

Hepatitis C virus infections affect 71 million people worldwide and cause severe chronic liver disease. Recently, efficient antiviral therapies have been established, with inhibitors of nonstructural protein NS5A as a cornerstone. NS5A is a central regulator of HCV replication and assembly but is still enigmatic in its molecular functions. It exists in two phosphoisoforms, p56 and p58. We identified a phosphopeptide exclusively found in p58 and analyzed the determinants involved in phosphorylation of this region. We found evidence for very different phosphorylation patterns resulting in p58. These results challenge the concept of p58 being a homogenous species of NS5A molecules phosphorylated at the same positions and argues for at least two independently phosphorylated variants showing the same electrophoretic mobility, likely serving different functions.
KEYWORDS: positive-strand RNA virus, hepatitis C virus, NS5A, phosphorylation, p58, p56, hyperphosphorylation, basal phosphorylation, PI4KA, PI4KIIIa
ABSTRACT
Hepatitis C virus (HCV) nonstructural protein 5A (NS5A) is a phosphoprotein with key functions in regulating viral RNA replication and assembly. Two phosphoisoforms are discriminated by their different apparent molecular weights: a basally phosphorylated (p56) and a hyperphosphorylated (p58) variant. The precise mechanisms governing p58 synthesis and specific functions of the isoforms are poorly understood. Our study aimed at a deeper understanding of determinants involved in p58 synthesis. We analyzed two variants of p56 and p58 of isolate JFH-1 separately by mass spectrometry using an expression model and thereby identified a threonine-rich phosphopeptide exclusively found in the hyperphosphorylated variant. Individual exchange of possible phosphoacceptor sites to phosphoablatant or -mimetic residues had little impact on HCV replication or assembly in cell culture. A phosphospecific antibody recognizing pT242 revealed that this position was indeed phosphorylated only in p58 and depended on casein kinase Iα. Importantly, phosphoablative mutations at positions T244 and S247 abrogated pT242 detection without substantial effects on global p58 levels, whereas mutations in the preceding serine-rich cluster dramatically reduced total p58 levels but had minor impact on pT242 levels, suggesting the existence of distinct subspecies of hyperphosphorylated NS5A. Mass spectrometry analyses of different genotypes showed variable phosphorylation patterns across NS5A and suggested that the threonine-rich region is also phosphorylated at T242 in gt4a and at S249 in gt1a, gt1b, and gt4a. Our data therefore indicate that p58 is not a single homogenously phosphorylated protein species but rather a population of various phosphoisoforms, with high variability between genotypes.
IMPORTANCE Hepatitis C virus infections affect 71 million people worldwide and cause severe chronic liver disease. Recently, efficient antiviral therapies have been established, with inhibitors of nonstructural protein NS5A as a cornerstone. NS5A is a central regulator of HCV replication and assembly but is still enigmatic in its molecular functions. It exists in two phosphoisoforms, p56 and p58. We identified a phosphopeptide exclusively found in p58 and analyzed the determinants involved in phosphorylation of this region. We found evidence for very different phosphorylation patterns resulting in p58. These results challenge the concept of p58 being a homogenous species of NS5A molecules phosphorylated at the same positions and argues for at least two independently phosphorylated variants showing the same electrophoretic mobility, likely serving different functions.
INTRODUCTION
Hepatitis C virus (HCV) is a positive-strand RNA virus and belongs to the family Flaviviridae. The viral genome encompasses about 9.6 kb and codes mainly for a polyprotein of about 3,000 amino acids (aa), which is flanked by nontranslated regions. The polyprotein is cleaved into ten mature proteins by cellular and viral proteases: core, envelope glycoprotein 1 (E1) and E2, p7, and the nonstructural (NS) proteins NS2, NS3, NS4A, NS4B, NS5A, and NS5B (reviewed in reference 1). The structural proteins core, E1, and E2 are physical components of the viral particle and, together with p7 and NS2, are mainly involved in the assembly of infectious virions, while NS3 to NS5B are mainly required for replication of the viral genome. NS3 exhibits helicase activity in the C-terminal part and an N-terminal protease responsible for cleavage of the nonstructural proteins in association with the cofactor NS4A. NS4B is a key factor in inducing membrane alterations that are required for viral replication, the so-called membranous web. NS5B is the viral RNA-dependent RNA polymerase.
NS5A is a phosphoprotein critically involved in the formation of the viral replication compartment (2) as well as in the morphogenesis of infectious virions (3–5). Due to this central role in various aspects of viral replication, NS5A inhibitors have become a cornerstone in recent antiviral therapies, simultaneously affecting different steps of the viral life cycle (6, 7). However, despite numerous studies, NS5A remains enigmatic in most of its functions regarding the underlying molecular mechanisms (reviewed in reference 8).
NS5A is built of an N-terminal amphipathic helix, essential for its membrane association (9), followed by three domains (DI, DII, and DIII), which are separated by two so-called low-complexity sequences (LCS-I and -II) (10). DI is structured, binds RNA (11), forms several dimeric isoforms (12–14), and is crucial for the formation of the membranous web (2). DII and DIII are intrinsically unstructured (15, 16). DII is widely dispensable for replication (3) but has recently been shown to dampen recognition of the virus by cytosolic pattern recognition receptors (17). DIII is involved in virion production by an interaction with the core, which is modulated by phosphorylation (3–5).
Two distinct NS5A phosphoisoforms have been described that are discriminated due to their different apparent molecular weights in SDS-PAGE: p56, the basally (hypo-)phosphorylated form, and p58, the hyperphosphorylated variant (18). P56 has been regarded as the major isoform involved in viral RNA replication, whereas p58 was supposed to inhibit replication and support assembly. This assumption was based mainly on the fact that LCS-I is a hot spot for adaptive mutations enhancing replication of many HCV genotypes in cell culture while at the same time abrogating p58 synthesis (19–21) and virus particle production (22). However, replication of HCV genotype 2a isolate JFH-1, which is still the only wild-type (WT) isolate efficiently replicating in cell culture (23), is impaired by the same adaptive mutations (24–26), challenging this concept. At this point, no distinct functions can be clearly assigned to p56 and p58.
Mass spectrometry (MS), reverse genetic studies, and proteomic analyses revealed phosphorylation of numerous serine and threonine residues within different domains of NS5A involving several serine/threonine kinases, thereby contributing to the functional diversity of NS5A (4, 5, 18–20, 24, 27–39). However, links between specific phosphorylation sites with particular kinases and specific functions have remained difficult and rare: phosphorylation at S356 (all numberings refer to NS5A of HCV JFH-1) is mediated by protein kinase A (PKA) and is necessary to facilitate interaction of NS5A with other SH3 domains, which is crucial for replication (40). Pharmacological and genetic approaches identified phosphorylation at S457 by casein kinase II (CKII) to be important for virus particle production (5, 27). In contrast, residue S222 has been identified as a major phosphorylation site by many proteomic studies (25, 26, 33, 41), but the phenotype of phosphoablative or phosphomimetic mutations at this site is weak (25, 33, 41).
Particularly enigmatic are the determinants of p58 synthesis. Generally, NS5A hyperphosphorylation requires the presence of NS3-5B as a polyprotein (39, 42–44) and is not found if NS5A is expressed as a single protein (39, 45, 46). In addition, completely unrelated mutations in other NS proteins (42, 43), as well as NS5A inhibitors, affect hyperphosphorylation (31, 47), overall suggesting that p58 synthesis requires a complex succession of events supported by other nonstructural proteins, likely due to serial interactions with different kinases.
A serine (Ser)-rich cluster in LCS-I is the most critical determinant of p58 synthesis, since phosphoablative mutations in this region strongly reduce hyperphosphorylation (e.g., S225, S228, and S232). Furthermore, pS232, pS235, and pS238 have been exclusively found in p58 using phosphospecific antibodies (34, 48). The main kinase known to be essential for the formation of p58 is CKIα, directly targeting residues within the Ser-rich cluster, e.g., S232 (49). Inhibition of CKIα abrogates NS5A hyperphosphorylation and strongly affects viral replication and particle production (26, 49). CKIα can perform multiple sequential phosphorylation steps, using already-existing phosphates as priming sites. Therefore, it has been speculated that p58 is a product of saltatory phosphorylation by CKIα within the Ser-rich cluster of LCS-I (25, 37). This hypothesis is particularly attractive, since several Ser residues within LCS-I follow the sequential phosphorylation pattern of CKIα (pS-X-X-S), e.g., S229-S232-S235-S238, and a recent study indeed provides proof in the case of 232-235-238 (48). However, the kinases involved in priming phosphorylation are not clear yet. Additional kinases involved in the synthesis of p58 are polo-like kinases (35) and calmodulin-dependent kinase II (CAMK2) γ and δ (28). In contrast, phosphorylation at S146 by a yet-to-be defined kinase has been shown to negatively regulate hyperphosphorylation of isolate JFH-1, likely by modulating the dimeric conformation of domain I (25). In addition, the lipid kinase phosphatidylinositol 4-kinase α (PI4KA or PI4KIIIα) has been shown to suppress p58 synthesis by an unknown mechanism (39). Overall, the succession of events involved in formation of p58, as well as distinct phosphoacceptors that are phosphorylated apart from S222, S232, S235, and S238, are still poorly understood.
NS5A is a central hub for interactions with host factors, and literally hundreds of cellular proteins have been identified to directly interact with it (reviewed in reference 8). Phosphorylation events are indeed a main suspect in the regulation of this plethora of protein-protein interactions, with a few examples suggesting specific functions of p58. The human vesicle-associated membrane protein A (hVAP-A) has been shown to be important for viral replication but only interacts with basally phosphorylated NS5A, whereas hyperphosphorylation disrupts the interaction (46). A recent study showed that a phosphoablative mutation at position S225, which was supposed to be phosphorylated in p58, impaired NS5A interactions with the nucleosome assembly protein 1-like protein 1 (NAP1L1), bridging integrator 1 (Bin1), and again hVAP-A (50), suggesting that they are regulated by phosphorylation at this site. The lipid kinase PI4KA furthermore, has been shown to interact with NS5A in multiple ways and also in a phosphorylation-dependent manner. PI4KA is a key factor in the biogenesis of the HCV replication compartment, supplying phosphatidylinositol 4-phosphate (PI4P) (51). PI4KA mainly interacts with a specific region adjacent to LCS-I, designated the PI4KA functional interaction site (PFIS), and its enzymatic activity is activated by this interaction (39). However, activation furthermore, requires phosphorylation at positions S225, S232, and S235 based on genetic evidence (21). In turn, PI4KA negatively affects p58 abundance by an unknown mechanism, since NS5A mutants with defects in PI4KA interaction show increased levels of hyperphosphorylated NS5A (39).
The aim of our study was a deeper understanding of the hyperphosphorylated p58 isoform of HCV NS5A. We therefore performed a phosphoproteomic analysis of NS5A, including the WT and a PFIS mutant with increased p58 levels, with a separate analysis of p56 and p58. We thereby identified a peptide exclusively in p58 representing the threonine (Thr)-rich cluster in LCS-I. While phosphoablatant and -mimetic mutations had no impact on replication and assembly, we could verify phosphorylation at position T242 using a phosphospecific antibody. Our data show that phosphorylation at T242 is widely independent of the preceding Ser-rich cluster and defines a minor subspecies of p58. Therefore, p58, so far defined only by its electrophoretic mobility in SDS-PAGE, seems to consist of several independently phosphorylated isoforms, probably serving different functions.
RESULTS
Phospho-mass spectrometry analysis of NS5A.
In our recent study we found that the lipid kinase PI4KA modulated NS5A phosphorylation, and we identified mutations in the PFIS interfering with PI4KA interaction, resulting in increased p58 levels (39). To identify specific phosphorylation sites modulated by PI4KA, we employed a mass spectrometry analysis, comparing the phosphorylation pattern of the WT and a mutated NS5A variant. Since all PFIS mutants were replication deficient (39), we transiently expressed NS3-5B JFH-1 under transcriptional control of the T7 promoter in Huh7-Lunet cells constitutively expressing T7 RNA polymerase. NS5A was immunoprecipitated, and bands corresponding to p58 or p56 were individually excised after SDS-PAGE, digested with trypsin, separated by high-performance liquid chromatography, and subjected to tandem mass spectrometry (MS/MS) analyses (Fig. 1A and Table 1; see also Table S1 in the supplemental material). Peptides and phosphorylation status were identified by their distinct mass and charge in the first mass spectrometry run. In the second run peptides were fragmented, allowing us to determine the position of the phosphorylation event within the peptide. We thereby identified a total of seven distinctly phosphorylated peptides located across all domains of NS5A (Fig. 1B and Table 1), four of which (II, IV, V, and VI) were identical to those of previous studies (25, 52). We could specifically determine phosphorylation sites at positions S146 (peptide II) and S360 (peptide VII) with an accuracy over 97%. In addition, the sites T14 (peptide I), T164 (peptide III), and T348 (peptide VI) could be clearly identified as phosphoacceptor sites inside the respective peptide and are therefore most likely phosphorylated, even though the PEP values are relatively high for these peptides. Peptide IV represents the well-characterized Ser-rich cluster where we identified single phosphorylations at positions S222 and S225 with a significant PEP score. We were not able to determine multiple phosphorylations within this peptide, although they are already known from other studies using reverse genetics. This would have required additional peptide enrichment and higher detection sensitivity, which was not included in our analysis. For the potential phosphoacceptor sites in peptide V, we could not conclusively determine which site exactly is phosphorylated (Table S1), but there was some evidence for phosphorylation at position T242 in sample C1 (Fig. 1C). We found only minor qualitative differences between the WT and mutant NS5A, not allowing us to draw conclusions on phosphoacceptor sites influenced by PI4KA. Interestingly, six of the phosphopeptides were found in both NS5A phosphoisoforms. In contrast, peptide I was only found in p56, whereas phosphorylation of peptide V was restricted to p58, although we cannot formally exclude minor amounts below the detection limit in p56. Peptide V is located at the very beginning of domain II in the interferon sensitivity-determining region (ISDR) adjacent to LCS-I and the serine-rich cluster (peptide IV), which has been identified before as a main determinant of hyperphosphorylation (25, 26, 34, 41, 48).
FIG 1.

Identification of NS5A phosphorylation sites. (A) Huh7-Lunet T7 cells were transfected with constructs encompassing NS3-5B JFH-1 with the NS5A WT or harboring a triple-alanine mutation in the PFIS, abrogating PI4KA interaction (mut5A [39]). Twenty-four h later, cells were lysed, NS5A was immunoprecipitated, and p58 and p56 bands were cut out after SDS-PAGE and subjected to mass spectrometry analysis to identify phosphorylated peptides. (B) Schematic of the different domains of NS5A. The location of the identified phosphopeptides is indicated below. All numberings on top refer to amino acid positions within NS5A JFH-1 (GenBank accession number KC164983.1). Residues shown in red are sites identified to be phosphorylated with high probability. Additional potential phosphoacceptor sites within each peptide that could not be identified as phosphorylated are shown in a smaller font size. Residues in italics of peptide V represent potential phosphoacceptor, where the exact site of phosphorylation could not be determined. Asterisks mark the residues already reported elsewhere: S146 (25), S222 (25, 41, 65), S225 (25), S232 (48), S235/238 (34), T249 (52), T348 (25), and T356 (40). (C) MSMS spectrum of peptide AT(ph)CTTHSNTYDVDMVDANLLM(ox)EGGVAQTEPESR with a mass of 3,723.5376 Da from NS3-3′ JFH-1 detected in sample C1. This peptide was identified with a MaxQuant score of 65.19. Annotated fragments are color coded. y ions, red; b ions, blue; internal fragments, purple; a, b, and y ions and internal fragments with additional loss of ammonia, water or phosphoric acid, yellow. MaxQuant protein database software annotated the threonine in position two as most likely to be phosphorylated by the relation of the nonphosphorylated singly charged internal fragment TTHSNTYDVDM(ox)VDANL (m/z = 1,793.76) and the phosphorylated internal fragment T(ph)CTTHSNTYD (m/z = 1,261.42).
TABLE 1.
Phosphopeptides of wild-type (C1 and C2) and mutated (C3 and C4) HCV NS5A proteinsa
| No. | Sequence | Phosphopeptide detection |
Mass (Da) | PEP | Score | |||
|---|---|---|---|---|---|---|---|---|
| C1 (p58) | C2 (p56) | C3 (p58) | C4 (p56) | |||||
| I | DVWDWVCT14ILTDFK | X | X | 1,876.8107 | 0.0077177 | 59.542 | ||
| II | IPCQLPS146PEFFSWVDGVQIHR | X | X | X | X | 2,591.2032 | 7.61E−12 | 156.39 |
| III | FAPT164PKPFFR | X | X | X | X | 1,286.6213 | 0.00053623 | 90.434 |
| IV | GS222PPS225EASSSVSQLSAPSLR | X | X | X | X | 2,022.9259 | 4.1E−135 | 393.39 |
| V | AT242CT244T245HS247NT249Y250DVDMVDANLLMEGGVAQTEPESR | X | X | 3,723.5376 | 1.67E−09 | 122.29 | ||
| VI | APT348PPPR (KAPT348PPPR, KAPT348PPPRR) | X | X | X | X | 814.3739 | 0.0004631 | 101.45 |
| VII | RRT356VGLS360ESTISEALQQLAIK | X | X | X | X | 2,379.2523 | 1.07E−14 | 178.99 |
| VII | T356VGLS360ESTISEALQQLAIK | X | X | X | X | 2,067.0501 | 1.82E−70 | 296.79 |
HCV NS5A proteins were identified by Orbitrap MS analysis and MaxQuant data analysis (Fig. 1A). Predominant phosphorylation sites identified by microsequencing are highlighted in boldface in the peptide sequences. Represented in italics are phosphoacceptors where the analysis could not clearly assign the phosphorylation to a specific site. Instances of phosphopeptides being detected in C1, C2, C3, and C4 are marked with an X. The mass of the phosphopeptides, the posterior error probability score (PEP; <0.02), and the individual peptide scores are presented for the most abundant MS scans. Note that all peptides identified contained only one phosphorylation event. Detection of multiply phosphorylated peptides would require previous enrichment, which was not done.
In summary, we identified seven phosphorylated peptides and eight distinct phosphorylation sites in different regions of NS5A and, for the first time, identified a phosphopeptide that exclusively mapped to the hyperphosphorylated form of NS5A.
Mutational analysis of potential phosphoacceptor sites in NS5A.
We next analyzed the impact of mutations in potential Ser/Thr phosphoacceptor sites identified by mass spectrometry on HCV replication, virion production, and NS5A phosphorylation using the reporter virus JcR2a, encoding renilla luciferase (Fig. 2A to C). We excluded the multiple serine residues in peptide IV at this point, since they have already been analyzed in multiple studies (25, 26, 41), as well as T14, since this was only found phosphorylated in p56 (Table 1). Each putatively phosphorylated serine or threonine residue was mutated to either alanine, to prevent phosphorylation, or to aspartic acid, mimicking phosphorylation by adding a negative charge. Interestingly, none of the individual mutations had a strong impact on viral replication (Fig. 2B) or virus production (Fig. 2C). In the case of T356 this was unexpected, since a recent study showed a lethal phosphoablative mutation at this site in the case of JFH-1 (40). Only a conversion of the five N-terminal Thr/Ser residues in peptide V to phosphomimetic aspartate strongly impaired replication (N-termD), whereas the phosphoablatant variant replicated with efficiency identical to that of the wild type, suggesting that phosphorylation events in this Thr-rich cluster are not essential for replication but that accumulation of negative charges is deleterious.
FIG 2.

Impact of phosphoablatant and -mimetic mutations at potential phosphoacceptor sites on replication and virus production. (A) Schematic representation of the experimental procedure. Huh7-Lunet cells were transfected with WT and mutant full-length monocistronic JcR2a reporter virus genomes encoding renilla luciferase, and RNA replication efficiency was determined by luciferase measurement. Supernatants of transfected cells were transferred to Huh7.5 cells to assess production of infectious virus, again by quantification of luciferase activity 72 h postinfection (hpi). hpe, hours postelectroporation. (B and C) Analysis of phosphoacceptor sites harboring alterations of serine and threonine residues to alanine (to A) or aspartate (to D). Mutant “N-term” comprises all five serine and threonine mutations from T242 to T249. Mutant “C-term” comprises substitutions at both positions T268 and S272. (B) Replication is represented in relative light units (RLU) measured 72 h posttransfection and normalized to 4 h posttransfection. A replication-deficient JcR2a variant harboring a deletion in NS5B was used as a negative control for replication (ΔGDD), indicated by the orange line. A green line highlights the replication level of JcR2a WT. The data shown are the mean values with standard deviations (SD) from three independent experiments with two technical replicates each. (C) Supernatants of transfected cells described for panel B were used for reinfection of naive Huh7.5 cells to assess production of infectious virus. Huh7.5 cells were lysed 72 h postinfection and analyzed for luciferase activity. Virus production efficiency is expressed in luciferase activity RLU 72 h postinfection relative to 72 h posttransfection in order to normalize for defects in RNA replication. An assembly-deficient JcR2a variant harboring a deletion in E1E2 proteins was used as a negative control for reinfection (ΔE1E2), indicated by the red line. A green line indicates the virus production of JcR2a WT. The data shown are the mean values with standard deviations from two independent experiments with two technical replicates each.
The phosphorylation pattern of the different mutants was comparable between cells replicating a full-length viral genome (Fig. 3A) or expressing NS3-NS5B (Fig. 3B), suggesting that transient expression of NS3-5B was indeed a feasible model to analyze determinants of NS5A phosphorylation, as shown before (39). The p58/p56 ratio (Fig. 3C) validated this observation. Except for reduced p58 levels in the case of S146D, which has been observed previously (25), no gross changes in the relative abundance of the phosphoisoforms were observed for any other individual mutation. However, the apparent molecular weight of p56 was clearly increased for the phosphomimetic mutants at positions T242, T244, S247, and N-term, suggesting that phosphorylation at these sites contributes to NS5A hyperphosphorylation. Furthermore, mutant N-termD only gave rise to a single band of a size comparable to that of p58. These data were in agreement with the observation that the phosphopeptide encompassing these positions was the only one exclusively found in p58. In addition, the Thr-rich cluster in LCS-I was directly adjacent to S235 and S238, which have been found to be exclusively phosphorylated in p58 as well (34). We therefore decided to focus our further studies on the contribution of the Thr-rich cluster in peptide V to the hyperphosphorylation of NS5A.
FIG 3.

Comparison of electrophoretic mobility of NS5A phosphoisoforms upon virus replication and NS3-5B expression. Huh7-Lunet cells were electroporated with RNA encoding the full-length reporter virus JcR2a (A), or Huh7-Lunet T7 cells were transfected with pTM vectors expressing NS3-5B of isolate JFH-1 (B), either the WT or a mutant harboring the indicated phosphoablative alanine (A) or the phosphomimetic aspartic acid (D) mutations. Cells were lysed 72 h (A) or 24 h after transfection (B) and subjected to Western blotting using NS5A-specific antibody 9E10. The data shown are from one representative experiment (n = 2). (C) Quantification of the p58/p56 ratios comparing JcR2a (gray bars) and pTM expression (black bars) based on the Western blots shown in panels A and B. n.d., not done, either due to replication defects (JcR2a) or due to the lack of separation between p56 and p58. The quantifications shown are from two experiments (n = 2). Note for this and subsequent figures that the phosphoisoforms of NS5A of isolate JFH-1 have higher apparent molecular weights than 56 and 58 kDa but are still referred to as p56 and p58, respectively.
A phosphospecific antibody reveals phosphorylation of T242 exclusively in p58.
To verify phosphorylation events in the Thr-rich cluster of peptide V, we aimed to generate phosphospecific antibodies, which would further allow studying determinants modulating phosphorylation. All HCV genotypes encode several potential phosphoacceptor sites in this region, but only position T242 is fully conserved. In addition, there was evidence by mass spectrometry indicating phosphorylation of this site (Fig. 1C), and T242 was in the neighborhood of the bona fide phosphorylation site S238. We therefore used a peptide encompassing amino acids 236 to 251 of JFH-1 NS5A, with a single phosphorylation at position T242 to generate phosphospecific antibodies targeting pT242. Indeed, after positive selection against the phosphorylated and negative selection with the unphosphorylated peptide, only the phosphorylated peptide was recognized with high specificity (Fig. 4A). These results overall indicated that the pT242-specific antibodies indeed could be used to characterize phosphorylation events at this site, with the limitation that proteins with multiple phosphorylations at neighboring positions might be missed.
FIG 4.
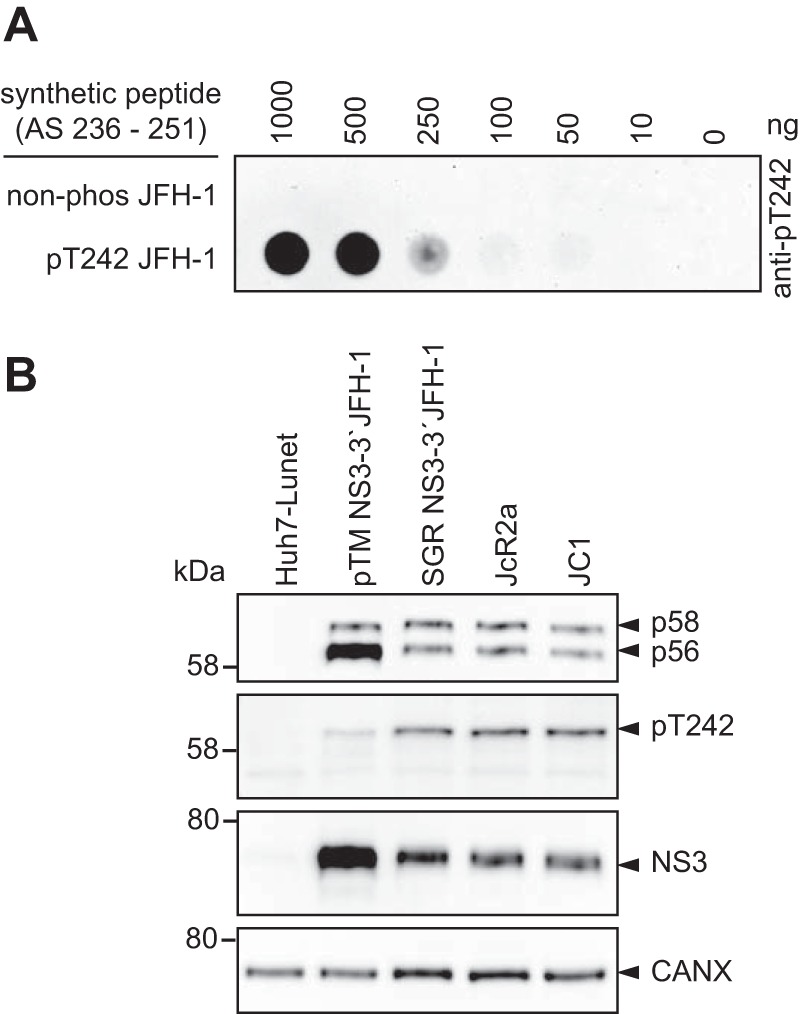
T242 is exclusively phosphorylated in the hyperphosphorylated form of NS5A. (A) Different concentrations of peptide either phosphorylated at position T242 or nonphosphorylated were titrated on PVDF membrane and incubated with purified, polyclonal anti-pT242 antibodies. (B) Huh7-Lunet T7 cells were either transfected with a pTM vector encoding JFH-1 NS3-3′ or electroporated with in vitro transcripts encoding a bicistronic reporter replicon (SGR JFH-1), a full-length reporter virus genome (JcR2a), or an unmodified full-length virus genome (JC1). The cells were lysed 24 h posttransfection or 72 h postelectroporation, respectively, and analyzed by 7.5% SDS-PAGE/Western blotting using anti-NS5A (monoclonal 9E10)-, anti-NS5A-pT242-, anti-NS3-, and anti-calnexin-specific antibodies. Shown is one representative experiment (n = 2). Note the discrepancy between the 58-kDa molecular weight (MW) marker band (left) and the band referred to as p58/pT242 (right), which is due to a higher apparent MW of the NS5A phosphoisoforms in the case of JFH-1.
We next analyzed the phosphorylation of T242 in cells either expressing NS3-5B or replicating a NS3-3′ JFH-1 subgenomic replicon (SGR JFH-1) or full-length viral genomes (JcR2a and JC1) (Fig. 4C). In all cases a clear band was detectable corresponding to p58, demonstrating that T242 indeed was phosphorylated upon HCV replication and expression of NS3-5B. However, pT242 seemed to be slightly more abundant in replicating HCV than in the pTM expression model (Fig. 4B). This effect might be caused by the higher expression level in the case of using pTM vectors, probably exceeding the phosphorylation capacity of kinases involved in p58 synthesis. The restriction of the signal to the p58 band was in line with the results from mass spectrometry, indicating that the entire peptide V, comprising T242, is only phosphorylated in the p58 fraction (Table 1). However, a minor reactivity of the antibodies to p56 was found in some samples that could be due either to residual antibodies binding to the nonphosphorylated epitope or to a minor fraction of p56 phosphorylated at T242.
Taking our findings together, we were able to generate antibodies specific to pT242 in NS5A, demonstrating that T242 was indeed phosphorylated upon HCV replication only in the p58 isoform.
Subcellular localization of NS5A phosphorylated at T242.
After having validated the genotype and phosphorylation specificity of pT242 detection by the antibody, we next studied the kinetics of T242 phosphorylation and the subcellular localization of NS5A phosphorylated at T242 (Fig. 5) upon transfection of a JFH-1 subgenomic reporter replicon. Luciferase activity already peaked after 24 h, while NS5A became clearly detectable only after 48 h (Fig. 5A and B). However, no clear kinetic difference was observed in a Western blot comparing the pT242-specific antibody and the monoclonal antibody 9E10, detecting both phosphoisoforms of NS5A, indicating that pT242 phosphorylation was not delayed (Fig. 5B). In immunofluorescence analysis the pT242 signal again was barely detectable at 24 h, even in cells with clear 9E10 signal, likely due to the limited sensitivity and the slight background signal generated by this antibody. However, at 48 h and 72 h the pT242 signal was found in the same dot-like distribution and at all sites with a strong total NS5A signal, suggesting that NS5A phosphorylated at T242 has no particularly different localization compared to that of total NS5A (Fig. 5C). This result was confirmed by a quantification of colocalization upon expression of NS3-5B in Huh7-Lunet T7 cells (Fig. 6). Here we also included a replication-deficient NS5A mutant with decreased PI4KA interaction, giving rise to increased p58 levels (PPH) (39). The pT242 signal widely and significantly colocalized with the total NS5A signal (Fig. 6A and C). In contrast, not all NS5A colocalized with pT242, as expected for a p58-specific antibody. For further control we used a pS235-specific antibody, which was previously shown to be an efficient marker for hyperphosphorylated NS5A (Fig. 6B and D) (34), and obtained a result similar to that for pT242. In the case of NS5A mutPPH, colocalization was even more prominent, likely due to the higher signal intensity and to the more focused, clustered perinuclear localization of the NS5A signal (Fig. 6A and B) (39).
FIG 5.

NS5A phosphorylated at T242 has no distinct subcellular localization compared to total NS5A. Huh7-Lunet cells were transfected with in vitro transcripts encoding a JFH-1 reporter replicon (A to C) and analyzed for RNA replication (A), NS5A phosphorylation (B), and pT242 localization (C) at the indicated time points. (A) RNA replication of a WT replicon compared to a replication-deficient mutant (ΔGDD) is represented in relative light units (RLU) measured 24 h, 48 h, and 72 h posttransfection and shown as fold relative to values at 4 h posttransfection to normalize for transfection efficiency. (B) Transfected cells from panel A were lysed and analyzed by Western blotting for NS5A phosphorylation using anti-NS5A (monoclonal antibody 9E10)-, anti-NS5A-pT242-, and anti-calnexin (CAXN)-specific antibodies, as indicated on the right. (C) Cells were seeded on coverslips and fixed at the indicated time points postelectroporation. Total NS5A and NS5A phosphorylated at T242 were detected by immunofluorescence analysis using monoclonal antibody 9E10 (red) and anti-NS5A-pT242 (green), respectively. Nuclei were stained with DAPI. Scale bar, 10 µm. Shown is a representative experiment of two with comparable outcomes.
FIG 6.
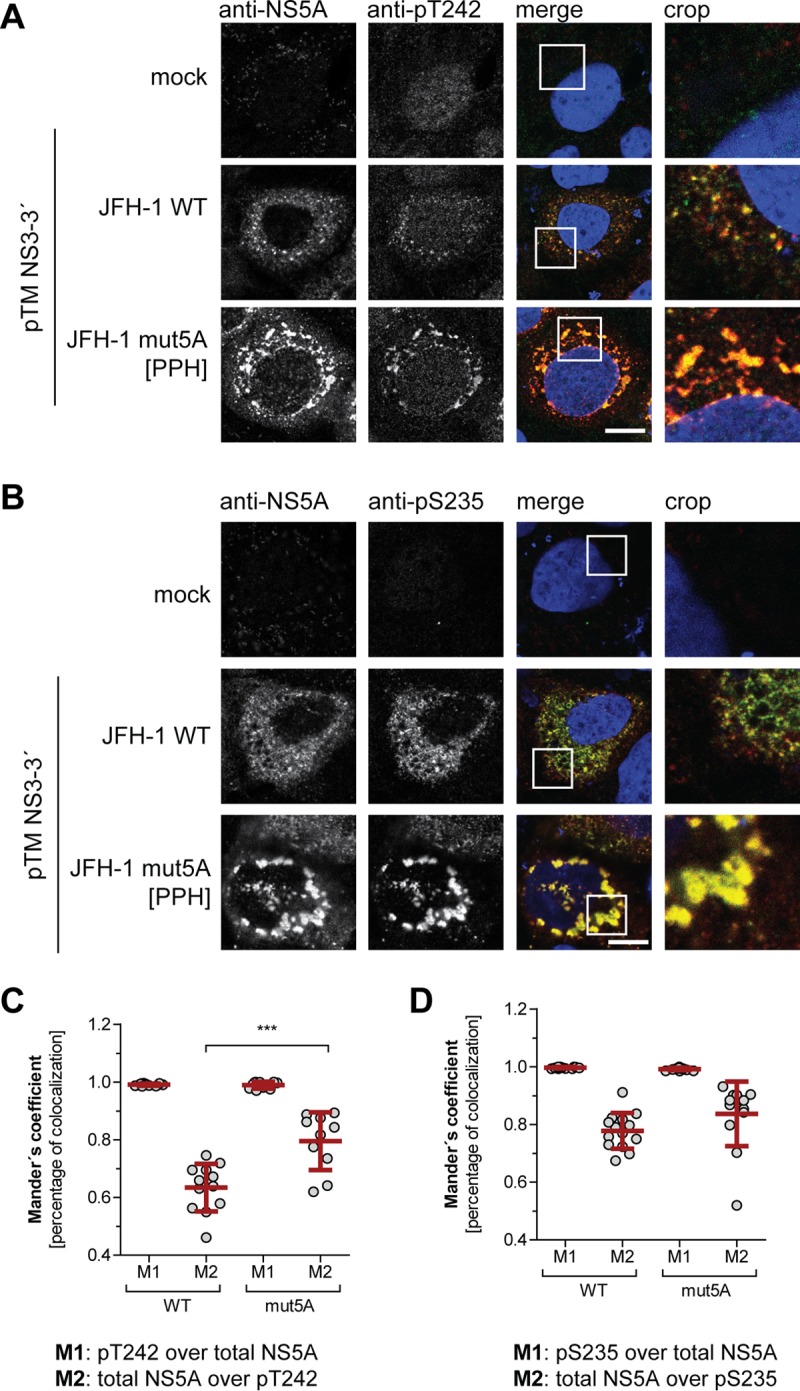
Subcellular localization of WT NS5A compared to NS5A mutant with impaired PI4KA activation. (A and B) Huh7-Lunet T7 cells were transfected with a pTM vector encoding NS3-5B of JFH-1 WT or mutPPH, seeded on coverslips, and fixed 24 h posttransfection. Total NS5A and NS5A phosphorylated at T242 (A) or S235 (B) was detected by immunofluorescence analysis using monoclonal antibody 9E10 (red) and anti-NS5A-pT242 or anti-NS5A-pS235 (both green), respectively. Nuclei were stained with DAPI. Scale bar, 10 µm. (C and D) Mander’s coefficient representing colocalization of pT242 with NS5A (M1) (C) or pS235 with NS5A (D) and vice versa (M2) was calculated for the pTM-transfected cells using Fiji. At least 10 cells per condition were analyzed. ***, P < 0.01 (homoscedastic, two-tailed t test).
In summary, NS5A phosphorylated at T242 is localized in a dot-like pattern indistinguishable from total NS5A.
Impact of kinase inhibitors on T242 phosphorylation.
Several kinases have been implicated in the regulation of NS5A phosphorylation. CKIα appears to be the main kinase involved in p58 formation, whereas CKII mainly contributes to basal phosphorylation (26, 38). In addition, polo-like kinases have been shown to be involved in hyperphosphorylation (35) and CAMK2 γ and δ redundantly modulate S235 phosphorylation (28), whereas PI4KA is a negative regulator of p58 synthesis (39). To identify kinases involved in phosphorylation of T242, we tested respective inhibitors regarding their impact on p58 synthesis in general and on the pT242 signal in particular, both relative to total NS5A (Fig. 7). Inhibition of CKIα abrogated p58 synthesis completely, including a total loss of pT242 signal, suggesting that this kinase either directly phosphorylates T242 or generates critical phosphorylation events essential for T242 phosphorylation. The PI4KA inhibitor increased the relative amount of total p58 and of pT242. In contrast, inhibition of CKII, PLK-1, PLK-2/3, GSK-3, CAMK2D, and PKA had only a minor impact on total p58 or pT242 (Fig. 7).
FIG 7.
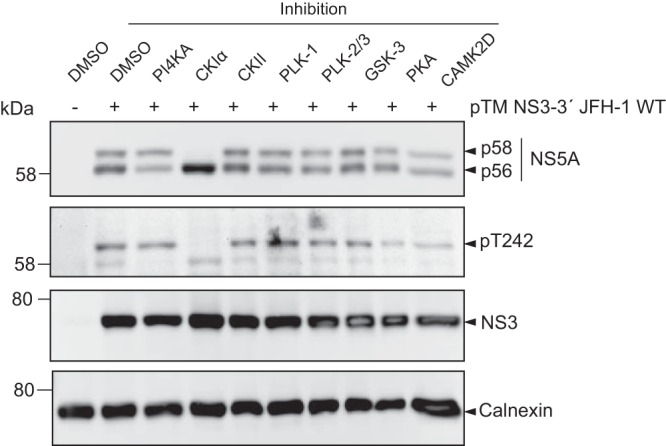
Impact of kinase inhibitors on phosphorylation of T242. Huh7-Lunet T7 cells were transfected with a pTM vector encoding NS3-5B of JFH-1. Four hours after transfection the medium was replaced with new medium containing drugs targeting the indicated kinases that have been described to modulate NS5A phosphorylation. Cells were lysed 24 h after transfection and analyzed by Western blotting for NS5A phosphorylation using anti-NS5A (monoclonal antibody 9E10)-, anti-NS5A-pT242-, anti-NS3-, and anti-calnexin-specific antibodies as indicated. One representative of three experiments is shown.
In conclusion, our data indicate that CKIα is involved in phosphorylation at position T242. In contrast, inhibition of other kinases previously linked to p58 synthesis did not substantially reduce total p58 or pT242 levels.
Sequence determinants of T242 phosphorylation.
Several studies suggest that p58 synthesis is based on saltatory phosphorylation events driven by CKIα and further involving priming phosphorylation by other kinases (25, 37). In particular, pS232, pS235, and pS238 are very likely products of saltatory phosphorylation by CKIα (34, 37, 48). Phosphorylation of S232 in addition seems to be a main regulator of p58 synthesis, since phosphoablatant mutations at this site apparently abrogate hyperphosphorylation completely (19, 20). To understand the succession of phosphorylation events underlying phosphorylation at position T242, we mutated a number of potential phosphoacceptor sites N and C terminally of this residue and analyzed the impact of these mutations on the abundance of pT242 (Fig. 8 and 9). We included S146, since this site has been found to modulate NS5A hyperphosphorylation (25), as well as T164A, which has been identified as a potential phosphorylation site in our study (Table 1). Interestingly, none of the phosphoablatant mutations of serine residues N terminal of T242 abrogated T242 phosphorylation, although some of them strongly impaired overall p58 abundance (Fig. 8A and B). Even in the case of mutant S232A, which generated barely detectable amounts of p58, the amount of pT242 phosphorylation relative to total NS5A was not significantly reduced compared to that of the WT (Fig. 8B). In contrast, mutations T242A and N-termA did not give rise to any pT242 signal, further proving the specificity of the antibody, since neither mutant contains the specific phosphoacceptor site. In addition, T244A and S247A strongly impaired T242 phosphorylation. While we cannot exclude at this point that T244A interfered with pT242 recognition by the antibody, due to the close proximity of both residues, this seems unlikely in the case of S247A, since T245A generated a visible pT242 signal. Therefore, it seems likely that phosphorylation at S247 (and probably T244) is a priming event crucial for T242 phosphorylation.
FIG 8.

Impact of phosphoablative mutations of potential phosphoacceptor sites on T242 phosphorylation. Huh7-Lunet T7 cells were transfected with pTM vectors expressing NS3-5B of isolate JFH-1, either WT or harboring the indicated phosphoablative alanine mutations. Mutant “N-termA” comprises all five serine and threonine mutations from T242 to T249. Mutant “C-termA” comprises both substitutions at positions T268 and S272. (A) Cells were lysed 24 h after transfection and analyzed by Western blotting using anti-NS5A (monoclonal antibody 9E10, upper)-, anti-NS5A-pT242-, anti-NS5A-pS235-, anti-NS3, and anti-β-actin (ACTB)-specific antibodies. (B) Ratio of p58 and pT242 to total NS5A. Intensities of the p58 bands, the pT242 bands, and the sum of p56 and p58 (total NS5A) as shown in panel A were quantified and used to calculate the indicated ratios. An additional staining of NS3 was performed subsequently on the same membranes previously used for detection of NS5A or pT242. These NS3 signals were used to obtain relative NS5A or pT242 levels for each membrane, which built the basis for pT242/NS5A levels. Bars represent mean values and SD from one representative experiment with three technical replicates. A second biological replicate was performed with a similar outcome (n = 2). n.d., not detectable. ***, P < 0.01 (homoscedastic, two-tailed t test).
FIG 9.

Impact of phosphomimetic mutations of potential phosphoacceptor sites on T242 phosphorylation. Huh7-Lunet T7 cells were transfected with pTM vectors expressing NS3-5B of isolate JFH-1, either WT or harboring the indicated phosphomimetic aspartic acid mutations. Mutant “N-termD” comprises all five serine and threonine mutations from T242 to T249. Mutant “C-termD” comprises both substitutions at positions T268 and S272. (A) Cells were lysed 24 h after transfection and analyzed by Western blotting using anti-NS5A (monoclonal antibody 9E10, upper)-, anti-NS5A-pT242-, anti-NS5A-pS235-, anti-NS3-, and anti-β-actin (ACTB)-specific antibodies. (B) Ratio of p58 and pT242 to total NS5A. Intensities of the p58 bands, the pT242 bands, and the sum of p56 and p58 (total NS5A) as shown in panel A were quantified and used to calculate the indicated ratios. An additional staining of NS3 was performed subsequently on the same membranes previously used for detection of NS5A or pT242. These NS3 signals were used to obtain relative NS5A or pT242 levels for each membrane, which built the basis for pT242/NS5A levels. Bars represent mean values and SD from one representative experiment with three technical replicates. A second biological replicate was performed with a similar outcome (n = 2). n.d., not detectable. ***, P < 0.01 (homoscedastic, two-tailed t test). (C) Cells were lysed 24 h after transfection, and the cleared lysate was treated with lambda phosphatase. Phosphatase-treated lysates were analyzed by Western blotting using anti-NS5A (monoclonal antibody 9E10)- and anti-calnexin-specific antibodies (n = 1). Note that dephosphorylation of NS5A by lambda phosphatase results in only one remaining band with an apparent MW similar to that of p56, but that different mutants show a consistently increased apparent molecular size.
Taken together, these results so far suggested independence of T242 phosphorylation from upstream phosphorylation in the Ser-rich cluster and vice versa. To further validate this hypothesis, we analyzed the same samples with anti-pS235 (Fig. 8A). Interestingly, p58 species phosphorylated at position S235 were found in all mutants, even in the absence of detectable levels of total p58 (S232A), and in most cases correlated with total p58 levels (34). This result indicated that T242 phosphorylation still depends on phosphorylation events in the preceding Ser-rich cluster and might explain the dependency of T242 phosphorylation on CKIα. In contrast, lack of phosphorylation of the Thr-rich cluster (N-termA) did not strongly impact the level of total p58 or pS235, supporting the assumption that total p58 is mainly composed of phosphorylation events in the Ser-rich cluster and that T242 phosphorylation only marks a minor species of hyperphosphorylated NS5A.
We next assessed the impact of phosphomimetic mutations on T242 phosphorylation (Fig. 9). Interestingly, S146D, which has been shown before to negatively regulate p58 synthesis of JFH-1 NS5A (25), also reduced the relative amounts of pT242, suggesting that phosphorylation at this site indeed is a master regulator of p58. All other phosphomimetic mutations showed effects on total p58 synthesis very similar to those of the phosphoablatant mutations: S232D again strongly reduced total p58 but not pT242. T242D and N-termD did not react with the pT242-specific antibodies, likely due to the loss of antigenicity. Surprisingly, T244D and S247D also negatively affected T242 phosphorylation, as did the phosphoablatant mutations. In case of pS235, phosphorylation abundance was widely similar for the phosphomimetic (Fig. 9A) and ablatant mutations (Fig. 8A). Altogether, the similarity of p58 phenotypes among phosphoablatant and phosphomimetic mutations indicated that replacement of the phosphoacceptor site with a charged residue in fact did not rescue the priming function of a phosphorylation event but rather interrupted the phosphorylation cascades up to this point involved in p58 synthesis. This interpretation was confirmed by phosphatase treatment of cleared cell lysates. While some of the aspartate mutations (S232D, S235D, S238D, and T242D) induced an apparent decrease of the electrophoretic mobility of NS5A p56, which has been interpreted before as a functional priming toward p58 synthesis (25), the same shift was still observed after treatment with lambda phosphatase (Fig. 9C). Overall these data demonstrated that a single mutation in this part of LCS-I can strongly affect the electrophoretic mobility of NS5A. These results further suggested that the increase in the apparent molecular weight of p56 in the case of S232D, S235D, S238D, and T242D indeed was not due to further phosphorylation events but based on the mutation itself. Interestingly, a similar shift in electrophoretic mobility was found for phosphoablatant mutations of the same residues, S232A, S235A, S238A, and T242A (Fig. 8A), indicating that the differences in the apparent molecular weight are independent of the negative charge.
Taken together, the mutational analysis of phosphoacceptor sites N and C terminal of T242 allowed several important conclusions. First, NS5A phosphorylated at pT242 is a minor subspecies of p58, since its abundance only marginally changed, even in cases where total p58 levels were dramatically reduced (e.g., S229A and S232A). Second, pT242 synthesis is regulated by C-terminal phosphorylation events, likely at position S247 and potentially at T244. Third, phosphorylation of the p58 subspecies harboring pS235/pS238, identified in previous studies (34), is regulated independently of pT242 and has only a minor impact on T242 phosphorylation. Fourth, some mutations of phosphoacceptor sites in the LCS-I have a direct impact on the electrophoretic mobility of NS5A p56, but the phosphomimetics most likely do not act as priming sites for saltatory phosphorylation events. Overall, these data suggest that p58 is composed of differentially phosphorylated subsets of phosphoisoforms, with identical electrophoretic mobility.
Assessment of phosphorylation events in the Thr-rich cluster for other HCV genotypes.
Finally, we aimed to clarify whether T242 or other residues in the Thr-rich cluster are phosphorylated in HCV genotypes other than 2a/JFH-1. We first assessed the genotype specificity of the pT242 antibodies by expressing NS3-5B of gt1a, gt1b, gt2a, gt3a, gt4a, and gt5a (isolates H77, Con1, JFH-1, S52, ED43, and SA1, respectively) in Huh7 Lunet-T7 cells (Fig. 10A). A signal was obtained only for JFH-1/gt2a. To analyze whether or not T242 was phosphorylated in genotypes other than 2a or whether the pT242-specific antibodies were not able to bind to corresponding epitopes from other genotypes due to high sequence divergence in this region (Fig. 10B), we synthesized peptides for each genotype encompassing the region used for immunization, including phosphorylation at T242. Indeed, the pT242 antibodies did not detect any genotype other than 2a (Fig. 10C). We therefore adapted the peptide sequence in NS3-5B expression constructs of all isolates to the JFH-1 sequence and analyzed T242 phosphorylation again by Western blotting (Fig. 10D). We found an anti-pT242 positive signal corresponding to p58 of the mutated ED43 (gt4a). This result provided the first evidence that T242 also is phosphorylated in genotypes other than gt2a. However, matching the surroundings of T242 to JFH-1 required 6 to 8 mutations (Fig. 10B), which could result in false-positive or -negative outcomes by interfering with potentially varying phosphorylation cascades.
FIG 10.

Analysis of T242 phosphorylation in different HCV genotypes. (A) Huh7-Lunet T7 cells were transfected with pTM vectors coding for the NS3-5B nonstructural proteins of the indicated HCV isolates. The cells were lysed 24 h after transfection and analyzed by 7.5% SDS-PAGE/Western blotting for total NS5A (monoclonal antibody 9E10), NS5A-pT242, and calnexin (CAXN). One representative experiment is shown (n = 2). (B) Alignment of NS5A sequence from 236 to 251 (numbering according to NS5A JFH-1) from HCV isolates H77 (gt1a), Con1 (gt1b), JFH-1 (gt2a), S52 (gt3a), ED43 (gt4a), and SA1 (gt5a). Highlighted in boldface are the amino acids that are different from those of JFH-1. Numbers on the right represent the homology of the respective sequences to JFH-1. Note that the peptide sequence corresponding to JFH-1 was used to generate the pT242-specific antiserum. (C) Indicated amounts of peptides corresponding to the sequence of the indicated isolates and phosphorylated at position T242 were spotted on PVDF membrane and incubated with purified, polyclonal anti-pT242 antibodies. The nonphosphorylated peptide of JFH-1 served as a negative control for staining. (D) Huh7-Lunet T7 cells were transfected with pTM vectors coding for the NS3-5B nonstructural proteins of the indicated HCV isolates, either the WT or an NS5A mutated version. All mutated genotypes have amino acid sequence changed from their respective WT sequence to the sequence of JFH-1 in NS5A positions 236 to 251 (compare to panel B). In addition, a Con1 point mutant, K240R, was included to test if this change alone would allow antigen recognition. The experiments were performed twice with a comparable outcome.
We chose an additional, unbiased approach and analyzed the immunoprecipitated wild-type NS5A of isolates H77, Con1, S52, ED43, and SA1 by mass spectrometry (Fig. 11). We again used the NS3-5B expression model (Fig. 10A), since the WT variants of these isolates are not replication competent and cell culture-adaptive mutations reduce the abundance of p58 (21). Due to lower expression levels compared to those of our previous experiment on JFH-1 (Fig. 1), we could not analyze p56 and p58 separately but isolated the respective molecular weight range corresponding to both isoforms from a gel (not shown) and subjected the contained tryptic peptides to MS/MS analysis (Fig. 11 and Table S2). We identified phosphorylated peptides in all cases carrying one or two phosphate groups (Table S2). Interestingly, consistent phosphorylation for all genotypes analyzed was found only within the Ser-rich cluster in LCS-I and at the N terminus of domain III, albeit at various positions between 222 to 235 (Fig. 11B) and 364 to 399 (Table S2). In addition, we found phosphorylation sites with high probability in domain I for gt1a at the same position as those for JFH-1 (146) (Table 1) (25) and for gt3a (positions 87 and 117). Regarding the Thr-rich cluster, a double-phosphorylated peptide was identified for gt4a with high probability of T242 phosphorylation (Fig. 11B and Table S2). S249 was found phosphorylated in all genotypes, with a phosphoacceptor site at this position (gt1a, gt1b, and gt4a), and S274, adjacent to the Thr-rich cluster, carried a phosphate group in genotypes 1a, 1b, and 3a (Table S2). Only in the case of gt5a/isolate SA1 did we find no indication of phosphorylation in the region following the Ser-rich cluster, although nonphosphorylated peptides were detectable (Table S2). In addition to these high-probability hits, several phosphorylated peptides were found without clearly assignable phosphorylation sites, suggesting a mixed population of peptides with variable phosphorylation events (Fig. 11A and Table S2). In addition, we cannot exclude that many phosphorylation events remained undetected in this analysis due to limited sensitivity, multiple and variable phosphorylation events in one peptide, etc.
FIG 11.

Phospho-mass spectrometry reveals additional phosphorylation events within the Thr-rich peptide. (A) Schematic of the different domains of NS5A. The location of the identified phosphopeptides is indicated below for each isolate analyzed here, as given on the right. All numberings refer to amino acid positions within the respective genotype. (B) Phospho-(STY)-probability scores of different potential phosphoacceptor sites within the Ser-rich and Thr-rich cluster. Shown are only values above 0.75 that can be considered high-probability phosphorylation events at the respective site. The numbers below refer to all possible Ser/Thr phosphoacceptor sites existing in all analyzed genotypes. Note that phosphorylation at S249 was detected for all genotypes with a phosphoacceptor site at this position and that S52 (gt3a) and SA1 (gt5a) harbor a histidine (H) at position 249. The phospho-mass spectrometry was performed once.
Taken together, our data suggest that the Thr-rich cluster is phosphorylated in the case of JFH-1, and at least in genotypes 1a, 1b, and 4a, it is phosphorylated at different positions. Combined evidence from genetics and mass spectrometry suggests that T242 is phosphorylated in gt4a/ED43.
DISCUSSION
Our study aimed at a deeper understanding of NS5A phosphorylation, starting with a phospho-mass spectrometry analysis of NS5A of isolate JFH-1 expressed in the context of NS3-5B in Huh-7 cells, analyzing p56 and p58 separately. We thereby identified seven phosphopeptides, with some of them having multiple potential phosphoacceptor sites. Overall, this result was well comparable with those of previous studies, which already identified the peptides II, IV, V, VI, and VII (25, 33, 41).
The reason to use an expression system rather than a replication model in the first place was to allow the analysis of replication-deficient mutants and inhibitors interfering with HCV replication. By comparing NS5A phosphorylation patterns from the replicating full-length HCV and based on expression of NS3-5B, we observed overall very similar results, as judged by p58/p56 ratios of numerous mutants. We only observed a slight overrepresentation of the basal phosphorylated form in the expression system compared to replicating HCV, probably due to the higher expression level of NS5A; therefore, limited expression of cellular kinases might restrict the amount of p58 synthesis. These data overall confirmed that expression of NS3-5B indeed is an excellent model to study NS5A phosphorylation, as already shown in previous studies by us and others (21, 34, 39, 42, 48, 51, 53). In particular, we aimed to include an NS5A mutant with strongly reduced PI4KA interaction, giving rise to enhanced p58 levels, to understand the mechanism underlying the modulation of NS5A phosphorylation by this lipid kinase. However, the pattern of phosphorylated peptides between the WT and the NS5A mutant was comparable in mass spectrometry, suggesting that PI4KA does not alter the overall phosphorylation pattern of NS5A but rather reduces the number of hyperphosphorylated NS5A molecules by an unknown mechanism.
Interestingly, we identified for the first time a phosphopeptide restricted to the hyperphosphorylated isoform of NS5A (peptide V), which was therefore the focus of our study. This Thr-rich cluster in LCS-I was also found previously (25) and contains eight potential phosphoacceptor sites, with one (T242) conserved throughout all genotypes. A mutational analysis of all seven Ser/Thr residues individually revealed no impact on RNA replication or particle production in cell culture, and a combination of only 5 phosphomimetic mutations in this region almost abrogated RNA replication (N-termD). This result is in contrast to a very recent study demonstrating a strong reduction of RNA replication for phosphomimetic mutations at positions T242 and T244, which was partly restored by a triple mutation (54). The reason for these discrepant results is not clear so far, but it might rely on the use of different Huh-7 cell populations with various expression levels of cellular kinases regulating NS5A phosphorylation. Of note, both studies agree on the lack of strong phenotypes for phosphoablative mutations, including position T249 (52). Therefore, phosphorylation of individual residues within the Thr-rich cluster in LCS-1 seems not to have an essential function in RNA replication or virus production.
By generating phosphospecific antibodies against pT242, we were able to demonstrate that this residue is indeed phosphorylated in a subspecies of NS5A molecules in p58, thereby confirming the mass spectrometry data. T242 was phosphorylated upon transient NS3-5B expression and virus replication, following the same kinetics as total p58. The pT242-specific antibodies further allowed assessment of the determinants of T242 phosphorylation. We used different inhibitors against the most prominent kinases known to be involved in NS5A phosphorylation and monitored their impact on T242 phosphorylation. Only inhibition of CKIα completely abrogated p58 and pT242 detection, in line with previous studies (26, 37, 39, 49). This overall suggests that regulation of phosphorylation at T242 is governed by the same mechanisms as total p58. However, it seems unlikely that T242 is directly phosphorylated by CKIα, since the only phosphoablative mutations found to abrogate pT242 phosphorylation, therefore being suspect priming sites for CKIα, are C terminal of this site (T244 and S247), whereas CKIα requires an N-terminal priming site (pS-X-X-S) (55). Therefore, we cannot precisely define the kinase responsible for T242 phosphorylation at this point.
Our data strongly indicate the existence of several independent subspecies of p58 with very different phosphorylation patterns. (i) Phosphoablative mutations at positions S225, S229, and S232 almost entirely abrogate global p58 synthesis, as shown before (19, 20, 25, 26), but have only a minor impact on T242 phosphorylation. Previous studies furthermore, have shown that pS222, pS232, pS235, and pS238 are exclusively found in p58 (25, 34, 48). Surprisingly, we found that even in the phophoablative mutants S225A, S228A, and S229A, the p58-specific phosphosignal for S235 seems only slightly affected, and even in S232A, lacking detectable levels of p58, small amounts of pS235 were detectable. This suggests that phosphorylation at position S235 occurs even if global p58 levels are strongly impaired. (ii) In striking contrast, mutations T244A and S247A abrogate T242 phosphorylation but hardly affect global p58 levels. Together, these data suggest that pT242 defines a minor population of hyperphosphorylated NS5A dependent on phosphorylation events in the N-terminal Ser-rich cluster, even though we could not identify specific sites priming for downstream T242 phosphorylation. These results challenge the concept of p58 as a homogenous NS5A isoform with defined functions and rather suggest that p58 is a heterogeneous set of molecules that are only defined by their homogenous apparent molecular weight. Individual phosphomimetic mutations at positions S235, S238, and T242 substantially increased the apparent molecular weight of p56, resistant to phosphatase treatment, suggesting that this increase was induced by the mutations itself, not by further phosphorylation. An accumulation of five phosphomimetic mutations in the Thr-rich cluster even resulted in a homogenous, phosphatase-resistant NS5A species indistinguishable from p58. Altogether these data demonstrate that accumulation of mutations in LCS-I, particularly adding negative charges, increases the apparent molecular weight of NS5A toward p58, which can be achieved by a limited number of mutations at various sites in LCS-I. However, these negative charges seem not to act as priming phosphorylation sites, as suggested previously (25, 26), thereby challenging the use of these mutants in studies addressing the function of distinctly phosphorylated NS5A subspecies. This observation is supported by in vitro phosphorylation studies demonstrating that the phosphomimetic mutation S229E was unable to prime CKIα activity compared to the pS229 peptide (37). Still, these phosphomimetic mutations demonstrate that a few changes in this region suffice to generate p58. Therefore, p58 likely contains a heterogeneous mixture of different phosphorylation patterns, either in the Ser-rich and/or in the Thr-rich cluster of LCS-I. This model further explains the difficulties of identifying distinct NS5A phosphorylation sites in mass spectrometry analyses, but why all these various phosphoisoforms culminate in a distinct single p58 band in SDS-PAGE remains puzzling.
What is the function of p58 in general and phosphorylation in the Thr-rich cluster of LCS-I in particular? P58 has been regarded as the NS5A isoform inhibiting replication and supporting assembly based on the fact that LCS-I is a hot spot for mutations enhancing RNA replication of many HCV genotypes in cell culture but at the same time abrogating or strongly reducing p58 synthesis (e.g., S225, S232, and S235) (19, 20, 56–60) and virus particle production (61). However, recent data argue for an essential role of p58 in both virus production (26) and RNA replication (21). Replication-enhancing mutations in LCS-I reduce p58 levels and abrogate activation of PI4KA, which is an essential host factor of HCV RNA replication (51, 62–64) but too abundant in Huh-7 cells (21). Therefore, the same mutations enhancing replication of many HCV isolates by dampening PI4KA activity strongly decrease JFH-1 replication. JFH-1 also requires PI4KA activation in Huh-7 cells, thereby reflecting more closely the WT behavior of HCV in vivo. However, p232 and pS235, two of the phosphorylation events required for PI4KA activation, are restricted to p58 (34). Even pS222, the major NS5A phosphorylation identified in multiple studies (24, 25, 27, 65) and located at the N terminus of the Ser-rich cluster in LCS-I, is mainly found in p58 (25). In addition, mutations in the Ser-rich cluster have recently been shown to regulate multiple host factor interactions required for replication and affect the membranous HCV replication organelle (24, 46, 66). Taken together, these data strongly argue for essential functions of distinctly hyperphosphorylated p58 subspecies of NS5A in HCV RNA replication.
We can only speculate on functions of phosphorylation events in the Thr-rich cluster, since we found no impact of phosphoablative mutations on HCV replication and virus production in cell culture. Previous studies have already shown that wide parts of NS5A domain II (aa 246 to 304), encompassing the C-terminal half of the Thr-rich cluster, can be deleted, with little impact on HCV replication or assembly (3). However, this deletion mutant was strongly impaired in vivo due to increased activation of cytosolic pattern recognition receptors (17). In addition, the entire Thr-rich cluster in LCS-I is part of the so-called interferon sensitivity-determining region (ISDR; aa 237 to 276), which has been linked to the interferon response of HCV patients (67, 68). Therefore, it is tempting to speculate that phosphorylation events in the Thr-rich cluster in LCS-I play a role in the interference with innate immune responses in vivo but that these are not relevant for replication in cell culture due to the limited capacity of Huh-7 cells to mount an innate immune response (17, 52). Our mass spectrometry data on different HCV genotypes suggest at least a certain degree of conservation of phosphorylation events in this region. T242 was found phosphorylated not only in gt2a but also in gt4a, S249 in gt1a, gt1b (also found in reference 52), and gt4a, and S274 in gt1a, gt1b and gt3a. These data, together with the complete conservation of T242, suggest a functional relevance of phosphorylation events in the Thr-rich cluster.
In summary, our study identifies a novel minor subspecies of hyperphosphorylated NS5A, marked by T242 phosphorylation. Our results challenge the concept of p58 being a population of homogenously phosphorylated molecules but rather suggest a variety of phosphoisoforms contained in p58 with identical electrophoretic mobility, likely serving multiple functions. We thereby add a new level of complexity to NS5A phosphorylation and argue for a more differentiated view on p58.
MATERIALS AND METHODS
Cell lines.
The Huh-7 cell clone Huh7-Lunet, highly permissive for HCV RNA replication (69), was used for electroporation assays. Huh7.5 cells (70) were used for infection experiments. Huh7-Lunet cells transduced with a T7 polymerase (Huh7-Lunet T7) (71) were used for transient expression of pTM plasmids coding for HCV proteins analyzed in immunofluorescence and SDS-PAGE/Western blotting assays.
Plasmid constructs.
All plasmids used in this study were generated with standard molecular cloning techniques (72). All sequencing reactions were performed by GATC Biotech, Cologne, Germany.
pTM vectors are composed of a T7 promoter, an encephalomyocarditis virus internal ribosomal entry site followed by the coding sequence to be expressed, in our case the HCV nonstructural NS3-5B proteins. This plasmid allows robust expression regardless of HCV replication competence. The vector pTM mut5A expresses NS3-5B with a mutation in the PI4KA functional interaction site (PFIS) of NS5A. Mutations in this region of nine amino acids at the very C terminus of domain I of NS5A impair replication, PI4KA binding, and PI4P induction and lead to a relative increase in hyperphosphorylated NS5A (39). In this study, we used a mutation of PPH to AAA within the PFIS domain.
All amino acid numbers refer to the position of the corresponding amino acid in the NS5A protein of JFH-1 (GenBank accession no. AB047639). All mutants in this study were generated by overlapping PCR (primer sequences are available on request) and ligated into the pTM NS3-3′ JFH-1 WT vector via the restriction sites NsiI (C terminus of NS3) and SfiI (NS5B). Afterwards the mutated fragments were moved from the pTM vector to the different plasmid vectors encoding genomic and subgenomic replicons (monocistronic pFK i389-JcR2a [51] or pFK JC1 [73] and the bicistronic subgenomic replicon pFK i389 Luc NS3-3′ JFH-1 [74]) by using NsiI/SfiI. A replication-deficient variant harboring a deletion in NS5B (ΔGDD) was used as a negative control for replication in the monocistronic pFK i389-JcR2a and the bicistronic pFK i389 Luc NS3-3′ JFH-1. In addition, a replication-competent but assembly-deficient variant (ΔE1E2) lacking the structural proteins E1 and E2 was used as a negative control for particle production.
PTM vectors expressing NS3-5B of other HCV genotypes have been described before (21), namely, H77 WT (gt1a; provided by Jens Bukh) (GenBank accession no. AF009606.1), Con1 WT (gt1b; GenBank accession no. AJ238799.1), JFH-1 WT (gt2a; provided by Takaji Wakita; GenBank accession no. AB047639.1), S52 WT (gt3a; GenBank accession no. GU814263.1), ED43 WT (gt4a; GenBank accession no. GU814265.1), and SA1 WT (gt5a; GenBank accession no. KJ925146.1; all provided by Charles M. Rice).
Pharmacological inhibition of NS5A phosphorylation.
Huh7-Lunet T7 cells were seeded at a density of 2 × 106 cells per 6-well dish and transfected 24 h later with LT1 transfection agent (Mirus Bio LLC, Madison, WI, USA) according to the manufacturer’s instructions. For drug treatment the supernatant was replaced 4 h after transfection with complete Dulbecco’s modified Eagle’s medium (DMEM) containing either 5 µM the PI4KA inhibitor G1 (GlaxoSmithKline, Munich, Germany) (75), 10 µM the CKIα inhibitor H479 (76), 500 nM the CKII inhibitor TBBz (Sigma-Aldrich, Darmstadt, Germany) (77), 30 µM the Polo-like kinase 1 inhibitor Poloxin (Sigma-Aldrich, Darmstadt, Germany) (78),100 nM the Polo-like kinase 2/3 inhibitor TC-S 7005 (Tocris, Wiesbaden-Nordenstadt, Germany), 1 µM GSK3 inhibitor BIO (Sigma-Aldrich, Darmstadt, Germany) (79),10 µM PKA inhibitor PKI5 to PKI24 (Tocris, Wiesbaden-Nordenstadt, Germany) (80), or 5 µM CamK2D inhibitor KN93 (28).
Phospho-mass spectrometry analysis of NS5A.
Huh7-Lunet T7 cells were seeded at a density of 2 × 106 cells in three 10-cm dishes. The next day, cells were transfected with pTM constructs coding for NS3-5B or NS3-5B mutPPH (mut5A). Twenty-four h after transfection, cells were scraped in phosphate-buffered saline (PBS) and lysed in 1 ml radioimmunoprecipitation assay (RIPA) buffer. NS5A was immunoprecipitated using monoclonal 9E10 NS5A antibodies and Protein G Sepharose beads. Immunoprecipitated proteins were resuspended in 50 µl SDS sample buffer, and the whole reaction mixture was loaded for 8% SDS-PAGE. Proteins were stained using colloidal Coomassie staining, and protein bands corresponding to NS5A p56 or p58 of each sample were cut out individually, in the case of isolate JFH-1, or the region of the gel encompassing the expected size range of NS5A was cut out entirely for the other HCV isolates. Proteins in the individual gel slices were reduced, alkylated, and in-gel digested with trypsin (Promega, Madison, WI). Gel pieces were desiccated and peptides were enriched by acetonitrile and water extraction steps. Desiccated peptide samples were redissolved in water containing 2.5% hexafluoro-2-propanol (HFIP) (10522; Sigma-Aldrich) and 0.1% formic acid (069141A8; Biosolve, Valkenswaard, Netherlands) prior to MS analysis.
Liquid chromatography and mass spectrometry.
Peptides from trypsin digestions were either analyzed by a linear ion trap quadrupole LTQ Orbitrap-XL mass spectrometer (Thermo Fisher Scientific) coupled to a nanoAcquity ultrahigh-performance liquid chromatography (UPLC) system (Waters) or a Q Exactive HF-X mass spectrometer (Thermo Fisher Scientific) coupled to an UltiMate 3000 RSLCnano system (Thermo Fisher Scientific).
For LTQ Orbitrap XL measurements, peptides were trapped with a constant flow of 5 μl/min on a 180-μm by 20-mm nanoAcquity UPLC 2 G trap column containing a Symmetry C18 column, using 5-μm particles. Sample separation was performed on a 100-μm by 100-mm nanoAcquity BEH C18, 1.7-μm analytical column at a constant flow of 400 nl/min using a 90-min stepped linear gradient of solvent A (96.9% water, 3% dimethyl sulfoxide [DMSO], 0.1% formic acid) and solvent B (99.9% acetonitrile and 0.1% formic acid) in the following sequence: from 4% to 30% B in 80 min and from 30% to 45% B in 10 min. The nanoUPLC was coupled online to a nanoelectrospray source of a linear ion trap quadrupole LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific). A Pico-Tip Emitter tip (360-μm outer diameter, 20-μm inner diameter; 10 μm; New Objective) was used for sample ionization and introduction into the MS. The MS was operated in sensitive mode with the following parameters: electrospray ionization voltage was set to 2,400 V, the capillary temperature was 200°C, and the normalized collision energy was 35 V. The Orbitrap filling time was set at a maximum of 500 ms. Full-scan MS spectra were acquired in a mass-to-charge ratio (m/z) range from 350 to 2,000 in the profile mode with a mass resolution of 60,000 at m/z 400. Simultaneously, the six most abundant precursor ions from the full-scan MS were selected for MS/MS fragmentation in the LTQ. MS/MS data were acquired in centroid mode. Only multiply charged (2+, 3+, …) precursor ions were selected for MS/MS. The dynamic exclusion list was restricted to 500 entries, with a maximum retention period of 30 s and a relative mass window of 10 ppm.
For Q-Exactive-HF-X measurements, digested samples were loaded on a cartridge trap column, packed with an Acclaim PepMap300 C18 column (5 µm, 300-Å-wide pore; Thermo Scientific), and separated on a nanoEase MZ peptide analytical column (300 Å, 1.7 µm, 75 µm by 200 mm; Waters) in a 120-min stepped linear gradient of solvent A (99.9% water, 0.1% formic acid) and solvent B (79.9% water, 20% ACN, 0.1% formic acid) in the following sequence: from 2% to 8% in 10 min, from 8% to 25% in 100 min, and from 25% to 40% in 20 min (from 3% to 40% ACN).
Data processing and validation/protein identification and quantification.
Phosphopeptide identification was performed with the MaxQuant software 1.6.0.16 (81), wherein peptide identification was performed using the Andromeda (82) search engine integrated into the MaxQuant environment against a single protein database of the HCV NS5A protein. The peptide mass tolerance for database searches was set to 4.5 ppm and fragment mass tolerance to 0.4 Da for spectra recorded in the ion trap and 20 ppm for Orbitrap spectra. Cysteine carbamidomethylation was set as a fixed modification. Variable modifications included oxidation of methionine, deamidation of asparagine and glutamine, protein N-terminal acetylation, and serine, threonine, and tyrosine phosphorylation. Two missed cleavage sites in the case of incomplete trypsin hydrolysis were allowed.
Transient HCV replication.
Transient HCV RNA replication assays were performed as described previously (83). In brief, replicon-encoding plasmid DNAs harboring hepatitis delta virus ribozymes to ensure proper generation of 3′ ends were used for in vitro transcription. Purified RNA was transfected in 4 × 106 Huh7-Lunet cells by electroporation. Cells were resuspended in 12 ml DMEM, 2-ml aliquots were seeded per well of a 6-well plate, and replication was determined by measuring luciferase activity at 4 h, 24 h, 48 h, and 72 h postelectroporation. Values obtained 4 h after transfection were used to normalize for transfection efficiency.
To determine the ability to produce infectious particles, the supernatants of full-length pFK i389-JcR2a electroporated cells were filtered (0.22 µm) and transferred to approximately 1 × 105 Huh7.5 cells, seeded 1 day prior to infection in a 6-well dish. Values were obtained 72 h postinfection and normalized to values for 72 h postelectroporation to normalize particle production to replication efficiency.
Luciferase assay.
Cells electroporated or infected with the different reporter replicons were harvested at time points described by aspirating the medium and washing once with PBS. Cells were lysed in 350 µl luciferase lysis buffer (1% Triton X-100, 25 mM Gly-Gly, pH 7.8, 15 mM MgSO4, 4 mM EGTA, and 1 mM dithiothreitol [DTT] added directly prior to use) and frozen at −20°C to ensure complete lysis. After collecting all time points, 100 µl of each lysate was mixed with 360 µl luciferase assay buffer (25 mM Gly-Gly, 15 mM K3PO4, pH 7.8, 15 mM MgSO4, 4 mM EGTA, 1 mM DTT, and 2 mM ATP added directly prior to use) and 200 µl luciferin substrate solution for Firefly luciferase (0.2 mM luciferin in 25 mM Gly-Gly) or 50 µl coelenterazine substrate solution for renilla luciferase (1.5 µM coelanterazine in luciferase assay buffer without ATP and DTT) (both from PJK, Kleinblittersdorf, Germany) and then injected automatically by a tube luminometer (Lumat LB9507; Berthold Technologies). Luciferase activity was measured for 20 s, with all wells measured in triplicate.
Immunofluorescence analysis.
For expression of HCV NS3-5B proteins, 4 × 104 Huh7-Lunet T7 cells per well, in a 24-well plate, were seeded 24 h prior to transfection with LT1 transfection agent (Mirus Bio LLC, Madison, WI, USA) according to the manufacturer’s instructions and were fixed 18 h posttransfection. Cells were fixed in 4% PFA for 20 min and permeabilized with 0.5% Triton X-100–TBS for 15 min. The permeabilized cells were then blocked for 1 h in 5% BSA-TBS prior to the incubation with the primary antibodies in 3% BSA-TBS again for 1 h at room temperature (RT). NS5A was detected by using either the NS5A-specific monoclonal mouse antibody 9E10 (generous gift from Charles M. Rice) at a dilution of 1:500 or the polyclonal anti-pT242, which specifically detects only the T242 phosphorylated isoforms of NS5A (Davids Bio, Regensburg, Germany), at a dilution of 1:50. In addition, the polyclonal anti-pS235, specific for NS5A phosphorylated at position S235, was used at a dilution of 1:50 (generous gift from Ming-Jiun Yu [34]). Alexa 488- or 546-conjugated secondary antibodies (Invitrogen, Molecular Probes) were incubated in 3% BSA–TBS for 45 min at RT with a dilution of 1:500. Nuclei were stained using 4′,6-diamidino-2-phenylindole (DAPI) for 1 min at a dilution of 1:4,000. Cells were mounted with Fluoromount G (Southern Biotechnology Associates, Birmingham, USA), and pictures were acquired with a Leica SP8 confocal laser-scanning microscope. For colocalization analysis, samples were evaluated quantitatively for Mander’s coefficient by using the integrated function in Fiji.
Western blotting.
For SDS-PAGE/Western blotting, approximately 1 × 106 cells were lysed in 50 µl ice-cold lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 15 mM NaCl2, 1% Triton X-100) containing an EDTA-free protease inhibitor cocktail (Roche) and phosphatase inhibitors (60 mM β-glycerophosphate, 15 mM 4-nitrophenylphosphate, 1 mM sodium orthovanadate, 1 mM sodium fluoride) for 1 h on ice.
In the case of the lambda phosphatase treatment, the lysis buffer lacked the phosphatase inhibitors, and the cleared lysate was incubated with 400 U lambda phosphatase in a total volume of 100 µl for 1 h at 30°C according to the manufacturer’s instruction (New England Biolabs, MA, USA).
The cleared supernatants were then denatured in 50 µl or 100 µl, respectively, and 2× Laemmli buffer, and lysates of approximately 1 × 105 cells were loaded on a 7.5% polyacrylamide–SDS gel. The color-prestained protein standard, broad range (11 to 245 kDa) (NEB), was used to determine the apparent molecular weight of the proteins; positions of the respective bands are indicated on the left of each blot. After separation, transfer to a polyvinylidene difluoride (PVDF) membrane, and blocking for 1 h in 5% BSA–TBS with 0.5% Tween 20 (TBST), the primary antibody was diluted in 3% BSA–TBST and incubated overnight at 4°C. For detection of total NS5A, a monoclonal mouse antibody (9E10; generous gift from Charles M. Rice) was used. In addition, we used polyclonal rabbit anti-pT242 antibodies (Davids Bio, Regensburg, Germany) detecting only the T242 phosphorylated isoform of NS5A, polyclonal rabbit anti-pS235 antibodies (generous gift from Ming-Jiun Yu [34]) detecting the S235 phosphorylated isoform of NS5A, a polyclonal rabbit anti-NS3 serum (71), polyclonal rabbit anti-calnexin antibodies (Enzo Life Sciences, Lörrach, Germany), and monoclonal mouse anti-ACTB antibodies (Sigma-Aldrich, Darmstadt, Germany). After three washing steps with TBST, the secondary antibody was diluted in 3% BSA–TBST for 1 h at RT. The detection was done using Clarity ECL blotting substrate (Bio-Rad, Hercules, CA) and the Advanced ECL imaging system (Intas Science Imaging Instruments, Goettingen, Germany). The signal was quantified using Fiji.
Phosphospecific antibodies and peptides.
PT242-specific antibodies were generated by Davids Biotechnology GmbH (Regensburg, Germany), using the peptide sequence APSLRATCTTHSNTYD with a phosphorylation at the first threonine residue (representing T242, indicated by a boldface letter) to immunize two rabbits. This sequence originates from NS5A of gt2a JFH-1 from positions 236 to 251. This phosphopeptide was injected 5 times over 56 days. The final bleeding of each rabbit was after day 63. The antiserum was purified first by affinity purification against the phosphorylated peptide and second by depletion of antibodies binding to the unphosphorylated version of the peptide.
Peptides of isolates H77, Con1, SA1, ED43, and S52, phosphorylated at position T242, were synthesized on a 433A peptide synthesizer (Applied Biosystems) using Fmoc chemistry on a Rink amide resin (TentaGel RAM, Rapp Polymere). The amino acids were coupled with standard protocols using the coupling agent N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate. The incorporation of phosphothreonine residues was achieved with N-α-Fmoc-O-benzyl-L-phosphothreonine (Merck KGaA) with 1-hydroxybenzotriazole (anhydrous) added to the coupling mixture. Using a cleavage protocol of 3 h with 94% trifluoroacetic acid, 2.5% H2O, 2.5% 1,2-ethanedithiol, 1% Triisopropylsilane afforded the crude peptides that were purified by reverse-phase high-performance liquid chromatography to a purity of >98% and lyophilized. Identity of the finals products was confirmed by LC-MS.
Statistical analysis and software.
Statistical analyses, as indicated in the corresponding figures, were performed using Microsoft Excel and GraphPad Prism software. Fluorescence images were analyzed using ImageJ/Fiji. Figures were arranged with Adobe Photoshop and Adobe Illustrator.
Supplementary Material
ACKNOWLEDGMENTS
We especially thank Rahel Klein and Ulrike Herian for excellent technical assistance. We thank Ramona Mayer, Martin Schneider, and Bernd Hessling from the DKFZ Genomics and Proteomics Core Facility (GPCF) for MS measurements and data analysis. We are grateful to Ming-Jiun (Brad) Yu for the generous gift of a phosphospecific antibody detecting pS235, Charles M. Rice for antibody 9E10, Huh7.5 cells, and plasmids encoding isolates S52, ED43, and SA1, Takaji Wakita for the JFH-1 isolate, and Jens Bukh for the H77 isolate. We further thank GSK for the PI4KA inhibitor G1 and Raffaele De Francesco and Petra Neddermann for the generous gift of inhibitor H479. This project was funded by grants from the Deutsche Forschungsgemeinschaft (LO 1556/4-1) to V.L.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00737-18.
REFERENCES
- 1.Lohmann V. 2013. Hepatitis C virus RNA replication. Curr Top Microbiol Immunol 369:167–198. doi: 10.1007/978-3-642-27340-7_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Romero-Brey I, Berger C, Kallis S, Kolovou A, Paul D, Lohmann V, Bartenschlager R. 2015. NS5A domain 1 and polyprotein cleavage kinetics are critical for induction of double-membrane vesicles associated with hepatitis C virus replication. mBio 6:e00759. doi: 10.1128/mBio.00759-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Appel N, Zayas M, Miller S, Krijnse-Locker J, Schaller T, Friebe P, Kallis S, Engel U, Bartenschlager R. 2008. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog 4:e1000035. doi: 10.1371/journal.ppat.1000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tellinghuisen TL, Foss KL, Treadaway JC, Rice CM. 2008. Identification of residues required for RNA replication in domains II and III of the hepatitis C virus NS5A protein. J Virol 82:1073–1083. doi: 10.1128/JVI.00328-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masaki T, Suzuki R, Murakami K, Aizaki H, Ishii K, Murayama A, Date T, Matsuura Y, Miyamura T, Wakita T, Suzuki T. 2008. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J Virol 82:7964–7976. doi: 10.1128/JVI.00826-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berger C, Romero-Brey I, Radujkovic D, Terreux R, Zayas M, Paul D, Harak C, Hoppe S, Gao M, Penin F, Lohmann V, Bartenschlager R. 2014. Daclatasvir-like inhibitors of NS5A block early biogenesis of hepatitis C virus-induced membranous replication factories, independent of RNA replication. Gastroenterology 147:1094–1105. doi: 10.1053/j.gastro.2014.07.019. [DOI] [PubMed] [Google Scholar]
- 7.McGivern DR, Masaki T, Williford S, Ingravallo P, Feng Z, Lahser F, Asante-Appiah E, Neddermann P, De FR, Howe AY, Lemon SM. 2014. Kinetic analyses reveal potent and early blockade of hepatitis C virus assembly by NS5A inhibitors. Gastroenterology 147:453–462. doi: 10.1053/j.gastro.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ross-Thriepland D, Harris M. 2015. Hepatitis C virus NS5A: enigmatic but still promiscuous 10 years on! J Gen Virol 96:727–738. doi: 10.1099/jgv.0.000009. [DOI] [PubMed] [Google Scholar]
- 9.Penin F, Brass V, Appel N, Ramboarina S, Montserret R, Ficheux D, Blum HE, Bartenschlager R, Moradpour D. 2004. Structure and function of the membrane anchor domain of hepatitis C virus nonstructural protein 5A. J Biol Chem 279:40835–40843. doi: 10.1074/jbc.M404761200. [DOI] [PubMed] [Google Scholar]
- 10.Tellinghuisen TL, Marcotrigiano J, Gorbalenya AE, Rice CM. 2004. The NS5A protein of hepatitis C virus is a zinc metalloprotein. J Biol Chem 279:48576–48587. doi: 10.1074/jbc.M407787200. [DOI] [PubMed] [Google Scholar]
- 11.Huang L, Hwang J, Sharma SD, Hargittai MR, Chen Y, Arnold JJ, Raney KD, Cameron CE. 2005. Hepatitis C virus non-structural protein 5A (NS5A) is a RNA-binding protein. J Biol Chem 280:36417–36428. doi: 10.1074/jbc.M508175200. [DOI] [PubMed] [Google Scholar]
- 12.Tellinghuisen TL, Marcotrigiano J, Rice CM. 2005. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 435:374–379. doi: 10.1038/nature03580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Love RA, Brodsky O, Hickey MJ, Wells PA, Cronin CN. 2009. Crystal structure of a novel dimeric form of NS5A domain I protein from hepatitis C virus. J Virol 83:4395–4403. doi: 10.1128/JVI.02352-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lambert SM, Langley DR, Garnett JA, Angell R, Hedgethorne K, Meanwell NA, Matthews SJ. 2014. The crystal structure of NS5A domain 1 from genotype 1a reveals new clues to the mechanism of action for dimeric HCV inhibitors. Protein Sci 23:723–734. doi: 10.1002/pro.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanoulle X, Badillo A, Wieruszeski JM, Verdegem D, Landrieu I, Bartenschlager R, Penin F, Lippens G. 2009. Hepatitis C virus NS5A protein is a substrate for the peptidyl-prolyl cis/trans isomerase activity of cyclophilins A and B. J Biol Chem 284:13589–13601. doi: 10.1074/jbc.M809244200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verdegem D, Badillo A, Wieruszeski JM, Landrieu I, Leroy A, Bartenschlager R, Penin F, Lippens G, Hanoulle X. 2011. Domain 3 of NS5A protein from the hepatitis C virus has intrinsic alpha-helical propensity and is a substrate of cyclophilin A. J Biol Chem 286:20441–20454. doi: 10.1074/jbc.M110.182436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hiet MS, Bauhofer O, Zayas M, Roth H, Tanaka Y, Schirmacher P, Willemsen J, Grunvogel O, Bender S, Binder M, Lohmann V, Lotteau V, Ruggieri A, Bartenschlager R. 2015. Control of temporal activation of hepatitis C virus-induced interferon response by domain 2 of nonstructural protein 5A. J Hepatol 63:829–837. doi: 10.1016/j.jhep.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 18.Kaneko T, Tanji Y, Satoh S, Hijikata M, Asabe S, Kimura K, Shimotohno K. 1994. Production of two phosphoproteins from the NS5A region of the hepatitis C viral genome. Biochem Biophys Res Commun 205:320–326. doi: 10.1006/bbrc.1994.2667. [DOI] [PubMed] [Google Scholar]
- 19.Blight KJ, Kolykhalov AA, Rice CM. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972–1974. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- 20.Appel N, Pietschmann T, Bartenschlager R. 2005. Mutational analysis of hepatitis C virus nonstructural protein 5A: potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J Virol 79:3187–3194. doi: 10.1128/JVI.79.5.3187-3194.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harak C, Meyrath M, Romero-Brey I, Schenk C, Gondeau C, Schult P, Esser-Nobis K, Saeed M, Neddermann P, Schnitzler P, Gotthardt D, Perez-Del-Pulgar S, Neumann-Haefelin C, Thimme R, Meuleman P, Vondran FW, De FR, Rice CM, Bartenschlager R, Lohmann V. 2016. Tuning a cellular lipid kinase activity adapts hepatitis C virus to replication in cell culture. Nat Microbiol 2:16247. doi: 10.1038/nmicrobiol.2016.247. [DOI] [PubMed] [Google Scholar]
- 22.Pietschmann T, Zayas M, Meuleman P, Long G, Appel N, Koutsoudakis G, Kallis S, Leroux-Roels G, Lohmann V, Bartenschlager R. 2009. Production of infectious genotype 1b virus particles in cell culture and impairment by replication enhancing mutations. PLoS Pathog 5:e1000475. doi: 10.1371/journal.ppat.1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, Mizokami M, Wakita T. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808–1817. doi: 10.1053/j.gastro.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 24.Eyre NS, Hampton-Smith RJ, Aloia AL, Eddes JS, Simpson KJ, Hoffmann P, Beard MR. 2016. Phosphorylation of NS5A serine-235 is essential to hepatitis C virus RNA replication and normal replication compartment formation. Virology 491:27–44. doi: 10.1016/j.virol.2016.01.018. [DOI] [PubMed] [Google Scholar]
- 25.Ross-Thriepland D, Harris M. 2014. Insights into the complexity and functionality of hepatitis C virus NS5A phosphorylation. J Virol 88:1421–1432. doi: 10.1128/JVI.03017-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Masaki T, Matsunaga S, Takahashi H, Nakashima K, Kimura Y, Ito M, Matsuda M, Murayama A, Kato T, Hirano H, Endo Y, Lemon SM, Wakita T, Sawasaki T, Suzuki T. 2014. Involvement of hepatitis C virus NS5A hyperphosphorylation mediated by casein kinase I-alpha in infectious virus production. J Virol 88:7541–7555. doi: 10.1128/JVI.03170-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tellinghuisen TL, Foss KL, Treadaway J. 2008. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog 4:e1000032. doi: 10.1371/journal.ppat.1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee KY, Chen YH, Hsu SC, Yu MJ. 2016. Phosphorylation of serine 235 of the hepatitis C Virus non-structural protein NS5A by multiple kinases. PLoS One 11:e0166763. doi: 10.1371/journal.pone.0166763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reed KE, Xu J, Rice CM. 1997. Phosphorylation of the hepatitis C virus NS5A protein in vitro and in vivo: properties of the NS5A-associated kinase. J Virol 71:7187–7197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fridell RA, Valera L, Qiu D, Kirk MJ, Wang C, Gao M. 2013. Intragenic complementation of hepatitis C virus NS5A RNA replication-defective alleles. J Virol 87:2320–2329. doi: 10.1128/JVI.02861-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fridell RA, Qiu D, Valera L, Wang C, Rose RE, Gao M. 2011. Distinct functions of NS5A in hepatitis C virus RNA replication uncovered by studies with the NS5A inhibitor BMS-790052. J Virol 85:7312–7320. doi: 10.1128/JVI.00253-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim S, Welsch C, Yi M, Lemon SM. 2011. Regulation of the production of infectious genotype 1a hepatitis C virus by NS5A domain III. J Virol 85:6645–6656. doi: 10.1128/JVI.02156-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chong WM, Hsu SC, Kao WT, Lo CW, Lee KY, Shao JS, Chen YH, Chang J, Chen SS, Yu MJ. 2016. Phosphoproteomics identified an NS5A phosphorylation site involved in hepatitis C virus replication. J Biol Chem 291:3918–3931. doi: 10.1074/jbc.M115.675413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsu SC, Lo CW, Pan TC, Lee KY, Yu MJ. 2017. Serine 235 is the primary NS5A hyperphosphorylation site responsible for hepatitis C virus replication. J Virol 91:e00194-17. doi: 10.1128/JVI.00194-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen YC, Su WC, Huang JY, Chao TC, Jeng KS, Machida K, Lai MM. 2010. Polo-like kinase 1 is involved in hepatitis C virus replication by hyperphosphorylating NS5A. J Virol 84:7983–7993. doi: 10.1128/JVI.00068-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coito C, Diamond DL, Neddermann P, Korth MJ, Katze MG. 2004. High-throughput screening of the yeast kinome: identification of human serine/threonine protein kinases that phosphorylate the hepatitis C virus NS5A protein. J Virol 78:3502–3513. doi: 10.1128/JVI.78.7.3502-3513.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quintavalle M, Sambucini S, Summa V, Orsatti L, Talamo F, De FR, Neddermann P. 2007. Hepatitis C virus NS5A is a direct substrate of casein kinase I-alpha, a cellular kinase identified by inhibitor affinity chromatography using specific NS5A hyperphosphorylation inhibitors. J Biol Chem 282:5536–5544. doi: 10.1074/jbc.M610486200. [DOI] [PubMed] [Google Scholar]
- 38.Kim J, Lee D, Choe J. 1999. Hepatitis C virus NS5A protein is phosphorylated by casein kinase II. Biochem Biophys Res Commun 257:777–781. doi: 10.1006/bbrc.1999.0460. [DOI] [PubMed] [Google Scholar]
- 39.Reiss S, Harak C, Romero-Brey I, Radujkovic D, Klein R, Ruggieri A, Rebhan I, Bartenschlager R, Lohmann V. 2013. The lipid kinase phosphatidylinositol-4 kinase III alpha regulates the phosphorylation status of hepatitis C virus NS5A. PLoS Pathog 9:e1003359. doi: 10.1371/journal.ppat.1003359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cordek DG, Croom-Perez TJ, Hwang J, Hargittai MR, Subba-Reddy CV, Han Q, Lodeiro MF, Ning G, McCrory TS, Arnold JJ, Koc H, Lindenbach BD, Showalter SA, Cameron CE. 2014. Expanding the proteome of an RNA virus by phosphorylation of an intrinsically disordered viral protein. J Biol Chem 289:24397–24416. doi: 10.1074/jbc.M114.589911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lemay KL, Treadaway J, Angulo I, Tellinghuisen TL. 2013. A hepatitis C virus NS5A phosphorylation site that regulates RNA replication. J Virol 87:1255–1260. doi: 10.1128/JVI.02154-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koch JO, Bartenschlager R. 1999. Modulation of hepatitis C virus NS5A hyperphosphorylation by nonstructural proteins NS3, NS4A, and NS4B. J Virol 73:7138–7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neddermann P, Clementi A, De FR. 1999. Hyperphosphorylation of the hepatitis C virus NS5A protein requires an active NS3 protease, NS4A, NS4B, and NS5A encoded on the same polyprotein. J Virol 73:9984–9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanji Y, Kaneko T, Satoh S, Shimotohno K. 1995. Phosphorylation of hepatitis C virus-encoded nonstructural protein NS5A. J Virol 69:3980–3986. doi: 10.1016/j.virol.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Badillo A, Receveur-Brechot V, Sarrazin S, Cantrelle FX, Delolme F, Fogeron ML, Molle J, Montserret R, Bockmann A, Bartenschlager R, Lohmann V, Lippens G, Ricard-Blum S, Hanoulle X, Penin F. 2017. Overall structural model of NS5A protein from hepatitis C virus and modulation by mutations conferring resistance of virus replication to cyclosporin A. Biochemistry 56:3029–3048. doi: 10.1021/acs.biochem.7b00212. [DOI] [PubMed] [Google Scholar]
- 46.Evans MJ, Rice CM, Goff SP. 2004. Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication. Proc Natl Acad Sci U S A 101:13038–13043. doi: 10.1073/pnas.0405152101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qiu D, Lemm JA, O'Boyle DR, Sun JH, Nower PT, Nguyen V, Hamann LG, Snyder LB, Deon DH, Ruediger E, Meanwell NA, Belema M, Gao M, Fridell RA. 2011. The effects of NS5A inhibitors on NS5A phosphorylation, polyprotein processing and localization. J Gen Virol 92:2502–2511. doi: 10.1099/vir.0.034801-0. [DOI] [PubMed] [Google Scholar]
- 48.Hsu SC, Tsai CN, Lee KY, Pan TC, Lo CW, Yu MJ. 2018. Sequential S232/S235/S238 phosphorylation of the hepatitis C virus nonstructural protein 5A. J Virol 92:e01295-18. doi: 10.1128/JVI.01295-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quintavalle M, Sambucini S, Di PC, De FR, Neddermann P. 2006. The alpha isoform of protein kinase CKI is responsible for hepatitis C virus NS5A hyperphosphorylation. J Virol 80:11305–11312. doi: 10.1128/JVI.01465-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goonawardane N, Gebhardt A, Bartlett C, Pichlmair A, Harris M. 2017. Phosphorylation of serine 225 in hepatitis C virus NS5A regulates protein-protein interactions. J Virol 91:e00805-17. doi: 10.1128/JVI.00805-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reiss S, Rebhan I, Backes P, Romero-Brey I, Erfle H, Matula P, Kaderali L, Poenisch M, Blankenburg H, Hiet MS, Longerich T, Diehl S, Ramirez F, Balla T, Rohr K, Kaul A, Buhler S, Pepperkok R, Lengauer T, Albrecht M, Eils R, Schirmacher P, Lohmann V, Bartenschlager R. 2011. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9:32–45. doi: 10.1016/j.chom.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nordle GA, Griffin S, Harris M. 2010. Identification of a novel phosphorylation site in hepatitis C virus NS5A. J Gen Virol 91:2428–2432. doi: 10.1099/vir.0.023614-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harak C, Radujkovic D, Taveneau C, Reiss S, Klein R, Bressanelli S, Lohmann V. 2014. Mapping of functional domains of the lipid kinase phosphatidylinositol 4-kinase type III alpha involved in enzymatic activity and hepatitis C virus replication. J Virol 88:9909–9926. doi: 10.1128/JVI.01063-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goonawardane N, Ross-Thriepland D, Harris M. 2018. Regulation of hepatitis C virus replication via threonine phosphorylation of the NS5A protein. J Gen Virol 99:62–72. doi: 10.1099/jgv.0.000975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Flotow H, Roach PJ. 1989. Synergistic phosphorylation of rabbit muscle glycogen synthase by cyclic AMP-dependent protein kinase and casein kinase I. Implications for hormonal regulation of glycogen synthase. J Biol Chem 264:9126–9128. [PubMed] [Google Scholar]
- 56.Lohmann V, Hoffmann S, Herian U, Penin F, Bartenschlager R. 2003. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J Virol 77:3007–3019. doi: 10.1128/JVI.77.5.3007-3019.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saeed M, Scheel TK, Gottwein JM, Marukian S, Dustin LB, Bukh J, Rice CM. 2012. Efficient replication of genotype 3a and 4a hepatitis C virus replicons in human hepatoma cells. Antimicrob Agents Chemother 56:5365–5373. doi: 10.1128/AAC.01256-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim S, Date T, Yokokawa H, Kono T, Aizaki H, Maurel P, Gondeau C, Wakita T. 2014. Development of hepatitis C virus genotype 3a cell culture system. Hepatology 60:1838–1850. doi: 10.1002/hep.27197. [DOI] [PubMed] [Google Scholar]
- 59.Wose Kinge CN, Espiritu C, Prabdial-Sing N, Sithebe NP, Saeed M, Rice CM. 2014. Hepatitis C virus genotype 5a subgenomic replicons for evaluation of direct-acting antiviral agents. Antimicrob Agents Chemother 58:5386–5394. doi: 10.1128/AAC.03534-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu M, Peng B, Chan K, Gong R, Yang H, Delaney W, Cheng G. 2014. Robust and persistent replication of the genotype 6a hepatitis C virus replicon in cell culture. Antimicrob Agents Chemother 58:2638–2646. doi: 10.1128/AAC.01780-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pietschmann T, Lohmann V, Rutter G, Kurpanek K, Bartenschlager R. 2001. Characterization of cell lines carrying self-replicating hepatitis C virus RNAs. J Virol 75:1252–1264. doi: 10.1128/JVI.75.3.1252-1264.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li Q, Brass AL, Ng A, Hu Z, Xavier RJ, Liang TJ, Elledge SJ. 2009. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc Natl Acad Sci U S A 106:16410–16415. doi: 10.1073/pnas.0907439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tai AW, Benita Y, Peng LF, Kim SS, Sakamoto N, Xavier RJ, Chung RT. 2009. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 5:298–307. doi: 10.1016/j.chom.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Trotard M, Lepere-Douard C, Regeard M, Piquet-Pellorce C, Lavillette D, Cosset FL, Gripon P, Le SJ. 2009. Kinases required in hepatitis C virus entry and replication highlighted by small interference RNA screening. FASEB J 23:3780–3789. doi: 10.1096/fj.09-131920. [DOI] [PubMed] [Google Scholar]
- 65.Katze MG, Kwieciszewski B, Goodlett DR, Blakely CM, Neddermann P, Tan SL, Aebersold R. 2000. Ser(2194) is a highly conserved major phosphorylation site of the hepatitis C virus nonstructural protein NS5A. Virology 278:501–513. doi: 10.1006/viro.2000.0662. [DOI] [PubMed] [Google Scholar]
- 66.Ross-Thriepland D, Mankouri J, Harris M. 2015. Serine phosphorylation of the hepatitis C virus NS5A protein controls the establishment of replication complexes. J Virol 89:3123–3135. doi: 10.1128/JVI.02995-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fukuma T, Enomoto N, Marumo F, Sato C. 1998. Mutations in the interferon-sensitivity determining region of hepatitis C virus and transcriptional activity of the nonstructural region 5A protein. Hepatology 28:1147–1153. doi: 10.1002/hep.510280433. [DOI] [PubMed] [Google Scholar]
- 68.Enomoto N, Sakuma I, Asahina Y, Kurosaki M, Murakami T, Yamamoto C, Ogura Y, Izumi N, Marumo F, Sato C. 1996. Mutations in the nonstructural protein 5A gene and response to interferon in patients with chronic hepatitis C virus 1b infection. N Engl J Med 334:77–81. doi: 10.1056/NEJM199601113340203. [DOI] [PubMed] [Google Scholar]
- 69.Koutsoudakis G, Herrmann E, Kallis S, Bartenschlager R, Pietschmann T. 2007. The level of CD81 cell surface expression is a key determinant for productive entry of hepatitis C virus into host cells. J Virol 81:588–598. doi: 10.1128/JVI.01534-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Backes P, Quinkert D, Reiss S, Binder M, Zayas M, Rescher U, Gerke V, Bartenschlager R, Lohmann V. 2010. Role of annexin A2 in the production of infectious hepatitis C virus particles. J Virol 84:5775–5789. doi: 10.1128/JVI.02343-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 73.Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, Bartenschlager R. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci U S A 103:7408–7413. doi: 10.1073/pnas.0504877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Binder M, Kochs G, Bartenschlager R, Lohmann V. 2007. Hepatitis C virus escape from the interferon regulatory factor 3 pathway by a passive and active evasion strategy. Hepatology 46:1365–1374. doi: 10.1002/hep.21829. [DOI] [PubMed] [Google Scholar]
- 75.Bojjireddy N, Botyanszki J, Hammond G, Creech D, Peterson R, Kemp DC, Snead M, Brown R, Morrison A, Wilson S, Harrison S, Moore C, Balla T. 2014. Pharmacological and genetic targeting of the PI4KA enzyme reveals its important role in maintaining plasma membrane phosphatidylinositol 4-phosphate and phosphatidylinositol 4,5-bisphosphate levels. J Biol Chem 289:6120–6132. doi: 10.1074/jbc.M113.531426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Neddermann P, Quintavalle M, Di Pietro C, Clementi A, Cerretani M, Altamura S, Bartholomew L, De, Francesco R. 2004. Reduction of hepatitis C virus NS5A hyperphosphorylation by selective inhibition of cellular kinases activates viral RNA replication in cell culture. J Virol 78:13306–13314. doi: 10.1128/JVI.78.23.13306-13314.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Duncan JS, Litchfield DW. 2008. Too much of a good thing: the role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochim Biophys Acta 1784:33–47. doi: 10.1016/j.bbapap.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 78.Ritter A, Friemel A, Kreis NN, Louwen F, Yuan J. 2016. Impact of Polo-like kinase 1 inhibitors on human adipose tissue-derived mesenchymal stem cells. Oncotarget 7:84271–84285. doi: 10.18632/oncotarget.12482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gozdz A, Nikolaienko O, Urbanska M, Cymerman IA, Sitkiewicz E, Blazejczyk M, Dadlez M, Bramham CR, Jaworski J. 2017. GSK3alpha and GSK3beta phosphorylate Arc and regulate its degradation. Front Mol Neurosci 10:192. doi: 10.3389/fnmol.2017.00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Narayana N, Cox S, Shaltiel S, Taylor SS, Xuong N. 1997. Crystal structure of a polyhistidine-tagged recombinant catalytic subunit of cAMP-dependent protein kinase complexed with the peptide inhibitor PKI(5-24) and adenosine. Biochemistry 36:4438–4448. doi: 10.1021/bi961947. [DOI] [PubMed] [Google Scholar]
- 81.Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 82.Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. 2011. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 83.Binder M, Quinkert D, Bochkarova O, Klein R, Kezmic N, Bartenschlager R, Lohmann V. 2007. Identification of determinants involved in initiation of hepatitis C virus RNA synthesis by using intergenotypic replicase chimeras. J Virol 81:5270–5283. doi: 10.1128/JVI.00032-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.