

Abstract
Alkyl chlorides are common functional groups in synthetic organic chemistry. However, the engagement of unactivated alkyl chlorides, especially tertiary alkyl chlorides, in transition-metal-catalyzed C–C bond formation remains challenging. Herein, we describe the development of a TiIII-catalyzed radical addition of 2° and 3° alkyl chlorides to electron-deficient alkenes. Mechanistic data are consistent with inner-sphere activation of the C–Cl bond featuring TiIII-mediated Cl atom abstraction. Evidence suggests that the active TiIII catalyst is generated from the TiIV precursor in a Lewis-acid-assisted electron transfer process.
Graphical Abstract

INTRODUCTION
Carbon–chlorine bonds are prevalent structural units in organic molecules. In particular, alkyl chlorides are frequently found in natural products1 and synthetic intermediates.2 These compounds can be readily prepared from common functional groups including alkenes,3 alcohols,4 ketones,5 epoxides,6 and alkanes.7 Nevertheless, methods that engage alkyl chlorides in organic synthesis are largely confined to the two-electron regime via canonical SN2 and Grignard reactions. New protocols that can selectively activate and functionalize alkyl chlorides may further expand the use of these electrophiles in complex target synthesis.8 Recent advances in radical chemistry have enabled the use of common functional groups (e.g., carboxylates,9 alcohols,10 alkenes,11 and alkanes12) in C–C bond forming reactions. Inspired by these seminal contributions and given our research interests in Ti radical catalysis, we aimed to develop a new approach for the activation and alkylation of unactivated 2° and 3° alkyl chlorides by employing the rich redox chemistry of Ti complexes.
In principle, three strategies may be envisioned for the activation of alkyl chlorides to form the corresponding carbon-centered radicals or their equivalents (Scheme 1A). First, metal oxidative insertion13 to the C–Cl bond can form metal–alkyl intermediates. In this context, first-row transition metals including Ni,14 Co,15 Fe,16 and Cu17 have been shown to be capable of engaging alkyl chlorides in C–C bond forming reactions. However, these methods are not suitable for transforming 3° R–Cl18 because the corresponding metal–alkyl species are susceptible to unproductive side reactions (e.g., via β-H elimination). Indeed, current examples using such a strategy are limited to the use of special 3° alkyl chlorides that cannot undergo β-H elimination due to geometric constraints. Recently, Fu et al. reported a Cu-catalyzed photochemical cyanation reaction,17 which constitutes a rare example in which a simple, unactivated 3° R–Cl was employed toward C–C formation. However, it was noted in the report that this method currently cannot be generalized to other tertiary substrates.
Scheme 1.

Ti-Catalyzed Alkylation of Unactivated Alkyl Chlorides: Challenges and Rationale
A second strategy relies on the direct, single-electron reduction of the alkyl chloride electrophile using a chemical reductant or a photocatalyst.19 This possibility is hampered by the very negative reduction potential of unactivated R–Cl (E ≤ −2.5 V vs Fc+/0).
The third strategy entails metal-promoted Cl atom abstraction20 to form the corresponding carbon-centered free radical. This pathway is the reversal of atom transfer radical addition but is challenged by the relatively high dissociation energy of C–Cl bonds (e.g., BDE of tBu–Cl is ca. 85 kcal/mol).
We were interested in using the third strategy for the activation of alkyl chlorides, as it could allow the engagement of 3∘ R–C, an underexplored class of electrophiles,21 in C–C forming reactions (Scheme 1B). In theory, TiIII is a well-suited catalyst for such a process because (1) TiIII compounds are versatile catalysts22 in the reductive activation of common functional groups (e.g., carbonyls23 and epoxides24) and (2) TiIV shows great affinity with highly electronegative, “hard” anions, making Cl atom abstraction thermodynamically favorable (e.g., BDE of TiIV–Cl in TiCl4 is 96 kcal/mol).25 This activation would lead to the formation of a carbon-centered radical that can participate in subsequent reactions in the presence of a radical acceptor. In a related study, Kambe achieved an elegant alkene carbomagnetization through the activation of 3∘ R–Cl.26 This reaction, however, relies on the formation of highly reducing TiII species generated in the presence of nBuMgCl, thus limiting the reaction scope to only unfunctionalized substrates. Huang recently reported the reductive addition of α-hydroxylactams to Michael acceptors using the Nugent–RajanBabu reagent.20b This reaction was proposed to undergo the intermediacy of α-chlorolactams, a highly activated class of electrophiles; it also required the use of Mg0 to achieve high efficiency, which limited the functional group compatibility.
In this article, we report the development of a Ti-catalyzed alkylation of unactivated alkyl chlorides using Michael acceptors. In particular, 3° R–Cl, a largely untapped class of electrophiles in transition metal catalysis, was successfully engaged in C–C bond formation. Our new method displays a reaction scope and functional group compatibility that are complementary to existing protocols for accessing similar types of products.
RESULTS AND DISCUSSION
A. Reaction Discovery and Catalyst Optimization
Our initial attempts to achieve the radical activation and addition of 3° alkyl chloride 1 to acrylate 2 proved challenging using the Nugent–RajanBabu reagent (Cp2TiCl2) or its derivative, Cp*2TiCl2. We hypothesize that the active site of these titanocenes is likely hindered by the pair of cyclopentadienyl ligands, rendering it difficult for the sterically demanding 3° R–Cl to approach (Scheme 2). We recently demonstrated the use of Cp*TiCl3—a catalyst frequently employed in polymerization reactions27 but is underexplored in radical chemistry— in the [3+2] cycloaddition of alkenes with N-acylaziridines28a or cyclopropyl ketones.23b The removal of a Cp* ligand decreased the steric profile of these catalysts but maintained their redox properties. As such, the cyclization of a carbon-centered radical onto the Ti-bound (aza)enolate can occur smoothly (see Scheme 2). In contrast, bulkier Cp*2TiCl2 provided the open-chain product instead.28b
Scheme 2.

Rationale for Catalyst Optimization
Indeed, the use of Cp*TiCl3 as the catalyst provided a positive lead result in the reductive C–C coupling between 1 and acrylate 2 (Table 1). Upon optimization, the desired alkylation product 3 was obtained in 70% yield using Zn as the stoichiometric reductant, Et3N⋅HCl as the proton source in toluene, and a reaction time of 1 h (entry 1). The unsubstituted CpTiCl3 complex proved inferior (entry 2), presumably due to the less negative reducing potential of the corresponding TiIII active catalyst (E1/2 = −0.79 V, compared with −1.12 V with Cp*TiCl3; see discussion below and the SI). As previously discussed, titanocene complexes Cp*2TiCl2 (E1/2 = −1.55 V) and Cp2TiCl2 (E1/2 = −1.19 V) were substantially less reactive (entries 3, 4).
Table 1.
Ti-Catalyzed Alkylation and Control Experimentsa
 | ||
|---|---|---|
| entry | variation from standard conditions | yield (%) |
| 1 | none | 70 |
| 2 | CpTiCl3 instead of Cp*TiCl3 | 10 |
| 3 | Cp2TiCl2 instead of Cp*TiCl3 | 22 |
| 4 | Cp*2TiCl2 instead of Cp*TiCl3 | 21 |
| 5 | 12 h reaction time | 98 |
| 6b | no Ti catalyst | <5 |
| 7b | no Zn or Et3N⋅HCl | <5 |
| 8b | Col⋅HCl instead of Et3N⋅HCl | 96 |
| 9b | Mn instead of Zn | <5 |
| 10b | DCM instead of toluene | 65 |
| 11b | EtOAc instead of toluene | 97 |
| 12b | MeCN or THF instead of toluene | <5 |
All reactions were conducted on a 0.1 mmol scale with NMR yields reported.
Reaction time: 12 h.
Extending the reaction time to 12 h led to the quantitative conversion of 1 to 3 (entry 5). Using the optimal conditions, we conducted control experiments to elucidate the role of each reaction component. As expected, the exclusion of the Ti catalyst completely shut down the C–C coupling reactivity (entry 6). Both the reductant and the ammonium salts are required to obtain appreciable amounts of 3 (entry 7). Collidine⋅HCl salt instead of Et3N⋅HCl as a proton source provided the product in nearly identical yield (entry 8). Notably, Mn instead of Zn is inactive (entry 9). Although Mn is more reducing than Zn, we observed no color change from red (TiIV) to green (TiIII), which was apparent in the Zn-promoted reaction. This finding indicated that Mn is incapable of reducing the precatalyst in our reaction medium (see discussion below). Nonpolar and poorly coordinating solvents, such as dichloromethane (DCM) or ethyl acetate, were compatible (entries 10 and 11), whereas MeCN and tetrahydrofuran (THF) strongly inhibited the formation of 3 (entry 12).
B. Substrate Scope
We then explored the reaction scope under the optimal conditions. Substituted acrylates (Table 2, entries 1–8) were transformed to their corresponding products smoothly. Various functional groups, including trifluoromethyl (4) and tertiary amine (12) motifs, were also tolerated. Importantly, our protocol was compatible with aryl bromide (14) and aryl boronate (16), functional groups that would likely induce catalyst promiscuity under previous conditions reported for alkyl chloride activation (e.g., Ni catalysis14). Notably, primary alkyl chloride (6) remained untouched, presumably because primary carbon-centered radicals are more difficult to generate than their tertiary congeners. Various other Michael acceptors (entries 9–13) provided the corresponding products in good to excellent yields. Interestingly, bicyclobutanes (28, 30)29 also underwent strain-relieving radical addition to provide disubstituted cyclobutenes in useful yields. Enones are currently incompatible presumably due to competing reductive transformations30 that do not involve the alkyl chloride.
Table 2.
Alkene Scope of Ti-Catalyzed Alkylationa
 |
All reactions were conducted on a 0.1 mmol scale unless otherwise noted with isolated yields reported.
EtOAc as the solvent.
40 instead of 1.
With 2 equiv of Zn, Et3N⋅HCl, and alkene.
40 °C.
1.0 mmol scale.
AlCl3 (1 equiv) used as Lewis acid additive.
The Ti-catalyzed alkylation proceeded smoothly with an array of structurally diverse 3° alkyl chlorides (Table 3, entries 1–11). In particular, alkylated furanoindoline 45 was obtained in a synthetically useful yield from the corresponding organochloride, and the structure was identified with X-ray crystallography. Secondary alkyl chlorides also proved to be suitable substrates (entries 12–18). Achieving high yields, however, sometimes required the use of higher loadings of Zn, Et3N⋅HCl, or Ti catalyst as well as prolonged catalyst preactivation before substrate addition. We also investigated secondary benzyl chloride and primary alkyl chloride. (1-Chloroethyl)benzene was fully consumed, yielding 2,3-diphenylbutane via radical dimerization as the major product (ca. 50%) along with a small amount of desired alkylation adduct (ca. 15%; see SI). (3-Chloropropyl)benzene was largely unreactive and afforded the alkylation product in ca. 10% yield. Primary alkyl chlorides are frequently suitable electrophilies for metal-catalyzed C–C formation in the literature.14–17 Therefore, our Ti-catalyzed reaction offers complementary selectivity and can be used for the selective functionalization of 2° and 3° R–Cl in the presence of their primary counterparts (see Table 2, entry 3).
Table 3.
Alkyl Chloride Scope of Ti-Catalyzed Alkylationa
 |
All reactions were conducted on 0.1 mmol scale with isolated yields reported.
With 2 equiv of alkene.
In the major product, 2 is added trans to Ph.
With 15 mol % Ti catalyst and 5 equiv of 2.
Cis-fused bicycle was formed as the observable stereoisomer.
Using alkene 14 instead of 2.
Using 14 as the limiting agent with 4 equiv of R–Cl.
With 20 mol % Ti catalyst for 72 h.
Using 3 equiv of Zn and Et3N⋅HCl.
Using 14 as the limiting agent with 2 equiv of R–Cl.
Only one stereoisomer was isolated; the stereochemistry at C2 is tentatively assigned as exo using 2D NMR.
Using 4 equiv of 2.
The configuration of C1 in the major product is S.
1 mmol scale with 20 mol % Ti catalyst.
Using 20 mol% Ti catalyst, 3 equiv Zn, Et3N⋅HCl, and 2; relative stereochemistry of the major product could not be determined.
The configuration of C3 in the major product is S.
C. Tandem Chlorination and Ti-Catalyzed Alkylation.
We then demonstrated the reductive alkylation reaction on synthetically relevant scales. Four alkyl chlorides were readily prepared from common functionalities using established protocols. Deoxychlorination of alcohols4 and hydrochlorination3c and chlorofunctionalization3g,h of alkenes led to structurally diverse alkyl chlorides in a single step in high efficiency (Scheme 3). Various other literature methods for the preparation of alkyl chlorides are provided in the SI. The resulting intermediates were then subjected to the described Ti-catalyzed conditions on a 1 mmol scale to afford the alkylation products with minimal changes in yield from the 0.1 mmol scale. In particular, owing to the large variety of alkene chlorofunctionalization reactions available in the literature, the two-step procedure comprising tandem chlorination and reductive alkylation constitutes a convenient and versatile method for the synthesis of vicinally difunctionalized products (e.g., 43, 45, 51). We also conducted gram-scale synthesis of 27, which resulted in a slightly decreased but synthetically useful yield (68%) likely as a result of the heterogeneity of the reaction system. Efforts are underway to improve the efficiency of gram-scale synthesis.
Scheme 3.
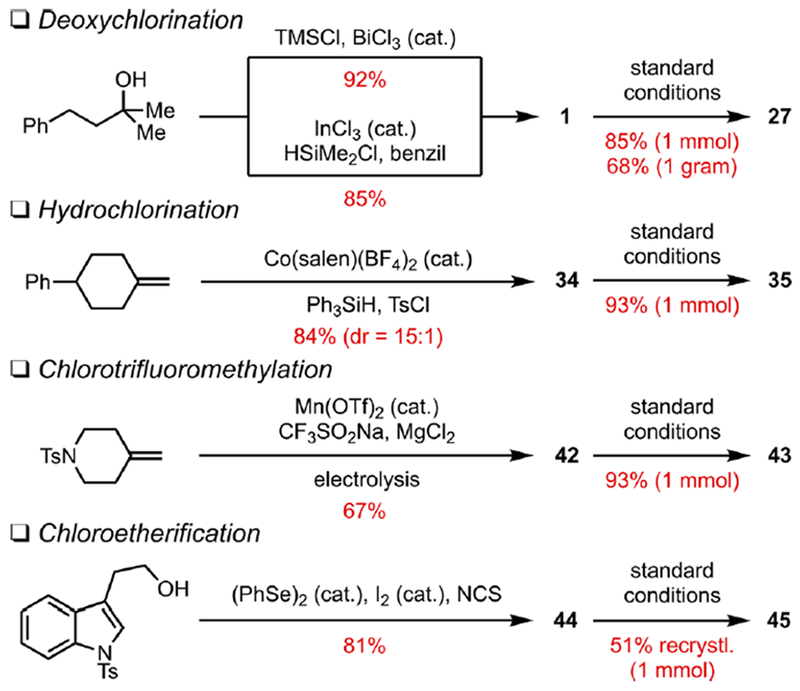
Synthetic Scale (1 mmol or Greater) Preparation and Ti-Catalyzed Alkylation of Alkyl Chlorides
To gain further insight into the compatibility of various functional groups with our reaction conditions, we examined the impact of additives (1.0 equiv) on the efficiency of the coupling process (Table 4). We found that adding benzofuran, N-Me indole, 4-phenylbutene, 4-octyne, 2-bromoethylbenzene, cyanobenzene, or phenyl methyl sulfide has no adverse impact on the yield of the reaction, with the additives recovered after the reaction. An aliphatic ketone has a moderate inhibition effect on the reaction, with product 3 isolated in 55% yield after 12 h. The presence of phenols, alcohols, epoxides, or pyridines, however, impedes the Ti catalysis, presumably by competitive coordination to the oxophilic metal center.
Table 4.
 |
Yields of product 3 were determined with 1H NMR.
Values in parentheses are recovery yields of the additives determined with GC or 1H NMR (see SI).
Alternative approaches to accessing the same type of products from alkyl chlorides frequently entail the generation of corresponding Grignard reagents followed by Michael addition31 or Sn-mediated radical processes,19a the latter of which are often carried out in an intramolecular fashion to avoid competitive hydrogen atom transfer (HAT) and other side reactions. Our Ti-catalyzed reaction thus provides an alternative platform for accessing these products with significantly improved ease of operation, substrate scope, and selectivity. Recently, several seminal contributions in the area of radical catalysis made it possible to obtain similar Giese-type products from common functional groups (e.g., carboxylates,9 alcohols,10 and alkenes11) other than alkyl chlorides. These methods, however, cannot provide a general access to vicinally difunctionalized products as described previously. In addition, our reaction displays a different scope compared to many existing methods in terms of functional group tolerance.32 Therefore, our Ti-catalyzed activation of alkyl chlorides provides a complementary approach to the catalytic radical reactions currently in place and allows an additional class of common functional groups (R–Cl) to be engaged in radical C–C bond formation.
D. Mechanistic Understanding of Potential Reaction Pathways.
Spin trapping experiments support the intermediacy of carbon-centered radicals in the alkylation reaction. In the presence of 5,5-dimethyl-1-pyrroline N-oxide (DMPO) and 1,4-cyclohexadiene (CHD), persistent nitroxyl radical 80 and HAT product 82 were formed, respectively (Scheme 4). The HAT reaction also constitutes an efficient alternative approach to the reduction of alkyl chlorides under Sn-/heavy metal-mediated conditions. The stereochemical infidelity of the reaction involving diastereomerically pure 66 also accords with the radical mechanism. An alternative, nonradical mechanism entails the in situ formation of an alkylzinc species from Zn and the alkyl chloride followed by Ti-catalyzed Michael addition. This possibility was ruled out using a reaction with stoichiometric Ti, wherein the Zn dust remaining after catalyst reduction was removed via filtration before addition of the alkyl chloride and acrylate. Product 3 was still formed in 40% yield.33 Currently, a pathway involving Ti-catalyzed alkylzinc formation via a transmetalation process34 cannot be ruled out. This mechanism, however, cannot account for the observed hydrodehalogenation in the presence of CHD.
Scheme 4.

Spin Trapping Experiments
Drastically different reactivity between Mn and Zn as the terminal reductant (see Table 1, entry 9) suggested that these metals do not serve simply as a reducing agent. As previously discussed, Mn is incapable of reducing Cp*TiCl3 to TiIII in the reaction medium. Given Mn’s more negative potential than Zn, we reasoned that the byproduct of the reduction process, Zn2+, likely plays a crucial role in the activation of the Ti catalyst.35 Indeed, in a control experiment using Mn as the stoichiometric reductant, the addition of Zn(OTf)2 restored the alkylation reactivity (Table 5). Interestingly, other Lewis acids such as Sc(OTf)3 and AlCl3 can also promote the desired reaction.
Table 5.
Lewis Acid Effect on the Ti-Catalyzed Alkylationa
| Lewis acid (1 equiv) | NMR yield (%) of 3 |
|---|---|
| none | <10 |
| Zn(OTf)2 | 70 |
| AlCl3 | 30 |
| Sc(OTf)2 | 46 |
Reaction conditions: see Table 1, entry 9 with a reaction time of 3 h.
Data from UV–vis experiments revealed that a strong Lewis acid can indeed accelerate the reduction of Cp*TiCl3 by Mn. In the presence of 10 equiv of AlCl3 (with respect to Ti), this reduction was nearly completed within 15 min (Figure 1). In stark contrast, with Mn alone, catalyst reduction was sluggish.
Figure 1.

Reduction of Cp*TiCl3 by Mn in the presence or absence of AlCl3. Top: UV–vis spectra showing the reduction in the presence of AlCl3; the peaks at 450 and 330 nm are assigned to Cp*TiCl3 and its corresponding reduced form, respectively. Bottom: Change in absorption at 450 nm showing the reduction of Cp*TiCl3.
1H NMR experiments showed that in the presence of AlCl3 a new Ti complex emerged. Cp*TiCl3 in toluene-d8 displays two CH3 resonances (2.12 and 1.95 ppm) presumably arising from different aggregation states. We tentatively assign these peaks to the monomeric and Cl-bridged dimeric36 Ti complexes.38 Upon addition of AlCl3, both resonances converged into a single signal at 1.93 ppm. We tentatively assign this species to the Cl-bridged heterometallic dimer with a composition of [Cp*TiCl3][AlCl3].37 The strong Lewis acidity of Al3+ can thus facilitate the reduction of the Ti center. The speciation of the active Ti complexes is currently underway.
The mechanism of C–Cl activation by TiIII was investigated using density functional theory (DFT) computation (see SI). Cyclic voltammetry data revealed that the potential required for the reduction of 1 was at least 1.1 V more negative than that of Cp*TiCl3 (Figure 2). As such, an outer-sphere electron transfer from the TiIII catalyst to 1 is highly unlikely. Owing to the higher dissociation energy of the TiIV–Cl bond relative to the C–Cl bond, an inner-sphere Cl atom abstraction by TiIII displays an energy barrier of only 6 kcal/mol. This mechanism provides an alternative means for the activation of strong bonds that are conventionally inert to single-electron transfer.
Figure 2.

Cyclic voltammetry of Cp*TiCl3 (2 mM) and tBuCl (2 mM) in DCM (0.2 M BU4NPF6) with a scan rate of 100 mV/s.
Taken together, a catalytic cycle was proposed (Scheme 5). The Cp*TiCl3 first undergoes a Lewis-acid-assisted electron transfer mediated by complex I to generate active Cp*TiIIICl2.38 This lower valent complex then abstracts a Cl atom from the alkyl chloride, furnishing R• and closing the catalytic cycle. The nascent R• then adds to the Michael acceptor to form electrophilic radical II, which is subsequently reduced and protonated to deliver the product, likely in a Ti-mediated process. An alternative pathway for the conversion of II to the product involves Cl atom transfer from the Ti catalyst to II followed by reduction of the resulting C–Cl bond. This pathway is less likely to operate on thermodynamic grounds because the Ti–Cl bond is substantially stronger than the C–Cl bond α to a carbonyl group (typically <80 kcal/mol).39
Scheme 5.
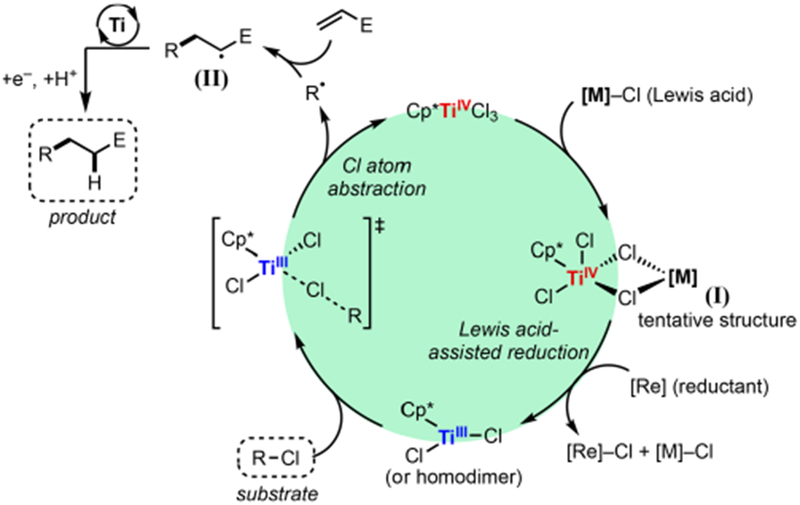
Proposed Catalytic Cycle
CONCLUSION
To summarize, we report the Ti-catalyzed radical alkylation of unactivated secondary and tertiary alkyl chlorides with Michael acceptors. This method provides alternative means to generate carbon-centered radicals and offers a new route to C–C bond formation that is complementary to existing protocols. Future efforts are directed toward (1) understanding the structure of the active Ti catalyst and its action in activating C–Cl and other strong chemical bonds and (2) expanding the new radical reactivity of alkyl chlorides to other types of synthetically valuable reactions.
Supplementary Material
ACKNOWLEDGMENTS
Funding support was provided by Cornell University. S.L. thanks NSF for a CAREER award (CHE-1751839). This study made use of the NMR facility (CHE-1531632) supported by NSF and the ESR facility supported by NIGMS (P41GM103521). We thank Dr. Terry McCallum for reproducing experimental results, Gregory Sauer for assistance with DFT calculations, Dr. Ivan Keresztes for help with NMR spectral analysis, Dr. Samantha MacMillan for help with X-ray crystal structure determination, and Dr. Boris Dzikovski for assistance with ESR data acquisition.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b08605.
Experimental procedures and characterization data (PDF)
Crystallography data for 45 (CIF)
The authors declare no competing financial interest.
Crystallography data for 45 were deposited in the Cambridge Structural Database (CCDC 1833922).
REFERENCES
- (1).(a) Gribble GW Acc. Chem. Res 1998, 31, 141–152. [Google Scholar]; (b) Gal B; Bucher C; Burns NZ Mar. Drugs 2016, 14, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).For examples, see:; (a) Weires NA; Styduhar ED; Baker EL ; Garg NK J. Am. Chem. Soc 2014, 136, 14710–14713. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Reyes JR; Xu J; Kobayashi K; Bhat V; Rawal VH Angew. Chem., Int. Ed 2017, 56, 9962–9966. [DOI] [PubMed] [Google Scholar]; (c) Okano K; Tokuyama H; Fukuyama T J. Am. Chem. Soe 2006, 128, 7136–7137. [DOI] [PubMed] [Google Scholar]
- (3).For reviews, see:; (a) Chung W; Vanderwal CD Angew. Chem., Int. Ed 2016, 55, 4396–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Denmark SE; Kuester WE; Burk MT Angew. Chem, Int. Ed 2012, 51, 10938–10953 [DOI] [PMC free article] [PubMed] [Google Scholar]; For examples, see: .; (c) Gaspar B; Carreira EM Angew. Chem., Int. Ed 2008, 47, 5758–5760. [DOI] [PubMed] [Google Scholar]; (d) Wilger DJ; Grandjean JMM; Lammert TR; Nicewicz DA Nat. Chem 2014, 6, 720–726. [DOI] [PubMed] [Google Scholar]; (e) Schevenels FT; Shen M; Snyder SA J. Am. Chem. Soc 2017, 139, 6329–6337. [DOI] [PubMed] [Google Scholar]; (f) Sakurada I; Yamasaki S; Gottlich R; Iida T; Kanai M; Shibasaki M J. Am. Chem. Soc 2000, 122, 1245–1246. [Google Scholar]; (g) Horibe T; Ohmura S; Ishihara K Org. Lett 2017, 19, 5525–5528. [DOI] [PubMed] [Google Scholar]; (h) Ye K; Pombar G; Fu N; Sauer GS; Keresztes I; Lin S J. Am. Chem. Soc 2018, 140, 2438–2441. [DOI] [PubMed] [Google Scholar]
- (4).For examples, see:; (a) Yasuda M; Yamasaki S; Onishi Y; Baba A J. Am. Chem. Soc 2004, 126, 7186–7187. [DOI] [PubMed] [Google Scholar]; (b) Labrouillere M; Le Roux C; Gaspard-Iloughmane H; Dubac J Synlett 1994, 1994, 723–724. [Google Scholar]; (c) Bendall JG; Payne AN; Screen TEO; Holmes AB Chem. Commun 1997, 1067–1068. [Google Scholar]; (d) Kelly BD; Lambert TH J. Am. Chem. Soc 2009, 131, 13930–13931. [DOI] [PubMed] [Google Scholar]; (e) Su JY; Grunenfelder DC; Takeuchi K; Reisman SE Org. Lett 2018, 20, 4912–4916. [DOI] [PubMed] [Google Scholar]
- (5).For examples, see:; (a) Yasuda M; Yamasaki S; Onishi Y; Baba A J. Am. Chem. Soc 2004, 126, 13690–13691. [Google Scholar]; (b) Reyes JR; Rawal VH Angew. Chem., Int. Ed 2016, 55, 3077–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).For an example, see:; Pu X; Qi X; Ready JM J. Am. Chem. Soc 2009, 131, 10364–10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).For examples, see:; (a) Quinn RK; Konst ZA; Michalak SE; Schmidt Y; Szklarski AR; Flores AR; Nam S; Horne DA; Vanderwal CD; Alexanian EJ J. Am. Chem. Soc 2016, 138, 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ozawa J; Kanai M Org. Lett 2017, 19, 1430–1433. [DOI] [PubMed] [Google Scholar]; (c) Short MA; Blackburn JM; Roizen JL Angew. Chem. Int. Ed 2018, 57, 296–299. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Li G; Dilger AK; Cheng PT; Ewing WR; Groves JT Angew. Chem. Int. Ed 2018, 57, 1251–1255. [DOI] [PubMed] [Google Scholar]
- (8).For a review on Pd-catalyzed cross coupling involving alkyl chlorides (1° R-Cl, in most cases), see:; Kambe N; Iwasakia T; Terao, J. Chem. Soc. Rev 2011, 40, 4937–4947. [DOI] [PubMed] [Google Scholar]
- (9).For examples, see:; (a) Qin T; Malins LR; Edwards JT; Merchant RR; Novak AJE; Zhong JZ; Mills RB; Yan M; Yuan C; Eastgate MD; Baran PS Angew. Chem. Int. Ed 2017, 56, 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chu L; Ohta C; Zuo Z; MacMillan DWC J. Am. Chem. Soc 2014, 136, 10886–10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).For an example, see:; Nawrat CC; Jamison CR; Slutskyy Y; MacMillan DW; Overman LE J. Am. Chem. Soc 2015, 137, 11270–11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).For examples, see:; (a) Lo JC; Yabe Y; Baran PS J. Am. Chem. Soc 2014, 136, 1304–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Obradors C; Martinez RM; Shenvi RA J. Am. Chem. Soc 2016, 138, 4962–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lo JC; Kim D; Pan CM; Edwards JT; Yabe Y; Gui J; Qin T; Gutierrez S; Giacoboni J; Smith MW; Holland PL; Baran PS J. Am. Chem. Soc 2017, 139, 2484–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).For examples, see:; (a) Chu JCK; Rovis T Nature 2016, 539, 272–275. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Choi GJ; Zhu Q; Miller DC; Gu CJ; Knowles RR Nature 2016, 539, 268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Depending on the metal, this insertion mechanism can be different. In some cases, it has been postulated that the metal insertion occurs through first Cl atom abstraction (analogous to the scenario in strategy 3) followed by radical recombination.
- (14).For a review, see:; (a) Choi J; Fu GC Science 2017, 356, eaaf7230. [DOI] [PMC free article] [PubMed] [Google Scholar]; For examples, see: .; (b) Terao J; Watanabe H; Ikumi A; Kuniyasu H; Kambe N J. Am. Chem. Soc 2002, 124, 4222–4223. [DOI] [PubMed] [Google Scholar]; (d) Gonzalez-Bobes F; Fu GC J. Am. Chem. Soc 2006, 128, 5360–5361. [DOI] [PubMed] [Google Scholar]; (e) Lu Z; Fu GC Angew. Chem. Int. Ed 2010, 49, 6676–6678. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wang X; Wang S; Xue W; Gong H J. Am. Chem. Soc 2015, 137, 11562–11565. [DOI] [PubMed] [Google Scholar]; (g) Hofstra JL; Cherney AH; Ordner CM; Reisman SE J. Am. Chem. Soc 2018, 140, 139–142. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Csok Z; Vechorkin O; Harkins SB; Scopelliti R; Hu X J. Am. Chem. Soc 2008, 130, 8156–8157. [DOI] [PubMed] [Google Scholar]; (i) Borjesson M; Moragas T; Martin R J. Am. Chem. Soc 2016, 138, 7504–7507. [DOI] [PubMed] [Google Scholar]; (j) Zhou Y-Y; Uyeda C Angew. Chem., Int. Ed 2016, 55, 3171–3175. [DOI] [PubMed] [Google Scholar]; (k) Erickson LW; Lucas EL; Tollefson EJ; Jarvo ER J. Am. Chem. Soc 2016, 138, 14006–14011. [DOI] [PubMed] [Google Scholar]; (l) Anka-Lufford LL; Huihui KMM; Gower NJ; Ackerman LKG; Weix D J. Chem. - Eur. J 2016, 22, 11564–11567. [DOI] [PubMed] [Google Scholar]
- (15).For examples, see:; (a) Qian X; Auffrant A; Felouat A; Gosmini C Angew. Chem., Int. Ed 2011, 50, 10402–10405. [DOI] [PubMed] [Google Scholar]; (b) Ikeda Y; Nakamura T; Yorimitsu H; Oshima K J. Am. Chem. Soc 2002, 124, 6514–6515. [DOI] [PubMed] [Google Scholar]
- (16).(a) Hatakeyama T; Hashimoto T; Kondo Y; Fujiwara Y; Seike H; Takaya H; Tamada Y; Ono T; Nakamura M J. Am. Chem. Soc 2010, 132, 10674–10676. [DOI] [PubMed] [Google Scholar]; (b) Ghorai SK; Jin M; Hatakeyama T; Nakamura M Org. Lett 2012, 14, 1066–1069. [DOI] [PubMed] [Google Scholar]
- (17).Ratani TS; Bachman S; Fu GC; Peters JC J. Am. Chem. Soc 2015, 137, 13902–13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ref 17 shows one example of an unactivated 3° alkyl chloride. Ref 14e shows one example of a 3° benzyl chloride, the alkyl-metal complexes of which also have substantial geometric constraints to undergo β-H elimination. Refs 14h, 15b, and 16b each shows one example of 1-chloroadamantane, whose alkyl-metal complexes cannot undergo β-H elimination.
- (19).Organotin hydrides as reductants:; (a) Hanessian S; Di Fabio R; Marcoux JF; Prud’homme M J. Org. Chem 1990, 55, 3436–3438 [Google Scholar]; Examples of photoredox alkyl bromide activation: .; (b) Zhang P; Le CC; MacMillan DW J. Am. Chem. Soc 2016, 138, 8084–8087. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Staveness D; Bosque I; Stephenson CR J. Acc. Chem. Res 2016, 49, 2295–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).(a) Agapie T; Diaconescu PL; Cummins CC J. Am. Chem. Soc 2002, 124, 2412–2413. [DOI] [PubMed] [Google Scholar]; A recent work shows that TiIII can catalyze reactions involving highly activated C-Cl bonds in α-chlorolactams:; (b) Zheng X; Dai X-J; Yuan H-Q; Ye C-X; Ma J ; Huang P-Q Angew. Chem. Int. Ed 2013, 52, 3494–3498. [DOI] [PubMed] [Google Scholar]
- (21).A recent example shows that Mn can catalyze the C-B bond formation of tertiary alkyl chlorides in the presence of EtMgBr:; (a) Atack TC; Cook SP J. Am. Chem. Soc 2016, 138, 6139. [DOI] [PubMed] [Google Scholar]; Amination of electronically activated, tertiary α-chloroamides:; (b) Kainz QM; Matier CD; Bartoszewicz A; Zultanski SL; Peters JC; Fu GC Science 2016, 351, 681–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).For representative reviews, see:; (a) Davis-Gilbert ZW; Tonks IA Dalton Trans 2017, 46, 11522–11528. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cuerva JM; Juan JC; Justicia J; Oller-Lopez JL; Oltra JE Top. Curr. Chem 2006, 264, 63–91. [Google Scholar]; (c) Streuff J Chem. Rec 2014, 14, 1100–1113. [DOI] [PubMed] [Google Scholar]; (d) Streuff J; Gansauer A Angew. Chem. Int. Ed 2015, 54, 14232–14242. [DOI] [PubMed] [Google Scholar]
- (23).For examples, see:; (a) Bensari A; Renaud J-L; Riant O Org. Lett 2001, 3, 3863–3865. [DOI] [PubMed] [Google Scholar]; (b) Hao W; Harenberg JH; Wu X; MacMillan SN; Lin S J. Am. Chem. Soc 2018, 140, 3514–3517. [DOI] [PubMed] [Google Scholar]; (c) Leijendekker LH; Weweler J; Leuther TM; Streuff J Angew. Chem., Int. Ed 2017, 56, 6103–6106. [DOI] [PubMed] [Google Scholar]; (d) Kablaoui NM; Hicks FA; Buchwald SL J. Am. Chem. Soc 1997, 119, 4424–4431. [Google Scholar]
- (24).For examples, see:; (a) Gansauer A; Behlendorf M; von Laufenberg D; Fleckhaus A; Kube C; Sadasivam DV; Flowers RA II Angew. Chem., Int. Ed 2012, 51, 4739–4742. [DOI] [PubMed] [Google Scholar]; (b) Gansauer A; Hildebrandt S; Michelmann A; Dahmen T; von Laufenberg D; Kube C; Fianu GD; Flowers RA II Angew. Chem., Int. Ed 2015, 54, 7003–7006. [DOI] [PubMed] [Google Scholar]; (c) Zhao Y; Weix DJ J. Am. Chem. Soc 2015, 137, 3237–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Luo Y-R Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, 2007. [Google Scholar]
- (26).Nii S; Terao J; Kambe N J. Org. Chem 2004, 69, 573–576. [DOI] [PubMed] [Google Scholar]
- (27).Nomura K, Liu J In Organometallic Reactions and Polymerization. Lecture Notes in Chemistry, Osakada K, Ed.; Springer: Berlin, 2014; Vol 85, pp 51–88. [Google Scholar]
- (28).(a) Hao W; Wu X; Sun JZ; Siu JC; MacMillan SN; Lin S J. Am. Chem. Soc 2017, 139, 12141–12144. [DOI] [PubMed] [Google Scholar]; (b) Zhang Y-Q; Vogelsang E; Qu Z-W; Grimme S; Gansauer A Angew. Chem. Int. Ed 2017, 56, 12654–12657. [DOI] [PubMed] [Google Scholar]
- (29).(a) Lopchuk JM; Fjelbye K; Kawamata Y; Malins LR; Pan C-M; Gianatassio R; Wang J; Prieto L; Bradow J; Brandt TA; Collins MR; Elleraas J; Ewanicki J; Farrell W; Fadeyi OO; Gallego GM; Mousseau JJ; Oliver R; Sach NW; Smith JK; Spangler JE; Zhu H; Zhu J; Baran PS J. Am. Chem. Soc 2017, 139, 3209–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Noyori R; Suzuki T; Kumagai Y; Takaya H J. Am. Chem. Soc 1971, 93, 5894–5896. [Google Scholar]
- (30).Streuff, J. Chem. - Eur. J 2011, 17, 5507–5510. [DOI] [PubMed] [Google Scholar]
- (31).For an example, see:; Chai G; Lu Z; Fu C; Ma S Adv. Synth. Catal 2009, 351, 1946–1954. [Google Scholar]
- (32).For example, aryl bromides (14), alkyl bromides (76), and aryl boronates (16) might not be compatible with Ni-catalyzed conditions, tertiary amines (12) might not be compatible with photoredox conditions, alkenes (66, 71) might not be compatible with Fe-catalyzed hydroalkylation conditions, and alkyl halides (6, 76) and ketones (74) might not be compatible with conditions involving Mg or Grignard reagents.
- (33).If the reaction goes through the proposed mechanism, the theoretical yield of reaction using 1 equiv of Cp*TiCl3 is 50%.
- (34).(a) Fleury LM; Kosal AD; Masters JT; Ashfeld BL J. Org. Chem 2013, 78, 253–269. [DOI] [PubMed] [Google Scholar]; (b) Fleury LM; Ashfeld BL Org. Lett 2009, 11, 5670–5673. [DOI] [PubMed] [Google Scholar]
- (35).Effect of metal reductants on the electrochemical behavior of titanocenes: Enemærke RJ; Larsen J; Skrydstrup T; Daasbjerg K J. Am. Chem. Soc 2004, 126, 7853–7864. [DOI] [PubMed] [Google Scholar]
- (36).For the observation of a similar dimeric half-titanocene complex, see:; Paisner SN; Lavoie GG; Bergman RG Inorg. Chim. Acta 2002, 334, 253–275. [Google Scholar]
- (37).Cp*TiIIICl2 can exist as a dimer in solution, as has been observed with Cp2TiIIICl (ref 35).
- (38).For studies on heterometallic dimers involving Ti, see:; (a) Sekutowski DJ; Stucky GD Inorg. Chem 1975, 14, 2192–2199. [Google Scholar]; (b) Folting K; Huffman JC; Bansemer RL; Caulton KG Inorg. Chem 1984, 23, 3289–3292. [Google Scholar]; Higher aggregation numbers have been reported, and their formation in our reaction cannot be ruled out at this stage.
- (39).By analogy, BDE(C–Cl) in 2-chloropropionic acid is 72.1 kcal/ mol. See:; Lagoa ALC; Diogo HP; Minas da Piedade ME; Amaral LMPF; Guedes RC; Costa Cabral BJ; Kulikov DV; Verevkin SP; Siedler M; Epple M J. Phys. Chem. A 2002, 106, 9855–9861. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.