ([GSH]C = 1.0 mM) |
| Hu et al. 2012 | NMR in vitro | Rats ♂, SD |
Chronic | 5–40 mg/kg x 14d IP, escalating dose, i48 h |
GSH ↓prefrontal ctx; ↔ cn/nac/hippocampus (the metabonomic study did not report GSH levels or percentage of changes) |
| Deng et al. 2012 | NMR in vitro |
Monkeys ♀♂, rhesus |
Chronic | 3×3–15 mg/kg x 90d SC, escalating dose, i8d |
GSH ↓hippocampus; ↔ prefrontal ctx (the metabonomic study did not report GSH levels or percentage of changes) |
| Joshi et al. 2014 | DTNB | Rats ♂, Wistar |
Acute Chronic |
1 and 5 mg/kg IP 1 and 5 mg/kg x 15d |
GSH ↑57% and 105% WB by acute morphine, respectively; GSH ↔ and ↓51% WB by chronic morphine, respectively (Note: morphine reversed GSH depletion induced by restraint stress). |
| Singh et al. 2015 | DTNB | Mice ♀♂, Swiss- albino |
Chronic | 2×5 mg/kg x 6d IP, i2h ± NAL 8 mg/kg |
GSH ↔ WB by morphine alone (Note: non-significant 41% loss) GSH ↓65% with naloxone (NAL) challenge ([GSH]C = 3.4 mM) |
| Yun et al. 2015 | DTNB | Mice ♂, C57BL/6 |
Chronic | 10 mg/kg x 7d IP, i6h + NAL 5 mg/kg |
GSH ↓36% frontal ctx ([GSH]C = 8 mM) (non-naloxone- challenged condition was not assessed) |
| Motaghinejad et al. 2015a,b | OxisResearc h kit (?) |
Rats ♂, Wistar |
Chronic | 15–45 mg/kg x 21d, i24h + NAL 3mg/kg [a]; 45 mg/kg x 28d SC [b] |
GSH ↓61–65% hippocampal mitochondria preparation (Note: the method of GSH assay employed was not clear; high GSSG levels were also reported). |
| Famitafreshi et al. 2017 | DTNB | Rats ♂, SD |
Chronic | 5 mg/kg x 14d IP | GSH ↓30% hippocampus (ns) ↓22% prefrontal ctx (ns) (isolation stress exacerbated GSH loss to −54–76%; [GSH]C = 2.1–3.2 mM) |
| Traudt et al. 2012 | MRS in vivo | Rat pups ♀♂, SD |
Chronic | 2×2 mg/kg x 5d IP, i24h |
GSH ↑43% hippocampus on P8 following morphine on P3- P7 (LCModel for spectra analysis; a volume of interest of 5 µl) |
| Cocaine: | |||||
| Wiener et al. 1990 | HPLC | Mice ♂, C57BL/6 ByJ |
Acute Chronic | 25 mg/kg x 14d IP, i24h ± 50 mg/kg |
GSH ↔ cn/ctx by either acute or chronic cocaine, with or without challenge ([GSH]C = 1.5 mM) |
| Macedo et al. 2010 | DTNB | Mice ♂, Swiss |
Acute | 90 mg/kg IP, i1 h [SE] or 5–15 min [death] |
GSH ↔ after status epilepticus [SE] or ↑28–50% prefrontal ctx/cn after death ([GSH]C = 0.0025 mM; note possible error in unit) |
| Muriach et al. 2010 | FDNP- HPLC |
Rats ♂, Wistar |
Chronic | 15 mg/kg x 20d IP, i24h |
GSH ↓22% hippocampus; ↔ frontal ctx ([GSH]C = 2–2.2 mM) |
| Uys et al. 2011 | UPLC-MS | Rats ♂ | Acute Chronic | 15–30 mg/kg x 7d IP, i21d ± 15 mg/kg |
GSH ↓23% by 7d cocaine; ↑62% by acute cocaine in nac; but ↔ in cocaine challenged rats ([GSH]C = 0.55 mM) |
| López-Pedrajas et al. 2015 | FDNP- HPLC |
Rats ♂, Wistar |
Chronic | 15 mg/kg x 18d IP | GSH ↔ cerebellum although GSSG ↑56% so the ratio of GSH vs GSSG decreased. |
| Vitcheva et al. 2015 | DTNB | Rats ♂, Wistar |
Chronic | 15 mg/kg x 7d IP, i24h |
GSH ↓44% WB; also GSH ↓55% in mitochondria preparations ([GSH]C = 0.0017 mM; note possible error in unit) |
| Zhang et al. 2016 | HPLC- IMMS |
Rats ♂, SD |
Chronic | SAD FR1 × 3d + FR3 × 7d, i24h and i21d |
GSH ↓45% and ↓57% cn at i24h and i21d, respectively; GSH ↔ prefrontal ctx (the metabonomic study did not report GSH levels or the cumulative extent of cocaine exposure) |
| Hu et al. 2016 | DTNB | Rats ♂, SD |
Chronic | 10 mg/kg x 6d IP, i15d |
GSH ↓47% hippocampus; ↔ prefrontal ctx ([GSH]C unit unclear) |
| Zhu et al. 2016, 2017 | DTNB | Rats ♂, SD P28 |
Chronic | 15 mg/kg x 15d IP, i35–38d |
GSH ↔ hippocampus, medial prefrontal ctx (adolescent cocaine exposure; GSH measured in adults; [GSH]C unit unclear) |
DTNB, referring to a variety of GSH assay using Ellman’s reagent 5,5′-dithiobis(2-nitrobenzoic acid); OPT = o-phthaldehyde; ECD = electrochemical detection; FDNP = Sanger reactant 1-fluoro-2,4-dinitrobencene; IMMS = ion mobility mass spectrometry;
SD = Sprague-Dawley. Adult animals were used unless otherwise indicated;
The dose regimen shows daily single or multiple doses by total days, administration route (IP = intra-peritoneal; SC = subcutaneous; ICV = intracerebroventricle; IT = intrathecal; SAD = intravenous self-administration; FR = fixed ratio), the interval (i) between final drug administration and sacrifice or drug challenge, if reported (see the cited references for more details);
Brain regions are caudate nucleus or striatum (cn), nucleus accumbens (nac), cortex (ctx), cerebellum (cereb), or whole brain (WB); ns = non-significant; [GSH]C denotes reported concentrations of GSH in the control group, with 1 mM = 1 µmol/g wet tissue or 307 µg/g wet tissue or 20 nmol/mg protein or 6.1 µg/mg protein by assuming a protein/tissue ratio of 0.05 (Tong et al., 2016) and a brain unit weight of 1 g/mL.
Investigations of GSH in central nervous system of human users of heroin and cocaine appear to be limited to a postmortem brain study in heroin users reporting markedly below normal GSH throughout the brain (Gutowicz et al., 2011) and a preliminary report of low GSH in cerebrospinal fluid of two (of three examined) patients receiving intracerebroventricular doses of morphine for intractable cancer pain (Goudas et al., 1999). Given the sparse literature on whether the two widely used drugs of abuse might cause oxidative stress in human brain, as suggested by animal data, our objective was to establish whether levels of GSH (primary outcome measure) are lower than normal (and by inference oxidative stress above-normal) in a regionally extensive sampling of well-characterized autopsied human brain of chronic cocaine and chronic heroin users (Wilson et al., 1996; Kish et al., 1999, 2001; Kalasinsky et al., 2000; McLeman et al., 2000; Worsley et al., 2000; Siegal et al., 2004; Frankel et al., 2008). For comparison, using an HPLC-electrochemical procedure, we measured the following compounds as secondary outcome measures which also appeared on the chromatogram: GSSG (oxidized GSH), GSH-Cysteine (GSH-CYS; the mixed disulfide), uric acid (UA), a xanthine catabolite and potential antioxidant and neuroprotective agent, reported to act via GSH (Mirecki et al., 2004; Bakshi et al., 2015), and methionine, the latter as an index sensitive to postmortem time (Mirecki et al., 2004). Our working hypothesis was that, based on the above-mentioned animal data, GSH levels would be below normal throughout the brain of users of either heroin or cocaine.
2. Subjects and Methods
2.1 Subjects
Postmortem brain from a total of 11 chronic users of heroin (1 female), 9 cocaine (2 females) and 16 controls (2 females) was obtained from medical examiner offices in USA/Canada using a standardized protocol. The study was approved by the Research Ethics Board of the Centre for Addiction and Mental Health at Toronto. Subject information, drug histories, and brain drug and dopamine levels are summarized in Table 2, with the information previously reported (Wilson et al., 1996; Kish et al., 1999, 2001; Kalasinsky et al., 2000; McLeman et al., 2000; Siegal et al., 2004). There were no statistically significant differences in age (control, 37.0±3.1 years; heroin, 36.2±2.5 years; cocaine, 35.4±4.8 years; mean±SEM), postmortem intervals (PMI, interval between death and freezing of the brain; control, 14.6±1.8 hours; heroin, 13.4±2.0 hours; cocaine, 17.0±2.4 hours), or freezer storage time at the time when the biochemical assays were performed in 2001–2003 (control, 5.4±0.5 years; heroin, 6.5±0.4 years; cocaine, 7.0±0.6 years) between the control and drug users. At autopsy, one half-brain was fixed in formalin fixative for neuropathological analysis, whereas the other half was immediately frozen until dissection for neurochemical analysis. Blood samples were obtained from all of the drug users and controls for drug screening. Sequential scalp hair samples for drug analyses could be obtained from 14 of 16 controls, 10 of 11 heroin users, and five of 9 cocaine users. Levels of drugs of abuse in blood and other bodily fluids were measured by the local medical examiner whereas drug analyses in brain and hair samples were conducted at the Armed Forces Institute of Pathology (Washington, DC, USA). Heroin users met the following criteria: 1) presence of heroin metabolites (6-acetylmorphine, morphine, or morphine glucuronide) on toxicology screens in blood and autopsied brain; 2) absence of other drugs of abuse in bodily fluids with the exception of ethanol (see below) or other opioid drugs (two subjects #H5 and #H7 had blood samples positive for the opioid drug propoxyphene and its metabolite norpropoxyphene; see Table 2); 3) evidence from the case records of primary use of heroin for >1 year prior to death; and 4) absence of evidence of neurological illness or, at autopsy, brain pathology unrelated to use of the drug. Five of the heroin users had recently used alcohol as indicated by the presence of ethanol in blood (Table 2). Available hair analysis of the (10 of 11) heroin users revealed presence of only heroin metabolites in eight of the users. The suspected cause of death was heroin intoxication (seven), mixed drug intoxication (two), and cardiovascular disease with heroin as a contributing factors (two). Cocaine users met the following criteria: 1) presence of cocaine or metabolite benzoylecgonine in blood or (one subject) urine; autopsied brain, and, if available, scalp hair by GC-MS; 2) absence of other drugs of abuse in bodily fluids, with the exception of ethanol (see below), or in brain; 3) evidence from the case records or interview with next of kin of use of cocaine as the primary drug of abuse for >1 year prior to death; and 4) absence of neurological illness or, at autopsy, brain pathology unrelated to use of the drug. Two of the cocaine users had recently used alcohol as indicated by the presence of ethanol in blood or cocaethylene in brain. Available hair analysis of the (five of nine) cocaine users revealed presence of only cocaine and/or metabolites in four of the users. Known or suspected causes of death of the cocaine users were cocaine intoxication (five), cardiovascular disease with cocaine as a contributing factors (two), carotid artery aneurysm with cocaine as a contributing factor (one) and chest trauma (one).
Table 2.
Characteristics and drug use histories of the 11 heroin (H1-H11) and 9 cocaine (C1-C9) usersa.
| Age | Duratio n |
Toxicology | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| (yrs), | PMI | of use | Recent drug use pattern | Route of drug | Suspected/known | Blood drug | Brain drug | Caudate | ||
| Case | Sex | (h) | (yrs) | administration | cause of death | Hair | levelb | levelc | DA leveld |
|
| H1 | 36,M | 8 | 10 | $200 per month | Intravenous | CVD/Narcotic intoxication |
— | 1.93 | 1.79 | +0.1% |
| H2* | 43,M | 13 | 27 | Unknown | Intravenous | Narcotic intoxication | + | 0.60 | 1.79 | -11% |
| H3 | 34,M | 23 | >1 | Unknown | Intravenous | Narcotic intoxication | + | 0.67 | 1.19 | -30% |
| H4* | 34,M | 10.5 | >1 | Unknown | Intravenous | Narcotic intoxication | + | 0.42 | 0.80 | -21% |
| H5 | 40,M | 5 | 20 | Daily | Intravenous | Mixed drug intoxication | + | 0.32 | 0.61 | +34% |
| H6 | 44,F | 18.5 | 23 | Daily, sometimes 1 g per day |
Intravenous | CVD/Narcotic intoxication |
+ | 0.11 | 0.10 | -16% |
| H7 | 43,M | 21 | >1 | Daily | Intravenous | Mixed drug intoxication | + | 1.47 | 1.79 | -5% |
| H8* | 35,M | 19.5 | >1 | Unknown | Unknown | Narcotic intoxication | ND | 1.09 | 1.80 | -47% |
| H9* | 28,M | 8 | 4 | Daily | Intravenous | Narcotic intoxication | + | 0.39 | 0.46 | -0.4% |
| H10 | 19,M | 9.5 | >1 | Unknown | Unknown | Narcotic intoxication | ND | 0.39 | 0.37 | +69% |
| H11 * |
42,M | 11 | 10 | Unknown | Intravenous | Narcotic intoxication | + | 0.70 | 0.76 | +10% |
| C1 | 26,M | 18 | 1–2 | Unknown | Oral; smoked | Cocaine intoxication | — | 424.0 | 179.7 | -53% |
| C2 | 21,M | 6 | 2–3 | $150/mo, weekend binges | Nasal | CVD/cocaine intoxication | + | ND | 20.7 | -38% |
| C3* | 26,F | 18 | 8 | Binge/limited only by funds |
Oral; smoked | Cocaine intoxication | ND | 0.26 | 12.4 | -19% |
| C4* | 36,M | 24 | 3 | Binge/limited only by funds |
Nasal | Cocaine intoxication | — | 27.0 | 157.6 | -79% |
| C5 | 39,M | 26 | 8 | Considered heavy user | Intravenous; nasal |
Cocaine intoxication | + | 0.43 | 11.0 | +9% |
| C6 | 31,M | 22 | >2 | Limited only by funds | Nasal; smoked | Cocaine intoxication | — | 39.2 | 164.4 | -31% |
| C7 | 40,M | 9 | >10 | Binge every 2–3 wk | Nasal; smoked | Gunshot wound to chest | + | 12.34 | 40.4 | -54% |
| C8 | 70,M | 10 | 55 | $60/mo, 1st wk of a month | Smoked | CVD | — | 19.44 | 15.7 | +50% |
| C9 | 30,F | 20 | >1 | Unknown | Smoked | CVD/cocaine intoxication | + | 17.16 | 19.4 | +5% |
M = male; F = female; PMI = postmortem interval; DA = dopamine; CVD = cardiovascular disorder.
Cases with ethanol detected in blood. + Drug hair analyses confirmed. ND = not detected. For cases H1, H6, C2 and C9, heroin or cocaine toxicity was considered to be a possible contributing factor to the cause of death; high levels of propoxyphene (0.63 and 1.2 mg/L, respectively) and norpropoxyphene (1.26 and 3.8 mg/L, respectively) were also detected in blood of cases H5 and H7 and could have contributed to the death.
Information on the cases including brain drug levels has been published previously in (Wilson et al., 1996; Kish et al., 1999; Kalasinsky et al., 2000; McLeman et al., 2000; Kish et al., 2001; Siegal et al., 2004);
Measured in µM of levels of the heroin metabolite morphine or cocaine plus metabolite benzoylecgonine;
Measured in nmol/g tissue of total levels of heroin (morphine plus 6-acetylmorphine plus morphine glucuronide) in occipital cortex or cocaine (cocaine plus metabolites benzoylecgonine, ecgonine methyl ester, norcocaine, cocaethylene) in caudate;
Measured as percentage decrease of the control mean (6.62 ng/mg wet tissue; see (Wilson et al., 1996)) with the exception of case#C8, for which a control mean of 4.20 ng/mg wet tissue for aged subjects (mean 69 y; see (Haycock et al., 2003)) was used.
All control subjects (for which brain GSH levels have been previously reported in (Mirecki et al., 2004)) were neurologically normal and had no evidence of brain pathology on neuropathological examination. All had no history of drug use and tested negative for drugs of abuse in blood, autopsied brain, and in sequential scalp hair samples where available. The cause of death for the controls were electrocution (n=1), morbid obesity (n=1), trauma (n=3), pulmonary embolism (n=2), and cardiovascular disease (n=9).
2.2 GSH, GSSG, GSH-Cys, UA and Methionine Analysis
Brain regions were dissected as previously described (Kish et al., 1988), using the Atlas of Riley (Riley, 1943) for the caudate, hippocampal Ammon’s horn and medial pulvinar thalamus and Brodmann classification for frontal (BA9), temporal (BA21) and insular cortices. Levels of GSH, GSSG, GSH-Cys, methionine, and UA in tissue homogenates were measured by a coulometric method using HPLC and electrochemical detection with coulometric cells as previously described (Fitzmaurice et al., 2003; Mirecki et al., 2004; Tong et al., 2016) (see Supplementary Methods for more details). Protein concentration was determined using the Bio-Rad Protein Assay Kit (Bio-Rad, Hercules, CA, USA) with bovine plasma albumin as the standard.
2.3 Statistical Analyses
Statistical analyses were performed using StatSoft STATISTICA 7.1 (Tulsa, Oklahoma, USA). Differences in levels of GSH, GSSG, GSSG-Cys, UA and methionine among controls and drug groups in brain regions examined were conducted using ANCOVA (p<0.05) with age and PMI as the covariates, given influences of age and PMI on some of the outcome measures (Mirecki et al., 2004; Tong et al., 2016), followed by post-hoc Bonferroni adjustments (p<0.05). Correlations were examined by Pearson product moment correlation or Spearman ranking order correlation as indicated in the text.
3. Results
A one-way ANCOVA with age and PMI as the covariates disclosed that GSH, GSSG, GSH-Cys, UA and methionine levels (Table 3) were normal in all examined brain regions of the two drug user groups versus the controls (p>0.05), with the exception of significantly lower levels of methionine in the caudate nucleus of users of heroin (−26%) and cocaine (−33%). Scatter plots of GSH levels (Figure 1) and those of GSSG, GSH-Cys, UA, and methionine (see Supplementary Figures 1–4)1 showed overlapped data range between drug users and control subjects.
Table 3.
Levels of glutathione (reduced [GSH], oxidized [GSSG], and cysteine-bound [GSH-cysteine]), uric acid, and methionine in brain of users of heroin and cocaine and control subjects.
| Region/Group | GSH | GSSG | GSH-cysteine | Uric acid | Methionine |
|---|---|---|---|---|---|
| Caudate nucleus | |||||
| Controls | 7.46 ± 1.32 (16) | 0.38 ± 0.04 (16) | 0.67 ± 0.08 (16) | 0.47 ± 0.06 (16) | 1.37 ± 0.08 (16) |
| Heroin | 6.86 ± 1.04 (11) | 0.26 ± 0.04 (11) | 0.63 ± 0.07 (11) | 0.42 ± 0.07 (11) | 1.01 ± 0.09* (11) |
| Cocaine | 6.60 ± 0.57 (9) | 0.24 ± 0.06 (9) | 0.85 ± 0.06 (9) | 0.59 ± 0.11 (9) | 0.92 ± 0.06* (9) |
| F | 0.17 (2, 31) | 3.06 (2, 31) | 3.01 (2, 31) | 0.76 (2, 31) | 9.13 (2, 31) |
| p | 0.85 | 0.06 | 0.06 | 0.48 | 0.001 |
| Hippocampus, ammon’s horn | |||||
| Controls | 6.60 ± 0.80 (16) | 0.06 ± 0.01 (16) | 0.30 ± 0.04 (16) | 0.44 ± 0.07 (16) | 2.03 ± 0.27 (16) |
| Heroin | 4.58 ± 0.44 (11) | 0.05 ± 0.02 (11) | 0.48 ± 0.13 (11) | 0.44 ± 0.07 (11) | 1.70 ± 0.21 (11) |
| Cocaine | 4.61 ± 0.99 (9) | 0.03 ± 0.01 (9) | 0.34 ± 0.12 (9) | 0.41 ± 0.13 (8) | 1.33 ± 0.24 (9) |
| F | 2.32 (2, 31) | 0.94 (2, 31) | 1.40 (2, 31) | 0.03 (2, 30) | 1.85 (2, 31) |
| p | 0.12 | 0.40 | 0.26 | 0.97 | 0.17 |
| Thalamus, medial pulvinar nucleus |
|||||
| Controls | 6.44 ± 1.64 (16) | 0.24 ± 0.08 (16) | 1.78 ± 0.28 (16) | 0.74 ± 0.11 (16) | 3.24 ± 0.40 (16) |
| Heroin | 5.71 ± 2.03 (11) | 0.18 ± 0.07 (9) | 1.27 ± 0.48 (11) | 1.05 ± 0.29 (11) | 3.53 ± 0.70 (11) |
| Cocaine | 4.89 ± 0.80 (9) | 0.09 ± 0.05 (8) | 0.97 ± 0.21 (9) | 0.72 ± 0.12 (9) | 2.79 ± 0.27 (9) |
| F | 0.19 (2, 31) | 1.27 (2, 28) | 1.28 (2, 31) | 0.84 (2, 31) | 0.37 (2, 31) |
| p | 0.83 | 0.30 | 0.29 | 0.44 | 0.69 |
| Frontal cortex | |||||
| Controls | 7.24 ± 1.07 (15) | 0.25 ± 0.06 (15) | 0.59 ± 0.07 (14) | 0.61 ± 0.14 (15) | 1.56 ± 0.16 (15) |
| Heroin | 6.85 ± 0.62 (11) | 0.11 ± 0.05 (9) | 0.66 ± 0.12 (11) | 0.32 ± 0.05 (11) | 1.46 ± 0.23 (11) |
| Cocaine | 6.94 ± 0.93 (9) | 0.15 ± 0.04 (9) | 0.62 ± 0.11 (9) | 0.31 ± 0.06 (9) | 1.19 ± 0.15 (9) |
| F | 0.05 (2, 30) | 1.56 (2, 28) | 0.47 (2, 29) | 2.36 (2, 30) | 0.69 (2, 30) |
| p | 0.95 | 0.23 | 0.63 | 0.11 | 0.51 |
| Temporal cortex | |||||
| Controls | 5.60 ± 0.91 (16) | 0.25 ± 0.08 (16) | 0.23 ± 0.05 (16) | 0.60 ± 0.14 (15) | 0.94 ± 0.16 (15) |
| Heroin | 7.45 ± 1.60 (11) | 0.42 ± 0.15 (11) | 0.21 ± 0.05 (11) | 1.00 ± 0.29 (11) | 1.65 ± 0.42 (11) |
| Cocaine | 4.62 ± 0.43 (9) | 0.17 ± 0.03 (9) | 0.15 ± 0.02 (9) | 0.43 ± 0.05 (9) | 0.77 ± 0.10 (9) |
| F | 1.04 (2, 31) | 1.54 (2, 31) | 0.52 (2, 31) | 1.98 (2, 30) | 3.02 (2, 30) |
| p | 0.36 | 0.23 | 0.60 | 0.16 | 0.06 |
| Insular cortex | |||||
| Controls | 6.14 ± 1.25 (16) | 0.36 ± 0.09 (15) | 0.37 ± 0.06 (16) | 0.78 ± 0.13 (16) | 1.76 ± 0.29 (16) |
| Heroin | 4.38 ± 0.68 (11) | 0.40 ± 0.04 (11) | 0.31 ± 0.03 (11) | 0.80 ± 0.09 (11) | 1.87 ± 0.10 (11) |
| Cocaine | 5.08 ± 0.81 (8) | 0.31 ± 0.05 (8) | 0.30 ± 0.05 (8) | 0.68 ± 0.10 (8) | 1.48 ± 0.15 (8) |
| F | 0.69 (2, 30) | 0.48 (2, 29) | 0.59 (2, 30) | 0.17 (2, 30) | 0.46 (2, 30) |
| p | 0.51 | 0.62 | 0.56 | 0.84 | 0.63 |
Data (mean ± SEM) are in µg/mg protein with the number of cases in parentheses. One-way analysis of co-variance [ANCOVA, F (df)] with age and postmortem interval as the covariates did not reveal any significant difference (p > 0.05) among heroin, cocaine users and control subjects except that of methionine in the caudate (*p < 0.05, heroin or cocaine users vs. controls, following post hoc Bonferroni adjustment).
Figure 1.
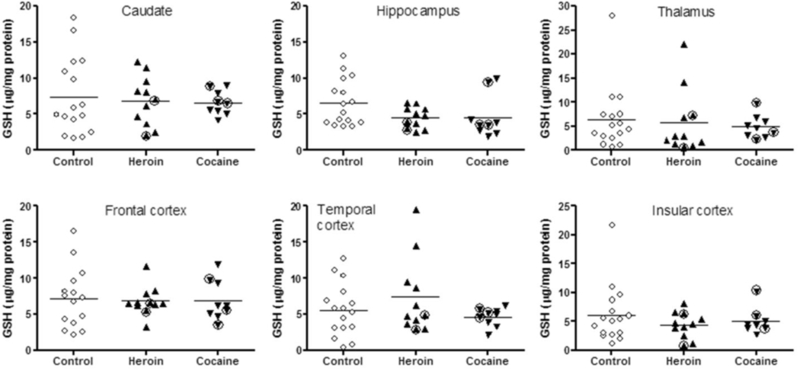
Scatter plots of levels of reduced glutathione (GSH) in brain of users of heroin and cocaine and control subjects. Circled up-triangles identify two heroin users with high blood levels of propoxyphene and metabolite norpropoxyphene; circled down-triangles identify three cocaine users with high levels of cocaine and metabolites in brain.
No significant correlation was observed between levels of GSH, GSSG, GSH-Cys, UA or methionine and available drug use parameters of the heroin and cocaine users including duration of use (Pearson) and composite blood and brain drug levels (Spearman). Three cocaine users (#C1, #C4 and #C6 in Table 2) demonstrated a markedly higher level of cocaine and metabolites in brain than other cocaine users (>150 versus <50 nmol/g tissue); however, the three cocaine users did not have abnormally low levels of GSH (Figure 1) or out-of-range values of GSSG, GSH-Cys, UA or methionine (see Supplementary Figures 1–4)2. Two heroin users (Table 2) had high blood levels of the opioid drug propoxyphene and its metabolite norpropoxyphene (5.76 µM and 15.21 µM for #H5 and #H7, respectively); however, the two were not particularly affected with respect to brain levels of GSH (see Figure 1), GSSG, GSH-Cys, UA or methionine (see Supplementary Figures 1–4)3.
Blood tested positive for ethanol in five heroin users (0.05–0.12%; #H2, #H4, #H8, #H9 and #H11 in Table 2) and two cocaine users (0.01% and 0.02% for #C3 and #C4, respectively, in Table 2); however, the presence or absence of ethanol did not differentiate the outcome measures of brain GSH, GSSG, GSH-Cys, UA or methionine. Cocaine users showed a moderate loss of striatal dopamine (Wilson et al., 1996) whereas dopamine levels were normal in heroin users (Kish et al., 2001); however, dopamine levels in the caudate were not significantly correlated (Pearson) with those of GSH, GSSG, GSH-Cys, UA or methionine in the drug users.
In this sample of controls and/or drug users, we found no significant correlation (Pearson) between levels of GSH, GSSG, GSH-Cys, UA or methionine and age or PMI of the subjects (all subjects included or in individual groups).
4. Discussion
The main finding of our study is that levels of GSH are normal in autopsied brain of chronic users of heroin and cocaine. Further, we found no correlation between recent drug exposure (as suggest by brain drug levels) and levels of the tri-peptide antioxidant.
4.1 Limitations
There are many limitations to postmortem human brain studies. Because, as we have shown previously (Mirecki et al., 2004), levels of both reduced and oxidized glutathione are decreased in autopsied vs. biopsied human brain, postmortem time must have influenced to some degree concentration of our major outcome measures. Nevertheless, we feel it reasonable to expect that qualitative differences or lack thereof, found in the autopsied brain would be generally similar to those occurring in living brain. Further, there were no statistically significant differences amongst mean PMI for the drug and control groups (note: PMI was included as a covariate in the statistical analysis), and mean levels of the amino acid methionine, which increase quite markedly after death (Mirecki et al., 2004), were also mostly similar in the three groups. Further, we did not observe any significant negative correlations between levels of GSH and methionine among brain regions and groups; including methionine levels as a covariate did not change any statistical outcomes of the other analytes.
Little information on drug doses, precise duration of chronic use, medication history, or history of alcohol consumption, which is known to be able to deplete brain GSH (Uys et al., 2014), was available for the subjects of our study. In this regard, the possibility cannot be excluded that unknown medications or other drugs used by the subjects of our study might have influenced GSH levels in the brain. However, we can be certain that the users of cocaine and heroin must have used these drugs, at least as recently as 72 hours before death as they all tested positive for the drug in the brain and/or blood.
Arguing against the notion that we might not have been able to detect a small/modest change in postmortem brain GSH concentrations are our previous findings demonstrating a modest GSH reduction (19 to 30%) in autopsied substantia nigra of patients with three different degenerative Parkinsonian conditions (n=10–16 per group; Fitzmaurice et al., 2003) and the observation of slightly decreased (by 17%) GSH levels in striatum of rodents exposed to a binge dose of methamphetamine (Moszczynska et al., 1998). Previously we reported a trend for a modest reduction (by 35%) in autopsied brain of human methamphetamine users, but which was restricted to the subgroup having severe loss of the neurotransmitter dopamine (Mirecki et al., 2004).
4.2 Comparison with Literature (Human)
To our knowledge, there have been no previous studies of glutathione in postmortem or living brain of human cocaine users. However, in autopsied brain of users of heroin, Gutowicz and colleagues (Gutowicz et al., 2011) reported markedly (by about 20–40%) lower GSH in the cerebral cortex, hippocampus, brain stem and white matter. Assuming in their report that brain “heroin level” means total concentration in brain of the major heroin metabolites (6-acetylmorphine, morphine, morphine glucuronide), the brain drug levels in heroin users of the Gutowicz study were generally similar to those in our investigation, suggesting that the extent of recent drug exposure to the heroin users was also similar in both studies. Possibly the discrepancy might be explained by differences in methodologies for glutathione (HPLC with electrochemical detection in our study vs. colorimetric assay; see Tong et al., 2016 for discussions), differences in drug history of the heroin users, or be related to the very long PMI (two-four days) in the Gutowicz study vs. a much shorter mean of 14 hours for both heroin users and controls in our investigation, and uncertainty whether drug and control groups were matched for PMI in the earlier investigation. In this regard, GSH levels reported by Gutowicz study in control brains (2.3–3.4 mM, assuming a protein/tissue ratio of 0.05 (Tong et al., 2016) and a brain unit weight of 1 g/mL) were higher than those reported in the literature for autopsied human brain (generally < 2 mM) (Perry et al., 1982; Slivka et al., 1987; Sofic et al., 1992; Sian et al., 1994) including our studies (Fitzmaurice et al., 2003; Mirecki et al., 2004; Tong et al., 2016) despite much longer PMI (>48 hrs. vs ≤26 hrs.). It is also possible that the autopsied brains in the Gutowicz study had suffered from more severe pathology (not reported), e.g., hypoxic/ischaemic lesions (Andersen and Skullerud, 1999; Buttner et al., 2000), than those in our study as some animal data suggest that prolonged ischemia might cause glutathione depletion in brain (Rehncrona et al., 1980; Slivka and Cohen, 1993). In this respect, qualitative brain neuropathological examination in our cases did not reveal obvious abnormalities (cell loss or gliosis) in the drug users with the exception of some hypoxic/ischaemic neuronal changes in CA1 of hippocampus (#C3), mild diffuse gliosis in midbrain (#C4) and acute subarachnoid hemorrhage (#C9) in three cocaine users, respectively, and mild ventricular dilatation (#H5) and mild diffuse gliosis in diencephalon and lower brainstem (#H11) in two heroin users, respectively.
4.3 Why Did Animal GSH Findings on Morphine/Cocaine Not Translate into The Human?
Although, as mentioned above, the experimental animal literature is generally consistent (below normal GSH following chronic heroin or cocaine exposure; Table 1), we found no significant change in GSH levels in brain of humans chronically exposed to either of the drugs. Possibly the difference could be explained by different extent of drug exposure in the animal studies vs. that in our human investigation and by different redox response in animals vs humans as exemplified by reported GSH depletion in animal ischemia (Slivka and Cohen, 1993) versus compensatory GSH elevation in human brain stroke (An et al. 2012). The possibility has to be considered that acute/sub-chronic exposure to the drugs, e.g., as suggested by experimental reports of GSH depletion by morphine and cocaine (Table 1) and by a report of depletion of cerebrospinal fluid GSH levels in the human after acute intracerebroventricular morphine for cancer pain (Goudas et al., 1999), might have resulted in excessive GSH utilization and depletion, but that tolerance (compensatory increase in GSH synthesis) occurred in the users of our investigation who likely had been exposed to the drugs for years. Most animal studies employed passive drug administration, and it is conceivable that this might have produced a different profile of brain redox response and disturbance as compared to that of drug self-administration (e.g., see Pomierny-Chamiolo et al., 2013), more relevant to the human condition. The study by Uys et al. (2011) shows that rats repeatedly exposed to cocaine had decreased levels of GSH in brain (nucleus accumbens) at three weeks withdrawal versus saline-treated animals whereas an acute challenging dose of cocaine restored brain GSH levels to that of the controls, suggesting a possible effect of abstinence although some of the studies examined GSH within hours of final drug administration (Abdel-Zaher et al., 2010, 2013a,b). The age of initial drug exposure could be another variable as cocaine exposure during adolescence, which is common in many human users, was not associated with GSH loss in rats later in adults (Zhu et al., 2016, 2017). Interestingly, a recent study (Joshi et al., 2014) showed that morphine could counteract chronic restraint stress-induced GSH depletion in rat brain, suggesting some interactions between drugs of abuse and stress-induced redox disturbance (Madrigal et al., 2001; Ahmad et al., 2010; Kumar et al., 2011; Moretti et al., 2012; Filho et al., 2015; Bouvier et al., 2017; Famitafreshi and Karimian, 2017). Perhaps some of the above factors might help to explain why available preclinical animal data demonstrating a brain GSH reduction following morphine or cocaine exposure do not translate to the human.
4.4 Glutathione and Oxidative Stress in Heroin and Cocaine Users
We emphasize that the “negative” results of our investigation do not imply that chronic use of the abused drugs cocaine and heroin do not cause oxidative stress in human brain, but rather that exposure to the drugs at the doses used by the subjects of our study, which are probably within the dose regimen employed in experimental animal studies (e.g., see Nayak et al., 1976; Djurendic-Brenesel et al., 2010), might not cause oxidative stress of a magnitude that produces some brain depletion of the antioxidant glutathione. Here we caution also that the extent of oxidative stress necessary to cause GSH depletion in human brain is not known and that mild to moderate oxidative stress could induce compensatory increase in GSH synthesis (cf. Tong et al., 2016). Recently, clinical trials of the antioxidant n-acetylcysteine, a prodrug to the rate-limiting GSH precursor cysteine, were performed in a variety of human addiction conditions including cocaine with mixed outcomes (see Berk et al., 2013; McClure et al., 2014; Deepmala et al., 2015; Trivedi and Deth, 2015; Duailibi et al., 2017; Nocito Echevarria et al., 2017; Schulte et al., 2017 for reviews). However, N-acetylcysteine was employed primarily as a modulator of glutamate neurotransmission, with its antioxidant property as a possible secondary mechanism (LaRowe et al., 2013; McClure et al., 2014). In retrospect, our findings of normal brain GSH in heroin and cocaine users provide no support to use of this GSH prodrug to address a GSH deficiency in brain, at least during early withdrawal when the drugs of abuse were tested positive. Future trials of N-acetylcysteine in opiate and cocaine dependence, aiming at redox homeostasis (Trivedi and Deth, 2015), might take our autopsied brain finding of normal brain GSH levels into consideration.
5. Conclusions
The main finding of our study is that, in contrast to results of animal studies, levels of glutathione were found to be normal in autopsied brain of chronic users of heroin and of cocaine, suggesting that any oxidative stress caused by the drugs might not be sufficient to deplete substantially tissue stores of the antioxidant. Our findings, although suggestive, must be considered preliminary especially given the limitations associated with autopsied brain investigations including large variability of GSH levels and a small sample size. Future studies might also consider measurement of GSH in living human brain using a magnetic resonance imaging approach in which the influence of heroin and cocaine (and also opiates for therapeutic purposes) can more easily, e.g., longitudinally, be examined.
Supplementary Material
Highlights.
Effects of morphine and cocaine on brain GSH in animal studies are reviewed.
Exposure to morphine or cocaine can deplete brain GSH in animals.
GSH was measured in autopsied brains of chronic heroin and cocaine users.
Extensive toxicology confirmed chronic use of heroin or cocaine.
Human chronic heroin and cocaine users have normal levels of brain GSH.
Acknowledgments
Role of Funding Source
This study was supported in part by the US NIDA/NIH DA07182 (SK), the New Zealand Institute of Environmental Science and Research, Ltd. (PF, SK), and the Centre for Addiction and Mental Health Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary material can be found by accessing the online version of this paper at http://dx.doi.org and by entering doi: …
Author Disclosures
Contributors
SJK, JT and PSF designed the study; JT, PSF, AM and TM conducted experiments, data collection and analysis; LCA performed neuropathology; JT, PSF and SJK wrote initial drafts; AM, GR, JHM, RM, IB, YF and NS contributed to revisions; all authors approved the final manuscript.
Conflict of Interest
No conflict declared.
References
- Abdel-Zaher AO, Abdel-Rahman MS, FM EL, 2010. Blockade of nitric oxide overproduction and oxidative stress by Nigella sativa oil attenuates morphine-induced tolerance and dependence in mice. Neurochem. Res 35, 1557–1565. [DOI] [PubMed] [Google Scholar]
- Abdel-Zaher AO, Mostafa MG, Farghaly HS, Hamdy MM, Abdel-Hady RH, 2013a. Role of oxidative stress and inducible nitric oxide synthase in morphine-induced tolerance and dependence in mice. Effect of alpha-lipoic acid. Behav. Brain Res 247, 17–26. [DOI] [PubMed] [Google Scholar]
- Abdel-Zaher AO, Mostafa MG, Farghly HM, Hamdy MM, Omran GA, Al-Shaibani NK, 2013b. Inhibition of brain oxidative stress and inducible nitric oxide synthase expression by thymoquinone attenuates the development of morphine tolerance and dependence in mice. Eur. J. Pharmacol 702, 62–70. [DOI] [PubMed] [Google Scholar]
- Ahmad A, Rasheed N, Banu N, Palit G, 2010. Alterations in monoamine levels and oxidative systems in the frontal cortex, striatum, and hippocampus of the rat brain during chronic unpredictable stress. Stress 13, 355–364. [DOI] [PubMed] [Google Scholar]
- An L, Dani KA, Shen J, Warach S; Natural History of Stroke Investigators, 2012. Pilot results of in vivo brain glutathione measurements in stroke patients. J. Cereb. Blood Flow Metab 32, 2118–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen SN, Skullerud K, 1999. Hypoxic/ischaemic brain damage, especially pallidal lesions, in heroin addicts. Forensic Sci. Int 102, 51–59. [DOI] [PubMed] [Google Scholar]
- Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, Kalivas PW, 2003a. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat. Neuroscience 6, 743–749. [DOI] [PubMed] [Google Scholar]
- Baker DA, McFarland K, Lake RW, Shen H, Toda S, Kalivas PW, 2003b. N-acetyl cysteine-induced blockade of cocaine-induced reinstatement. Ann. N.Y. Acad. Sci 1003, 349–351. [DOI] [PubMed] [Google Scholar]
- Bakshi R, Zhang H, Logan R, Joshi I, Xu Y, Chen X, Schwarzschild MA, 2015. Neuroprotective effects of urate are mediated by augmenting astrocytic glutathione synthesis and release. Neurobiol. Dis 82, 574–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk M, Malhi GS, Gray LJ, Dean OM, 2013. The promise of N-acetylcysteine in neuropsychiatry. Trends Pharmacol. Sci 34, 167–177. [DOI] [PubMed] [Google Scholar]
- Bien E, Vick K, Skorka G, 1992. Effects of exogenous factors on the cerebral glutathione in rodents. Arch.Toxicol 66, 279–285. [DOI] [PubMed] [Google Scholar]
- Bouvier E, Brouillard F, Molet J, Claverie D, Cabungcal JH, Cresto N, Doligez N, Rivat C, Do KQ, Bernard C, Benoliel JJ, Becker C, 2017. Nrf2-dependent persistent oxidative stress results in stress-induced vulnerability to depression. Mol. Psychiatry 22, 1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttner A, Mall G, Penning R, Weis S, 2000. The neuropathology of heroin abuse. Forensic Sci. Int 113, 435–442. [DOI] [PubMed] [Google Scholar]
- Cunha-Oliveira T, Rego AC, Oliveira CR, 2008. Cellular and molecular mechanisms involved in the neurotoxicity of opioid and psychostimulant drugs. Brain Res. Rev 58, 192–208. [DOI] [PubMed] [Google Scholar]
- Deepmala, Slattery J, Kumar N, Delhey L, Berk M, Dean O, Spielholz C, Frye R, 2015. Clinical trials of N-acetylcysteine in psychiatry and neurology: A systematic review. Neurosci. Biobehav. Rev 55, 294–321. [DOI] [PubMed] [Google Scholar]
- Deng Y, Bu Q, Hu Z, Deng P, Yan G, Duan J, Hu C, Zhou J, Shao X, Zhao J, Li Y, Zhu R, Zhao Y, Cen X, 2012. (1) H-nuclear magnetic resonance-based metabonomic analysis of brain in rhesus monkeys with morphine treatment and withdrawal intervention. J. Neurosci. Res 90, 2154–2162. [DOI] [PubMed] [Google Scholar]
- Di Monte DA, Chan P, Sandy MS, 1992. Glutathione in Parkinson’s disease: A link between oxidative stress and mitochondrial damage? Ann. Neurol 32, S111–115. [DOI] [PubMed] [Google Scholar]
- Djurendic-Brenesel M, Mimica-Dukic N, Pilija V, Tasic M, 2010. Gender-related differences in the pharmacokinetics of opiates. Forensic Sci. Int 194, 28–33. [DOI] [PubMed] [Google Scholar]
- Dringen R, 2000. Metabolism and functions of glutathione in brain. Prog. Neurobiol 62, 649–671. [DOI] [PubMed] [Google Scholar]
- Duailibi MS, Cordeiro Q, Brietzke E, Ribeiro M, LaRowe S, Berk M, Trevizol AP, 2017. N-acetylcysteine in the treatment of craving in substance use disorders: Systematic review and meta-analysis. Am. J. Addict 26, 660–666. [DOI] [PubMed] [Google Scholar]
- Famitafreshi H, Karimian M, 2017. Socialization alleviates burden of oxidative-stress in hippocampus and prefrontal cortex in morphine addiction period in male rats. Curr. Mol. Pharmacol [Epub ahead of print] [DOI] [PubMed]
- Filho CB, Jesse CR, Donato F, Giacomeli R, Del Fabbro L, da Silva Antunes M, de Gomes MG, Goes AT, Boeira SP, Prigol M, Souza LC, 2015. Chronic unpredictable mild stress decreases BDNF and NGF levels and Na(+), K(+)-ATPase activity in the hippocampus and prefrontal cortex of mice: Antidepressant effect of chrysin. Neuroscience 289, 367–380. [DOI] [PubMed] [Google Scholar]
- Fitzmaurice PS, Ang L, Guttman M, Rajput AH, Furukawa Y, Kish SJ, 2003. Nigral glutathione deficiency is not specific for idiopathic Parkinson’s disease. Mov. Disord 18, 969–976. [DOI] [PubMed] [Google Scholar]
- Frankel PS, Alburges ME, Bush L, Hanson GR, Kish SJ, 2008. Striatal and ventral pallidum dynorphin concentrations are markedly increased in human chronic cocaine users. Neuropharmacology 55, 41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudas LC, Carr DB, Maszczynska I, Marchand JE, Wurm WH, Greenblatt DJ, Kream RM, 1997. Differential effect of central versus parenteral administration of morphine sulfate on regional concentrations of reduced glutathione in rat brain. Pharmacology 54, 92–97. [DOI] [PubMed] [Google Scholar]
- Goudas LC, Langlade A, Serrie A, Matson W, Milbury P, Thurel C, Sandouk P, Carr DB, 1999. Acute decreases in cerebrospinal fluid glutathione levels after intracerebroventricular morphine for cancer pain. Anesth. Analg 89, 1209–1215. [PubMed] [Google Scholar]
- Gu F, Chauhan V, Chauhan A, 2015. Glutathione redox imbalance in brain disorders. Curr. Opin. Clin. Nutr. Metab. Care 18, 89–95. [DOI] [PubMed] [Google Scholar]
- Gutowicz M, Kazmierczak B, Baranczyk-Kuzma A, 2011. The influence of heroin abuse on glutathione-dependent enzymes in human brain. Drug Alcohol Depend 113, 8–12. [DOI] [PubMed] [Google Scholar]
- Guzman DC, Brizuela NO, Alvarez RG, Garcia EH, Mejia GB, Olguin HJ, 2009a. Cerebrolysin and morphine decrease glutathione and 5-hydroxyindole acetic acid levels in fasted rat brain. Biomed. Pharmacother 63, 517–521. [DOI] [PubMed] [Google Scholar]
- Guzman DC, Osnaya-Brizuela N, Garcia-Alvarez R, Hernandez-Garcia E, Juarez-Olguin H, 2009b. Oxidative stress induced by morphine in brain of rats fed with a protein deficient diet. Hum. Exp. Toxicol 28, 577–582. [DOI] [PubMed] [Google Scholar]
- Guzman DC, Vazquez IE, Brizuela NO, Alvarez RG, Mejia GB, Garcia EH, Santamaria D, de Apreza M, Olguin HJ, 2006. Assessment of oxidative damage induced by acute doses of morphine sulfate in postnatal and adult rat brain. Neurochem. Res 31, 549–554. [DOI] [PubMed] [Google Scholar]
- Haycock JW, Becker L, Ang L, Furukawa Y, Hornykiewicz O, Kish SJ, 2003. Marked disparity between age-related changes in dopamine and other presynaptic dopaminergic markers in human striatum. J. Neurochem 87, 574–585. [DOI] [PubMed] [Google Scholar]
- Hissin PJ, Hilf R, 1976. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal. Biochem 74, 214–226. [DOI] [PubMed] [Google Scholar]
- Hu P, Zhu W, Zhu C, Jin L, Guan Y, Guan X, 2016. Resveratrol fails to affect cocaine conditioned place preference behavior, but alleviates anxiety-like behaviors in cocaine withdrawn rats. Psychopharmacology 233, 1279–1287. [DOI] [PubMed] [Google Scholar]
- Hu Z, Deng Y, Hu C, Deng P, Bu Q, Yan G, Zhou J, Shao X, Zhao J, Li Y, Zhu R, Xu Y, Zhao Y, Cen X, 2012. (1)H NMR-based metabonomic analysis of brain in rats of morphine dependence and withdrawal intervention. Behav. Brain Res 231, 11–19. [DOI] [PubMed] [Google Scholar]
- Jenner P, Olanow CW, 1996. Oxidative stress and the pathogenesis of Parkinson’s disease. Neurology 47, S161–170. [DOI] [PubMed] [Google Scholar]
- Joshi JC, Ray A, Gulati K, 2014. Differential modulatory effects of morphine on acute and chronic stress induced neurobehavioral and cellular markers in rats. Eur. J. Pharmacol 729, 17–21. [DOI] [PubMed] [Google Scholar]
- Kalasinsky KS, Bosy TZ, Schmunk GA, Ang L, Adams V, Gore SB, Smialek J, Furukawa Y, Guttman M, Kish SJ, 2000. Regional distribution of cocaine in postmortem brain of chronic human cocaine users. J. Forensic Sci 45, 1041–1048. [PubMed] [Google Scholar]
- Kish SJ, 2014. Chapter 08: The pathology of methamphetamine use in the human brain, in Madras BK, Kuhar M (Eds.), The effects of drug abuse on the human nervous system Elsevier Inc., Amsterdam, pp. 203–297. [Google Scholar]
- Kish SJ, Kalasinsky KS, Derkach P, Schmunk GA, Guttman M, Ang L, Adams V, Furukawa Y, Haycock JW, 2001. Striatal dopaminergic and serotonergic markers in human heroin users. Neuropsychopharmacology 24, 561–567. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Kalasinsky KS, Furukawa Y, Guttman M, Ang L, Li L, Adams V, Reiber G, Anthony RA, Anderson W, Smialek J, DiStefano L, 1999. Brain choline acetyltransferase activity in chronic, human users of cocaine, methamphetamine, and heroin. Mol. Psychiatry 4, 26–32. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Shannak K, Hornykiewicz O, 1988. Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease. Pathophysiologic and clinical implications. N. Engl. J. Med 318, 876–880. [DOI] [PubMed] [Google Scholar]
- Kumar B, Kuhad A, Chopra K, 2011. Neuropsychopharmacological effect of sesamol in unpredictable chronic mild stress model of depression: Behavioral and biochemical evidences. Psychopharmacology 214, 819–828. [DOI] [PubMed] [Google Scholar]
- LaRowe SD, Kalivas PW, Nicholas JS, Randall PK, Mardikian PN, Malcolm RJ, 2013. A double-blind placebo-controlled trial of N-acetylcysteine in the treatment of cocaine dependence. Am. J. Addict 22, 443–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Pedrajas R, Ramirez-Lamelas DT, Muriach B, Sanchez-Villarejo MV, Almansa I, Vidal-Gil L, Romero FJ, Barcia JM, Muriach M, 2015. Cocaine promotes oxidative stress and microglial-macrophage activation in rat cerebellum. Front. Cell. Neurosci 9, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macedo DS, Vasconcelos SM, Andrade-Neto M, Belchior LD, Honorio Junior JE, Goncalves DO, Fonteles MM, Silva MI, Aguiar LM, Viana GS, de Sousa FC, 2010. Cocaine-induced status epilepticus and death generate oxidative stress in prefrontal cortex and striatum of mice. Neurochem. Int 56, 183–187. [DOI] [PubMed] [Google Scholar]
- Madrigal JL, Olivenza R, Moro MA, Lizasoain I, Lorenzo P, Rodrigo J, Leza JC, 2001. Glutathione depletion, lipid peroxidation and mitochondrial dysfunction are induced by chronic stress in rat brain. Neuropsychopharmacology 24, 420–429. [DOI] [PubMed] [Google Scholar]
- McClure EA, Gipson CD, Malcolm RJ, Kalivas PW, Gray KM, 2014. Potential role of N-acetylcysteine in the management of substance use disorders. CNS Drugs 28, 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeman ER, Warsh JJ, Ang L, Li PP, Kalasinsky KS, Ross BM, Tong J, Schmunk G, Adams V, Kish SJ, 2000. The human nucleus accumbens is highly susceptible to G protein down-regulation by methamphetamine and heroin. J. Neurochem 74, 2120–2126. [DOI] [PubMed] [Google Scholar]
- Mirecki A, Fitzmaurice P, Ang L, Kalasinsky KS, Peretti FJ, Aiken SS, Wickham DJ, Sherwin A, Nobrega JN, Forman HJ, Kish SJ, 2004. Brain antioxidant systems in human methamphetamine users. J. Neurochem 89, 1396–1408. [DOI] [PubMed] [Google Scholar]
- Moretti M, Colla A, de Oliveira Balen G, dos Santos DB, Budni J, de Freitas AE, Farina M, Severo Rodrigues AL, 2012. Ascorbic acid treatment, similarly to fluoxetine, reverses depressive-like behavior and brain oxidative damage induced by chronic unpredictable stress. J. Psychiatr. Res 46, 331–340. [DOI] [PubMed] [Google Scholar]
- Moszczynska A, Turenne S, Kish SJ, 1998. Rat striatal levels of the antioxidant glutathione are decreased following binge administration of methamphetamine. Neurosci. Lett 255, 49–52. [DOI] [PubMed] [Google Scholar]
- Motaghinejad M, Karimian M, Motaghinejad O, Shabab B, Yazdani I, Fatima S, 2015a. Protective effects of various dosage of Curcumin against morphine induced apoptosis and oxidative stress in rat isolated hippocampus. Pharmacol. Rep 67, 230–235. [DOI] [PubMed] [Google Scholar]
- Motaghinejad M, Karimian SM, Motaghinejad O, Shabab B, Asadighaleni M, Fatima S, 2015b. The effect of various morphine weaning regimens on the sequelae of opioid tolerance involving physical dependency, anxiety and hippocampus cell neurodegeneration in rats. Fundam. Clin. Pharmacol 29, 299–309. [DOI] [PubMed] [Google Scholar]
- Moussawi K, Pacchioni A, Moran M, Olive MF, Gass JT, Lavin A, Kalivas PW, 2009. N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat. Neurosci 12, 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muriach M, Lopez-Pedrajas R, Barcia JM, Sanchez-Villarejo MV, Almansa I, Romero FJ, 2010. Cocaine causes memory and learning impairments in rats: involvement of nuclear factor kappa B and oxidative stress, and prevention by topiramate. J. Neurochem 114, 675–684. [DOI] [PubMed] [Google Scholar]
- Nayak PK, Misra AL, Mule SJ, 1976. Physiological disposition and biotransformation of (3H) cocaine in acutely and chronically treated rats. J. Pharmacol. Exp. Ther 196, 556–569. [PubMed] [Google Scholar]
- Ng F, Berk M, Dean O, Bush AI, 2008. Oxidative stress in psychiatric disorders: evidence base and therapeutic implications. Int. J. Neuropsychopharmacol 11, 851–876. [DOI] [PubMed] [Google Scholar]
- Nocito Echevarria MA, Andrade Reis T, Ruffo Capatti G, Siciliano Soares V, da Silveira DX, Fidalgo TM, 2017. N-acetylcysteine for treating cocaine addiction -A systematic review. Psychiatry Res 251, 197–203. [DOI] [PubMed] [Google Scholar]
- Ozmen I, Naziroglu M, Alici HA, Sahin F, Cengiz M, Eren I, 2007. Spinal morphine administration reduces the fatty acid contents in spinal cord and brain by increasing oxidative stress. Neurochem. Res 32, 19–25. [DOI] [PubMed] [Google Scholar]
- Perry TL, Godin DV, Hansen S, 1982. Parkinson’s disease: a disorder due to nigral glutathione deficiency? Neurosci. Lett 33, 305–310. [DOI] [PubMed] [Google Scholar]
- Pomierny-Chamiolo L, Moniczewski A, Wydra K, Suder A, Filip M, 2013. Oxidative stress biomarkers in some rat brain structures and peripheral organs underwent cocaine. Neurotox. Res 23, 92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiusheng Z, Yuntao Z, Rongliang Z, Dean G, Changling L, 2005. Effects of verbascoside and luteolin on oxidative damage in brain of heroin treated mice. Pharmazie 60, 539–543. [PubMed] [Google Scholar]
- Rehncrona S, Folbergrova J, Smith DS, Siesjo BK, 1980. Influence of complete and pronounced incomplete cerebral ischemia and subsequent recirculation on cortical concentrations of oxidized and reduced glutathione in the rat. J. Neurochem 34, 477–486. [DOI] [PubMed] [Google Scholar]
- Ren X, Zou L, Zhang X, Branco V, Wang J, Carvalho C, Holmgren A, Lu J, 2017. Redox signaling mediated by thioredoxin and glutathione systems in the central nervous system. Antioxid. Redox Signa 27, 989–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley RA, 1943. An atlas of the basal ganglia, brain stem and spinal cord The Williams and Wilkins Company, Baltimore, MD. [Google Scholar]
- Sajja RK, Rahman S, Cucullo L, 2016. Drugs of abuse and blood-brain barrier endothelial dysfunction: A focus on the role of oxidative stress. J. Cereb. Blood Flow Metab 36, 539–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte MHJ, Wiers RW, Boendermaker WJ, Goudriaan AE, van den Brink W, van Deursen DS, Friese M, Brede E, Waters AJ, 2017. The effect of N-acetylcysteine and working memory training on cocaine use, craving and inhibition in regular cocaine users: Correspondence of lab assessments and Ecological Momentary Assessment. Addict. Behav 79, 24–31. [DOI] [PubMed] [Google Scholar]
- Sen CK, Packer L, 2000. Thiol homeostasis and supplements in physical exercise. Am. J. Clin. Nutr 72, 653S–669S. [DOI] [PubMed] [Google Scholar]
- Sharma HS, Sjoquist PO, Ali SF, 2007. Drugs of abuse-induced hyperthermia, blood-brain barrier dysfunction and neurotoxicity: Neuroprotective effects of a new antioxidant compound H-290/51. Curr. Pharm. Des 13, 1903–1923. [DOI] [PubMed] [Google Scholar]
- Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, Jenner P, Marsden CD, 1994. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol 36, 348–355. [DOI] [PubMed] [Google Scholar]
- Siegal D, Erickson J, Varoqui H, Ang L, Kalasinsky KS, Peretti FJ, Aiken SS, Wickham DJ, Kish SJ, 2004. Brain vesicular acetylcholine transporter in human users of drugs of abuse. Synapse 52, 223–232. [DOI] [PubMed] [Google Scholar]
- Singh P, Sharma B, Gupta S, Sharma BM, 2015. In vivo and in vitro attenuation of naloxone-precipitated experimental opioid withdrawal syndrome by insulin and selective KATP channel modulator. Psychopharmacology 232, 465–475. [DOI] [PubMed] [Google Scholar]
- Slivka A, Cohen G, 1993. Brain ischemia markedly elevates levels of the neurotoxic amino acid, cysteine. Brain Res 608, 33–37. [DOI] [PubMed] [Google Scholar]
- Slivka A, Spina MB, Cohen G, 1987. Reduced and oxidized glutathione in human and monkey brain. Neurosci. Lett 74, 112–118. [DOI] [PubMed] [Google Scholar]
- Sofic E, Lange KW, Jellinger K, Riederer P, 1992. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson’s disease. Neurosci. Lett 142, 128–130. [DOI] [PubMed] [Google Scholar]
- Sumathi T, Nathiya VC, Sakthikumar M, 2011. Protective Effect of Bacoside-A against Morphine-Induced Oxidative Stress in Rats. Indian J. Pharm. Sci 73, 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J, Fitzmaurice PS, Moszczynska A, Mattina K, Ang LC, Boileau I, Furukawa Y, Sailasuta N, Kish SJ, 2016. Do glutathione levels decline in aging human brain? Free Rad. Biol. Med 93, 110–117. [DOI] [PubMed] [Google Scholar]
- Traudt CM, Tkac I, Ennis KM, Sutton LM, Mammel DM, Rao R, 2012. Postnatal morphine administration alters hippocampal development in rats. J. Neurosci. Res 90, 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi MS, Deth R, 2015. Redox-based epigenetic status in drug addiction: a potential contributor to gene priming and a mechanistic rationale for metabolic intervention. Front. Neurosci 8, 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uys JD, Knackstedt L, Hurt P, Tew KD, Manevich Y, Hutchens S, Townsend DM, Kalivas PW, 2011. Cocaine-induced adaptations in cellular redox balance contributes to enduring behavioral plasticity. Neuropsychopharmacology 36, 2551–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uys JD, Mulholland PJ, Townsend DM, 2014. Glutathione and redox signaling in substance abuse. Biomed. Pharm 68, 799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitcheva V, Simeonova R, Kondeva-Burdina M, Mitcheva M, 2015. Selective nitric oxide synthase inhibitor 7-nitroindazole protects against cocaine-induced oxidative stress in rat brain. Oxid. Med. Cell. Longev 2015, 157876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener HL, Reith ME, 1990. Differential effects of daily administration of cocaine on hepatic and cerebral glutathione in mice. Biochem. Pharmacol 40, 1763–1768. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Levey AI, Bergeron C, Kalasinsky K, Ang L, Peretti F, Adams VI, Smialek J, Anderson WR, Shannak K, Deck J, Niznik HB, Kish SJ, 1996. Striatal dopamine, dopamine transporter, and vesicular monoamine transporter in chronic cocaine users. Ann. Neurol 40, 428–439. [DOI] [PubMed] [Google Scholar]
- Won SJ, Kim JE, Cittolin-Santos GF, Swanson RA, 2015. Assessment at the single-cell level identifies neuronal glutathione depletion as both a cause and effect of ischemia-reperfusion oxidative stress. J. Neurosci 35, 7143–7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worsley JN, Moszczynska A, Falardeau P, Kalasinsky KS, Schmunk G, Guttman M, Furukawa Y, Ang L, Adams V, Reiber G, Anthony RA, Wickham D, Kish SJ, 2000. Dopamine D1 receptor protein is elevated in nucleus accumbens of human, chronic methamphetamine users. Mol. Psychiatry 5, 664–672. [DOI] [PubMed] [Google Scholar]
- Yamamoto BK, Moszczynska A, Gudelsky GA, 2010. Amphetamine toxicities: Classical and emerging mechanisms. Ann. N.Y. Acad. Sci 1187, 101–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, Lee Y, Yun K, Oh S, 2015. Bergenin decreases the morphine-induced physical dependence via antioxidative activity in mice. Arch. Pharmacal. Res 38, 1248–1254. [DOI] [PubMed] [Google Scholar]
- Zhang X, Chiu VM, Todd RP, Sorg BA, Hill HH Jr., 2016. Neuronal metabolomics by ion mobility mass spectrometry in cocaine self-administering rats after early and late withdrawal. Anal. Bioanal. Chem 408, 4233–4245. [DOI] [PubMed] [Google Scholar]
- Zhou W, Kalivas PW, 2008. N-acetylcysteine reduces extinction responding and induces enduring reductions in cue-and heroin-induced drug-seeking. Biol. Psychiatry 63, 338–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Ge X, Gao P, Li M, Guan Y, Guan X, 2017. Adolescent cocaine exposure induces prolonged synaptic modifications in medial prefrontal cortex of adult rats. Brain Struct. Funct 223, 1829–1838. [DOI] [PubMed] [Google Scholar]
- Zhu W, Mao Z, Zhu C, Li M, Cao C, Guan Y, Yuan J, Xie G, Guan X, 2016. Adolescent exposure to cocaine increases anxiety-like behavior and induces morphologic and neurochemical changes in the hippocampus of adult rats. Neuroscience 313, 174–183. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.