

Abstract
Keratinocyte-derived chemokine (KC or mCXCL1) and macrophage inflammatory protein 2 (MIP2 or mCXCL2) play nonredundant roles in trafficking blood neutrophils to sites of infection and injury. The functional responses of KC and MIP2 are intimately coupled to their interactions with glycosaminoglycans (GAGs). GAG interactions orchestrate chemokine concentration gradients and modulate receptor activity, which together regulate neutrophil trafficking. Here, using NMR, molecular dynamics (MD) simulations, and isothermal titration calorimetry (ITC), we characterized the molecular basis of KC and MIP2 binding to the GAG heparin. Both chemokines reversibly exist as monomers and dimers, and the NMR analysis indicates that the dimer binds heparin with higher affinity. The ITC experiments indicate a stoichiometry of two GAGs per KC or MIP2 dimer and that the enthalpic and entropic contributions vary significantly between the two chemokine–heparin complexes. NMR-based structural models of heparin–KC and heparin–MIP2 complexes reveal that different combinations of residues from the N-loop, 40s turn, β3-strand, and C-terminal helix form a binding surface within a monomer and that both conserved residues and residues unique to a particular chemokine mediate the binding interactions. MD simulations indicate significant residue-specific differences in their contribution to binding and affinity for a given chemokine and between chemokines. On the basis of our observations that KC and MIP2 bind to GAG via distinct molecular interactions, we propose that the differences in these GAG interactions lead to differences in neutrophil recruitment and play nonoverlapping roles in resolution of inflammation.
Keywords: glycosaminoglycan, nuclear magnetic resonance (NMR), isothermal titration calorimetry (ITC), molecular dynamics, heparin, chemokine
Introduction
Chemokines, a large family of small molecular weight proteins, play crucial roles in the pathophysiology of infectious and inflammatory diseases (1–3). Common to these diverse functions is the directed movement of various leukocytes to distal and remote locations. Cellular trafficking must be highly coordinated to elicit the required biological function, and a dysregulation in this process has been implicated in acute and chronic inflammatory diseases. Chemokines are broadly classified on the basis of their conserved cysteines into two major (CXC and CC) 2 and two minor (CX3C and C) subfamilies (4, 5). In mice, the CXC chemokines KC/mCXCL1 (keratinocyte-derived chemokine/mouse CXCL1) and MIP2/mCXCL2 (macrophage inflammatory protein 2/mouse CXCL2) 3 play a prominent role in recruiting neutrophils in response to infection and tissue injury.
Animal models have shown that coordinated activity of both chemokines is required for optimal neutrophil function, their local and systemic concentrations vary, and disrupting KC expression results in impaired neutrophil recruitment and greater susceptibility to infection (6–14). KC and MIP2 recruit neutrophils by activating the CXCR2 receptor, which is regulated by glycosaminoglycan (GAG) interactions. GAGs are acidic and contain carboxylate and sulfate moieties. Chemokines are basic or contain clusters of basic residues, which implies that electrostatic/H-bonding interactions play an important role in the binding process. GAG interactions dictate chemokine concentration gradients, which orchestrate directed neutrophil migration to the target tissue (15–17). GAGs, such as heparan sulfate (HS), are highly sulfated polysaccharides covalently attached to core proteins called proteoglycans (PGs). PGs are expressed by most cell types including endothelial cells and are also an integral component of the extracellular matrix where they exist as noncovalent macromolecular complexes with matrix proteins (18–20).
In this study, we characterized the molecular bases, stoichiometry, and thermodynamics of heparin binding to KC and MIP2 using solution NMR spectroscopy, molecular dynamics (MD) simulations, and isothermal titration calorimetry (ITC). Our studies indicate that the molecular bases of how KC and MIP2 bind GAG are quite distinct. On the basis of our data, we propose that GAG interactions play nonredundant roles in regulating KC- and MIP2-mediated neutrophil recruitment and that these differences play important roles in successful resolution of inflammation.
Results
Relative GAG affinities of the chemokine monomer and dimer
KC and MIP2 reversibly exist as monomers and dimers (dissociation constants of ∼35 and 15 μm, respectively). To characterize whether the monomer or dimer binds GAG with higher affinity, we characterized binding of heparin to 30 μm KC and 20 μm MIP2 samples using solution NMR spectroscopy. At these concentrations, for both chemokines, peaks corresponding to both the monomer and dimer could be observed in the HSQC spectrum. For both chemokines, upon adding heparin dp26, peaks corresponding to the monomer disappeared, indicating that the dimer is the high-affinity GAG-binding form (Fig. 1).
Figure 1.

The dimer is the high-affinity GAG ligand. A and B, sections of the 1H-15N HSQC spectra showing the overlay of KC (A) and MIP2 (B) in the free (black) and heparin-bound (red) forms. Dimer and monomer peaks are indicated as D and M. The monomer peaks disappear in both spectra on heparin dp26 binding, indicating that the dimer is the high-affinity GAG ligand. The spectra were collected using 20 μm KC and 15 μm MIP2 samples in 50 mm sodium phosphate, pH 6.0, at 25 °C.
Thermodynamics and stoichiometry
A complete understanding of chemokine–GAG interactions requires knowledge of the thermodynamics of the binding process. Classically, enthalpy provides insight into how favorable hydrophobic, H-bonding, and electrostatic interactions mediate binding, whereas entropy arises because of changes in restriction/freedom of the backbone and side-chain atoms and rearrangement or release of solvent water and counter ions. ITC can provide all of the thermodynamic parameters from a single experiment: enthalpy (ΔH), entropy (ΔS), and free energy (ΔG) that is also related to the dissociation constant (Kd) and stoichiometry (21, 22). The binding isotherm of heparin binding to WT MIP2 is shown in Fig. 2A. The data fit best to a single-binding site model, yielding a stoichiometry of two GAG chains per MIP2 dimer and binding affinity (Kd) of ∼25 μm (Table 1). For WT KC, the data also fit best to a single-binding site model, yielding a stoichiometry of two GAG chains per KC dimer and a Kd of 4 μm (Fig. 2B). Because some of the protein exists as monomers under the experimental conditions, we also measured binding of heparin to the trapped dimer to eliminate any contribution from binding to the monomer (Fig. 2, C and D). For MIP2, binding affinity for the trapped dimer was ∼6-fold higher, but otherwise the stoichiometry and relative enthalpy and entropy contributions were similar. For KC, stoichiometry and thermodynamic parameters were essentially the same for the trapped dimer. ITC data indicate that binding of MIP2 to heparin is driven by favorable enthalpic and entropic contributions, whereas binding of KC to heparin is predominantly driven by enthalpic interactions and is entropically disfavored. In both cases, the enthalpic contribution outweighs the entropic contribution.
Figure 2.

Thermodynamics of chemokine–heparin interactions. A–D, isotherms corresponding to the binding of heparin dp8 to WT MIP2 (A), WT KC (B), MIP2 dimer (C), and KC dimer (D). The titrations and the integrated data obtained after subtracting the heat of dilution are shown in the upper and lower panels, respectively. The titrations were performed in 50 mm sodium phosphate, pH 6.0, at 25 °C.
Table 1.
Thermodynamic parameters for the binding of mouse chemokines to heparin
The thermodynamic parameters for the binding of mouse chemokines to heparin dp8 were measured by ITC in 50 mm sodium phosphate, pH 6.0 buffer at 25 °C. The values reported are the means of two experiments.
| n | Kd | ΔH | −TΔS | ΔG | |
|---|---|---|---|---|---|
| μm | kcal/mol | kcal/mol | kcal/mol | ||
| WT KC | 2.0 | 3.8 ± 0.1 | −11.2 ± 0.1 | 3.9 ± 0.1 | −7.4 ± 0.1 |
| KC dimer | 2.0 | 4.2 ± 0.1 | −12.6 ± 0.1 | 5.3 ± 0.1 | −7.3 ± 0.1 |
| WT MIP2 | 1.8 | 24.8 ± 0.1 | −4.3 ± 0.1 | −2.0 ± 0.1 | −6.3 ± 0.1 |
| MIP2 dimer | 2.0 | 3.9 ± 0.1 | −5.4 ± 0.1 | −1.9 ± 0.1 | −7.4 ± 0.1 |
Heparin-binding interactions
We characterized the structural basis of mouse chemokines binding to heparin oligosaccharides using solution NMR spectroscopy. For all titrations we observed only one set of peaks, indicating that binding is in the fast-exchange regime on the NMR time scale (Fig. S2).
MIP2–heparin interactions
The CSP profile on heparin dp8 binding is shown in Fig. 3. Perturbed residues are located in the N-loop, 40s turn, and the C-helix. In particular, the basic residues Arg-17 and Lys-21 from the N-loop, Lys-45 from the 40s turn, and Lys-K61, Lys-65, and Lys-69 from the C-helix show significant perturbation. In addition, chemical shifts of several nonbasic residues, Val-18, Asp-19, and Phe-20 in N-loop; Gly-46 from the 40s loop; and Ile-66 and Asn-68 in the C-helix were also perturbed. A previous NMR study of heparin disaccharide binding to MIP2 had reported Asp-19, Lys-21, Lys-61, Lys-65, and Lys-69 as potential GAG-binding residues (23). Lack of perturbation for Arg-17 and Lys-45 could be attributed to the weak binding of the disaccharide compared with longer octasaccharides. We previously observed for hCXCL8 that chemical shift perturbation of a disaccharide is significantly less compared with longer oligosaccharides (24).
Figure 3.

Binding of MIP2 to the heparin dp8. A, sections of the 1H-15N HSQC spectra showing overlay of MIP2 in the free (black) and dp8-bound (red) forms. Arrows indicate direction of the peak movement. B, histogram of heparin dp8 binding-induced chemical shift changes as a function of MIP2 residue number. Basic residues are in blue, and nonbasic residues are in red. The spectra were collected using a 100 μm MIP2 sample in 50 mm sodium phosphate, pH 6.0, at 35 °C.
KC–heparin interactions
We previously characterized the binding of heparin dp8 to the KC trapped dimer using NMR spectroscopy (25). We now characterized the binding of heparin dp14 and observed that the binding profile is essentially the same as that observed for dp8. The CSP profiles are shown in Fig. 4. Because chemical shift assignments of KC WT were challenging, we characterized heparin binding using the trapped dimer. Perturbed basic residues include His-20 and Lys-22 from the N-loop, Lys-46 from the 40s turn, Arg-49 from the β3-strand, and Lys-62 and Lys-66 from the C-helix. Several nonbasic residues, Leu-20 in the N-loop; Asn-23 and Ile-24 in the β1-strand; Leu-45, Asn-47, and Ala-51 in the 40s loop; and Leu-59, Val-60, Gln-61, Val-64, Gln-65, Val-66, Met-67 and Leu-68 in the C-helix, were also perturbed.
Figure 4.

Binding of KC to the heparin dp14. A, sections of the 1H-15N HSQC spectra showing the overlay of KC in the free (black) and dp14-bound (red) forms. Arrows indicate direction of the peak movement. B, histogram of heparin dp14 binding-induced chemical shift changes as a function of KC residue number. Basic residues are in blue, and nonbasic residues are in red. The spectra were collected using a 100 μm KC sample in 50 mm sodium phosphate, pH 6.0, at 35 °C.
Previous GAG-binding studies for the human chemokines CXCL1, CXCL5, CXCL7, and CXCL8 have shown that several GAG-binding basic residues are conserved, and at the same time, basic residues that are unique to a chemokine are also involved in binding. These studies identified eight conserved basic residues that mediate GAG interactions (Fig. 5), and interestingly, these studies show that not all conserved residues are involved in binding all chemokines (24, 26–29). Comparison of KC and MIP2 and human sequences also reveals that several perturbed basic residues are conserved and, at the same time, that residues unique to MIP2 also mediate binding interactions (Fig. 5). The KC sequence reveals seven of the eight conserved basic residues (except B6), whereas MIP2 shows only five (except B2, B5, and B6). Significant perturbation of Arg-17, which is unique to MIP2, is striking. Comparison of the mouse and human sequences indicates that only hCXCL8 has a lysine (Lys-15) at a comparable location (24). KC and MIP2 structures reveal that hydrophobic and acidic residues that show perturbation are either in the proximity of basic residues and/or are buried, indicating that they are unlikely to be involved in direct binding interactions.
Figure 5.

Sequences of mouse and human neutrophil activating chemokines. Heparin-binding basic residues of KC and MIP2 are highlighted in red, shaded in gray, and numbered. Arg-17 and Lys-69 of MIP2 are highlighted in blue. Conserved basic residues of human neutrophil activating chemokines implicated in heparin binding are labeled B1–B8, highlighted in red, and shaded in gray. Similar to mouse chemokines, chemokine-specific residues involved in heparin binding are in blue.
Structural models of heparin-bound chemokine complexes
MIP2–GAG complex
The MIP2 structure reveals that the basic residues identified from NMR experiments form a contiguous surface within a monomer. To better understand how these residues are involved in complex formation, we carried out HADDOCK-based docking. We performed three different HADDOCK runs to ensure that the input constraints did not bias specific structural models and that all possible binding geometries within a monomer and across the dimer interface were considered. In run 1, we modeled binding of one heparin with constraints given to both monomers of the dimer. In run 2, we modeled the binding of one heparin with constraints given to only one monomer of the dimer. In run 3, we modeled binding of two heparins with constraints given to both monomers of the dimer. Run 1 resulted in one major and one minor cluster. In the major cluster, heparin bound to one monomer of the dimer with no evidence of interactions across the dimer interface (defined as model I; Fig. S1). In the minor cluster, heparin spanned the dimer interface and bound both monomers of the dimer (defined as model II). Run 2 resulted in only one major cluster with heparin binding to one monomer of a dimer similar to model I. Run 3 also resulted in only one major cluster with two heparins binding to two monomers of the dimer similar to model I (Fig. 6).
Figure 6.

MIP2-heparin binding models. A–D, structural depiction of model I. In A and B, the MIP2 dimer is shown in ribbon representation, and GAG is shown as sticks. A shows the binding of heparin to one of the monomers of the MIP2 dimer. B shows how the individual basic residues engage the heparin acidic groups. C highlights the GAG-binding residues (blue) in a surface presentation. In D, GAG-binding residues are labeled on the electrostatic surface.
NMR and ITC data are consistent with only model I. In model I, heparin engages all of the basic and polar residues identified from NMR CSP measurements. Model II shows interactions from residues of the N-loop of one monomer and the C-helix of both monomers of the dimer but none from the 40s turn residues. Most importantly, only model I allows binding of two heparins per dimer, whereas model II allows binding of only one heparin per dimer that is inconsistent with the ITC data. In model I, the heparin chain adopts a specific directionality in ∼70% of the structures, with the other 30% running in the opposite orientation with both showing similar energies.
KC–heparin complex
The chemical shift-based structural model reveals that the basic residues identified from NMR experiments form a contiguous surface within a monomer. We carried out HADDOCK based calculations as described for MIP2. Like for MIP2, run 1 resulted in one major and one minor cluster. In the major cluster (model I), heparin bound to one monomer of the dimer with no evidence of interactions across the dimer interface (Fig. 7). In the minor cluster, heparin spanned the dimer interface and bound both monomers of the dimer (model II). Run 2 also resulted in only one major cluster with heparin binding to a monomer of a dimer similar to run 1. Run 3 resulted in only one major cluster with two heparins binding to two monomers of the dimer similar to model I. As described for MIP2, NMR and ITC data are consistent with only model I, and further, only model I allows binding of two heparins per dimer. In model I, the heparin chain adopts a specific directionality in ∼80% of the structures with the other 20% running in the opposite orientation, with both showing similar energies. Our previous NMR-based modeling showed a preference for model II over model I (25). At that time, neither the stoichiometry nor the involvement of the 40s residues in GAG binding were known. These studies highlight a limitation of modeling based on backbone chemical shifts alone and the importance of the stoichiometry of the chemokine–GAG complex.
Figure 7.

KC-heparin binding models. A–D, structural depictions of model I. In A and B, the KC dimer is shown in ribbon representation, and GAG is shown as sticks. A shows the binding of heparin to one monomer of the KC dimer. B shows a close-up of how the individual basic residues engage the heparin acidic groups. C highlights the GAG-binding residues (blue) in a surface presentation. In D, GAG-binding residues are labeled on the electrostatic surface.
Comparison of the MIP2-heparin and KC-heparin models indicates that six basic residues mediate binding of both chemokines. In MIP2, the conserved basic residues B3 (Lys-21), B4 (Lys-45), B7 (Lys-61), and B8 (Lys-65) and the specific residues Arg-17 and Lys-69, mediate binding. In the case of KC, B2 (His-20), B3 (Lys-22), B4 (Lys-46), B5 (Arg-49), B7 (Lys-61), and B8 (Lys-65) mediate binding interactions. In both proteins, these residues form a contiguous surface spanning one face of the protein (Figs. 6 and 7).
MD simulations of heparin-bound chemokine complexes
To understand residue-specific binding contributions, we carried out MD simulations of the heparin dp8-bound chemokine complexes. HADDOCK models that were consistent with NMR and ITC data were used as the starting structures. Simulations were carried out for 100 ns in NPT ensemble to explore the conformational behavior of heparin bound KC and MIP2 in the presence of explicit solvent. Throughout the simulation, basic side chains were observed to be dynamic, and a given residue could engage more than one sulfate/carboxylate group of the neighboring sugar residues. The heparin chain was also observed to undergo considerable translational motion within the observable span of 100 ns (Fig. S3). To understand stability and specificity, residues participating in stable intermolecular H-bonds were calculated using the cpptraj program with a donor-acceptor distance of 3.5 Å and an angle cutoff of 45° (30). Our MD data also indicate water-mediated interactions for both the complexes. Crystal structures of GAG–protein complexes show evidence for water-mediated interactions, and computational analysis and MD studies have also shown that water-mediated interactions stabilize the binding interface (27, 31–33). A convergence of the sampling was observed for both the chemokine–GAG complexes (Fig. S3). For calculating binding free-energy calculations of the chemokine–GAG complexes, the MM/PB(GB)SA method was used because of its ease and efficiency. However, it is important to note that the free-energy values derived from these calculations may be overestimated because of inherent limitations. Nevertheless, free-energy differences between two conditions offer valuable insights into interaction changes, as also described previously (34). Further, the predicted higher binding energy for KC compared with MIP2 is in agreement with the experimental values (Table 1).
MIP2–heparin complexes
The occupancy of intermolecular H-bonds is shown in Fig. 8A. The N-loop residues Arg-17 and Lys-21 displayed occupancy of 100% or more, suggesting at least one H-bond throughout the simulation. Residues Lys-45, Lys-61, Lys-65, and Lys-69 displayed occupancy of ∼50–80% implying at least one H-bond across most of the simulation. A summary of water-mediated interactions is shown in Fig. 8B. Lys-21 showed the highest occupancy (∼120%), and residues Arg-17, Lys-45, Lys-61, Lys-65, and Lys-69 showed occupancy in the ∼40–60% range. Other positively charged residues did not (or only minimally) participate in these interactions. Of all the residues, Lys-21 exhibited the strongest interaction that persisted from the initial to final state of simulation. The single-residue energy decomposition values indicated that direct H-bond–forming residues contributed relative free energy in the range of −2.8 to −7.5 to kcal/mol (Fig. 9A). The rank order in terms of stability can be clustered into N-loop residues contributing the most, C-helix residues moderate, and 40s turn the least for binding interactions.
Figure 8.

H-bonding properties from MD simulations. A and C, occupancy of direct H-bond interactions between MIP2 and heparin (A) and between KC and heparin (C). B and D, occupancy of water-mediated H-bond interactions between MIP2 and heparin (B) and KC and heparin (D).
Figure 9.
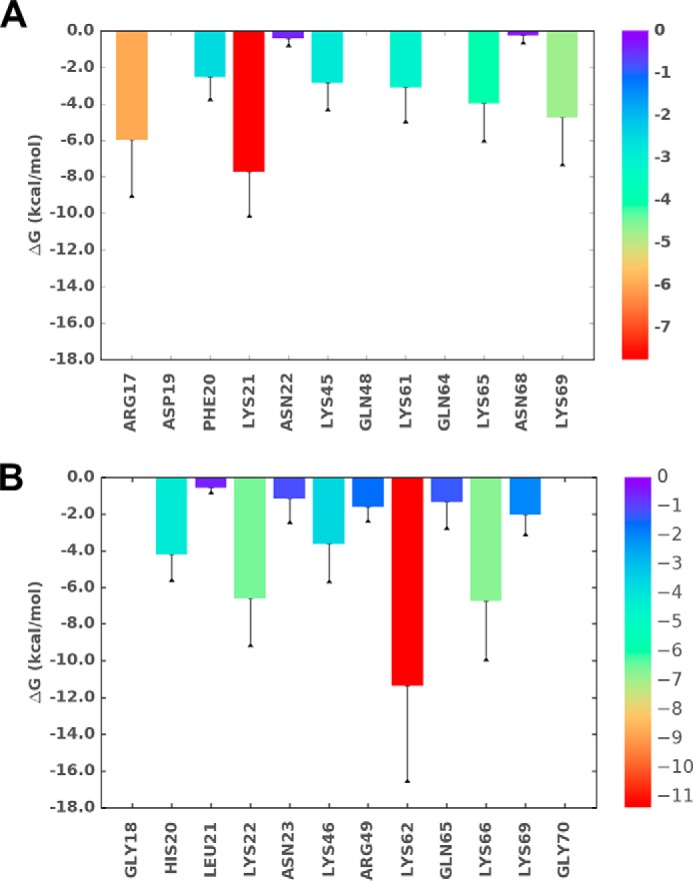
Binding energetics from MD simulations. A and B, single-residue energy decomposition values for MIP2–heparin (A) and KC–heparin (B) complexes. The highest to lowest energy are shown from red to blue. The error bar represents the standard deviation.
KC–heparin complexes
Occupancy of intermolecular H-bonds is shown in Fig. 8C. Residues His-20, Lys-22, Lys-62, and Lys-66 displayed occupancy of 100% or more, suggesting at least one H-bond throughout the simulation. Asn-23 and Lys-46 displayed occupancy of ∼50% implying one H-bond approximately half the time during the simulation. A summary of water-mediated interactions is shown in Fig. 8D. Lys-22 alone showed an occupancy of ∼80%, and Asn-23, Lys-46, Lys-62, Gln-65, and Lys-66 displayed ∼30–50% occupancy. Other basic residues either did not (or only minimally) participate in these interactions. Of the interacting residues, Lys-62 alone exhibited the strongest interaction persisting from the initial to final state of simulation. We calculated single-residue energy decomposition values to understand and also rank the contribution of the individual residues for heparin interactions. The direct H-bond–forming residues contributed relative free energy in the range of −1.6 to −11.4 kcal/mol (Fig. 9B). In terms of stability, C-helical residues provided the highest, N-loop residues moderate, and the 40s turn and β3-strand the least. Interestingly, the polar residues Asn-23 and Gln-65 that were shown to be involved in direct H-bonding and/or water-mediated interactions contributed very little to binding energy, suggesting that they likely play a more prominent role in defining specificity (35).
Comparison of the MD data of KC and MIP2 complexes provides several interesting and striking differences, including differences in the rank order of how different residues contribute to the stability of the GAG complex that could be attributed to differences in sequences. Residues corresponding to Arg-17 and Lys-69 in MIP2 are absent in KC, and the conserved N-loop histidine that is present in KC is absent in MIP2. The arginine guanidinium group allows more extensive H-bonding interactions compared with the histidine imidazole side chain (36), and this could be the reason for the prominent role of N-loop residues for MIP2 compared with KC. On the other hand, both N-loop and C-helical residues are involved in KC-binding interactions. For both chemokines, MD studies also show that rapid, transitory, and plastic conformational fluctuations mediate heparin binding that is further enhanced by water-mediated interactions with surface-exposed residues.
Can GAG-bound KC and MIP2 bind the receptor?
Our observation that the N-loop residues are involved in heparin binding has a direct impact on function, because these residues have also been implicated in receptor binding. Functional studies have shown that receptor activation involves interactions between the chemokine N-loop/β3-strand and receptor N-domain residues (defined as site I) and between the ligand N-terminal and receptor extracellular/transmembrane residues (site II) (37). Site I functions as a critical docking site, and we have shown that the structural basis of site I interactions can be studied outside the context of the intact receptor by characterizing chemokine binding to N-domain peptides (26, 38–40). Therefore, we first characterized the binding of the mouse CXCR2 N-domain (CXCR2Nd) peptide to KC and MIP2 using NMR spectroscopy and then characterized whether GAG-bound chemokine can bind the receptor.
For MIP2, CXCR2Nd peptide titrations were carried out under conditions where the WT exists as a dimer. During titration, a new set of peaks emerged that can be attributed to the monomer, indicating that the receptor peptide binds the monomer with higher affinity. As expected, CSP data indicate that the binding site involves a groove defined by the N-loop and adjacent β-residues (not shown). In the case of KC, receptor titrations were carried out under conditions where WT KC exists as monomers and dimers. The peaks corresponding to the monomer gain intensity and are perturbed to a greater extent than the dimer, indicating that the monomer binds the receptor peptide with higher affinity. Because the chemical shifts of the WT monomer and dimer are not available, we also carried out the titration to the trapped dimer. As expected, significant CSP were observed for the N-loop and the adjacent β3 strand, indicating that these residues mediate the binding process (not shown).
We then characterized whether the heparin-bound chemokines can bind the CXCR2Nd using NMR spectroscopy. For each chemokine, the addition of the CXCR2Nd peptide did not alter the heparin-bound chemokine spectrum, indicating that the GAG-bound chemokines are unable to bind the receptor N domain (Fig. 10). Overlap between GAG-binding and receptor-binding domains and the absence of ternary complex formation on adding receptor peptide to heparin-bound chemokines has also been made for several human neutrophil-activating chemokines (NACs) using solution NMR spectroscopy (26–28, 40).
Figure 10.

Heparin-bound chemokine cannot bind the receptor. NMR data for MIP2 and KC are shown in the left and right panels, respectively. A and F show the overlay of a selected region of 1H-15N HSQC spectra showing free chemokine (black), CXCR2 N domain–bound form (green), heparin-bound form (blue), and on titrating CXCR2 N domain to the heparin-bound chemokine (red). Individual spectra of free MIP2 and KC (B and G), CXCR2 N domain–bound MIP2 and KC (C and H), heparin-bound MIP2 and KC (D and I), and on titrating the CXCR2 N domain to the heparin-bound (E and J) are also shown.
Discussion
KC and MIP2 play important roles in trafficking neutrophils for eliminating infection. Animal disease models have shown that both are essential for successful resolution but show several differences including in their levels and neutrophil trafficking profiles (6–14). Considering that GAG interactions orchestrate gradient formation, differences in recruitment can be attributed to differences in GAG interactions. Our current studies show that the structural basis and molecular features of KC–GAG and MIP2–GAG interactions are quite different.
NMR studies reveal that basic residues from the N-loop, 40s loop, and C-helix mediate KC and MIP2 binding. However, there are differences in their contribution to binding including the number of residues from each subdomain, the total number of lysines and arginines, and whether they are conserved or specific. MD studies show pronounced residue-specific differences in lifetimes of direct H-bonds, water-mediated interactions, and contributions to binding affinity. In particular, differences in rank order of different subdomains contributing to stability stands out.
NACs were thought to bind GAGs essentially via C-helical basic residues. Our current studies and previous studies of hNACs show that residues from the N-loop and 40s loop also determine binding, that their contributions can be quite significant, and that the binding geometries can be quite diverse (24, 26–29). Recent NMR studies of GAG binding of chemokines CXCL10, CXCL12, and CXCL13 also show diverse binding interfaces and geometries (41–44). Considering all chemokines bind GAGs, it is very likely that each chemokine shows distinct GAG interactions.
During the time frame from initial infection to eventual elimination, local chemokine concentration can vary by many fold and so can exist as monomers, dimers, or both. Animal models have shown that chemokine monomer–dimer equilibrium regulates neutrophil recruitment (45, 46). KC and MIP2 exist as monomers and dimers, but their dimerization constants vary. MIP2 forms stronger dimers, but for both chemokines, the dimer binds GAGs with higher affinity. Higher affinity of the dimer for GAGs has also been observed for human neutrophil-activating chemokines (24, 26–28). Both KC and MIP2 are potent agonists for the mouse neutrophil CXCR2 receptor, but MIP2, compared with KC, shows ∼10-fold higher activity (47).
GAG interactions determine haptotactic (GAG-bound) and chemotactic (soluble) gradients and also whether free or GAG-bound chemokine binds the receptor. We observe overlap between receptor-binding and GAG-binding domains and that the heparin-bound KC and MIP2 are unable to bind the receptor, suggesting that GAG-bound chemokines are unlikely to activate the receptor. However, in vivo conditions are complex, and proximity and geometry of GAGs within a PG and between PGs will determine how chemokines bind GAGs and are presented to the receptor (48–50). PGs carry multiple GAG chains, and all GAG chains will have the same orientation within a PG. Because the GAG-binding site lies within a monomer of the dimer, GAG orientation in the second monomer will be antiparallel because of 2-fold symmetry. Because of geometric restriction, only one site can be occupied, and the second monomer is available for receptor interactions. In principle, two GAG chains from the same PG can bind two monomers of a dimer if GAG interactions are not specific. Such a nonspecific interaction may be possible because of geometric proximity but would be a low-affinity interaction. Two GAGs in antiparallel orientation can bind a dimer if they are from two different PGs. Independent of the actual mode of receptor binding, we conclude that GAG interactions differentially regulate the makeup of KC and MIP2 gradients, which in turn regulate neutrophil trafficking for eliminating infection and restoration of homeostasis.
Materials and methods
HS has a modular structure with sulfated sequences (defined as NS domain) separated by nonsulfated regions containing acetylated sequences (defined as NA domain). Both HS and heparin share a repeating disaccharide unit composed of glucosamine and a hexuronic acid. Heparin is preferred for structural and biophysical studies because it is more uniformly sulfated and because of the availability of size-defined oligosaccharides. Previous studies have shown that heparin functions as a useful surrogate to understand endogenous HS interactions (45, 46, 51). However, heparin may not fully capture HS interactions, because endogenous HS is intrinsically heterogeneous because of differential sulfation in the NS domain. Heparin oligosaccharides were purchased from Iduron. According to the manufacturer, the oligosaccharides were purified using high-resolution gel-filtration chromatography, the main disaccharide unit is IdoA,2S-GlcNS,6S (∼75%), they show some variation in sulfation pattern, they contain uronic acid at the nonreducing end, and there is a C4–C5 double bond as a result of the heparinase endolytic action. Expression and purification of isotopically labeled KC and MIP2 were carried out as described (52).
NMR experiments
NMR spectra were recorded at 35 °C using Bruker Avance III 600 and 800 MHz spectrometers equipped with QCI and TCI cryoprobes, respectively. Titrations of heparin oligosaccharides to ∼100 μm 15N-labeled KC and MIP2 were carried out in 50 mm sodium phosphate, pH 6.0. Aliquots of 10 mm heparin oligosaccharides prepared in the same buffer were added to the protein sample, and a series of 1H-15N HSQC spectra was collected until essentially no changes in chemical shifts were observed. For all titrations, the final protein:ligand molar ratio was ∼1:6. The observed chemical shift perturbation (Δδobs) was calculated as a weighted average chemical shift change of 1H (ΔδH) and 15N(ΔδN). In the case of receptor titrations, a series of HSQC spectra was collected on adding aliquots of the 1 mm of CXCR2 N domain (CXCR2Nd) to ∼60–70 μm MIP2 and KC samples. The final chemokine:CXCR2Nd molar ratio was 1:4. The chemical shifts of MIP2 were determined using 15N NOESY and 15N total correlation spectroscopy experiments and were similar to previously reported assignments (53).
Isothermal titration calorimetry
Binding of heparin octasaccharide (dp8) to KC and MIP2 WT and trapped dimer was characterized at 25 °C using a Malvern PEAQ-ITC microcalorimeter (21). The protein and GAG solutions were centrifuged and degassed under vacuum before use. Titrations were performed by injecting 1 × 0.5-μl and 17 × 2-μl aliquots of 0.5 mm heparin oligosaccharides to 20–40 μm chemokine in 50 mm sodium phosphate, pH 6.0. All titrations were carried out at least twice. The raw data were corrected using buffer and protein controls and analyzed using the software supplied by the manufacturer.
Docking of the chemokine–GAG complexes
Molecular docking of heparin dp8 to the MIP2 dimer was carried out using High Ambiguity Driven biomolecular DOCKing (HADDOCK) (54, 55). The docking was carried out using NMR chemical shift perturbations (CSP) as ambiguous interaction restraints using MIP2 dimer (Protein Data Bank code 3N52) and heparin (Protein Data Bank code 1HPN) structures (23, 56). Residues with chemical shift perturbation greater than the average plus one standard deviation, and accessible surface areas of >30% were selected as “active” residues. Surface neighbors of “active” residues were selected as “passive” residues, and both active and passive residues were used as ambiguous interaction restraints. Optimized parameters for liquid simulation were used for the nonbonded interaction. The topology and parameter files for heparin oligosaccharides were generated using the PRODRG server (57). In total, 3000 complex structures were generated during the initial rigid body docking. The best 1000 structures in terms of intermolecular energies were further subjected to semiflexible simulated annealing. This step was followed by explicit solvent refinement where the heparin and the protein interface residues were allowed to be flexible. The pair-wise “ligand interface root-mean-square deviation matrix” over all structures was calculated, and final structures were clustered using a cut-off value of 7.5 Å. The clusters were sorted using root-mean-square deviation and HADDOCK score. The molecular docking of KC to heparin was carried out as essentially described above. The structure of KC is not available, so we used a structural model generated from NMR chemical shifts as described (25).
MD simulations
Structural models of chemokine-heparin complexes obtained from HADDOCK runs consistent with both NMR and ITC data were used as starting geometries for unrestrained MD simulations. Each docked complex was prepared using AMBER-ff12SB and GLYCAM_06J-1 force field parameters in the Leap module of AMBER14 for the chemokine and heparin dp8, respectively (58, 59). The total charge of the complex was then brought to zero by adding an adequate number of counter ions. The charge-neutralized complex was centered in a three-point water box (TIP3P) with a minimum distance of 12 Å between the wall and the nearest atom in a complex followed by generation of initial coordinates and parameters for the starting solvated complex (60). Initially, the system was relaxed to achieve a minimum energy state. Energy minimization was then carried out in two steps with the nonbonded cutoff of 10 Å. In the first step, the solute atoms including the counter ions were restrained by a harmonic potential with a force constant of 100 kcal/(mol. Å2). The water molecules were relaxed using 500 cycles of steepest descent and 2000 cycles of conjugate gradient methods. In the second step, the whole system was relaxed using conjugate gradient minimization of 2500 cycles without any restraints. Equilibration and the simulation process were validated using the physical observables of the system, which confirmed that the system obeyed NPT ensemble. The MD simulations were performed over 100 ns with snapshots captured every 2 ps. The sugar puckering for IdoA2S and GlcNS6S was maintained at 2SO and 4C1 conformations, respectively. Binding free energies of the heparin–chemokine complexes were computed using the post-processing MM/PB(GB)SA method using the reordered trajectories (61). MMGBSA and MMPBSA methods employed single-residue energy decomposition to estimate energy contribution of the individual chemokine residues that mediate GAG binding. For both complexes, 5000 snapshots from 50 to 100 ns at equal intervals of time (10 ps) were considered for these calculations. MMPBSA calculations were carried out using default solvent probe radius and surface parameters using the Amber Tools15 (58, 61), whereas modified model parameters “OBC” (62), including an ion concentration of 0.10, were used for MMGBSA calculations. Both methods gave essentially similar results (63), of which MMPBSA results are being reported for sake of brevity.
Author contributions
K. M. S., U. R. D., and K. R. conceptualization; K. M. S. and B. N. data curation; K. R. supervision; K. M. S. and B. N. investigation; K. M. S. and B. N. methodology; K. M. S. and K. R. writing-original draft; K. M. S. project administration; K. M. S., B. N., and K. R. writing-review and editing; S. K. M., B. N., U. R. D., and K. R. formal analysis; U. R. D. and K. R. resources; U. R. D. and K. R. funding acquisition.
Supplementary Material
Acknowledgments
We thank Dr. Wang and Dr. Holthauzen for technical support, Emily Lowry and Brigith Penaranda for protein purification, and Dr. Heather Lander for editorial assistance. We also acknowledge the Sealy Center for Structural Biology and Molecular Biophysics at the University of Texas Medical Branch at Galveston for providing research resources. The Sealy Center for Structural Biology and Molecular Biophysics was supported by a Sealy and Smith Foundation grant.
This work was supported by National Institutes of Health Grants P01 HL107152, R21AI124681, and S10OD023576. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3 and Movies S1 and S2.
Both mice and humans express chemokines labeled as CXCL1 and CXCL2. In humans, CXCL1 is also known as MGSA or Groα, and CXCL2 is also known as Groβ. In mice, CXCL1 is also known as KC, and CXCL2 is also known as MIP2. To minimize confusion between human and mouse chemokines and for clarity, we explicitly define that KC and MIP2 correspond to mCXCL1 and mCXCL2 and use KC and MIP2 nomenclature throughout the manuscript; NAC, neutrophil-activating chemokine.
- CXC
- CXC ligand
- CXCR
- CXC chemokine receptor
- GAG
- glycosaminoglycan
- HSQC
- heteronuclear single quantum coherence
- ITC
- isothermal titration calorimetry
- MD
- molecular dynamics
- HS
- heparan sulfate
- PG
- proteoglycan.
References
- 1. Griffith J. W., Sokol C. L., and Luster A. D. (2014) Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu. Rev. Immunol. 32, 659–702 10.1146/annurev-immunol-032713-120145 [DOI] [PubMed] [Google Scholar]
- 2. Kobayashi Y. (2008) The role of chemokines in neutrophil biology. Front. Biosci. 13, 2400–2407 10.2741/2853 [DOI] [PubMed] [Google Scholar]
- 3. Bonecchi R., Galliera E., Borroni E. M., Corsi M. M., Locati M., and Mantovani A. (2009) Chemokines and chemokine receptors: an overview. Front. Biosci. (Landmark Ed.) 14, 540–551 [DOI] [PubMed] [Google Scholar]
- 4. Zlotnik A., and Yoshie O. (2000) Chemokines: a new classification system and their role in immunity. Immunity 12, 121–127 10.1016/S1074-7613(00)80165-X [DOI] [PubMed] [Google Scholar]
- 5. Fernandez E. J., and Lolis E. (2002) Structure, function, and inhibition of chemokines. Annu. Rev. Pharmacol. Toxicol. 42, 469–499 10.1146/annurev.pharmtox.42.091901.115838 [DOI] [PubMed] [Google Scholar]
- 6. Call D. R., Nemzek J. A., Ebong S. J., Bolgos G. R., Newcomb D. E., Wollenberg G. K., and Remick D. G. (2001) Differential local and systemic regulation of the murine chemokines KC and MIP2. Shock 15, 278–284 10.1097/00024382-200115040-00005 [DOI] [PubMed] [Google Scholar]
- 7. Luscinskas F. W. (2013) CXCL1 excess stops neutrophils in their tracks. Blood 122, 3708–3710 10.1182/blood-2013-10-531905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zwijnenburg P. J., Polfliet M. M., Florquin S., van den Berg T. K., Dijkstra C. D., van Deventer S. J., Roord J. J., van der Poll T., and van Furth A. M. (2003) CXC-chemokines KC and macrophage inflammatory protein-2 (MIP-2) synergistically induce leukocyte recruitment to the central nervous system in rats. Immunol. Lett. 85, 1–4 10.1016/S0165-2478(02)00200-6 [DOI] [PubMed] [Google Scholar]
- 9. De Filippo K., Dudeck A., Hasenberg M., Nye E., van Rooijen N., Hartmann K., Gunzer M., Roers A., and Hogg N. (2013) Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood 121, 4930–4937 10.1182/blood-2013-02-486217 [DOI] [PubMed] [Google Scholar]
- 10. Wengner A. M., Pitchford S. C., Furze R. C., and Rankin S. M. (2008) The coordinated action of G-CSF and ELR + CXC chemokines in neutrophil mobilization during acute inflammation. Blood 111, 42–49 10.1182/blood-2007-07-099648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Craciun F. L., Schuller E. R., and Remick D. G. (2010) Early enhanced local neutrophil recruitment in peritonitis-induced sepsis improves bacterial clearance and survival. J. Immunol. 185, 6930–6938 10.4049/jimmunol.1002300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jin L., Batra S., Douda D. N., Palaniyar N., and Jeyaseelan S. (2014) CXCL1 contributes to host defense in polymicrobial sepsis via modulating T cell and neutrophil functions. J. Immunol. 193, 3549–3558 10.4049/jimmunol.1401138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shea-Donohue T., Thomas K., Cody M. J., Aiping Z., Detolla L. J., Kopydlowski K. M., Fukata M., Lira S. A., and Vogel S. N. (2008) Mice deficient in the CXCR2 ligand, CXCL1 (KC/GRO-alpha), exhibit increased susceptibility to dextran sodium sulfate (DSS)-induced colitis. Innate Immun. 14, 117–124 10.1177/1753425908088724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tanino Y., Coombe D. R., Gill S. E., Kett W. C., Kajikawa O., Proudfoot A. E., Wells T. N., Parks W. C., Wight T. N., Martin T. R., and Frevert C. W. (2010) Kinetics of chemokine-glycosaminoglycan interactions control neutrophil migration into the airspaces of the lungs. J. Immunol. 184, 2677–2685 10.4049/jimmunol.0903274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Massena S., Christoffersson G., Hjertström E., Zcharia E., Vlodavsky I., Ausmees N., Rolny C., Li J. P., and Phillipson M. (2010) A chemotactic gradient sequestered on endothelial heparan sulfate induces directional intraluminal crawling of neutrophils. Blood 116, 1924–1931 10.1182/blood-2010-01-266072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nourshargh S., and Alon R. (2014) Leukocyte migration into inflamed tissues. Immunity 41, 694–707 10.1016/j.immuni.2014.10.008 [DOI] [PubMed] [Google Scholar]
- 17. Monneau Y., Arenzana-Seisdedos F., and Lortat-Jacob H. (2016) The sweet spot: how GAGs help chemokines guide migrating cells. J. Leukoc. Biol. 99, 935–953 10.1189/jlb.3MR0915-440R [DOI] [PubMed] [Google Scholar]
- 18. Schaefer L., and Schaefer R. M. (2010) Proteoglycans: from structural compounds to signaling molecules. Cell Tissue Res. 339, 237–246 10.1007/s00441-009-0821-y [DOI] [PubMed] [Google Scholar]
- 19. Xu D., and Esko J. D. (2014) Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 83, 129–157 10.1146/annurev-biochem-060713-035314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li L., Ly M., and Linhardt R. J. (2012) Proteoglycan sequence. Mol Biosyst 8, 1613–1625 10.1039/c2mb25021g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dutta A. K., Rösgen J., and Rajarathnam K. (2015) Using isothermal titration calorimetry to determine thermodynamic parameters of protein-glycosaminoglycan interactions. Methods Mol. Biol. 1229, 315–324 10.1007/978-1-4939-1714-3_25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ladbury J. E., Klebe G., and Freire E. (2010) Adding calorimetric data to decision making in lead discovery: a hot tip. Nat. Rev. Drug Discov. 9, 23–27 10.1038/nrd3054 [DOI] [PubMed] [Google Scholar]
- 23. Rajasekaran D., Keeler C., Syed M. A., Jones M. C., Harrison J. K., Wu D., Bhandari V., Hodsdon M. E., and Lolis E. J. (2012) A model of GAG/MIP-2/CXCR2 interfaces and its functional effects. Biochemistry 51, 5642–5654 10.1021/bi3001566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Joseph P. R., Mosier P. D., Desai U. R., and Rajarathnam K. (2015) Solution NMR characterization of chemokine CXCL8/IL-8 monomer and dimer binding to glycosaminoglycans: structural plasticity mediates differential binding interactions. Biochem. J. 472, 121–133 10.1042/BJ20150059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Poluri K. M., Joseph P. R., Sawant K. V., and Rajarathnam K. (2013) Molecular basis of glycosaminoglycan heparin binding to the chemokine CXCL1 dimer. J. Biol. Chem. 288, 25143–25153 10.1074/jbc.M113.492579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sepuru K. M., and Rajarathnam K. (2016) CXCL1/MGSA is a novel glycosaminoglycan (GAG)-binding chemokine: structural evidence for two distinct non-overlapping binding domains. J. Biol. Chem. 291, 4247–4255 10.1074/jbc.M115.697888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sepuru K. M., Nagarajan B., Desai U. R., and Rajarathnam K. (2016) Molecular basis of chemokine CXCL5-glycosaminoglycan interactions. J. Biol. Chem. 291, 20539–20550 10.1074/jbc.M116.745265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brown A. J., Sepuru K. M., Sawant K. V., and Rajarathnam K. (2017) Platelet-derived chemokine CXCL7 dimer preferentially exists in the glycosaminoglycan-bound form: implications for neutrophil-platelet crosstalk. Front. Immunol. 8, 1248 10.3389/fimmu.2017.01248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rajarathnam K., Sepuru K. M., Joseph P. R. B., Sawant K. V., and Brown A. J. (2018) Glycosaminoglycan interactions fine-tune chemokine-mediated neutrophil trafficking: structural insights and molecular mechanisms. J. Histochem. Cytochem. 66, 229–239 10.1369/0022155417739864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roe D. R., and Cheatham T. E. 3rd (2013) PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 10.1021/ct400341p [DOI] [PubMed] [Google Scholar]
- 31. Sankaranarayanan N. V., Nagarajan B., and Desai U. R. (2018) So you think computational approaches to understanding glycosaminoglycan–protein interactions are too dry and too rigid? Think again! Curr. Opin. Struct. Biol. 50, 91–100 10.1016/j.sbi.2017.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Samsonov S. A., Teyra J., and Pisabarro M. T. (2011) Docking glycosaminoglycans to proteins: analysis of solvent inclusion. J. Comput. Aided Mol. Des. 25, 477–489 10.1007/s10822-011-9433-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Johnson D. J., Langdown J., and Huntington J. A. (2010) Molecular basis of factor IXa recognition by heparin-activated antithrombin revealed by a 1.7-A structure of the ternary complex. Proc. Natl. Acad. Sci. U.S.A. 107, 645–650 10.1073/pnas.0910144107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gandhi N. S., and Mancera R. L. (2009) Free energy calculations of glycosaminoglycan-protein interactions. Glycobiology 19, 1103–1115 10.1093/glycob/cwp101 [DOI] [PubMed] [Google Scholar]
- 35. Sarkar A., and Desai U. R. (2015) A simple method for discovering druggable, specific glycosaminoglycan-protein systems: elucidation of key principles from heparin/heparan sulfate-binding proteins. PLoS One 10, e0141127 10.1371/journal.pone.0141127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ojala W. H., Sudbeck E. A., Lu L. K., Richardson T. I., Lovrien R. E., and Gleason W. B. (1996) Complexes of lysine, histidine, and arginine with sulfonated azo dyes: model systems for understanding the biomolecular recognition of glycosaminoglycans by proteins. J. Am. Chem. Soc. 118, 2131–2142 10.1021/ja951121f [DOI] [Google Scholar]
- 37. Rajagopalan L., and Rajarathnam K. (2006) Structural basis of chemokine receptor function: a model for binding affinity and ligand selectivity. Biosci. Rep. 26, 325–339 10.1007/s10540-006-9025-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Joseph P. R., and Rajarathnam K. (2015) Solution NMR characterization of WT CXCL8 monomer and dimer binding to CXCR1 N-terminal domain. Protein Sci. 24, 81–92 10.1002/pro.2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ravindran A., Sawant K. V., Sarmiento J., Navarro J., and Rajarathnam K. (2013) Chemokine CXCL1 dimer is a potent agonist for the CXCR2 receptor. J. Biol. Chem. 288, 12244–12252 10.1074/jbc.M112.443762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Joseph P. R. B., Sawant K. V., and Rajarathnam K. (2017) Heparin-bound chemokine CXCL8 monomer and dimer are impaired for CXCR1 and CXCR2 activation: implications for gradients and neutrophil trafficking. Open Biol. 7, 170168 10.1098/rsob.170168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Monneau Y. R., Luo L., Sankaranarayanan N. V., Nagarajan B., Vivès R. R., Baleux F., Desai U. R., Arenzana-Seidedos F., and Lortat-Jacob H. (2017) Solution structure of CXCL13 and heparan sulfate binding show that GAG binding site and cellular signalling rely on distinct domains. Open Biol. 7, 170133 10.1098/rsob.170133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Panitz N., Theisgen S., Samsonov S. A., Gehrcke J. P., Baumann L., Bellmann-Sickert K., Köhling S., Pisabarro M. T., Rademann J., Huster D., and Beck-Sickinger A. G. (2016) The structural investigation of glycosaminoglycan binding to CXCL12 displays distinct interaction sites. Glycobiology 26, 1209–1221 [DOI] [PubMed] [Google Scholar]
- 43. Ziarek J. J., Veldkamp C. T., Zhang F., Murray N. J., Kartz G. A., Liang X., Su J., Baker J. E., Linhardt R. J., and Volkman B. F. (2013) Heparin oligosaccharides inhibit chemokine (CXC motif) ligand 12 (CXCL12) cardioprotection by binding orthogonal to the dimerization interface, promoting oligomerization, and competing with the chemokine (CXC motif) receptor 4 (CXCR4) N terminus. J. Biol. Chem. 288, 737–746 10.1074/jbc.M112.394064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Severin I. C., Gaudry J. P., Johnson Z., Kungl A., Jansma A., Gesslbauer B., Mulloy B., Power C., Proudfoot A. E., and Handel T. (2010) Characterization of the chemokine CXCL11-heparin interaction suggests two different affinities for glycosaminoglycans. J. Biol. Chem. 285, 17713–17724 10.1074/jbc.M109.082552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sawant K. V., Poluri K. M., Dutta A. K., Sepuru K. M., Troshkina A., Garofalo R. P., and Rajarathnam K. (2016) Chemokine CXCL1 mediated neutrophil recruitment: role of glycosaminoglycan interactions. Sci. Rep. 6, 33123 10.1038/srep33123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gangavarapu P., Rajagopalan L., Kolli D., Guerrero-Plata A., Garofalo R. P., and Rajarathnam K. (2012) The monomer–dimer equilibrium and glycosaminoglycan interactions of chemokine CXCL8 regulate tissue-specific neutrophil recruitment. J. Leukoc. Biol. 91, 259–265 10.1189/jlb.0511239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee J., Cacalano G., Camerato T., Toy K., Moore M. W., and Wood W. I. (1995) Chemokine binding and activities mediated by the mouse IL-8 receptor. J. Immunol. 155, 2158–2164 [PubMed] [Google Scholar]
- 48. Marki A., Esko J. D., Pries A. R., and Ley K. (2015) Role of the endothelial surface layer in neutrophil recruitment. J. Leukoc. Biol. 98, 503–515 10.1189/jlb.3MR0115-011R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Weinbaum S., Tarbell J. M., and Damiano E. R. (2007) The structure and function of the endothelial glycocalyx layer. Annu. Rev. Biomed. Eng. 9, 121–167 10.1146/annurev.bioeng.9.060906.151959 [DOI] [PubMed] [Google Scholar]
- 50. Iozzo R. V. (2005) Basement membrane proteoglycans: from cellar to ceiling. Nat. Rev. Mol. Cell Biol. 6, 646–656 10.1038/nrm1702 [DOI] [PubMed] [Google Scholar]
- 51. Meneghetti M. C., Hughes A. J., Rudd T. R., Nader H. B., Powell A. K., Yates E. A., and Lima M. A. (2015) Heparan sulfate and heparin interactions with proteins. J. R. Soc. Interface 12, 0589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sepuru K. M., Poluri K. M., and Rajarathnam K. (2014) Solution structure of CXCL5: a novel chemokine and adipokine implicated in inflammation and obesity. PLoS One 9, e93228 10.1371/journal.pone.0093228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shao W., Jerva L. F., West J., Lolis E., and Schweitzer B. I. (1998) Solution structure of murine macrophage inflammatory protein-2. Biochemistry 37, 8303–8313 10.1021/bi980112r [DOI] [PubMed] [Google Scholar]
- 54. Dominguez C., Boelens R., and Bonvin A. M. (2003) HADDOCK: a protein–protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 10.1021/ja026939x [DOI] [PubMed] [Google Scholar]
- 55. de Vries S. J., van Dijk A. D., Krzeminski M., van Dijk M., Thureau A., Hsu V., Wassenaar T., and Bonvin A. M. (2007) HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets. Proteins 69, 726–733 10.1002/prot.21723 [DOI] [PubMed] [Google Scholar]
- 56. Mulloy B., Forster M. J., Jones C., and Davies D. B. (1993) N.M.R., and molecular-modelling studies of the solution conformation of heparin. Biochem. J. 293, 849–858 10.1042/bj2930849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schüttelkopf A. W., and van Aalten D. M. (2004) PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363 10.1107/S0907444904011679 [DOI] [PubMed] [Google Scholar]
- 58. Case D. A., Darden T. A., Cheatham T. E. 3rd, Simmerling C. L., Wang J., Duke R. E., Luo R., Walker R. C., Zhang W., Merz K. M., Roberts B., Hayik S., Roitberg A., Seabra G., Swails J., et al. (2014) AMBER 14, University of California, San Francisco [Google Scholar]
- 59. Kirschner K. N., Yongye A. B., Tschampel S. M., González-Outeirino J., Daniels C. R., Foley B. L., and Woods R. J. (2008) GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 29, 622–655 10.1002/jcc.20820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 10.1063/1.445869 [DOI] [Google Scholar]
- 61. Miller B. R. 3rd, McGee T. D. Jr., Swails J. M., Homeyer N., Gohlke H., and Roitberg A. E. (2012) MMPBSA.py: an efficient program for end-state free energy calculations. J. Chem. Theory Comput. 8, 3314–3321 10.1021/ct300418h [DOI] [PubMed] [Google Scholar]
- 62. Onufriev A., Bashford D., and Case D. A. (2004) Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 55, 383–394 10.1002/prot.20033 [DOI] [PubMed] [Google Scholar]
- 63. Feig M., Onufriev A., Lee M. S., Im W., Case D. A., and Brooks C. L. 3rd. (2004) Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J. Comput. Chem. 25, 265–284 10.1002/jcc.10378 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.