

Abstract
The formation of a disulfide bond is a critical step in the folding of numerous secretory and membrane proteins and catalyzed in vivo. A variety of mechanisms and protein structures have evolved to catalyze oxidative protein folding. Those enzymes that directly interact with a folding protein to accelerate its oxidative folding are mostly thiol‐disulfide oxidoreductases that belong to the thioredoxin superfamily. The enzymes of this class often use a CXXC active‐site motif embedded in their thioredoxin‐like fold to promote formation, isomerization, and reduction of a disulfide bond in their target proteins. Over the past decade or so, an increasing number of substrates of the thiol‐disulfide oxidoreductases that are present in the ER of mammalian cells have been discovered, revealing that the enzymes play unexpectedly diverse physiological functions. However, functions of some of these enzymes still remain unclear due to the lack of information on their substrates. Here, we review the methods used by researchers to identify the substrates of these enzymes and provide data that show the importance of using trichloroacetic acid in sample preparation for the substrate identification, hoping to aid future studies. We particularly focus on successful studies that have uncovered physiological substrates and functions of the enzymes in the periplasm of Gram‐negative bacteria and the endoplasmic reticulum of mammalian cells. Similar approaches should be applicable to enzymes in other cellular compartments or in other organisms.
Keywords: thioredoxin superfamily member, oxidative protein folding, disulfide bond, thiol‐based redox regulation, protein disulfide isomerase, disulfide‐linked enzyme–substrate complex
Abbreviations
- DTT
dithiothreitol
- ER
endoplasmic reticulum
- IAM
iodoacetamide
- NEM
N‐ethylmaleimide
- PDI
protein disulfide isomerase
- SDS
sodium dodecyl sulfate
- TCA
trichloroacetic acid
Introduction
A protein disulfide bond is a covalent linkage formed by oxidation of two cysteines. The formation of disulfide bonds is an important step in the folding of many secretory and membrane proteins. Formation of disulfide bonds can occur spontaneously in vitro in the presence of appropriate electron acceptor such as O2. However, the process is catalyzed in vivo. To date, a variety of enzymes have been identified that promote the efficient oxidative folding of proteins.1, 2, 3, 4, 5, 6
Protein disulfide bond formation mainly takes place in the periplasm of Gram‐negative bacteria or in the endoplasmic reticulum (ER) of eukaryote. The process is intensively studied in a Gram‐negative bacterium Escherichia coli.
In the periplasm of E. coli, DsbA introduces disulfide bonds into newly translocated proteins by transferring a disulfide bond formed in the active site CXXC (two cysteines separated by two amino acids residues) embedded in its thioredoxin‐like fold to a pair of cysteines on the substrate.7, 8 After receiving two electrons from the substrate, the two cysteines in the active site of DsbA is reoxidized by a cytoplasmic membrane protein DsbB that passes electrons from DsbA to quinones in the respiratory chain.3, 9, 10, 11, 12 To repair a misoxidized protein, DsbC, another thioredoxin‐like protein in the periplasm, reduces the disulfide bond formed between non‐native pair of cysteines.13, 14 The active site cysteines of DsbC are kept reduced by a membrane protein DsbD that transfers electron directly from cytoplasmic thioredoxins to DsbC. Cytoplasmic thioredoxins are, in turn, kept reduced by thioredoxin reductase that uses NAPDH.15, 16
In the ER of eukaryote, protein disulfide isomerase (PDI) family members introduce disulfide bonds into folding proteins.1, 5 PDI family members are characterized by the presence of one or more thioredoxin‐like domain(s) with or without CXXC motif and their localization to the ER. After transfer of a disulfide bond(s) to its substrate, PDIs are in turn reoxidized by PDI oxidases such as Ero1α, Ero1β, Prx4, and VKOR.1, 5, 17 Whether or not a reductive system analogous to the DsbC–DsbD pathway exists in the ER of mammalian cells still remains to be uncovered although the formation of correct disulfide bonds in several proteins was impaired by inhibitors of cytosolic thioredoxin reductase.18, 19, 20
Importantly, accumulating evidence indicates that the functions of PDI family members are not limited to protein disulfide bond formation. For example, ERdj5, a PDI family member also known as JPDI, accelerates the ER‐associated degradation of misfolded proteins by cleaving disulfide bonds in the proteins, which is presumably required for the efficient retrotranslocation of the proteins from the ER to the cytosol and their subsequent degradation by proteasome.21, 22, 23, 24 Moreover, some PDI family members regulate ER stress response or Ca2+ homeostasis by altering the oxidative status of cysteines in ER stress sensors such as IRE1α and PERK or in calcium pumps such as SERCA2b, an isoform of sarcoplasmic/endoplasmic reticulum calcium ATPase, resulting in their activation or inactivation.25, 26, 27 Furthermore, a portion of PDI is secreted outside of cells and participates in the regulation of blood clotting.28
The ER of mammalian cells harbors more than 20 PDI family members.1, 5, 29 They are supposed to play some important roles within or outside of cells by controlling biological processes perhaps by their redox activities. However, the physiological functions of some members still remain largely unknown perhaps because of the lack of information on their substrates. Indeed, the identification of the substrates of enzymes of this class greatly contributed to our understanding of their physiological functions.30, 31, 32, 33, 34
Here, we will provide an overview of approaches used to identify the physiological substrates of thiol‐disulfide oxidoreductases that are present in the periplasm of E. coli or on the secretory pathway of mammalian cells. The use of these approaches will further help us define the roles of each enzyme in cells or tissues. Although, in this review, we focus on studies with thiol‐oxidoreductases localized in the cell surfaces, similar approached should be applicable to studies with enzymes localized in other cellular compartments such as the cytosol and mitochondria.
Chemistry of thiol‐disulfide exchange reactions
The two cysteines in the CXXC active site of the thiol‐disulfide oxidoreductases belonging to thioredoxin superfamily are mostly redox‐active. They usually exist in an oxidized (disulfide bonded) state or a reduced (unpaired) state. These enzymes use their redox‐active cysteines to introduce, break, or isomerize disulfide bonds, resulting in the changes in the redox state and/or conformation of their target proteins.3, 4, 5
The enzyme‐dependent formation of a disulfide bond in a substrate protein proceeds via two steps [Fig. 1(A)].35, 36, 37, 38, 39 First, a deprotonated cysteine of a substrate attacks the sulfur atom of the N‐terminal cysteine of the disulfide bond in the active site of the enzyme, leading to the formation of disulfide‐linked complex between the enzyme and the substrate. Second, one of the remaining cysteines of the substrate is deprotonated and attacks the sulfur atom of the substrate cysteine that is disulfide‐bonded to a cysteine of the enzyme. This reaction results in the formation and cleavage of a disulfide bond in the substrate and the enzyme, respectively.
Figure 1.
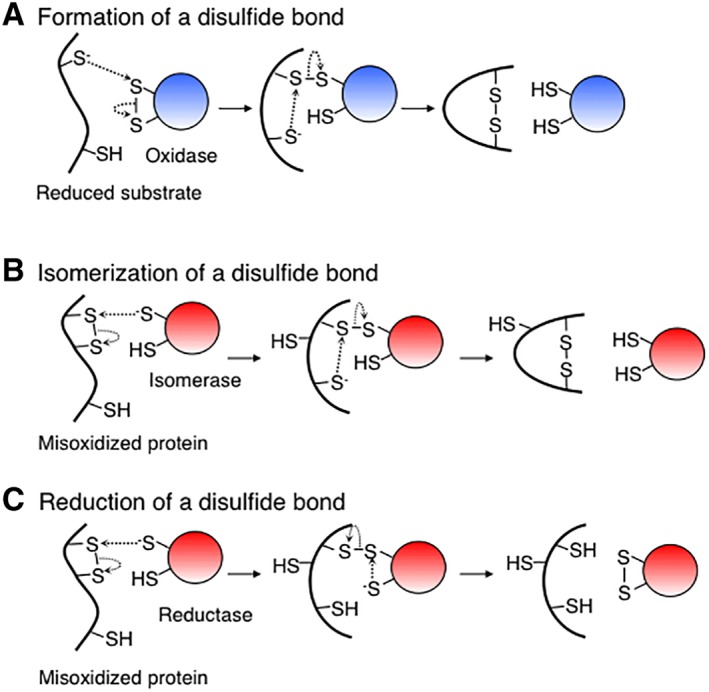
Mechanisms of thiol‐disulfide exchange reactions catalyzed by thiol‐disulfide oxidoreductases. Formation (A), isomerization (B), and reduction (C) of a disulfide bond.
The reduction or isomerization of a disulfide bond in a substrate protein also proceeds via two steps [Fig. 1(B and C)].4, 37, 38, 39, 40, 41 In both processes, the reaction starts with the nucleophilic attack by a deprotonated cysteine in the active site of the enzyme on the disulfide bond in the substrate protein. This reaction results in a mixed‐disulfide bond between the enzyme and its substrate, which can be resolved in two ways. In the first, the mixed disulfide is attacked by a third cysteine of the substrate, resolving the complex, allowing a more stable disulfide bond to form within the substrate and restoring the reduced form of the enzyme. In this case, the enzyme is acting as a disulfide isomerase. In the second, the mixed disulfide is attacked by the C‐terminal cysteine in the active site of the enzyme, generating a reduced substrate and an oxidized enzyme. In this case, the enzyme is acting as a disulfide reductase.
In these ways, a thiol‐disulfide exchange reaction catalyzed by this class of enzymes all proceed through a disulfide‐linked intermediate between the enzyme and the substrate. Thus, the stabilization, purification, and mass spectrometric analysis of such a complex may lead to the identification of the substrates of the enzyme. Using this property of thiol‐disulfide oxidoreductases, researchers have developed various methods to effectively identify their physiological substrates, as we will see below.
Stabilization of enzyme–substrate complexes by modification of free cysteines with alkylation reagents
To identify the substrates of a thiol‐disulfide oxidoreductase, it is often necessary to modify free cysteines in the sample. Since thiol groups of free cysteines are highly reactive and can attack disulfide bonds, their presence can cause the artifactual oxidation or reduction of proteins or breakage of mixed‐disulfide complexes.4, 42, 43, 44 Thus, to stabilize the disulfide‐linked complexes between the enzyme and its substrates, it is often essential to modify the thiol groups with alkylation reagents.
A variety of thiol‐reactive reagents are commercially available. Here, we discuss two of the typical thiol‐alkylation reagents: iodoacetamide (IAM) and N‐ethylmaleimide (NEM).42, 44 The derivatives of these reagents are commonly used in biochemical studies. IAM reacts specifically with exposed thiol group, leaving behind a carbamidomethyl (CH2CONH2) group irreversibly linked to the sulfur in cysteine. N‐Ethylmaleimide (NEM) also reacts with exposed thiols. The adduct it leaves behind is a C6NO2H8 group. Regrettably, although NEM is stable at pH lower than 6, this reagent is unstable at higher pH. At pH 7, the half‐time of hydrolysis is ~45 h; however, at pH 9, the half‐time is less than 1 h. At high pH, the maleimide ring of NEM is hydrolyzed to form N‐ethylmaleamic acid, resulting in the loss of thiol reactivity.44 Nevertheless, NEM has an advantage that it reacts with the thiols of cysteines much faster than IAM.4, 42 Because of this reason, NEM is especially suitable for the alkylation of free cysteines in mixed‐disulfide complexes that are highly unstable.
Both IAM and NEM require the presence of the thiolate anion and are specific to thiols at neutral pH values. At higher pH, undesirable side reactions with other amino acids including lysine and histidine may occur.42, 44 Since both reagents are small and uncharged, they can pass through cellular membranes. So, it is possible to alkylate free cysteines on a protein within cells by adding these reagents to the culture. However, it should be emphasized that both reagents react with surface‐exposed cysteines. Thus, buried or partially exposed cysteines in a protein are hard to be alkylated with these reagents unless the protein is denatured.
Stabilization of enzyme–substrate complexes by acid quenching
The reactive entities in thiol‐disulfide exchange reactions are deprotonated thiolate anions.43 Thus, protonation of the thiol groups greatly decreases its reactivity. In addition, the acid quenching is very fast, occurring with a rate constant greater than 109 sec−1 M−1, providing a prompt way to block thiol‐disulfide exchange reactions.42
Acidification not only protonates surface‐exposed protein thiols but also denatures proteins. Thus, almost all protein thiols are rendered accessible to protonation by acidification. Therefore, a rapid shift to low pH by addition of strong acid to protein solution provides an efficient way to quench protein thiols and, thereby, quickly stop thiol‐exchange reactions.43
Importantly, while most acid‐denatured proteins form aggregates, low‐molecular‐weight thiol compounds such as cysteine and gluthathione remain soluble. Thus, the denatured proteins can be separated from low‐molecular‐weight compounds by centrifugation.42
Trichloroacetic acid (TCA) is most often used to quench protein thiols. TCA is often used at a final concentration of 10% (W/V). When protein concentration is low, sodium deoxycholate should be added to the solution at a final concentration of 0.1% (W/V) to facilitate the protein precipitation by TCA.45
Substrate trapping using active‐site cysteine mutant
Thiol‐disulfide oxidoreductases catalyze disulfide bond formation, reduction, and isomerization. All these reactions go through disulfide‐linked intermediate between the enzyme and its substrate.4, 35, 36, 37, 38, 39, 40, 41 Thus, as discussed above, stabilization, purification, and mass spectrometric analysis of the complex may lead to the identification of substrates of each enzyme. However, it is often difficult to detect such intermediate complexes. We can think of two reasons for the difficulty. First, the collision of two molecules (enzyme and substrate) is required for the first reaction (the formation of the mixed‐disulfide complexes) to take place. However, as the second reaction (the resolution of the complex) is an intra‐molecular reaction, the local concentration of the free cysteine that attacks the mixed‐disulfide bond is so high that the resolution of the complexes “may” proceed extremely quickly. If this is the case, it will be hard to detect the intermediate complexes because of the shortness of their lifetimes. Second, although disulfide‐linked enzyme–substrate complexes may accumulate within cells at levels high enough to detect them, these complexes may be resolved during the processing of the sample, causing it difficult to detect them. Importantly, the latter could really be a case as we will show later in Figure 2.
Figure 2.
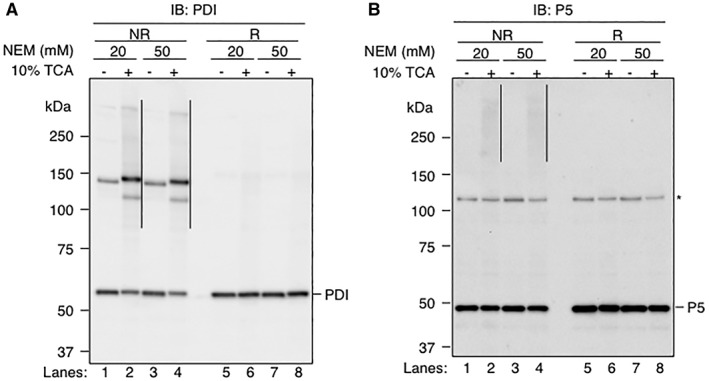
Importance of pretreatment of cells with TCA for stabilizing disulfide‐linked complexes using NEM. (A and B) HeLa cells were grown in DMEM medium supplemented with 10% FBS for 48 h, and washed twice with PBS. Following this step, free cysteines in the sample were modified using two different protocols. In the first protocol, the cells were directly lysed and alkylated in TritonX‐100 buffer [50 mM Tris–HCl (pH 7.4), 1% (v/v) TritonX‐100, 150 mM NaCl, 2 mM EDTA] containing 20 mM NEM (Lanes 1 and 5) or 50 mM NEM (Lanes 3 and 7). In the second protocol, the cells were first treated with 10% TCA, washed twice with ice‐cold acetone, and dissolved in SDS buffer [100 mM Tris–HCl (pH 6.8), 2% sodium dodecyl sulfate (SDS)] containing 20 mM NEM (Lanes 2 and 6) or 50 mM NEM (Lanes 4 and 8). Samples were separated by SDS‐polyacrylamide gel electrophoresis and analyzed by immunoblotting with either rabbit polyclonal antibody against PDI (Panel A) or rabbit polyclonal antibody against P5 (Panel B). Each lane contains 5 μg of proteins. Samples were reduced with 100 mM DTT before electrophoresis in Lanes 5–8. The positions of molecular mass standards are indicated on the left in kDa. The positions of the monomeric form of PDI and P5 are indicated on the right. Disulfide‐linked complexes involving either PDI or P5 are indicated on the right of Lanes 2 and 4 by vertical lines. A non‐specific band is indicated by asterisk on the right of the Panel B.
To increase the stability of the complexes, researchers have used the variants of enzymes that contained a CXXC‐to‐CXXA or CXXC‐to‐CXXS mutation (hereafter we will call it as a CXXC‐to‐CXXA/S mutation for simplicity) at each of their active sites. To understand how these mutants work to trap the substrate, we need to understand the role of each cysteine in the CXXC active site of wild‐type enzymes. If an enzyme is acting as a reductase toward a disulfide bond in its substrate, the N‐terminal cysteine (attacking cysteine) of the CXXC active site of the enzyme initiates the reaction by attacking the disulfide bond on the substrate protein, resulting in the formation of mixed‐disulfide complex between the enzyme and substrate. In the second step, the C‐terminal cysteine (resolving cysteine) of the CXXC active site attacks the mixed‐disulfide bond, resolving the complex and releasing the oxidized enzyme and reduced substrate. Next, we will think of the situation of a variant of the enzyme containing a CXXC‐to‐CXXA/S mutation. The mutant is still able to attack the disulfide bond on the substrate and form an intermolecular disulfide bond. However, due to the lack of the resolving cysteine, the mutant is unable to resolve the disulfide bond or is able to resolve the complex only very slowly and stay attached to the substrate proteins. This method has been successfully used previously to trap substrates in covalent complexes with enzymes such as ERdj533, ERp1831, ERp4631, ERp5730, P531, PDI34, 46, and TMX147.
Trapping substrates of a sulfenic acid reductase
Principally, the use of the resolving cysteine mutant is not limited to the identification of the substrates of disulfide reductase or isomerase.
DsbG is a periplasmic enzyme whose overall structure resembles DsbC, a periplasmic enzyme that repairs proteins containing non‐native disulfide bonds.3 However, the physiological function of DsbG remained unclear because of the lack of information on the substrates. Depuydet et al.48 adopted the resolving cysteine mutant of DsbG to find that DsbG preferentially interacts with three periplasmic enzymes. All of them belong to the same family of l,d‐transpeptidases that contain a sole cysteine essential for activity. Depydet et al.48 showed that the active site cysteine of these transpeptidases can be oxidized to a sulfenic acid in the periplasm and that DsbG can protect the cysteine from sulfenylation. Thus, these results indicate that even substrates of a sulfenic acid reductase such as DsbG can be identified if the reaction goes through a disulfide‐linked intermediate between the enzyme and its substrate protein.
As seen above, the use of the trapping mutant of a thiol oxidoreductase has proven to be effective in identification of its redox partners. However, there is a limitation with this strategy. This method is applicable to reductase or isomerase but not to oxidase, since the CXXC active site of oxidase needs to be disulfide bonded to interact with its substrate and initiate the oxidation reaction. To trap substrates of an oxidase in covalent complexes with enzymes, the use of a different approach is required.
Trapping substrates of an oxidase using a variant of the enzyme containing a mutation in a residue outside the CXXC active‐site motif
DsbA is a primary oxidase of secreted proteins in E. coli. We previously discovered that a mutation that alters the Pro151 of DsbA to threonine provides this enzyme with an intriguing feature. In this mutant, disulfide bond formation in secreted proteins was fairly efficient. Yet, this mutant accumulated intermediate complexes between DsbA and its substrates in vivo, suggesting that Pro151 is important for substrate release.49 Since this mutation allows the detection of complexes between DsbA and its substrates, we used this property of the enzyme to identify the substrates49 and to study the mechanisms of oxidative folding of cell surface proteins.39, 50 Thus, even with an enzyme that acts to oxidize secreted proteins, it is possible to isolate a mutant that accumulates the intermediate complexes with its substrates.
A model for the accumulation of enzyme–substrate complexes in DsbA P151T mutant
Then, why the resolution of the enzyme–substrate complexes is retarded in P151T mutant? Several lines of evidence allow us to speculate what is happening with the mutant. First, Pro151 takes a cis‐configuration and is highly conserved among the thioredoxin superfamily members,51 implying that this residue is important for the structure and functionality of this protein superfamily. Second, the loop containing Pro151 is distant to the CXXC active site in amino acid sequence, but close to it in three‐dimensional space.8 In the crystal structure of a complex between DsbA and its model substrate peptide, a few residues preceding the cis‐proline form an anti‐parallel β‐sheet with a part of the substrate, suggesting that the cis‐proline loop is important for substrate recognition.52 Third, when Pro151 of DsbA was changed to all of the other amino acids, only P151T mutation caused a defect in the resolution of enzyme–substrate complexes.53 Thus, we suspect that some structural feature resulting from this specific amino acid change prevents the second cysteine of the substrate from resolving the complex. Finally, in the solution structure of human thioredoxin complexed with Ref‐1 peptide, the ring of cis‐proline residue abuts the sulfur atom of the substrate cysteine involved in the mixed‐disulfide bond. Thus, this interaction may be important for the correct positioning of the mixed‐disulfide bond.54 The P151T mutation in DsbA may change this positioning, leading to slower resolution of the complexes.
Then, how could the positioning of the mixed‐disulfide bond affect the resolution of the complex by the second cysteine of the substrate? There is good evidence that thiol‐disulfide exchange reaction of the thioredoxin‐superfamily members uses an SN2 mechanism.35, 36, 38, 39, 40, 41 A reaction that uses an SN2 mechanism is predicted to be highly directional, proceeding through a transition state in which the three involved atoms form an ~180° angle.6, 37, 40 For example, in a reaction where a mixed‐disulfide complex between DsbA and its substrate is resolved by a nucleophilic attack by a deprotonated cysteine from the substrate, the three involved atoms are the two sulfur atoms that form the mixed disulfide, and the sulfur atom of the attacking cysteine from the substrate. Here, SN2 reaction theory predicts that these sulfur atoms need to form an ~180° angle. Thus, the P151T mutation in DsbA may prevent the proper positioning of these three sulfur atoms, possibly resulting in the slow resolution of the complex.
Conservation of cis‐proline residue in the thioredoxin superfamily members
Since cis‐proline residue corresponding to P151 of DsbA is conserved in most thioredoxin superfamily members,51, 55 mutational alternation of this residue in other superfamily members may allow detection of reaction intermediates. Indeed, Matsuo et al. substituted the cis‐proline residue of Tmx1, a PDI family member, to threonine and successfully trapped its substrates in covalent complexes with this enzyme in human cells.56
Nevertheless, as we have already discussed above, when Pro151 of DsbA was changed to all of the other amino acids, only P151T mutation caused a defect in the resolution of enzyme–substrate complexes,53 suggesting that some structural feature resulting from this specific amino acid change prevents the second cysteine of the substrate from resolving the complex. Thus, success in such attempts will depend on the precise positioning of cis‐proline residue near the active site and the nature of the amino acid substituted.53
Use of a variant of PDI containing a mutation in the intervening sequence of its CXXC active site to trap the substrates oxidized by the enzyme
In the ER of mammalian cells, PDI catalyzes the formation, reduction, and isomerization of disulfide bonds. Importantly, as explained above, a portion of PDI is secreted out of the cells and plays a critical role in thrombus formation. However, its extracellular substrates remained unknown.57 As discussed above, a variant of PDI containing a CXXC‐to‐CXXA/S mutation will capture the substrates that are reduced or isomerized by PDI, but not the substrates oxidized by this enzyme. Recently, Stopa et al.34 discovered a mutation in PDI that captures the substrates oxidized by this enzyme, which led to the identification of novel substrates of extracellular PDI. To find such a mutant, they introduced mutations into the highly conserved GH residues in the CGHC active site of PDI. When the oxidized form of a PDI variant with a CGHC‐to‐CGPC mutation was mixed with proteins released from platelets and on the platelet surface, the PDI variant formed disulfide‐linked complexes with four proteins. One of the identified proteins is cathepsin G, a protease that serves to regulate thrombosis by cleaving several proteins upon activation. Consistent with that cathepsin G may represent a substrate protein oxidized by PDI, the oxidized PDI, but not reduced PDI, significantly activated cathepsin G in an ex vivo assay using platelet releasate. Stopa et al. suggested that, since the CGHC motif is present in some members of PDI family including PDI, ERp57, and P5, the use of a similar variant may help identify their targets in biological systems.34
Efficient stabilization of the disulfide‐linked complexes involving endogenous enzymes using TCA and NEM
To irreversibly prevent the thiol‐disulfide rearrangement, it is essential to covalently modify free cysteines with alkylation reagent. Since NEM is a small and uncharged thiol‐alkylation reagent, it can penetrate into cells and react with thiols within cells. Thus, many researchers simply expose cells directly to NEM, and lyse them in the presence of mild detergent such as Triton X100 to modify free cysteines in the samples.30, 31, 33, 34, 46, 47
However, we routinely treat cells or tissues with TCA before alkylation of free cysteines with NEM because, as we have already discussed above, TCA can inhibit the thiol‐disulfide exchange reactions promptly.42 Following this step, proteins denatured by TCA treatment are collected by centrifugation, washed twice with acetone to remove acid, and then dissolved in freshly prepared 50 mM NEM in a denaturing buffer [100 mM Tris–HCl (pH 6.8), 1% sodium dodecyl sulfate (SDS)] containing appropriate protease inhibitors, incubated for 30 min at 37°C, and stored at −30°C until use.32 In the above procedure, TCA‐treated sample is washed with acetone twice to remove excess acid because alkylation of free cysteines with NEM occurs at neutral pH. Furthermore, alkylation under the denaturing conditions ensures that catalytic thiols are not active during the procedure, minimizing the chance of unwanted thiol‐disulfide exchange reactions.
Importance of pretreatment of cell culture with TCA for stabilizing mixed‐disulfide complexes using NEM
To know whether the pretreatment of cells with TCA improves the stability of mixed‐disulfide complexes between PDI family members and their substrates, we treated HeLa cells with or without 10% TCA before alkylation of the cellular proteins with NEM, and examined the effect of TCA treatment on the accumulation of mixed‐disulfide complexes. Antibody to PDI detected a band corresponding to the monomeric form of PDI at ~60 kDa (Fig. 2, Panel A, Lanes 1–4). In addition to this band, smeared bands were detected at positions higher than 80 kDa (indicated by vertical line) (Fig. 2, Panel A, Lanes 2 and 4). We suggest that the latter bands represent disulfide‐linked complexes containing PDI for two reasons. First, they ran slower than the monomeric form of this protein. Second, when the sample was reduced with a reducing agent, dithiothreitol (DTT), these band disappeared, giving rise to a band corresponding to the monomeric form of PDI (Fig. 2, Panel A, Lanes 6 and 8). Importantly, these complexes were detected at greatly reduced levels in the absence of TCA treatment (Fig. 2, Panel A, Lanes 1 and 3). The use of a higher concentration of NEM for alkylation did not alter the levels of the observed complexes [compare Lanes 1 and 2 (20 mM NEM) with Lanes 3 and 4 (50 mM NEM)], suggesting that NEM is present in the solutions at levels high enough to stabilize mixed‐disulfide complexes. In a similar manner, we observed that the pretreatment with 10% TCA also enhanced the accumulation of mixed‐disulfide complexes involving P5 (Fig. 2, Panel B). These results reveal the importance of TCA‐pretreatment in preparation of the disulfide‐linked complexes involving PDI family members.
Identification of endogenous enzymes that interact with a specific protein
Thus far, we have discussed methods to identify the substrates of a thiol‐disulfide oxidoreductase. However, an experiment in a reverse way—the identification of enzymes that interact with a specific protein—appears to be possible, as we will explain below.
Proinsulin has three disulfide bonds. Failure to form the disulfide bonds in a native conformation can lead to diabetes, underlining the importance of identifying the enzymes involved in disulfide bond formation in proinsulin. Using acid quenching and subsequent alkylation of free cysteines, we successfully stabilized disulfide‐linked intermediates between endogenous proinsulin and endogenous PDI family members in an insulin‐producing cell line. Subsequent purification of the complexes enabled us to identify five members of PDI family (PDI, PDIR, P5, ERp44, and ERp46) that directly interact with proinsulin.58 Thus, the method may be used to identify the enzymes that interact with a protein of interest.
Trapping enzyme–substrate complexes in tissue samples using acid and NEM
We have discussed the use of TCA and NEM to stabilize mixed‐disulfide complexes that arise in mammalian cells. We have found that a similar strategy can be used to stabilize disulfide‐linked intermediates that arise in animal tissues. The basic strategy is the same as the one used to stabilize disulfide‐linked complexes in culture cells. However, for the efficient quenching of the thiol‐disulfide exchange reactions by acid, we disrupted each tissue in 10% TCA using a homogenizer immediately after cutting the tissue from a mouse (Fig. 3). Following the step, proteins are collected by centrifugation, and washed with ice‐cold acetone. Free cysteines in the samples were then modified with NEM. In this way, we were able to stabilize the disulfide‐linked complexes involving ERdj5 or PDIp in mouse tissues, leading to the identification of the substrates of these enzymes.32, 59
Figure 3.
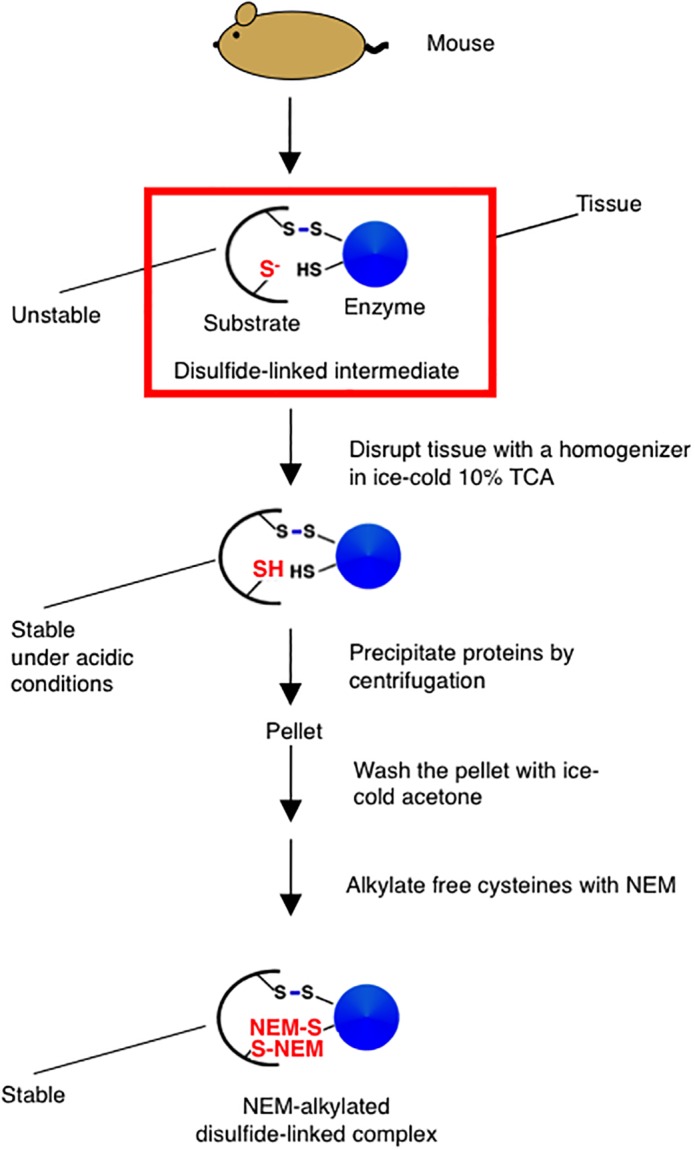
Stabilization of disulfide‐linked complexes in a tissue. See text for details.
Purification of the enzyme–substrate complexes for substrate identification by mass spectrometry
After stabilization of the enzyme–substrate complexes, the complexes need to be purified from cell or tissue lysate before identification of the substrates by mass spectrometry. Depending on the properties of the enzyme expressed in the cells, an appropriate purification protocol needs to be chosen. For example, to identify the substrates of an endogenous enzyme, an antibody specific to the enzyme is used to purify the enzyme–substrate complexes from the cell or tissue lysate. To reduce the amounts of contaminants, these complexes can be further purified using one of the following two methods before protein identification by mass spectrometry.
Purification and identification of the disulfide‐linked partners of an enzyme using two‐dimensional (2D) gel electrophoresis
The disulfide‐linked partners of the enzyme that exist in the purified protein sample can be further separated and thus purified by 2D gel electrophoresis in which the first dimension is non‐reducing and the second is reducing (Fig. 4).30, 31, 34, 48, 49 For example, a disulfide‐linked complex that migrates at Position B in the first dimension will be resolved into a partner protein at Position D, and the enzyme at Position E in the second dimension. Similarly, the enzyme released from other disulfide‐linked complexes will be observed as dots on a horizontal line at the size of the enzyme in the second dimension (black dots). In contrast, the partners of the enzyme will be observed as dots on an off‐diagonal line (faint blue dots) (Fig. 4). Proteins contained in each spot on the off‐diagonal line are then excised from the gel, digested by trypsin, and subjected to mass spectrometry for the identification of the potential disulfide‐linked partners of the enzyme.30, 31, 34, 48, 49
Figure 4.
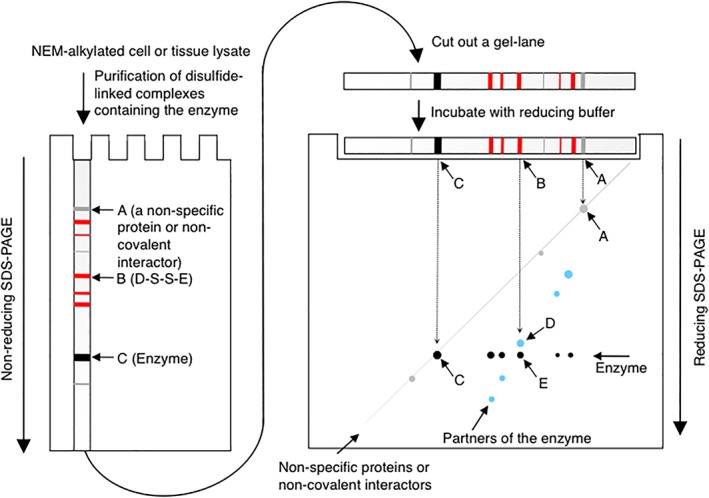
Separation of the partners of an enzyme by non‐reducing‐reducing 2D gel electrophoresis.30, 31, 34, 48, 49 Disulfide‐linked complexes containing an enzyme of interest are purified from NEM‐alkylated cell or tissue lysate and separated under non‐reducing conditions. The gel lane is then excised from the gel, incubated in SDS sample buffer containing 5% β‐mercaptoethanol, and placed on the top of the second gel. After electrophoresis, proteins are visualized with an appropriate method such as silver staining. Gray band or spot, non‐specific proteins or non‐covalent interactors of the enzyme; red band, disulfide‐linked complexes between the enzyme and its partners; black band or spot, monomeric enzyme; faint blue dot, disulfide‐linked partners of the enzyme. A non‐specific protein or non‐covalent interactor of the enzyme will migrate on a diagonal line (A). The enzyme (E) contained in a disulfide‐linked complex (B) will migrate at the size of the enzyme (C) in the second dimension. The disulfide‐linked partner (D) of the enzyme in the complex will migrate on the off‐diagonal line, allowing the easy identification of the spots corresponding to the partners of the enzyme.
Purification and identification of the disulfide‐linked partners of an enzyme using non‐reducing gel electrophoresis
In an alternative approach, the proteins, after the initial purification, can be separated and further purified simply by standard one‐dimensional non‐reducing gel electrophoresis.32, 46, 59 However, in this case, it is preferable to have a proper negative control (Fig. 5).46, 59 For example, in an experiment where the disulfide‐linked partners of an endogenous enzyme are purified from the cell or tissue lysate by immunopurification using an antibody to the enzyme, the control experiment will be the one performed using control IgG from the same species as the antibody to the enzyme. Using the latter sample as a negative control, one can distinguish non‐specific bands (gray bands in Fig. 5) from specific bands representing disulfide‐linked complexes containing the enzyme (red bands in Fig. 5). Proteins contained in the latter bands will then be excised from the gel and sent for protein identification by mass spectrometry.
Figure 5.

Identification of the partners of an endogenous enzyme after separation of disulfide‐linked complexes containing the enzyme by regular one‐dimensional non‐reducing gel electrophoresis. (A and B) Disulfide‐linked complexes containing an enzyme of interest are purified from NEM‐alkylated cell or tissue lysate using an antibody to the enzyme, separated by non‐reducing gel electrophoresis, and detected with an appropriate protein‐staining method such as silver staining (Panel A, Lane 2).32, 59 Lane 1 contains proteins purified from the same lysate using control IgG and serves as a negative control. Gray band, non‐specific proteins; black band, monomeric enzyme; red band, disulfide‐linked complexes between the enzyme and its partner proteins. (Panel B) Individual bands corresponding to the disulfide‐linked complexes (red band in Panel A) or a region of the gel containing the complexes (indicated by a black vertical line on the right of Lane 2 in Panel A) are excised from the gel, digested by trypsin, and analyzed by mass spectrometry. The obtained data is analyzed using an appropriate search program to identify the proteins contained in the sample. The same analysis can be performed with a sample obtained by immunopurification using control IgG to exclude non‐specific proteins from the list of the potential partners of the enzyme.59
If amounts of the purified disulfide‐linked complexes are small, a region containing the complexes (indicated by a vertical line on the right of Lane 2 in Fig. 5) may be excised from the gel, digested with trypsin and analyzed by mass spectrometry.59 In this case, the same analysis should be performed with proteins immuno‐purified using control IgG, and any proteins identified in the latter sample should be excluded from the list of potential partners of the enzyme to increase the chance to identify true partners of the enzyme.59
The substrates of the enzyme may be found in proteins identified using the above procedures. However, to confirm that an identified protein is a substrate of the enzyme, some validation experiments will be required.48, 49 For this purpose, researchers often test the direct interaction between the two proteins by immunoprecipitation followed by western blotting, and examine the effect of depleting the enzyme on the biosynthesis of the identified protein.33, 46
Use of a null mutant and differential thiol labeling of proteins to identify the substrates of a thiol‐disulfide oxidoreductase
Leichert and Jakob60 developed a differential thiol trapping technique, which allows to describe thiol‐disulfide state of cellular proteins on a global scale. In this method, free cysteines are labeled with nonradioactive IAM, while disulfide‐bonded cysteines are labeled with 14C‐IAM as follows.60 In the first step, cells were treated with TCA to quench thiol‐disulfide exchange reactions and free cysteines in the sample are blocked by IAM. In the second step, reducing agent such as DTT is added to break disulfide bonds. Finally, radioactive 14C‐IAM is added, which can react now with the previously oxidized cysteines. This leads to the complete alkylation of all thiol groups of a protein, regardless of their original oxidation state in vivo. Thus, oxidized and reduced forms of the same protein migrate to the identical spot in a 2D gel. Accordingly, the extent of thiol oxidation in a given protein can be expressed as the ratio of radioactivity to the protein on the spot.
Leichert and Jakob60 successfully applied this method to find the substrate proteins of thiol‐disulfide oxidoreductases in E. coli. For this purpose, they analyzed the in vivo redox state of proteins in cells deficient for an oxidoreductase of interest, and compared it to the redox state of proteins in wild‐type cells. In this way, they discovered a number of substrates of the periplasmic disulfide‐introducing enzyme DsbA and of the major cytoplasmic disulfide reductase Trx1 in E. coli. This method can potentially be used to find the substrates of thiol‐oxidoreductases of any organisms, given that wild‐type cell and a mutant deficient for the particular enzyme is available.
A major drawback of this approach will be a problem caused by functional redundancy. For example, there exist ~20 PDI family members in the ER of mammalian cells.1, 5, 29 While accumulating evidence suggests that each PDI family likely has its own preference for substrates, it is also true that another PDI family member can often compensate for the depletion of a particular PDI family member, albeit with reduced efficiency, minimizing the effect of depletion of the protein.61 Thus, in a case where there exists functional redundancy among related proteins, it could be difficult to find the physiological substrates of the protein using this approach.
Conclusion
In this article, we have reviewed methods used to identify the substrates of thiol‐disulfide oxidoreductases that are present in the periplasm of Gram‐negative bacteria or the ER of mammalian cells. Thus far, hunts for the substrates of thiol‐disulfide oxidoreductases in mammalian cells have been performed mostly by expressing a variant of the enzyme containing a CXXC‐to‐CXXA/S mutant in cultured mammalian cells such as HEK293T. In mammalian cells, this approach has proven to be effective in identifying the general substrates of a given enzyme if the enzyme is acting as a reductase or isomerase. Thus, this method will be the first choice for the identification of the general substrates of such enzymes. However, since some of disulfide bond‐containing proteins with specific physiological functions are expressed only in specialized cells or tissues (e.g., insulin is produced predominantly in the β‐cells of pancreas.), it will also be important to know the roles of ER‐localized thiol‐disulfide oxidoreductases in these cells or tissues. For such a purpose, the method shown in Figure 3 will be particularly useful. With the method, it was possible for us to detect, purify, and identify disulfide‐linked partners of thiol‐disulfide oxidoreductases from mouse tissues.32, 59 A great advantage of the method is that it does not require genetic engineering of mice for the identification of physiological substrates of an endogenous thiol‐disulfide oxidoreductase in a specific tissue or cells. Thus, we think that the approach will be applicable for the identification of the substrates of a wider range of thiol‐disulfide oxidoreductases.
Increasingly, it became apparent that thiol‐disulfide oxidoreductases play profound roles in the maintenance of the function of the ER in mammalian cells. Since defect in the function of thiol‐disulfide reductases in a specialized tissue can lead to disease conditions such as diabetes,58 the future challenge will be to use these methods to reveal the hidden functions of each of thiol‐disulfide reductases in various tissues associated with diseases. This should aid in understanding the mechanisms of disease progression and may eventually lead to the development of cures.
Conflicts of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgments
This work was supported by a JSPS KAKENHI grant JP15K07381 (to H.K.), and a MEXT KAKENHI grant JP26116005 (to H.K. and K.I.). This work was also supported, in part, by a grant from Noda Institute for Scientific Research (to H.K.).
References
- 1. Braakman I, Bulleid NJ (2011) Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem 80:71–99. [DOI] [PubMed] [Google Scholar]
- 2. Landeta C, Boyd D, Beckwith J (2018) Disulfide bond formation in prokaryotes. Nat Microbiol 3:270–280. [DOI] [PubMed] [Google Scholar]
- 3. Kadokura H, Beckwith J (2010) Mechanisms of oxidative protein folding in the bacterial cell envelope. Antioxid Redox Signal 13:1231–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hudson DA, Gannon SA, Thorpe C (2015) Oxidative protein folding: From thiol‐disulfide exchange reactions to the redox poise of the endoplasmic reticulum. Free Radic Biol Med 80:171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Okumura M, Kadokura H, Inaba K (2015) Structures and functions of protein disulfide isomerase family members involved in proteostasis in the endoplasmic reticulum. Free Radic Biol Med 83:314–322. [DOI] [PubMed] [Google Scholar]
- 6. Fass D, Thorpe C (2018) Chemistry and enzymology of disulfide cross‐linking in proteins. Chem Rev 118:1169–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bardwell JC, McGovern K, Beckwith J (1991) Identification of a protein required for disulfide bond formation in vivo. Cell 67:581–589. [DOI] [PubMed] [Google Scholar]
- 8. Martin JL, Bardwell JCA, Kuriyan J (1993) Crystal structure of the DsbA protein required for disulphide bond formation in vivo. Nature 365:464–468. [DOI] [PubMed] [Google Scholar]
- 9. Bardwell JC, Lee JO, Jander G, Martin N, Belin D, Beckwith J (1993) A pathway for disulfide bond formation in vivo. Proc Natl Acad Sci USA 90:1038–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kobayashi T, Kishigami S, Sone M, Inokuchi H, Mogi T, Ito K (1997) Respiratory chain is required to maintain oxidized states of the DsbA–DsbB disulfide bond formation system in aerobically growing Escherichia coli cells. Proc Natl Acad Sci USA 94:11857–11862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bader M, Muse W, Ballou DP, Gassner C, Bardwell JCA (1999) Oxidative protein folding is driven by the electron transport system. Cell 98:217–227. [DOI] [PubMed] [Google Scholar]
- 12. Inaba K, Murakami S, Suzuki M, Nakagawa A, Yamashita E, Okada K, Ito K (2006) Crystal structure of the DsbB–DsbA complex reveals a mechanism of disulfide bond generation. Cell 127:789–801. [DOI] [PubMed] [Google Scholar]
- 13. McCarthy AA, Haebel PW, Törrönen A, Rybin V, Baker EN, Metcalf P (2000) Crystal structure of the protein disulfide bond isomerase, DsbC, from Escherichia coli . Nat Struct Biol 7:196–199. [DOI] [PubMed] [Google Scholar]
- 14. Berkmen M, Boyd D, Beckwith J (2005) The nonconsecutive disulfide bond of Escherichia coli phytase (AppA) renders it dependent on the protein‐disulfide isomerase, DsbC. J Biol Chem 280:11387–11394. [DOI] [PubMed] [Google Scholar]
- 15. Rietsch A, Bessette P, Georgiou G, Beckwith J (1997) Reduction of the periplasmic disulfide bond isomerase, DsbC, occurs by passage of electrons from cytoplasmic thioredoxin. J Bacteriol 179:6602–6608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Katzen F, Beckwith J (2000) Transmembrane electron transfer by the membrane protein DsbD occurs via a disulfide bond cascade. Cell 103:769–779. [DOI] [PubMed] [Google Scholar]
- 17. Sato Y, Inaba K (2012) Disulfide bond formation network in the three biological kingdoms, bacteria, fungi and mammals. FEBS J 279:2262–2271. [DOI] [PubMed] [Google Scholar]
- 18. Poet GJ, Oka OB, van Lith M, Cao Z, Robinson PJ, Pringle MA, Arnér ES, Bulleid NJ (2017) Cytosolic thioredoxin reductase 1 is required for correct disulfide formation in the ER. EMBO J 36:693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ponsero AJ, Igbaria A, Darch MA, Miled S, Outten CE, Winther JR, Palais G, D'Autréaux B, Delaunay‐Moisan A, Toledano MB (2017) Endoplasmic reticulum transport of glutathione by Sec61 is regulated by Ero1 and Bip. Mol Cell 67:962–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ellgaard L, Sevier CS, Bulleid NJ (2018) How Are Proteins Reduced in the Endoplasmic Reticulum? Trends Biochem Sci 43:32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hosoda A, Kimata Y, Tsuru A, Kohno K (2003) JPDI, a novel endoplasmic reticulum‐resident protein containing both a BiP‐interacting J‐domain and thioredoxin‐like motifs. J Biol Chem 278:2669–2676. [DOI] [PubMed] [Google Scholar]
- 22. Ushioda R, Hoseki J, Araki K, Jansen G, Thomas DY, Nagata K (2008) ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 321:569–572. [DOI] [PubMed] [Google Scholar]
- 23. Hagiwara M, Maegawa K, Suzuki M, Ushioda R, Araki K, Matsumoto Y, Hoseki J, Nagata K, Inaba K (2011) Structural basis of an ERAD pathway mediated by the ER‐resident protein disulfide reductase ERdj5. Mol Cell 41:432–444. [DOI] [PubMed] [Google Scholar]
- 24. Maegawa K, Watanabe S, Noi K, Okumura M, Amagai Y, Inoue M, Ushioda R, Nagata K, Ogura T, Inaba K (2017) The highly dynamic nature of ERdj5 is key to efficient elimination of aberrant protein oligomers through ER‐associated degradation. Structure 25:846–857. [DOI] [PubMed] [Google Scholar]
- 25. Eletto D, Eletto D, Dersh D, Gidalevitz T, Argon Y (2014) Protein disulfide isomerase A6 controls the decay of IRE1α signaling via disulfide‐dependent association. Mol Cell 53:562–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Higa A, Taouji S, Lhomond S, Jensen D, Fernandez‐Zapico ME, Simpson JC, Pasquet J‐M, Schekman R, Chevet E (2014) Endoplasmic reticulum stress‐activated transcription factor ATF6 requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol Cell Biol 34:1839–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ushioda R, Miyamoto A, Inoue M, Watanabe S, Okumura M, Maegawa K, Uegaki K, Fujii S, Fukuda Y, Umitsu M, Takagi J, Inaba K, Mikoshiba K, Nagata K (2016) Redox‐assisted regulation of Ca2+ homeostasis in the endoplasmic reticulum by disulfide reductase ERdj5. Proc Natl Acad Sci USA 113:E6055–E6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bowley SR, Fang C, Merrill‐Skoloff G, Furie BC, Furie B (2017) Protein disulfide isomerase secretion following vascular injury initiates a regulatory pathway for thrombus formation. Nat Commun 8:14151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hatahet F, Ruddock LW (2009) Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal 11:2807–2850. [DOI] [PubMed] [Google Scholar]
- 30. Jessop CE, Chakravarthi S, Garbi N, Hämmerling GJ, Lovell S, Bulleid NJ (2007) ERp57 is essential for efficient folding of glycoproteins sharing common structural domains. EMBO J 26:28–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jessop CE, Watkins RH, Simmons JJ, Tasab M, Bulleid NJ (2009) Protein disulphide isomerase family members show distinct substrate specificity: P5 is targeted to BiP client proteins. J Cell Sci 122:4287–4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kadokura H, Saito M, Tsuru A, Hosoda A, Iwawaki T, Inaba K, Kohno K (2013) Identification of the redox partners of ERdj5/JPDI, a PDI family member, from an animal tissue. Biochem Biophys Res Commun 440:245–250. [DOI] [PubMed] [Google Scholar]
- 33. Oka OBV, Pringle MA, Schopp IM, Braakman I, Bulleid NJ (2013) ERdj5 is the ER reductase that catalyzes the removal of non‐native disulfides and correct folding of the LDL receptor. Mol Cell 50:793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stopa JD, Baker KM, Grover SP, Flaumenhaft R, Furie B (2017) Kinetic‐based trapping by intervening sequence variants of the active sites of protein‐disulfide isomerase identifies platelet protein substrates. J Biol Chem 292:9063–9074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Darby NJ, Creighton TE (1995) Catalytic mechanism of DsbA and its comparison with that of protein disulfide isomerase. Biochemistry. 34:3576–3587. [DOI] [PubMed] [Google Scholar]
- 36. Frech C, Wunderlich M, Glockshuber R, Schmid FX (1996) Preferential binding of an unfolded protein to DsbA. EMBO J 15:392–398. [PMC free article] [PubMed] [Google Scholar]
- 37. Fernandes PA, Ramos MJ (2004) Theoretical insights into the mechanism for thiol/disulfide exchange. Chem Eur J 10:257–266. [DOI] [PubMed] [Google Scholar]
- 38. Vlamis‐Gardikas A (2008) The multiple functions of the thiol‐based electron flow pathways of Escherichia coli: Eternal concepts revisited. Biochim Biophys Acta 1780:1170–1200. [DOI] [PubMed] [Google Scholar]
- 39. Kadokura H, Beckwith J (2009) Detecting folding intermediates of a protein as it passes through the bacterial translocation channel. Cell 138:1164–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wiita AP, Perez‐Jimenez R, Walther KA, Gräter F, Berne BJ, Holmgren A, Sanchez‐Ruiz JM, Fernandez JM (2007) Probing the chemistry of thioredoxin catalysis with force. Nature 450:124–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Perez‐jimenez R, Li J, Kosuri P, Sanchez‐romero I, Wiita AP, Rodriguez‐larrea D, Chueca A, Holmgren A, Miranda‐vizuete A, Becker K, Cho S, Beckwith J, Gelhaye E, Jacquot JP, Gaucher E, Sanchez‐ruiz JM, Berne BJ, Fernandez JM (2009) Diversity of chemical mechanisms in thioredoxin catalysis revealed by single‐molecule force spectroscopy. Nat Struct Mol Biol. 16:890–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zander T, Phadke ND, Bardwell JCA (1998) Disulfide bond catalysts in Escherichia coli . Methods Enzymol 290:59–74. [DOI] [PubMed] [Google Scholar]
- 43. Leichert LI, Jakob U (2006) Global methods to monitor thiol‐disulfide state of proteins in vivo. Antioxid Redox Signal 8:763–772. [DOI] [PubMed] [Google Scholar]
- 44. Hansen RE, Winther JR (2009) An introduction to methods for analyzing thiols and disulfides: Reactions, reagents, and practical considerations. Anal Biochem 394:147–158. [DOI] [PubMed] [Google Scholar]
- 45. Chevallet M, Diemer H, Van Dorssealer A, Villiers C, Rabilloud T (2007) Toward a better analysis of secreted proteins: the example of the myeloid cells secretome. Proteomics 7:1757–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zito E, Melo EP, Yang Y, Wahlander Å, Neubert TA, Ron D (2010) Oxidative protein folding by an endoplasmic reticulum‐localized peroxiredoxin. Mol Cell 40:787–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pisoni GB, Ruddock LW, Bulleid N, Molinari M (2015) Division of labor among oxidoreductases: TMX1 preferentially acts on transmembrane polypeptides. Mol Biol Cell 26:3390–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Depuydt M, Leonard SE, Vertommen D, Denoncin K, Morsomme P, Wahni K, Messens J, Carroll KS, Collet J (2009) Periplasmic reducing system protects single cysteine residues from oxidation. Science 326:1109–1112. [DOI] [PubMed] [Google Scholar]
- 49. Kadokura H, Tian H, Zander T, Bardwell JCA, Beckwith J (2004) Snapshots of DsbA in action: detection of proteins in the process of oxidative folding. Science 303:534–537. [DOI] [PubMed] [Google Scholar]
- 50. Chng S‐S, Xue M, Garner RA, Kadokura H, Boyd D, Beckwith J, Kahne D (2012) Disulfide rearrangement triggered by translocon assembly controls lipopolysaccharide export. Science 337:1665–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Martin JL (1995) Thioredoxin—a fold for all reasons. Structure 3:245–250. [DOI] [PubMed] [Google Scholar]
- 52. Paxman JJ, Borg NA, Horne J, Thompson PE, Chin Y, Sharma P, Simpson JS, Wielens J, Piek S, Kahler CM, Sakellaris H, Pearce M, Bottomley SP, Rossjohn J, Scanlon MJ (2009) The structure of the bacterial oxidoreductase enzyme DsbA in complex with a peptide reveals a basis for substrate specificity in the catalytic cycle of DsbA enzymes. J Biol Chem 284:17835–17845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kadokura H, Nichols L, Beckwith J (2005) Mutational alterations of the key cis proline residue that cause accumulation of enzymatic reaction intermediates of DsbA, a member of the thioredoxin superfamily. J Bacteriol 187:1519–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Qin J, Clore GM, Kennedy WP, Kuszewski J, Gronenborn AM (1996) The solution structure of human thioredoxin complexed with its target from Ref‐1 reveals peptide chain reversal. Structure 4:613–620. [DOI] [PubMed] [Google Scholar]
- 55. Biran S, Gat Y, Fass D (2014) The Eps1p protein disulfide isomerase conserves classic thioredoxin superfamily amino acid motifs but not their functional geometries. PLoS One 9:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Matsuo Y, Masutani H, Son A, Kizaka‐Kondoh S, Yodoi J (2009) Physical and functional interaction of transmembrane thioredoxin‐related protein with major histocompatibility complex class I heavy chain: redox‐based protein quality control and its potential relevance to immune responses. Mol Biol Cell 20:4552–4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Furie B, Flaumenhaft R (2014) Thiol isomerases in thrombus formation. Circ Res 114:1162–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tsuchiya Y, Saito M, Kadokura H, Miyazaki J, Tashiro F, Imagawa Y, Iwawaki T, Kohno K (2018) IRE1–XBP1 pathway regulates oxidative proinsulin folding in pancreatic β cells. J Cell Biol 217:1287–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fujimoto T, Nakamura O, Saito M, Tsuru A, Matsumoto M, Kohno K, Inaba K, Kadokura H (2018) Identification of the physiological substrates of PDIp, a pancreas‐specific protein disulfide isomerase family member. J Biol Chem 293:18421–18433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Leichert LI, Jakob U (2004) Protein thiol modifications visualized in vivo. PLoS Biol 2:1723–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rutkevich LA, Cohen‐doyle MF, Brockmeier U, Williams DB (2010) Functional relationship between protein disulfide isomerase family members during the oxidative folding of human secretory proteins. Mol Biol Cell 21:3093–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]