

Abstract
Neuropathic pain genesis is related to gene alterations in the dorsal root ganglion (DRG) following peripheral nerve injury. Transcription factors control gene expression. In this study, we investigated whether octamer transcription factor 1 (OCT1), a transcription factor, contributed to neuropathic pain caused by chronic constriction injury (CCI) of the sciatic nerve. CCI produced a time-dependent increase in the level of OCT1 protein in the ipsilateral L4/5 DRG, but not in the spinal cord. Blocking this increase through microinjection of OCT1 siRNA into the ipsilateral L4/5 DRG attenuated the initiation and maintenance of CCI-induced mechanical allodynia, heat hyperalgesia, and cold allodynia and improved morphine analgesia after CCI, without affecting basal responses to acute mechanical, heat, and cold stimuli as well as locomotor functions. Mimicking this increase through microinjection of recombinant adeno-associated virus 5 harboring full-length OCT1 into the unilateral L4/5 DRG led to marked mechanical allodynia, heat hyperalgesia and cold allodynia in naive rats. Mechanistically, OCT1 participated in CCI-induced increases in Dnmt3a mRNA and its protein and DNMT3a-mediated decreases in Oprm1 and Kcna2 mRNAs and their proteins in the injured DRG. These findings indicate that OCT1 may participate in neuropathic pain at least in part by transcriptionally activating Dnmt3a and subsequently epigenetic silencing of Oprm1 and Kcan2 in the DRG. OCT1 may serve as a potential target for therapeutic treatments against neuropathic pain.
Keywords: octamer transcription factor 1, DNMT3a, Kv1.2, μ opioid receptor, dorsal root ganglion, neuropathic pain
Introduction
Peripheral nerve injury-induced neuropathic pain often occurs after surgery14. Despite concerted efforts to study, treat, and prevent neuropathic pain, effective treatment of this disorder remains relatively unsuccessful5,14. Treating neuropathic pain often involves opioids, the most potent class of drugs used for relieving surgical pain21. Nearly 45% of patients prescribed opioids report inadequate pain relief, leading to the enhanced concerns in both patients and physicians for opioid addition15. In the United States, increases in opioid prescriptions over past decades have been accompanied by a huge increase in the incidence of addition and opioid-related mortality that constitutes the current “opioid crisis”15. Therefore, further understanding the mechanisms of how peripheral nerve injury produces the development and maintenance of neuropathic pain may open a new avenue in the management of this disorder.
Neuropahtic pain is characterized with pain in response to innocuous stimuli (allodynia), an enhanced rsposne to noxiou stimuli (hyperalgesia) and spontenaous pain. Abnormal hyperexcitability and ectopic discharges in neuronal cell bodies of the dorsal root ganglion (DRG) as well as neuroma at the sites of peripheral nerve injury are considered to be one of the primary casues of these pain hypersesntivitities under neuropahtic pain condiitons1,2. Abnormal hyperexcitablity is likely attributed to pheripheral nerve injury-caused alterations in gene transcription and translation in the DRG2,9,13. DNA methylation controls gene expression. Recently, accumulating evidence reveals that DNA methylation in the DRG participates in the mechanisms that underlie neuropathic pain genesis. An increase in the level of DNA methyltransferases 3a (DNMT3a) following unilateral fifth lumbar (L5) spinal nerve ligation (SNL) contributes to DNA hypermethylation at some CpG sites within the promoter regions of Oprm1 and Kcna2 genes, resulting in their expressional silencing in the DRG20,26,28. Blocking this increase through pharmacological inhibition or genetic knockdown of DRG DNMT3a attenuates nerve injury-induced pain hypersensitivities via rescuing the downregulation of DRG Oprm1 and Kcna2 genes20,26,28. DNMT3a in the DRG is likely an important player in the genesis of neuropathic pain.
A transcription factor octamer transcription factor 1 (OCT1) can bind to the “ATTTGCAT” sequence16. The promoter region of the Dnmt3a gene contains this consensus binding motif26. OCT1 increases the activity of this promoter in HEK-293 cells26. SNL increases the expression of OCT1 and the binding activity of OCT1 to the promoter region of the Dnmt3a gene in the ipsilateral L5 DRG26. OCT1 overexpression leads to an increase in the expression of DNMT3a in DRG neurons26. Given that DNMT3a is an endogenous trigger of neuropathic pain26, DRG OCT1 may contribute to neuropathic pain.
In this study, we first examined whether the level of OCT1 expression increased in the ipsilateral DRG after chronic constriction injury (CCI) of unilateral sciatic nerve. We then characterized cellular expression and distribution patterns of OCT1 in the DRG of normal and CCI rats. We also examined whether blocking the increased DRG OCT1 alleviated CCI-caused pain hypersensitivities and whether mimicking CCI-caused increase in DRG OCT1 led to neuropathic pain symptoms. Finally, we observed whether these effects were mediated through OCT1 regulation of DNMT3a-triggered Oprm1 and Kcna2 gene expression in the DRG.
Materials and Methods
Animal preparation
Adult male Sprague-Dawley rats (220 to 250 g; Charles River Lab., Wilington, MA) were prepared as described25,26. All animal procedures were approved by the Animal Care and Use Committee at the Zhengzhou University and Rutgers New Jersey Medical School and ethical guidelines of the International Association for the Study of Pain and the US National Institutes of Health. Efforts were made to ensure minimal animal use and discomfort. All experiments were performed by the investigators blinded to group assignments and treatment conditions.
Neuropathic pain model
Chronic constriction injury (CCI) model was established as described previously6,7. Briefly, 4–0 chromic gut thread was used to loosely ligate the left sciatic nerve at 4 sites with an interval of 1 mm around the nerve proximal to its trifurcation. Sham surgery was carried out by exposing the sciatic nerve without ligation. Only rats without any abnormality were included in this study.
Morphine-induced analgesia
Morphine analgesia was established in rats as described10,11. In brief, the morphine (1.5 mg/kg; WEST-WARD, Eatotown, NJ) was injected subcutaneously (s.c.) 3 days after CCI or sham surgery. Heat test as described below were carried out before surgery (baseline latency) and 30 min after morphine injection (response latency). The cut-off time was 20 s to avoid tissue damage. The percentage of maximal possible analgesic effect (% MPAE) was calculated as follow: % MPAE = [(response latency-baseline latency)/(cut-off time-baseline latency)] × 100%. Additionally, a peripheral MOR antagonist methylnaltrexone bromide (5 mg/kg, dissolved in saline; Medchemexpress USA, Monmouth Junction, NJ) was administered intraperitoneally (i.p.) 4 days after CCI or sham surgery. Heat test were carried out 1 day before CCI or sham surgery, 3 days post-CCI or sham surgery, and prior to and 30 min after drug or saline administration on day 4 after CCI or sham surgery.
DRG microinjection
DRG microinjection was performed according to our previous studies4,6,7,27. In brief, after the incision of lower lumbar back in the midline region was made, unilateral L4/5 DRG was exposed and received the injection of the virus (1–1.5 μl, 7.3× 1012), PBS (1–1.5 μl), OCT1 siRNA (Santa Cruz Biotechnology, Inc, Dallas, TX; 1–1.5 μl, 20 μM), or negative control siRNA (Santa Cruz Biotechnology, Inc; 1–1.5 μl, 20 μM) with a glass micropipette (tip diameter 20 – 40μm) connected to a Hamilton syringe. In order to avoid degradation and improve cell membrane penetration of siRNA, the delivery vehicle TurboFect in vivo transfection reagent (Thermo Scientific Inc, Pittsburgh, PA) was used as described4,6,7,27. Following injection, the glass micropipette was remained at least for 10 min. Finally, the wound clips were used to close the skin incision. The injected rats showing the signs of paresis or other abnormalities were excluded in the study.
Behaviors tests
Mechanical test was carried out as reported previously4,24,26,27. The up–down testing paradigm was used to measure paw withdrawal thresholds (PWTS) in response to mechanical stimuli. Based on a formula provided by Dixon3, paw withdrawal threshold was computed through conversion of the pattern of positive and negative responses into a 50% threshold value. Heat test was performed as described4,24,26,27. The Model 336 Analgesia Meter (IITC Inc. Life Science Instruments, Woodland Hills, CA, USA) was used to measure paw withdrawal latencies (PWLs) to noxious heat. To avoid a burn injury, 20 s was used as a cutoff time. Each animal had five trials with a 5-min interval per side. The mean value was calculated for the analysis. Cold test was performed as reported previously4,24,26,27. A thermometer-monitored cold aluminum plate was used to measure paw withdrawal latencies (PWLs) to noxious cold stimulation. Each animal had three trials with a 10-min interval on the ipsilateral side. To avoid tissue damage, 60 s was used as a cut-off time. The locomotor function test was performed based on the protocols reported previously4,24,26,27. The placing, righting, and grasping reflexes were carried out before tissue collection or rat euthanization. The counts of each reflex displayed in five trials were scored.
Western blot analysis
Protein extraction and Western blot analysis were performed as reported previously4,24,26,27. Briefly, the tissue was homogenized on the ice with the lysis buffer. The pellet (nuclear fraction) and the supernatant (membrane/cytosolic fractions) were collected after centrifugation at 4°C for 15 min at 1,000g. The lysis buffer containing 2% sodium dodecyl sulfate (SDS) and 0.1% Triton X-100 was used to dissolve the pellet. After being heated at 99 °C for 5 min, the samples (20 μg/sample) were loaded onto a 4% stacking/7.5% separating SDS-polyacrylamide gel (Bio-Rad Laboratories) and then electrophoretically transferred onto a polyvinylidene difluoride membrane (Bio-Rad Laboratories). The membranes were incubated first in the blocking buffer (3% nonfat milk plus 0.1% Tween-20 in the Tris-buffered saline) for 2 h at room temperature and then in the primary antibodies at 4oC overnight. The primary antibodies include: monoclonal rabbit anti-OCT1 (1:1,200, Abcam) mouse anti-Kv1.2 (1:1,000, NeuroMab), rabbit anti-DNMT3A (1:1,000, Cell Signaling), rabbit anti-MOR (1:500, Neuromics, Edina, MN), rabbit anti-phosphorylated extracellular signal-regulated kinase (p-ERK1/2, 1:1,000, Cell Signaling), rabbit anti- extracellular signal-regulated kinase (ERK1/2, 1:1,000, Cell Signaling), mouse anti-glial fibrillary acidic protein (GFAP, 1:1,000, Cell Signaling), rabbit anti–histone H3 (1:1,000, Cell Signaling), and rabbit anti-GAPDH (1:1,000, Santa Cruz, Dallas, Texas). After the membranes were incubated with the horseradish peroxidase–conjugated anti-rabbit or anti-mouse secondary antibody (1:3,000, Bio-Rad Laboratories) at room temperature for 1 h, the enhanced chemiluminescence reagent (Bio-Rad Laboratories) were used to visualize the proteins. ChemiDoc XRS and System with Image Lab software (Bio-Rad Laboratories) were performed to generate the images. System with Image Lab software (Bio-Rad Laboratories) was carried out to quantify to image intensities. The band intensities for cytosol proteins were normalized to GAPDH and those for nucleus proteins were normalized to H3.
Immunofluorescence histochemistry
The single- or double-label immunofluorescence histochemistry was carried out as described previously4,24,26,27. Briefly, after the rats were transcardially perfused, L4/5 DRG was collected. The DRGs were sectioned into 20 μm at the thickness. The sections were incubated in the blocking buffer (0.01M PBS containing 0.3% TritonX-100 and 3% goat serum) for 1 h at room temperature. For single-label immunofluorescence histochemistry, the sections were incubated alone with monoclonal rabbit anti-OCT1 (1:1,200, Abcam, Cambridge, MA) at 4°C overnight and then with goat anti-rabbit IgG conjugated to Cy3 (1: 200, Jackson ImmunoResearch, West Grove, PA) at room temperature for 1 h. For double-label immunofluorescence histochemistry, the sections were incubated with primary monoclonal rabbit anti-OCT1 and each of the following antibodies/regents: mouse anti-neurofilament 200 (NF200; 1:500, Sigma-Aldrich, St. Louis, MO), biotinylated isolectin B4 (IB4, 1:100, Sigma), mouse anti- calcitonin gene-related peptide (CGRP, 1:50, Abcam), mouse anti-NeuN (1:50, Gene Tex, USA), mouse anti-Glutamine synthetase (GS, 1:500, EMD Millipore), mouse anti-Kv1.2 (1:200, NeuroMab, Davis, CA), and guinea pig anti-MOR (1:1,000, EMD Millipore) overnight at 4 °C. The sections were then incubated at room temperature for 1 h with a mixture of goat anti-rabbit IgG conjugated with Cy3 (1:200, Jackson ImmunoResearch) plus donkey anti-guinea pig IgG or donkey anti-mouse IgG conjugated with Cy2 (1:200, Jackson ImmunoResearch). Control experiments were carried out in parallel as reported before4,24,26,27.
Quantitative real-time reverse transcription polymerase chain reaction (RT-PCR)
Quantitative real-time RT-PCR were carried out as described4,24,26,27. Briefly, the extracted RNA was reverse-transcribed with either specific reverse primers or the oligo (dT) primers (Supplemental Table 1). The cDNA (1 μl) was amplified by real-time PCR with the primers indicated in the Supplemental Table 1. GAPDH as an internal control was used. A 20 μl reaction containing 20 ng of cDNA, 10 μl of SsoAdvanced Universal SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA) and 200 nM forward and reverse primers was prepared. The triplicate reactions from each sample was carried out in a BIO-RAD CFX96 real-time PCR system. The PCR cycle parameters consist of an initial 3-min incubation at 95˚ C, followed by 35 cycles of 95˚ C for 10 s, 58˚C for 30 s, and 72˚ C for 30 s. All results were normalized to GAPDH as peripheral nerve injury does not alter its expression4,24,26,27. The △Ct method (2-△△Ct) was used to calculate the ratios of mRNA levels on the ipsilateral side to ones on the contralateral side.
Plasmid constructs and virus preparation
The SuperScript III One-Step RT-PCR System with the Platinum Taq High Fidelity Kit (Invitrogen) was used to reversely transcribe rat Oct1 full-length mRNA extracted from rat DRG using the primers listed in the Supplemental Table 1(Integrated DNA Technologies). After nested PCR using the primers listed in the Supplemental Table 1 and Platinum Pfx DNA Polymerase (Invitrogen), the OCT1 coding region was amplified and insert to the pENTR/D-TOPO vector (Invitrogen) and validated by sequencing. After digestion with BspEI and NotI (New England Biolabs, Beverly, Massachusetts, USA), the OCT1 cDNA was gel-purified and then was ligated into proviral plasmids. The control plasmid harboring enhanced green fluorescent protein (EGFP) was prepared in parallel. Recombinant adeno-associated virus 5 (AAV5) particles were packaged at the UNC Vector Core.
Statistical analysis
All data were analyzed by SigmaPlot 12.5 (USA). One-way or two-way analysis of variance (ANOVA) with the post hoc Tukey testing and paired or unpaired Student’s t-test were applied for normally distributed data and the Mann-Whitney U-test was used for non-parametric data. The level of significance was set at P < 0.05. Data are present as mean ± SEM.
Results
CCI increased OCT1 expression in the injured DRG
We first observed whether OCT1 expression was changed in dorsal root ganglion and spinal cord dorsal horn, two major pain-related regions, under CCI-induced neuropathic pain conditions. The expression of OCT1 protein was time-dependently increased in the ipsilateral L4/5 DRG following CCI, but not sham surgery (Fig. 1A and1B). As expected, CCI did not produce any alterations in the basal expression of OCT1 protein in the contralateral L4/5 DRGs (data not shown). CCI also did not significantly alter OCT1 protein expression in the ipsilateral L4/5 spinal cord dorsal horn (Fig. 1C).
Fig. 1.
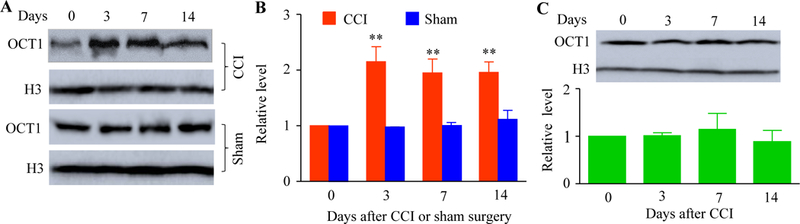
Peripheral nerve injury-caused increase in the OCT1 expression in the injured DRG. (A and B) The amounts of OCT1 protein in the ipsilateral L4/5 DRG at the different days post-CCI or sham surgery. A: Representative Western blots. B: A statistical summry of the densitometric analysis. n = 3 biological replicates (6 rats) per time point. Two-way ANOVA followed by post hoc Tukey test. **P < 0.01 versus the corresponding naïve group (0 day). (C) OCT1 protein expression in the ipsilateral L4/5 spinal cord at the different days after CCI. n = 3 biological replicates (6 rats) per time point. One-way ANOVA followed by post hoc Tukey test.
We further examined the distribution pattern of OCT1 in the DRG. OCT1 colocalized with NeuN (a specific neuronal marker, Fig. 2A) as well as with glutamine synthetase (a marker for satellite glial cells, Fig. 2B). Approximately 94.9% (335/353) OCT1-labeled cells are positive for NeuN. Across-sectional area observation of neuronal somata indicated that about 40.0% of OCT1-labeled neurons were large (> 1, 200 μm2 in area), 44.8% were medium (600 to 1,200 μm2 in area), and 21.2% were small (< 600 μm2 in area; Fig. 2C). Further analysis showed that approximately 43.5% of OCT1-labeled cells were labeled for NF200 (a marker for medium/large neurons and myelinated Aβ-fibers; Fig. 2D), 33.3% were labeled for IB4 (a marker for small nonpeptidergic neurons; Fiog. 2E), and 42.9% were labeled for CGRP (a marker for small DRG peptidergic neurons; Fig. 2F). Consistent with the observations by Western blot analysis above, on day 7 after CCI, the number of OCT1-labeled cells in the ipsilateral L4/5 DRG was 1.81-fold higher than one in the contralateral L3/4 DRG (Fig. 2G and2H).
Fig. 2.

Immunohistochemistry for OCT1 in the DRG. (A abd B) OCT1 is colocalized with NeuN (A) and glutamine synthetase (GS; Arrows; B) in the individual neurons of naïve DRG. Scale bar: 50 μm. (C) Distribution of OCT1-labeled neuronal somata in naïve DRG. Large, 40%; medium: 44.8%; small: 21.2%. (D-F) OCT1 is co-expressed with NF200 (D), IB4 (E), and CGRP (F) in the individual neurons of naïve DRG. Scale bar: 25 μm. (G and H) The cells labeled by OCT1 in the ipsilateral and contralalteral L5 DRG on day 7 post-CCI. G: Representative immunostaining images. H: A statistical summary of the analysi in number of OCT1-labeled cells. n = 3 rats. *P < 0.05 versus the contralateral side by two-tailed unpaired Student’s t-test.
Effect of blocking increased DRG OCT1 on the induction of CCI-caused pain hypersensitivity
Next, we investigated whether blocking the CCI-caused increase in DRG OCT1 through pre-microinjecting OCT1 siRNA into the ipsilateral L4/5 DRG changed the induction of CCI-caused pain hypersensitivities. As expected, the amounts of OCT1 mRNA and protein were markedly increased in the ipsilateral L4/5 DRG on day 5 after CCI in rats pre-microinjected with either vehicle or control negative siRNA (Fig. 3A and3B). These increases were absent in the CCI rats pre-microinjected with OCT1 siRNA (Fig. 2A and2B). No significant decreases in basal amounts of OCT1 mRNA and protein were seen in the ipsilateral L4/5 DRG of the sham rats pre-microinjected with OCT1 siRNA (Fig. 2A and2B). Behaviorally, the CCI rats pre-microinjected with the vehicle displayed the enhanced paw withdrawal responses to mechanical, heat and cold stimuli on the ipsilateral side (Fig. 2C–2E). On days 3 and 5 after CCI, the paw withdrawal thresholds in response to mechanical stimulation (Fig. 3C) and the paw withdrawal latencies in response to heat (Fig. 3D) and cold (Fig. 3E) stimuli, respectively, applied to the ipsilateral hindpaw were markedly reduced as compared to pre-surgical baseline values. In contrast, the CCI rats pre-microinjected with OCT1 siRNA failed to exhibit the augmented paw withdrawal responses to mechanical, heat and cold stimuli on days 3 and 5 after CCI as compared to pre-surgical baseline values (Fig. 3C–3E). As expected, pre-microinjection of OCT1 siRNA did not change basal paw responses to mechanical, heat, or cold stimuli on the ipsilateral side of sham mice (Fig. 3C–3E). Pre-microinjection of control negative siRNA did not alter CCI-caused enhanced paw withdrawal responses to mechanical, heat, and cold stimuli on the ipsilateral side (Fig. 3C–3E). No significant differences in paw withdrawal responses were seen between the vehicle-microinjected and negative control siRNA-microinjected groups (Fig. 3C–3E). Basal paw withdrawal responses to mechanical, heat, and cold stimuli on the contralateral side (Fig. 3F and3G) and locomotor activities (Supplemental Table 2) were not changed by pre-microinjection of either siRNA or vehicle.
Fig. 3.

Effect of DRG pre-microinjection of OCT1 siRNA on CCI-caused induction of pain hypersensitivities and dorsal horn central sensitization. (A and B) Effect of pre-microinjection of OCT1 siRNA (Si), vehicle (Veh), or negative control siRNA (NC) into the ipsilateral L4/5 DRG on the expression of Oct1 mRNA (A) and OCT1 protein (B) in the ipsilateral L4/5 DRG on day 5 after CCI or sham surgery. n = 3 biological replicates (6 rats) per group. One-way ANOVA followed by post hoc Tukey test. *P < 0.05 or **P < 0.01 versus the vehicle (Veh) plus sham group. #P < 0.05 versus the vehicle plus CCI group. (C-G) Effect of pre-micoinjection of OCT1 siRNA (Si), vehicle (Veh), or negative control siRNA (NC) into the ipsilateral L4/5 DRG on paw withdrawal threshold to mechanical stimulation (C and F), paw withdrawal latency to heat stimulation (D and G) and paw withdrawal latency to cold stimulation (E) on the ipsilateral (C-E) and contralateral (F and G) sides at the different days after CCI or sham surgery. n = 5 rats per group. Two-way ANOVA followed by post hoc Tukey test. **P < 0.01 versus the vehicle plus CCI group at the corresponding time point. (H) Effect of pre-microinjection of OCT1 siRNA (Si) or vehicle (Veh) into the ipsilateral L4/5 DRG on CCI-caused increases in the phosphorylation of ERK1/2 and expression of GFAP in the ipsilateral L4/5 dorsal horn on day 5 after CCI or sham surgery. Left: Represetaitive Western blots; Right: A statistical summary of densitometric analysis. n = 3 biological replicates (6 rats) per group. One-way ANOVA followed by post hoc Tukey test. *P < 0.05 or **P < 0.01 versus the corresponding vehicle plus sham group. ##P < 0.01 versus the corresponding vehicle plus CCI group.
We also determined whether pre-microinjection of OCT1 siRNA affected CCI-caused dorsal horn central sensitization, as revealed by the increases in the levels of p-ERK1/2 (a marker for neuronal hyperactivation) and GFAP (a marker for astrocyte hyperactivation) in dorsal horn7,8,25,26. Consistent with previous studies in the spinal nerve ligation-induced neuropathic pain model8,25,26, the levels of p-ERK1/2 (but not total ERK1/2) and GFAP were markedly increased in the ipsilateral L4/5 dorsal horn from the vehicle-microinjected CCI rats compared to those from the vehicle-microinjected sham rats (Fig. 3H). These increases were absent in the OCT1 siRNA-microinjected CCI rats (Fig. 3H).
Effect of blocking increased DRG OCT1 on the maintenance of CCI-caused pain hypersensitivity
We further examine whether DRG OCT1 was required for the maintenance of CCI-caused pain hypersensitivities. Vehicle or siRNA was given 7 days post-CCI, at this time point when CCI-induced pain hypersensitivity was robust and stable. Post-administration of OCT1 siRNA, but not control negative siRNA, attenuated CCI-caused the enhanced paw withdrawal responses to mechanical, heat, and cold stimuli on the ipsilateral side on days 7 till day 14 post-CCI (Fig. 4A–4C). In the vehicle-treated group, CCI led to a robust decrease in rat paw withdrawal threshold to mechanical stimulation and paw withdrawal latency to heat or cold stimulation on the ipsilateral side during the maintenance period (Fig. 4A–4C). OCT1 siRNA markedly reversed these decreases compared to the vehicle or control negative siRNA-treated group on days 12 and 14 post-CCI (Fig. 4A–4C). As expected, post-microinjection of vehicle, OCT1 siRNA, or control negative siRNA did not alter basal paw withdrawal responses on the contralateral side (Fig. 4D and4E).
Fig. 4.
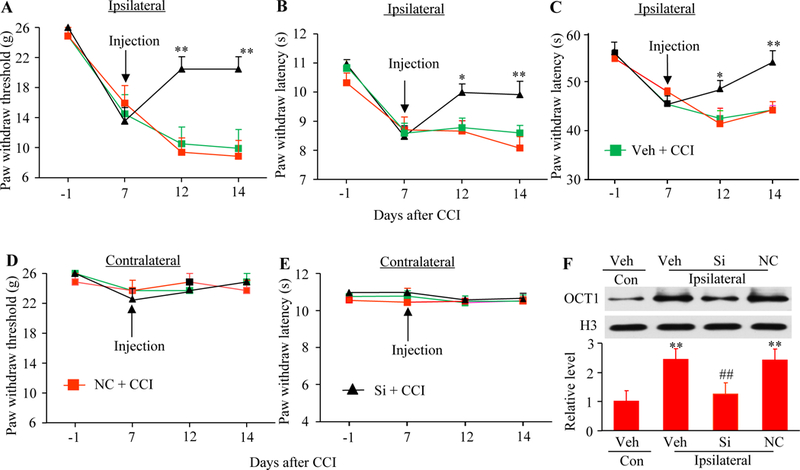
Effect of DRG post-microinjection of OCT1 siRNA on CCI-caused maintenance of pain hypersensitivities. (A-E) Effect of DRG microinjection of OCT1 siRNA (Si), vehicle (Veh) or negative control siRNA (NC) into the ipsilateral L4/5 DRG starting on day 7 after CCI on paw withdrawal threshold to mechanical stimulation (A and D), paw withdrawal latency to heat stimulatuon (B and E), and paw withdrawal latency to cold stimulation (C) on the ipsilateral (A-C) and contralateral (D and E) sides. n = 5 rats per group. Two-way ANOVA followed by post hoc Tukey test. *P < 0.05 or **P < 0.01 versus the vehicle plus CCI group at the corresponding time point. (F) Effect of DRG microinjection of OCT1 siRNA (Si), vehicle (Veh) or negative control siRNA (NC) into the ipsilateral L4/5 DRG starting on day 7 after CCI on the expression of OCT1 protein in the ipsilateral and contralaterl (Con) L4/5 DRG on day 14 after CCI. n = 3 biological replicates (6 rats) per group. One-way ANOVA followed by post hoc Tukey test. **P < 0.01 versus the vehicle (Veh) plus CCI group on the contralateral side. ##P < 0.01 versus the vehicle plus CCI group on the ipsilateral side.
Effect of DRG OCT1 overexpression on nociceptive thresholds in naive rats
We then determined whether mimicking the CCI-caused increase in DRG OCT1 via microinjection of AAV5 that expresses rat Oct1 full-length mRNA (AAV5-OCT1) into unilateral L4/5 DRG changed the nociceptive thresholds in naive rats. As expected, the amounts of OCT1 mRNA and protein in the microinjected DRG from the AAV5-OCT1-injected rats were increased significantly compared to those from control AAV5-EGFP-injected group 8 weeks post-microinjection (Fig. 5A and5B). Microinjection of AAV5-OCT1, but not control AAV5-EGFP, produced the augmented paw withdrawal responses to mechanical, heat, and cold stimuli on the ipsilateral side (Fig. 5C–5E). These enhanced responses occurred between 3- and 6-weeks and lasted for at least 8 weeks (Fig. 5C–5E). This phenomenon is in the line with the fact that AAV5 requires 3- to 4-weeks to become expressed and persists for at least 3 months4,6. On the contralateral side, the basal paw withdrawal responses were not altered in either virus-microinjected rats (Fig. 5F and5G).
Fig.5.

Effect of DRG OCT1 overexpression on nocicpetive thereshold in naive rats. (A and B) Oct1 mRNA (A) and OCT1 protein (B) expression in the injected L4/5 DRG 8 weeks after microinjection of AAV5-OCT1 or control AAV5-EGFP into unilateral L4/5 DRG. n = 3 biological replicates (3 rats) per group. **P < 0.01 versus the AAV5-EGFP group by two-tailed unpaired Student’s t-test. (C-G) Effect of micorinjection of AAV5-OCT1 or AAV5-EGFP into the unilateral L4/5 DRG on paw withdrawal threshold to mechanical stimulation (C and F), paw withdrawal latency to heat stimulation (D and G), and paw withdrawal latency to cold stimulation (E) on the ipsilateral (C-E) and controlateral (F and G) sides at the differenct weeks after microinjection. n = 5 rats/group. Two-way ANOVA followed by post hoc Tukey test. *P < 0.05 or **P < 0.01 versus the AAV5-EGFP group at the corrsponding time point.
Participation of OCT1 in CCI-induced increase in DNMT3a and downregulation of MOR and Kv1.2 in the injured DRG
How is OCT1, a transcription factor in the DRG, involved in CCI-caused pain hypersensitivity? It is evident that OCT1 promotes Dnmt3a gene transcription in DRG neurons20,26. Given that the increased DNMT3a in the injured DRG participates in neuropathic pain genesis through downregulation of DRG MOR and Kv1.220,26, we predicted that OCT1 contributed to neuropathic pain through DNMT3a/MOR/Kv1.2 pathway in the injured DRG. Like the fifth lumbar spinal nerve ligation20,26, CCI led to the increases in the amounts of Dnmt3a mRNA and DNMT3a protein and the decreases in the levels of MOR and Kv1.2 mRNAs and proteins in the ipsilateral DRG 7 days post-CCI in the vehicle- or control negative siRNA-microinjected rats (Fig. 6A and6B). These changes were not seen in the OCT1 siRNA-microinjected CCI rats (Fig. 6A and6B). Microinjection of OCT1 siRNA did not markedly change the basal amounts of Dnmt3a, Oprm1, and Kcna2 mRNAs and their corresponding proteins in the injected DRG of sham rats (Fig. 6A and6B). Additionally, DRG microinjection of AAV5-OCT1 produced the increases in the levels of Dnmt3a mRNA and its protein DNMT3a and the reductions in the levels of Oprm1 mRNA, Kcna2 mRNA and their encoding proteins compared to the AAV5-EGFP-microinjected group in the injected DRG of naive rats 8 weeks post-microinjection (Fig. 6C and6D). The double-labeled immunostaining assay showed the co-expression of OCT1 with MOR or Kv1.2 in some DRG neurons (Fig. 6E).
Fig. 6.

OCT1 transcriptionally activates Dnmt3a gene and represses DNMT3a-mediated MOR and Kv1.2 expression in the DRG. (A and B) The expression of Dnmt3a, Oprm1, and Kcna2 mRNA (A) and protein (B) in the ipsilateral L4/5 DRG on day 5 after sham surgery or CCI in the rats pre-microinjected with vehicle (Veh), negative control siRNA (NC), or OCT1 siRNA into the unilateral L4/5 DRG. n = 3–6 biological replicates (3–6 rats) per group. One-way ANOVA followed by post hoc Tukey test. *P < 0.05 or **P < 0.01 versus the corresponding vehicle plus sham group. #P < 0.05 or ##P < 0.01 versus the corresponding vehicle plus CCI group. (C and D) The expression of Dnmt3a, Oprm1 and Kcna2 mRNA (C) and protein (D) in the injected L4/5 DRG 8 weeks after microinjection of AAV5-EGFP or AAV5-OCT1 into the unilateral L4/5 DRG. n = 3–5 biological replicates (3–5 rats) per group. *P < 0.05 or **P < 0.01 versus the corresponding AAV5-EGFP group by two-tailed unpaired Student’s t-test. (E) Colocalization of OCT1 with MOR and Kv1.2 Double-label immunofluorescent staining of OCT1 (red) with MOR (green) or Kv1.2 (green) in the DRG neurons of naïve rats. Scale bar: 50 μm.
Effect of blocking increased DRG OCT1 on morphine analgesia under CCI-caused neuropathic pain conditions
We finally examined whether blocking the increase in DRG OCT1 improved morphine analgesia after CCI. Consistent with previous studies7,25, analgesia caused by systemically s.c injection of morphine 3 days after CCI was markedly decreased compared to that 3 days after sham surgery on the ipsilateral side of the vehicle-treated group (Fig. 7A). This decrease was rescued on the ipsilateral side of the OCT siRNA-treated group (Fig. 7A). As predicted, systemic morphine injection led to significant analgesic effects on the contralateral side of all treated rats (Fig. 7A). To further define whether anti-nociceptive effect in the OCT siRNA-microinjected rats was mediated by DRG MOR, we i.p. injected methylnaltrexone, a peripheral MOR antagonist, into the microinjected rats 4 days post-CCI or sham surgery. The OCT1 siRNA-microinjected rats displayed complete attenuation of CCI-caused thermal hyperalgesia on the ipsilateral side before systemic methylnaltrexone administration (Fig. 7B). Nevertheless, this effect was absent 30 min after i.p. injection of methylnaltrexone (Fig. 7B). Systemic methylnaltrexone at the dose used failed to alter basal paw withdrawal responses on the ipsilateral side of the vehicle-microinjected sham groups and on the contralateral side of all microinjected groups (Fig. 7B and7C).
Fig. 7.

Blocking CCI-caused increase in OCT1 in the injured DRG improves MOR-mediated analgesia under CCI-caused neuropathic pain conditions. (A) Effect of pre-microinjection of OCT1 siRNA or vehicle (Veh) into the unilateral L4/5 DRG on morphine analgesia on the ipsilateral and contralateral sides 3 days after CCI or sham surgery. n = 5 rats/group. Two-way ANOVA followed by post hoc Tukey test. **P < 0.01 versus the correspondung vehicle plus sham group. #P < 0.05 versus the correspondung vehicle plus CCI group. (B and C) Effect of intraperitoneal injection of methylnatrexone (M) on the OCT1 siRNA-produced antinociceptive effect 4 days post-CCI or sham surgery on the ipsilateral side. n = 5 rats/group. Two-way ANOVA followed by post hoc Tukey test. **P < 0.01 versus the vehicle plus sham group at the corresponding time point. #P < 0.05 versus the vehicle plus CCI group at the corresponding time point.
Discussion
CCI-caused neuropathic pain in the preclinical animal model imitates neuropathic pain caused by some surgeries (such as breast surgery, thoracotomy, limb amputation, and cardiac surgery) in the clinic5,23. Investigating how CCI leads to pain hypersensitivities may provide a new avenue for the management of this disorder. Here, we report that blocking increased OCT1 in the injured DRG alleviated CCI-caused mechanical allodynia, heat hyperalgesia, and cold allodynia during the induction and maintenance periods through rescuing DNMT3a-triggered downregulation of DRG MOR and Kv1.2. Given that acute pain and locomotor activity were not affected by this blockage, OCT1 may be a potential target for therapeutic management of neuropathic pain.
Like other transcription factors (such as myeloid zinc finger 1 and CCAAT/enhancer binding protein β)6,7, the expression of OCT1 can be regulated following peripheral nerve injury. We revealed that CCI produced a significant and time-dependent increase of OCT1 protein in the ipsilateral DRG, an observation that is in the line with our previous study in an SNL model26. This suggests that OCT1 is activated at the transcriptional and/or translational levels following peripheral nerve injury. Although the detailed mechanisms by which CCI causes the transcriptional and translational activations of Oct1 gene are still elusive, these activations are likely related to epigenetic modifications, posttranslational modifications, or increases in RNA stability, and/or triggered by other transcription factors under neuropathic pain conditions. These possibilities merit investigation in our future studies.
In this study, we used OCT1 siRNA to investigate the role of DRG OCT1 in neuropathic pain. We found that microinjection of OCT1 siRNA into the ipsilateral DRG attenuated the CCI-caused increase in DRG OCT1 and pain hypersensitivities during the development and maintenance periods. Given that OCT1 siRNA did not affect acute pain and locomotor functions, this suggests the specificity and selectivity of OCT1 siRNA effects. Interestingly, microinjection of OCT1 siRNA into the DRG failed to reduce basal expression of OCT1 in sham DRG, although this siRNA was effective in the cultured DRG. The mechanism by which OCT1 siRNA did not affect basal OCT1 expression in vivo is unknown but could be due to its low expression in the DRG under normal conditions. Additionally, the remaining OCT1 mRNA after its knockdown may have a high translational efficacy, which likely maintains normal level of basal OCT1 protein in sham DRG.
The increased OCT1 in the injured DRG may participate in the CCI-caused pain hypersensitivities by triggering Dnmt3a gene transcription in the injured DRG. The DNMT3a methylates DNA at some CpG locations of the promoter and 5-untralslated regions of the genes, resulting in gene transcription repression through physically blocking the interaction of transcriptional factors and/or functioning as docking locations for transcriptional repressors/corepressors9,17,18,22. DNMT3a is required for nerve injury-caused reduction of Kv1.2 and MOR in the injured DRG20,26. Given that the reduction of DRG Kv1.2 and MOR contributes to neuropathic pain genesis4,8,10,20,26,27, the increased DNMT3a in the injured DRG is thought as a trigger of neuropathic pain. The data from the present study and previous work26, demonstrated that OCT1 was required for nerve injury-caused transcriptional activation of DRG Dnmt3a gene, resulting in an increase in DNMT3a protein and in the reductions in MOR and Kv1.2 in the injured DRG. Moreover, OCT1 co-expresses with DNMT3a, MOR or Kv1.2 and DNMT3a with MOR or Kv1.2 in individual DRG neurons26. Therefore, the anti-nociceptive effect produced by blocking the CCI-caused increase in DRG OCT1 likely results from the failure to transcriptionally activate Dnmt3a gene in the injured DRG following CCI. Without an increase in DNMT3a protein, no alterations would occur in MOR and Kv1.2 expression, their gated neuronal excitability in the injured DRG neurons, and subsequent central sensitization in the ipsilateral dorsal horn. Indeed, blocking the CCI-caused increase of DRG OCT1 attenuated the CCI-caused increases in markers for central sensitization in the ipsilateral L4/5 dorsal horn and pain hypersensitivities. In addition, the antinociceptive effect produced by this blocking on CCI-caused pain hypersensitivities was abolished by antagonizing peripheral MOR. Given that the promoter regions of Oprm1 (encoding MOR) and Kcna2 (encoding Kv1.2) do not contain the consensus binding motif of OCT126, we conclude that the contribution of DRG OCT1 to neuropathic pain induction and maintenance may be mediated by epigenetic silencing of MOR and Kv1.2 caused by OCT1-triggered transcriptional activation of the Dnmt3a gene in the injured DRG. It is worth noting that other potential mechanisms by which DRG OCT1 participates in neuropathic pain cannot be excluded. DNMT3a may take part in nerve injury-caused alternations of other gene expression besides MOR and Kv1.2 in the DRG20,26. OCT1 as a transcriptional factor may also transcriptionally activate the expression of other genes in addition to Dnmt3a gene in the DRG under neuropathic pain conditions12,19. Moreover, we observed that a few OCT1 were expressed in the satellite cells of the DRG. Whether these possibilities participate in the role of DRG OCT1 in neuropathic pain remains to be further investigated.
Other OCT1-indepednent mechanisms may also contribute to nerve injury-caused increase in DRG DNMT3a. We recently reported that SNL-caused reduction of miR-143 participated in SNL-caused increases in DRG Dnmt3a mRNA and DNMT3a protein as rescuing DRG miR-143 reduction attenuated these increases, restored the SNL-caused reduction of DRG MOR and impaired neuropathic pain24. miR-143 is likely a negative modulator in DRG Dnmt3a gene expression following peripheral nerve injury. It appears that these mechanisms do not compensate each other, but how these mechanisms cooperate together to modulate Dnmt3a gene expression under neuropathic pain conditions merits to be further studied.
In summary, the present study revealed that the increased OCT1 contributed to the epigenetic silence of DRG Oprm1 and Kcna 2 genes through the transcriptional activation of DRG Dnmt3a gene under neuropathic pain conditions. It is well evident that DNMT3a, MOR, and Kv1.2 are required for the induction and maintenance of neuropathic pain. Blocking the increased DRG OCT1 impaired this disorder and improved morphine analgesia without affecting locomotor function and acute pain. Therefore, targeting OCT1 may have potential therapeutic value for neuropathic pain managements.
Supplementary Material
Acknowledgments
This work was supported by NIH grants (R01NS094664, R01NS094224, and R01DA033390) to Y.X.T., by the National Natural Science Foundation of China (81571082) to W.Z., by the National Natural Science Foundation of China (81701097) and the Henan Science and Technology project (172102310083) to J.Y.
Footnotes
Conflict of interest statement
The authors have no conflicts of interest to declare.
References
- [1].Campbell JN, Meyer RA. Mechanisms of neuropathic pain. Neuron 2006;52:77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci 2009;32:1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol 1980;20:441–62. [DOI] [PubMed] [Google Scholar]
- [4].Fan L, Guan X, Wang W, Zhao JY, Zhang H, Tiwari V, Hoffman PN, Li M, Tao YX. Impaired neuropathic pain and preserved acute pain in rats overexpressing voltage-gated potassium channel subunit Kv1.2 in primary afferent neurons. Mol Pain 2014;10:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kehlet H, Jensen TS, Woolf CJ. Persistent postsurgical pain: risk factors and prevention. Lancet 2006;367:1618–25. [DOI] [PubMed] [Google Scholar]
- [6].Li Z, Gu X, Sun L, Wu S, Liang L, Cao J, Lutz BM, Bekker A, Zhang W, Tao YX. Dorsal root ganglion myeloid zinc finger protein 1 contributes to neuropathic pain after peripheral nerve trauma. Pain 2015;156:711–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li Z, Mao Y, Liang L, Wu S, Yuan J, Mo K, Cai W, Mao Q, Cao J, Bekker A, Zhang W, Tao YX. The transcription factor C/EBPbeta in the dorsal root ganglion contributes to peripheral nerve trauma-induced nociceptive hypersensitivity. Sci Signal 2017;10:eaam5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Liang L, Gu X, Zhao JY, Wu S, Miao X, Xiao J, Mo K, Zhang J, Lutz BM, Bekker A, Tao YX. G9a participates in nerve injury-induced Kcna2 downregulation in primary sensory neurons. Sci Rep 2016;6:37704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Liang L, Lutz BM, Bekker A, Tao YX. Epigenetic regulation of chronic pain. Epigenomics 2015;7:235–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Liang L, Zhao JY, Gu X, Wu S, Mo K, Xiong M, Bekker A, Tao YX. G9a inhibits CREB-triggered expression of mu opioid receptor in primary sensory neurons following peripheral nerve injury. Mol Pain 2016;12:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Liaw WJ, Zhu XG, Yaster M, Johns RA, Gauda EB, Tao YX. Distinct expression of synaptic NR2A and NR2B in the central nervous system and impaired morphine tolerance and physical dependence in mice deficient in postsynaptic density-93 protein. Mol Pain 2008;4:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liu C, Jia X, Zou Z, Wang X, Wang Y, Zhang Z. VIH from the mud crab is specifically expressed in the eyestalk and potentially regulated by transactivator of Sox9/Oct4/Oct1. Gen Comp Endocrinol 2018;255:1–11. [DOI] [PubMed] [Google Scholar]
- [13].Lutz BM, Bekker A, Tao YX. Noncoding RNAs: new players in chronic pain. Anesthesiology 2014;121:409–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Macrae WA. Chronic post-surgical pain: 10 years on. Br J Anaesth 2008;101:77–86. [DOI] [PubMed] [Google Scholar]
- [15].Meyer R, Patel AM, Rattana SK, Quock TP, Mody SH. Prescription opioid abuse: a literature review of the clinical and economic burden in the United States. Popul Health Manag 2014;17:372–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Petryniak B, Staudt LM, Postema CE, McCormack WT, Thompson CB. Characterization of chicken octamer-binding proteins demonstrates that POU domain-containing homeobox transcription factors have been highly conserved during vertebrate evolution. Proc Natl Acad Sci U S A 1990;87:1099–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Poetsch AR, Plass C. Transcriptional regulation by DNA methylation. Cancer Treat Rev 2011;37 Suppl 1:S8–12. [DOI] [PubMed] [Google Scholar]
- [18].Siedlecki P, Zielenkiewicz P. Mammalian DNA methyltransferases. Acta Biochim Pol 2006;53:245–56. [PubMed] [Google Scholar]
- [19].Stauss D, Brunner C, Berberich-Siebelt F, Hopken UE, Lipp M, Muller G. The transcriptional coactivator Bob1 promotes the development of follicular T helper cells via Bcl6. EMBO J 2016;35:881–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sun L, Zhao JY, Gu X, Liang L, Wu S, Mo K, Feng J, Guo W, Zhang J, Bekker A, Zhao X, Nestler EJ, Tao YX. Nerve injury-induced epigenetic silencing of opioid receptors controlled by DNMT3a in primary afferent neurons. Pain 2017;158:1153–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Trang T, Al-Hasani R, Salvemini D, Salter MW, Gutstein H, Cahill CM. Pain and Poppies: The Good, the Bad, and the Ugly of Opioid Analgesics. J Neurosci 2015;35:13879–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Turek-Plewa J, Jagodzinski PP. The role of mammalian DNA methyltransferases in the regulation of gene expression. Cell Mol Biol Lett 2005;10:631–47. [PubMed] [Google Scholar]
- [23].Wu CL, Raja SN. Treatment of acute postoperative pain. Lancet 2011;377:2215–25. [DOI] [PubMed] [Google Scholar]
- [24].Xu B, Cao J, Zhang J, Jia S, Wu S, Mo K, Wei G, Liang L, Miao X, Bekker A, Tao YX. Role of MicroRNA-143 in Nerve Injury-Induced Upregulation of Dnmt3a Expression in Primary Sensory Neurons. Front Mol Neurosci 2017;10:350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhang J, Liang L, Miao X, Wu S, Cao J, Tao B, Mao Q, Mo K, Xiong M, Lutz BM, Bekker A, Tao YX. Contribution of the Suppressor of Variegation 3–9 Homolog 1 in Dorsal Root Ganglia and Spinal Cord Dorsal Horn to Nerve Injury-induced Nociceptive Hypersensitivity. Anesthesiology 2016;125:765–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhao JY, Liang L, Gu X, Li Z, Wu S, Sun L, Atianjoh FE, Feng J, Mo K, Jia S, Lutz BM, Bekker A, Nestler EJ, Tao YX. DNA methyltransferase DNMT3a contributes to neuropathic pain by repressing Kcna2 in primary afferent neurons. Nat Commun 2017;8:14712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhao X, Tang Z, Zhang H, Atianjoh FE, Zhao JY, Liang L, Wang W, Guan X, Kao SC, Tiwari V, Gao YJ, Hoffman PN, Cui H, Li M, Dong X, Tao YX. A long noncoding RNA contributes to neuropathic pain by silencing Kcna2 in primary afferent neurons. Nat Neurosci 2013;16:1024–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhou XL, Yu LN, Wang Y, Tang LH, Peng YN, Cao JL, Yan M. Increased methylation of the MOR gene proximal promoter in primary sensory neurons plays a crucial role in the decreased analgesic effect of opioids in neuropathic pain. Mol Pain 2014;10:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.