

Abstract
Introduction:
Acute intermittent porphyria (AIP) is a rare and challenging hereditary neurovisceral disease with no specific symptoms. Posterior reversible encephalopathy syndrome (PRES) is a clinicoradiological syndrome with bilateral reversible posterior gyriform lesions that can be associated with many different conditions, including AIP. Usually, peripheral neuropathy is considered the most common neurological manifestation of AIP. However, AIP should also be considered when seizures and PRES are associated with unexplained abdominal pain.
Case presentation:
Both the patients were presented with seizures and PRES on brain magnetic resonance imaging (MRI). Unexplained abdominal pain occurred before the onset of seizures. The AIP diagnosis was made after repeated Watson–Schwartz tests. Hematin was not available for these 2 patients. However, supportive treatment including adequate nutrition and fluid therapy as well as specific antiepileptic drugs aided the patient's recovery and no acute attacks had occurred by the 3-year follow-up.
Conclusion:
In contrast to other causes of PRES patients, seizure is the most common symptom in AIP patients with PRES. This is a strong diagnostic clue for AIP when ambiguous abdominal pain patients presented with seizures and PRES on brain MRI. A positive prognosis can be achieved with the combination of early recognition, supportive and intravenous hematin therapy, and withdrawal of precipitating factors, including some antiepileptic drugs.
Keywords: acute intermittent porphyria, inappropriate secretion of antidiuretic hormone, posterior reversible encephalopathy syndrome, seizure
1. Introduction
Posterior reversible encephalopathy syndrome (PRES) is a clinicoradiological syndrome presenting with headache, seizures, visual disturbances, or mental dysfunction associated with bilateral posterior white and/or gray matter lesions.[1] Since its first description in 1996, more and more cases related to PRES have been identified with the advent of modern neuroimaging techniques, especially magnetic resonance imaging (MRI). Several conditions can be associated with PRES, including puerperal eclampsia, renal failure, hypertensive crises, chemotherapeutic agents, immunosuppressants, organ transplantation, blood transfusion, infection, and autoimmune diseases.[2] The diagnosis of PRES is usually made on the basis of neuroradiologic findings and clinical presentations, especially in the presence of characteristic parieto-occipital gyriform lesions on T2-weighted MRI. The reversible neurologic deficits and neuroradiologic findings are thought to be factors of differentiation from other clinical settings.
Acute intermittent porphyria (AIP), the most common type of acute porphyrias in most of the countries worldwide, is a rare hereditary disease due to deficiencies of heme biosynthesis.[3] Although abdominal pain, neurological dysfunction, and psychiatric disturbances form the classic triad of AIP, the diagnosis is often delayed and challenging because of diverse clinical features from asymptomatic to life-threatening attacks. The major clinical manifestation of AIP is an acute attack involving severe abdominal pain and nervous system presentations.
As early as 1991, King et al[4] reported a case of AIP presented with seizures and hallucinations. The brain MRI showed transient abnormalities with multiple bilateral lesions. Although the nomenclature PRES had not been identified at that time, neurologists were aware of the association between AIP and reversible multiple lesions on MRI. Recently, more and more cases with neuroimaging suggestive of PRES have been identified in patients with AIP, although peripheral neuropathy is considered as the most common neurological manifestation of AIP. However, it is still a substantial challenge for neurologists to make a prompt correct diagnosis when presented with cases involving seizures and PRES-like neuroimaging. The association between PRES and AIP is still an unknown phenomenon and needs further studies. Here, we describe 2 cases with AIP presenting with seizures and PRES on MRI in our department, and we review the few existing studies to provide awareness among clinicians of this difficult-to-diagnose disease so that early accuracy diagnoses can be achieved.
2. Case report
2.1. Case 1
In December 2014, a 27-year-old man was admitted to our hospital for abdominal and lumbar pain lasting for 12 days after heavy food and alcohol intake before 1 week. He was diagnosed with acute pancreatitis with mild elevated urine amylase in his local hospital. No abnormalities were found in his abdominal computed tomography (CT) and X-ray. However, the paroxysmal pain was not relieved, and his blood pressure increased. Six days later, a generalized tonic-clonic seizure occurred. Then, partial seizures in the left limb continued. He was transferred to our hospital. No particular medical or family history was reported.
At the time of admission, he described recurrent severe paroxysmal abdominal and lumbar pain. His clinical examination was unremarkable, except for decreased tendon reflex in the 4 limbs. There was no complaint of visual disturbance. The patient had moderately increased blood pressure (159/101 mm Hg) and tachycardia (112/min). His creatine kinase was 246 U/L (normal, 0–170 U/L), aspartate transaminase was 42 U/L (15–40 U/L) and serum sodium was 130 mmol/L (normal, 137–147 mmol/L). Other biochemistry tests were not suggestive. Tests for connective tissue disorders, viral infection, heavy metal poisoning, thyroid function, and antithyroid antibodies were all negative. Brain MRI demonstrated multifocal lesions in the bilateral occipitoparietal and frontal lobes. The lesions were hypointense on T1-weighted images and were hyperintense on T2-weighted and fluid-attenuated inversion recovery images. Diffusion-weighted images (DWI) were hypointense, and some were patchy hyperintense, while apparent diffusion coefficient (ADC) maps were hyperintense, which was suggestive of vasogenic edema instead of cytotoxic edema in PRES (Fig. 1). There was mild contrast enhancement (data not shown). Electroencephalography showed diffuse slow waves without epileptiform discharges. The results of the cerebrospinal fluid analysis were normal. His severe recurrent abdominal pain without clear etiology was suspect of AIP. The diagnosis of AIP was made after 3 repeated positive urine Watson–Schwartz tests for porphobilinogen (PBG). His urine turned dark and red upon exposure to light (Fig. 2).
Figure 1.

Magnetic resonance imaging of case 1 shows hyperintense gyriform lesions on T2-weighted (A, E) and fluid-attenuated inversion recovery images (B, F) of bilateral occipitoparietal and frontal lobes. Hypointense and patchy hyperintense lesions were found on diffusion-weighted images (C, G), apparent diffusion coefficient maps (D, H) showed hyperintensity of these lesions.
Figure 2.

Urine samples of normal control (A) and case 1 after exposure to sunlight for a short (B) and long (C) time.
Glucose infusion and a high carbohydrate diet were given to the patient, as hematin was not available in our hospital. The patient responded well and recovered gradually. He was discharged with no seizure or abdominal pain. Upon review, he remained asymptomatic.
2.2. Case 2
An 18-year-old girl was admitted to our hospital following development of generalized tonic-clonic seizure in November 2016. Ten days ago, she was diagnosed with ileus because of abdominal pain as well as constipation in her local hospital. However, the symptoms were not relieved and a series of tests including abdominal X-ray, CT scan, and ultrasound were normal. Her brain MRI was performed in her local hospital indicating PRES (Fig. 3). MR angiography showed no abnormality (data not shown). However, the diagnosis was vague and the patient was transferred to the emergency department of our hospital due to seizures. Anticonvulsants including benzodiazepines and oxcarbazepine were used to control the seizures. The recurrent abdominal pain and seizures gave us reason to suspect of AIP, given the prognosis from case 1. She had no family history of similar symptomatology or other particularly notable medical history.
Figure 3.

Gyriform lesions on both the cortical and subcortical bilateral occipitoparietal and frontal lobes in case 2. These lesions were hyperintense on T2-weighted (A, E) and T2-fluid-attenuated inversion recovery (B, F) images. They were hypointense on diffusion-weighted images (C, G) images and were hyperintense on apparent diffusion coefficient maps (D, H).
At the time of admission, her blood pressure was 136/101 mm Hg, and her heart rate was 109/min. The general physical examination was normal and there was no focal neurological deficit. On the night of admission, the patient had hallucination. Except for remarkable hyponatremia of 104 mmol/L, her serum potassium was 3.0 mmol/L, aspartate transaminase 243 U/L, alanine transaminase 142 U/L, and total bilirubin 37 μmol/L. Tests for antinuclear and anticardiolipin antibodies were normal. The patient was sero-negative for human immunodeficiency virus, hepatitis B virus, and hepatitis C virus. The cerebral spinal fluid (CSF) analysis was normal. Tests for heavy metal toxins including lead, mercury, and arsenic were also negative. Brain MRI performed in our hospital 6 days later revealed partial resolution (Fig. 4). AIP was confirmed by 3 repeated urine Watson–Schwartz tests. The patient was given a high carbohydrate diet and intravenous dextrose. No hematin was available for her, but her status improved gradually when she was discharged. No seizures occurred, and her abdominal pain regressed. Two weeks later, a repeat brain MRI performed in her local hospital confirmed the total disappearance of the lesions (Fig. 5). One month later, she recovered completely and went back to work. No acute attacks have occurred to date.
Figure 4.
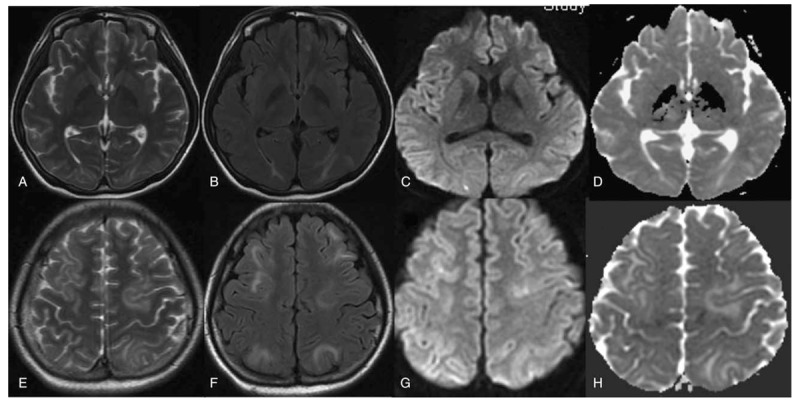
Six days later, gyriform lesions of case 2 resolve incompletely on T2 (A, E), T2-fluid-attenuated inversion recovery images (B, F), diffusion-weighted images (C, G), and apparent diffusion coefficient maps (D, H).
Figure 5.

Follow-up brain magnetic resonance images of case 2 show complete resolution on T2 (A, E), T2-fluid-attenuated inversion recovery images (B, F), diffusion-weighted images (C, G), and apparent diffusion coefficient maps (D, H).
Because this was cases report without any research involving human beings or experimental subjects, the ethical approval was not required in our institute. But patients have provided informed consent for publication of the case.
3. Discussion
We performed a systemic review of the existing literatures and published reports to compile a comprehensive review of AIP patients presenting with PRES. Case reports lacking MRI neuroimaging were excluded. Search keywords included posterior leukoencephalopathy syndrome, PRES, reversible posterior leukoencephalopathy syndrome, RPLS, posterior reversible leukoencephalopathy, reversible posterior leukoencephalopathy, PRES, reversible, neuroimaging, MRI, DWI, imaging, and AIP. Additional case reports were selected from article references.
A total of 22 cases (including our 2 cases) were included in the literature review (Table 1).[5–21] Of those, 20 (91%) patients were female, while only 2 patients (9%) were male. This result strongly indicates a female predominance of the disease in AIP patients with PRES. The average age at onset was 23 years old. Usually, attacks of AIP occurred in the third and fourth decades of life, and the mean age of PRES at presentation was 44 years. However, attacks in AIP patients presented with PRES in our case studies and in the review of the literatures suggested the disease most commonly occurred in the 20th decade, with earlier onset when these 2 conditions occurred simultaneously.
Table 1.
.

PRES-related clinical presentations include encephalopathy, seizure and status epilepticus, headache, visual disturbances, focal neurological deficits, nausea, and vomiting.[22] Both of our patients had seizures during the initial neurological presentation. The literature review showed that all of the AIP-associated PRES patients except for one presented with seizures. A total of 95% (21/22) of those patients presented with seizures during their acute attacks. This result clearly indicated a higher incidence of seizures in AIP patients with PRES compared with those AIP patients without PRES or other PRES conditions. The percentage of patients presenting with visual dysfunction and neuropsychiatric symptoms was 41% (9/22) and 55% (12/22), respectively. Patients presenting with neuropsychiatric symptoms comprised of confusion, lethargy, drowsy, somnolent, stuporous, agitation, hallucination, and even coma. Headache was not prominent in AIP patients with PRES. Among 22 patients, only 2 patients (9%) presented with headache. According to a review from Granata et al,[2] the most common symptoms of PRES with any comorbid conditions include seizure, headache, and visual dysfunction. However, headache is not a common symptom in patients with AIP according to our literature review. Seizures are still the most common symptom in AIP patients with PRES. Consciousness changes range from drowsiness to coma, which is similar to reports of other PRES case studies. The frequency of visual dysfunction in AIP patients with PRES is not different from other PRES patients. This result may indicate that an acute attack of AIP may increase the occurrence of seizure but not visual dysfunction or headache. In addition, headache becomes less frequent when AIP patients present with PRES. Severe recurrent abdominal pain may surpass the headache pain in these patients.
The most common presenting finding among patients with AIP is ambiguous abdominal pain. Among those reviewed patients, 86% (19/22) suffered from abdominal pain, sometimes accompanied by lumbar or leg pain. Constipation is also not uncommon (9/22). However, patients with abdominal pain are usually misdiagnosed or delayed because of variable and nonspecific symptoms. Both of our patients had suffered from recurrent abdominal pain without a definite diagnosis. There are still 3 patients without complaint of abdominal pain, which makes the diagnosis more challenging. However, neurological symptoms combined with abdominal pain may be helpful for early diagnosis, including peripheral flaccid paralysis, seizures, and neuropsychiatric symptoms. The most common neurologic feature of AIP is peripheral neuropathy presenting as generalized paresthesia and muscle weakness.[23] Seizures could be the presentation of both AIP and PRES. Both our patients presented with seizures as the prominent initial neurological manifestation. Compared with 9% of seizures in AIP without PRES,[24] most of the reviewed cases (95%) presented with seizures, which are the most common neurological manifestation of AIP patients with PRES. This result contradicted previous assessments of the less common prevalence of epileptic seizures in patients with AIP.[25] From this review, we know that seizures are not uncommon in AIP patients with PRES. In contrast, seizure is the most common presentation in AIP patients with PRES.
The pathogenesis of seizures may be related to metabolic imbalance such as hyponatremia or to the intrinsic epileptogenic role of some porphyrins such as delta-aminolevulinic acid (ALA). ALA has been shown to interact with gamma-aminobutyric acid and glutamate receptors. Further, neural damage may occur following an acute attack, indicating that these lesions can be epileptogenic such as symmetric or asymmetric cortical gyriform lesions shown on MRI. Thus, all of these reviewed cases with PRES except one presented with seizures. Taken together, seizures are likely the most prominent dysfunction of nervous system during acute attack of AIP patients with PRES.
For the most common neurological deficit in AIP, our patients did not present with muscle weakness or peripheral neuropathy. However, as reviewed in the literature, 9 (41%) patients suffered from quadriparesis during the acute attack, suggesting that both the peripheral and central nervous systems could be involved in the same AIP patients. Aside from the peripheral and central nervous systems, the autonomic nervous system could also be involved, including tachycardia, hypertension, tremors, and diaphoresis. Most of the patients had tachycardia (13/22) and hypertension (18/22) during the acute attack, indicating overactivity of the sympathetic nervous system. Some patients showed nonspecific symptoms of mildly increased creatinine and transaminases. Hyponatremia, as seen in our 2 cases, can occur in half of the literature reviewed cases (50%, 11/22), which is more common compared with the 18% reported previously.[26] Inappropriate secretion of antidiuretic hormone (SIADH) from the hypothalamus, or increased gastrointestinal and renal sodium losses may be the causes of hyponatremia. A case report with histopathology from Suarez et al[26] showed a significant decrease in the number of hypothalamic cells. These results indicate abnormal electrolyte concentration during AIP exacerbations as a result from the damage of hypothalamic hypophyseal tracts, leading to an increase in circulating antidiuretic hormones. Therefore, hypothalamic damage resulting in SIADH may be the most likely explanation for hyponatremia. After review of the morphologic studies of 35 patients, Suarez[26] found that both the central and peripheral nervous systems, including muscular system and autonomic nervous system, were involved. This outcome may explain the heterogeneous and diverse clinical characteristics of AIP. Although most of the patients recovered after acute attacks, 1 patient died and 6 patients needed mechanical ventilation. This result means that irreversible or even fatal conditions may occur when the reversible PRES occurs with AIP. Most of the ventilation was due to respiratory failure of severe motor polyneuropathy presenting with quadriparesis. Bulbar palsy and respiratory muscle paralysis are the most dreaded complications that make AIP become fatal. Clinicians should be aware of this rare condition to avoid debilitating and potentially life-threatening outcomes.
In all 22 reviewed cases with MRI, bilateral parietal lobes (91%, 20/22) were the most common regions involved. The occipital (18/22) and frontal (17/22) lobes were the other common regions involved either bilaterally or unilaterally. Other sites including the cerebellum, splenium of the corpus callosum, pons, midbrain, thalamus, basal ganglia, and corona radiate were detected in some cases. Most of those lesions were not enhanced or were mildly enhanced. However, contrast-enhanced MRI was not performed in most of the cases. More recent advances in MR technology allow the distinction between cytotoxic edema and vasogenic edema. A bright signal on DWI can reflect either diffusion weighting caused by ischemic injury or some underlying T2 effects caused by T2 shine-through from vasogenic edema. However, ADC maps serve to highlight free unrestricted water by removing the underlying T2 signal contribution. ADC maps show a bright signal in vasogenic edema but a dark signal in cytotoxic edema.
The pathophysiology of PRES is still unclear and controversial. Several theories have been postulated, including disruption of the blood–brain barrier, endothelial dysfunction due to circulating toxins, focal vasospasm, and less sympathetic innervation of blood vessels in posterior regions.[27] The most favored hypothesis holds that PRES manifests as vasogenic rather than cytotoxic edema. Among patients (5/22) with DWI hyperintensity, only 1 patient showed decreased signal on ADC maps, which means cytotoxic edema was present. However, a follow-up MRI of this patient showed high signal on DWI and ADC maps, which suggested predominant vasogenic edema.[9]
One report showed that if the precipitating factors were not removed promptly, lesions of vasogenic edema in PRES would switch to cytotoxic edema.[5] The presence of increased signal intensity on DWI can also stem from T2 effects (also known as T2 shine through), which can be eliminated by additional postprocessing to form ADC maps. Our case 1 patient showed such a phenomenon, which also indicated vasogenic edema. Other patients with high signals on both DWI and ADC maps also represented T2 shine-through rather than cerebral infarction. The combination of DWI and ADC maps can confirm the vasogenic pattern of edema as shown in this case review. Unfortunately, only few cases provided both DWI and ADC maps. However, most of the authors indicated vasogenic edema as a possible mechanism in their cases. CSF was usually normal in our cases and in the cases presented in the literature review. However, CSF could help us to exclude many differential diseases such as encephalitis, meningitis, multiple sclerosis, and acute disseminated demyelinating disease.
Although AIP is an autosomal dominant disease, approximately 80% to 90% of carriers of the mutated gene are asymptomatic. Our 2 patients had no family history of AIP, and only 3 patients (14%) had definite or probable family history in these reviewed patients. Most of these patients did not have extensive family history. Unfortunately, our 2 patients and their families were reluctant to have the gene tests. In fact, the 3 common types of porphyria including AIP, hereditary coproporphyria, and variegate porphyria, can characteristically cause neurologic diseases. All of these porphyrias are under the classification of acute porphyrias. Hereditary coproporphyria, which is less common than AIP, can produce attacks of gastrointestinal and neuropsychiatric symptoms (similar to AIP but typically milder) as well as skin changes. Variegate porphyria always has skin manifestations such as photosensitivity, and one-third of the cases of hereditary coproporphyria also had cutaneous findings.[27] Our 2 patients had no skin manifestations. Although the attacks are very similar in these 3 types of porphyria, such characteristic presentations and genetic tests would be helpful for differentiation. Quantitative measurements of PBG and ALA in urine are more reliable but are not available in our hospital. More than 3 repeated Watson–Schwartz tests using Ehrlich's aldehyde reagent were positive in our patients, further confirming the diagnosis. Although the test may occasionally be associated with false-positive findings, it is quick and quite sufficient for the rapid diagnosis of an acute attack of AIP.[28] The Watson–Schwartz test had been widely used as a screening test for urinary PBG before more specific and quantitative methods were introduced.[29] Burgundy red discoloration of long stored urine is also useful to help make the diagnosis. Except for the most common type of AIP, the other 2 types of acute porphyria could still not be definitely excluded in our 2 patients due to a lack of genetic tests.
Management of patients with AIP includes acute attack and prophylactic therapy. Intravenous hematin is the most effective therapy during attack. The placebo-controlled trial of human hemin did not show a statistically significant effect, but patients with hemin treatment have a faster resolution of symptoms.[30] However, heme preparations are not available in most conditions. Therefore, supportive treatment including adequate nutrition and fluid therapy becomes very important during the acute attack. Our patients and some reviewed patients gradually recovered with the supportive therapy, without available hemin. Some antiepileptic drugs that may exacerbate acute attacks including barbiturates, diazepam, phenytoin, and carbamazepine should be avoided in AIP patients with PRES because seizures are the most common and initial symptoms.
It is imperative to be aware of this seemingly rare condition. Early recognition, supportive and intravenous hematin therapy, and withdrawal of the precipitating factors are the key steps in the management of acute attacks of AIP. This “little imitator” is often missed or wrongly diagnosed because of its heterogeneous symptoms. AIP should always be suspected in the setting of MRI presentation of PRES in patients with unexplained abdominal pain and seizures.
Author contributions
Conceptualization: Xinbao Yin.
Data curation: Xuejun Liu, Lintao Qu.
Formal analysis: Yan Wang, Lintao Qu.
Funding acquisition: Xueping Zheng, Xinbao Yin.
Investigation: Haitao Pei, Yong Zhang, Liou Tang.
Methodology: Haitao Pei, Xiaojun Ji.
Project administration: Renliang Zhao, Liou Tang.
Resources: Yuqiang Song.
Software: Yong Zhang.
Supervision: Yan Wang, Miao Tuo, Xiaojun Ji.
Validation: Renliang Zhao, Yuqiang Song.
Visualization: Miao Tuo, Hongyun Li.
Writing – original draft: Xueping Zheng.
Writing – review & editing: Xuejun Liu, Hongyun Li.
Footnotes
Abbreviations: ADC = apparent diffusion coefficient, AIP = acute intermittent porphyria, ALA = aminolevulinic acid, CSF = cerebral spinal fluid, CT = computed tomography, DWI = diffusion-weighted images, MRI = magnetic resonance imaging, PBG = porphobilinogen, PRES = posterior reversible encephalopathy syndrome, SIADH = inappropriate secretion of antidiuretic hormone.
Xueping Zheng and Xuejun Liu have contributed equally to this article.
This work was supported by grants from Natural Science Foundation of China (30901324, 81202025).
The authors have no conflicts of interest to disclose.
References
- [1].Hinchey J, Chaves C, Appignani B, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med 1996;334:494–500. [DOI] [PubMed] [Google Scholar]
- [2].Granata G, Greco A, Iannella G, et al. Posterior reversible encephalopathy syndrome—insight into pathogenesis, clinical variants and treatment approaches. Autoimmun Rev 2015;14:830–6. [DOI] [PubMed] [Google Scholar]
- [3].Solis C, Martinez-Bermejo A, Naidich TP, et al. Acute intermittent porphyria: studies of the severe homozygous dominant disease provides insights into the neurologic attacks in acute porphyrias. Arch Neurol 2004;61:1764–70. [DOI] [PubMed] [Google Scholar]
- [4].King PH, Bragdon AC. MRI reveals multiple reversible cerebral lesions in an attack of acute intermittent porphyria. Neurology 1991;41:1300–2. [DOI] [PubMed] [Google Scholar]
- [5].Kupferschmidt H, Bont A, Schnorf H, et al. Transient cortical blindness and bioccipital brain lesions in two patients with acute intermittent porphyria. Ann Intern Med 1995;123:598–600. [DOI] [PubMed] [Google Scholar]
- [6].Utz N, Kinkel B, Hedde JP, et al. MR imaging of acute intermittent porphyria mimicking posterior leukoencephalopathy syndrome. Neuroradiology 2001;43:1059–62. [DOI] [PubMed] [Google Scholar]
- [7].Yen PS, Chen CJ, Lui CC, et al. Diffusion-weighted magnetic resonance imaging of porphyric encephalopathy: a case report. Eur Neurol 2002;48:119–21. [DOI] [PubMed] [Google Scholar]
- [8].Celik M, Forta H, Dalkilic T, et al. MRI reveals reversible lesions resembling posterior reversible encephalopathy in porphyria. Neuroradiology 2002;44:839–41. [DOI] [PubMed] [Google Scholar]
- [9].Gürses C, Durukan A, Sencer S, et al. A severe neurological sequela in acute intermittent porphyria: presentation of a case from encephalopathy to quadriparesis. Br J Radiol 2008;81:e135–40. [DOI] [PubMed] [Google Scholar]
- [10].Soysal A, Dogan P, Dayan C, et al. Reversible MRI findings of porphyric encephalopathy. A report of two cases. Neuroradiol J 2008;21:655–9. [DOI] [PubMed] [Google Scholar]
- [11].Kang SY, Kang JH, Choi JC, et al. Posterior reversible encephalopathy syndrome in a patient with acute intermittent porphyria. J Neurol 2010;257:663–4. [DOI] [PubMed] [Google Scholar]
- [12].Bicknell SG, Stewart JD. Neuroimaging findings in acute intermittent porphyria. Can J Neurol Sci 2011;38:656–8. [DOI] [PubMed] [Google Scholar]
- [13].Zhao B, Wei Q, Wang Y, et al. Posterior reversible encephalopathy syndrome in acute intermittent porphyria. Pediatr Neurol 2014;51:457–60. [DOI] [PubMed] [Google Scholar]
- [14].Bhuyan S, Sharma AK, Sureka RK, et al. Sudden bilateral reversible vision loss: a rare presentation of acute intermittent porphyria. J Assoc Physicians India 2014;62:432–4. [PubMed] [Google Scholar]
- [15].Lakhotia M, Pahadiya HR, Singh J, et al. Posterior reversible encephalopathy syndrome as a rare presenting feature of acute intermittent porphyria. Neurol India 2015;63:607–9. [DOI] [PubMed] [Google Scholar]
- [16].Guéniat J, Delpont B, Garnier L, et al. Posterior reversible encephalopathy syndrome revealing acute intermittent porphyria. Rev Neurol (Paris) 2016;172:402–3. [DOI] [PubMed] [Google Scholar]
- [17].Dagens A, Gilhooley MJ. Acute intermittent porphyria leading to posterior reversible encephalopathy syndrome (PRES): a rare cause of abdominal pain and seizures. BMJ Case Rep 2016;2016: [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Silveira DC, Bashir M, Daniel J, et al. Acute intermittent porphyria presenting with posterior reversible encephalopathy syndrome and lateralized periodic discharges plus fast activity on EEG. Epilepsy Behav Case Rep 2016;6:58–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mohanlal S, Ghildiyal RG, Kondekar A, et al. A commonly missed well known entity-acute intermittent porphyria: a case report. J Clin Diagn Res 2016;10:SD01–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yang J, Yang H, Chen Q, et al. Reversible MRI findings in a case of acute intermittent porphyria with a novel mutation in the porphobilinogen deaminase gene. Blood Cells Mol Dis 2017;63:21–4. [DOI] [PubMed] [Google Scholar]
- [21].Takata T, Kume K, Kokudo Y, et al. Acute intermittent porphyria presenting with posterior reversible encephalopathy syndrome, accompanied by prolonged vasoconstriction. Intern Med 2017;56:713–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fugate JE, Rabinstein AA. Posterior reversible encephalopathy syndrome: clinical and radiological manifestations, pathophysiology, and outstanding questions. Lancet Neurol 2015;14:914–25. [DOI] [PubMed] [Google Scholar]
- [23].Stein JA, Tschudy DP. Acute intermittent porphyria: a clinical and biochemical study of 46 patients. Medicine 1970;49:1–6. [PubMed] [Google Scholar]
- [24].Bonkovsky HL, Maddukuri VC, Yazici C, et al. Acute porphyrias in the USA: features of 108 subjects from porphyrias consortium. Am J Med 2014;127:1233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bylesjö I, Forsgren L, Lithner F, et al. Epidemiology and clinical characteristics of seizures in patients with acute intermittent porphyria. Epilepsia 1996;37:230–5. [DOI] [PubMed] [Google Scholar]
- [26].Suarez JI, Cohen ML, Larkin J, et al. Acute intermittent porphyria: clinicopathologic correlation. Report of a case and review of the literature. Neurology 1997;48:1678–83. [DOI] [PubMed] [Google Scholar]
- [27].Roth C, Ferbert A. The posterior reversible encephalopathy syndrome: what's certain, what's new? Pract Neurol 2011;11:136–44. [DOI] [PubMed] [Google Scholar]
- [28].Sassa S. Modern diagnosis and management of the porphyrias. Br J Haematol 2006;135:281–92. [DOI] [PubMed] [Google Scholar]
- [29].Watson CJ, Schwartz S. A simple test for urinary porphobilinogen. Proc Soc Exp Biol Med 1941;47:393–4. [Google Scholar]
- [30].Herrick AL, McColl KE, Moore MR, et al. Controlled trial of haem arginate in acute hepatic porphyria. Lancet 1989;1:1295–7. [DOI] [PubMed] [Google Scholar]