

Abstract
Background
Prostaglandin E2 has long been known to be an immune modulator. It is released after tissue injury and plays a role in modulating macrophage activities, which are essential for tissue regeneration. However, the involvement of prostaglandin E2 receptor 2 (EP2)–dependent regulation of macrophages in postischemic heart is unclear. This study aims to evaluate the role of EP2 in damaged heart.
Methods and Results
The effect of EP2 in postischemic heart was evaluated using EP2‐deficient transgenic mice. We demonstrated that cardiac function was worse after myocardial injury on loss of EP2. Furthermore, EP2 deficiency also altered proinflammatory response and resulted in a defect in macrophage recruitment to the injured myocardium. Transcriptome analysis revealed that the expression of erythroid differentiation regulator 1 (Erdr1) was significantly induced in EP2‐deficient macrophages. Knocking down Erdr1 expression restored migration ability of EP2‐deficient cells both in vitro and in vivo. By using a genetic fate‐mapping approach, we showed that abolishment of EP2 expression effectively attenuated cell replenishment.
Conclusions
The EP2‐dependent signaling pathway plays a critical role in regulating macrophage recruitment to the injured myocardium, thereby exerting a function in modulating the inflammatory microenvironment for cardiac repair.
Keywords: EP2, inflammation, ischemia, macrophage, myocardial, therapy
Subject Categories: Basic Science Research, Inflammation, Ischemia, Mechanisms, Myocardial Regeneration
Clinical Perspective
What Is New?
Knocking down of the prostaglandin E2 receptor 2 results in an attenuated cardiac function as well as a reduction in the proinflammatory response and macrophage population present in the heart after infarction.
Macrophage migration ability is affected on prostaglandin E2 receptor 2 deficiency.
Erythroid differentiation regulator 1 expression that is significantly increased in prostaglandin E2 receptor 2–deficient macrophages impairs cell migration ability, and knocking down of erythroid differentiation regulator 1 rescues cell function.
What Are the Clinical Implications?
An adequate level of macrophage‐dependent inflammatory response is critical at an early stage of myocardial infarction.
Prostaglandin E2 receptor 2 agonist may serve as an immune modulator to regulate early inflammatory response after injury.
Introduction
The signaling molecule prostaglandin E2 (PGE2) is released after tissue injury and has long been known to be an immune modulator. PGE2 works synergistically with interleukin‐23 or tumor necrosis factor‐α to promote the proinflammatory phenotype of immune effectors and secretion of inflammatory cytokines.1, 2 However, PGE2 also attenuates the immune response by augmenting production of anti‐inflammatory cytokines and inhibiting release of proinflammatory cytokines.3, 4 Collectively, these findings imply that PGE2 can exert both proinflammatory and anti‐inflammatory effects to fine‐tune immune responses, depending on the microenvironmental context.
In response to inflammatory stimuli, PGE2 is synthesized sequentially by cyclooxygenase 2 and the terminal prostaglandin synthase, microsomal prostaglandin E synthase‐1.5 Inhibition of cyclooxygenase 2 is reported to be associated with increased risk of cardiovascular diseases.6 Furthermore, deletion of the Ptges gene encoding microsomal prostaglandin E synthase‐1 significantly impairs cardiac function after myocardial infarction (MI) in mice.7 These studies point to the importance of the PGE2 signaling axis in the heart. To exert its function, PGE2 acts through EP1 to EP4, which are G‐protein–coupled receptors.8 Numerous studies have shown that modulating the activity of EP3 or EP4 receptors, via either genetic manipulation or pharmacological intervention, can affect heart function after injury.9, 10, 11 In addition, PGE2 can act through the PGE 2 receptor 2 (EP2) to improve stem cell–dependent cardiomyocyte repopulation after MI.12
Activation of the EP2 signaling pathway has diverse inflammatory effects under different physiological conditions. In response to myocardial injury, up‐regulated EP2 expression after PGE2 treatment is associated with enhanced efficiency of cardiomyocyte regeneration and increased number of anti‐inflammatory M2 type macrophages in the myocardium.12 Consistent with this study, results from a sepsis animal model revealed that PGE2 acts on macrophages through EP2 to promote release of anti‐inflammatory cytokine interleukin‐10.4 However, it has also been observed that the EP2‐dependent pathway is associated with innate immunity–induced oxidative damage to the brain.13 EP2 has been shown to be necessary for activation of brain‐resident microglia.14 Furthermore, suppression of EP2 signaling significantly reduces production of proinflammatory cytokines by the peripheral macrophages and microglia.15 These findings suggest the importance of EP2 in modulating the inflammatory microenvironment by regulating macrophage activities in response to tissue injuries. The role of macrophages in efficient tissue repair has been documented in some organs, such as the liver and skeletal muscle.16, 17, 18 We, therefore, speculated that EP2 may also be involved in cardiac repair through modulation of macrophage activities. To test this hypothesis, in this study, EP2 knockout transgenic mice were used to examine the association between EP2 and cardiac repair after heart injury.
Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Animal Model
The investigation conforms to the Guide for the Use and Care of Laboratory Animals, and all of the animal protocols were approved by the Institutional Animal Care and Use Committees of National Cheng Kung University and Academia Sinica.
The α‐MHC promoter‐driven MerCreMer, Z/EG transgenic mice, and the EP2 heterozygous (EP2+/−) mice in the C57BL/6 background were purchased from the Jackson Laboratory. The EP2+/− mice were mated to generate EP2‐null (EP2−/−) mice. The double‐transgenic MCM/ZEG (MZ) mice were generated by mating the MCM to Z/EG. The MZ females were mated with EP2−/− males to generate MCM and Z/EG heterozygous for EP2. The triple transgenic EP2−/−/MZ mice were generated by mating the EP2+/− MCM and EP2+/− Z/EG mice. The wild‐type (WT) mice in the C57BL/6J background were purchased from the National Laboratory Animal Center (Taiwan). Adult male and female animals of 12 to 18 weeks of age were used for the experiments conducted in this study.
Surgery and Echocardiography
To induce cardiac injury in double and triple transgenic MZ mice, surgery was performed 1 month after the last tamoxifen injection. To generate acute MI, the mice were anesthetized with 2% isoflurane by inhalation and the left anterior descending coronary artery at 2 to 3 mm distal to the left atrial appendage was permanently ligated. To take organs, the animals were anesthetized with tiletamine/zolazepam (Zoletil), >50 mg/kg, supplemented with xylazine (Rompun) at 10 mg/kg via peritoneal injection. The adequacy of anesthesia during euthanasia was monitored by toe‐pinch reflex. At day 56 after MI, the mice were anesthetized with 2% isoflurane by inhalation for cardiac performance assessment by echocardiography using a Vevo 770 device (Visualsonics, Toronto, ON, Canada).
Drug Treatment
Tamoxifen (Sigma) was prepared by dissolving in sunflower oil (Sigma) at a concentration of 5 mg/mL. The mice were injected daily at a dosage of 40 μg per 1 g body weight for 14 days.
Immunohistochemistry and Immunofluorescence Microscopy
The hearts were harvested and fixed with 4% paraformaldehyde and embedded in paraffin. The sections were then immunostained with the rabbit anti–green fluorescent protein (GFP) antibody (GeneTex) and rabbit anti–β‐Gal antibody (Invitrogen). For visualization, a diaminobenzidine substrate kit (Vector Laboratories) was used. To evaluate the degree of fibrosis, Masson's trichrome staining was performed and the infarct size was quantified using the midline length measurement, as previously reported.19 In brief, infarct size was evaluated from 3 sections per sample. To measure the infarct size, the sum of midline infarct lengths from all sections was divided by the sum of midline circumferences from all sections. The infarct size value is presented as a percentage. For immunofluorescent staining of macrophages, the collected hearts were processed for frozen sectioning by embedding in the OCT media. The sections were stained with the mouse anti‐cTnT (DSHB) and rat anti‐F4/80 (Abcam). Appropriate secondary antibodies (Invitrogen) were used for visualization under a fluorescence microscope. Nuclei were stained with 4,6‐diamidino‐2‐phenylindole (1 μg/mL; Sigma) for visualization.
Cytokine Screening
The infarcted region of the heart was excised at day 3 after injury. Proteins were extracted by homogenizing in 500 μL of extraction buffer, which was composed of 0.05 mol/L HEPES, 0.14 mol/L NaCl, 0.001 mol/L EDTA, 0.01% Triton X‐100, and 0.1% SDS. After homogenization, the samples were centrifuged at 16200 G for 10 minutes at 4°C. The supernatant was then collected for analysis using the Plex mouse ProcartaPlex panel (EPX170‐26087‐901; Invitrogen).
Flow Cytometric Analysis
To analyze immune cells in the injured myocardium, the infarct region was excised and digested with 50 mg/mL liberase (Roche) and 10 U DNase (Sigma) at 37°C for 30 minutes. A 40‐μm filter was then used to remove any undigested tissue. The cells were then stained with fluorochrome‐conjugated antibodies against CD45, CD11b, F4/80, Gr‐1, CD206, and EP2 (BD Biosciences or AbD Serotec) on ice for 30 minutes. For analysis of monocytes, the bone marrow cells were flushed out of the tibia and femurs with Hanks’ balanced salt solution. The peripheral blood was collected from the retro‐orbital sinus, and the red blood cells were lysed by ACK lysing buffer (Lonza). To harvest the splenocytes, the spleen was torn into small pieces with forceps, and the larger tissue pieces were removed by filtering through a 40‐μm filter. The cells were then stained with fluorochrome‐conjugated antibodies against Ly6C and CD115 (BD Biosciences) on ice for 30 minutes. Flow cytometry was performed using FACSCanto II, and the data were processed using FlowJo software.
Production and Purification of Recombinant Adenoviral Vectors
The erythroid differentiation regulator 1 (Erdr1) small hairpin RNA (shRNA) sequence was amplified from TRCN0000246782 plasmid from the RNA interference core with Phusion High‐Fidelity PCR Master Mix (New England Biolabs). The amplified fragment was cloned into the site adjacent to IRES‐EGFP of the pENTR plasmid. The shRNA‐embedded pENTR plasmids were then recombined into pAd/PL‐DEST plasmids using a pAd/PL‐DEST Gateway Vector Kit (Invitrogen). Viral particles were concentrated by CsCl2 gradient centrifugation.
In Vivo Migration Assay
The macrophages were collected from the peritoneal cavity of WT and EP2−/− mice 3 days after intraperitoneal injection with 3% thioglycollate (BD Biosciences). Two days after viral transduction, 4×106 cells were intravenously injected into the WT mice at day 2 after surgery‐induced MI. Cardiomyocyte‐depleted cardiac small cells were collected at day 3 after MI. The cells were prepared by digesting the heart with 0.1% collagenase B (Roche Molecular Biochemicals), 2.4 U/mL dispase II (Roche Molecular Biochemicals), and 2.5 mmol/L CaCl2 at 37°C for 30 minutes and then filtered through a 40‐μm filter. The cells were then stained with fluorochrome‐conjugated antibodies against F4/80 and GFP (BD Biosciences or AbD Serotec) on ice for 30 minutes. Flow cytometric analysis was performed using LSR II, and the data were processed using FlowJo software.
Transcriptomic Analysis
The macrophages of WT and EP2−/− mice collected from the peritoneal cavity 3 days after intraperitoneal injection with 3% thioglycollate (BD Biosciences) were hybridized to a Mouse Oligo Microarray (Agilent), according to the manufacturer's procedure, and the arrays were scanned with a Microarray Scanner System (Agilent). All data were analyzed by GeneSpring GX software (Agilent), and gene ontology analysis was conducted using DAVID software. The macrophages from the WT mice served as the baseline for quantile normalization and median polish probe summarization. The expression levels in the first quantile were filtered out to remove noise, and genes that were not detected in at least 2 of the 3 biological replicates were further removed from additional analyses. Genes were defined as differentially expressed if they had a fold change of at least ±2 combined with the Student t test (P<0.05), with the Benjamini‐Hochberg adjustment for false discovery rate.
Statistical Analysis
All statistical data were analyzed in GraphPad Prism and shown as mean±SEM. Unpaired t test, nonparametric test, 1‐way ANOVA, and 2‐way ANOVA with Dunnette's or Sidak's post hoc tests were applied for statistical comparisons, and P<0.05 was considered significant.
For Supplemental Methods, please refer to Data S1.
Results
Loss of EP2 Exacerbates Cardiac Dysfunction
To assess whether ablation of EP2 affected the heart after injury, mice with targeted disruption of the EP2 gene were subjected to surgery‐induced acute MI. In response to sham surgery, cardiac performance in the EP2−/− mice was not different from that of the WT and EP2+/− littermates (Figure S1A). Echocardiographic analyses at 1 and 2 months after MI revealed that the heart function was significantly worse in the EP2−/− mice. Furthermore, chamber dilation was more profound in homozygous null mice, as evidenced by increased left ventricular diastolic dimension and left ventricular systolic dimension (Figure 1A and Figure S1B). However, cardiac performance was not significantly affected in the heterozygous littermates compared with the WT mice (Figure 1A). In addition to cardiac function, further evaluation revealed that infarct region was significantly larger in the injured myocardium of EP2−/− mice compared with the WT and EP2+/− mice at 2 months after injury (Figure 1B and 1C). Collectively, these findings suggest that EP2 has a role in the heart after injury.
Figure 1.

Loss of prostaglandin E2 receptor 2 (EP2) expression worsens cardiac function after injury. A, At 2 months after myocardial infarction, cardiac function of EP2‐null (EP2−/−) mice (n=15) and their heterozygous (EP2+/−; n=13) and wild‐type (WT; n=14) littermates was evaluated by echocardiography. Statistical analysis was performed using 1‐way ANOVA with Dunnett's test. **P=0.0023 for ejection fraction (EF); **P=0.0026 for fractional shortening (FS); **P=0.0081 for left ventricular diastolic dimension (LVDd); **P=0.0042 for left ventricular systolic dimension (LVDs). B and C, The infarct region at mid‐LV was evaluated by Masson trichrome staining. WT, n=7; EP2+/−, n=7; EP2−/−, n=9. Statistical analysis was performed using 1‐way ANOVA with Dunnett's test. Bar=1 mm. n.s. indicates not significant. **P=0.005.
EP2 Signaling Regulates the Inflammatory Microenvironment After Infarction
The participation of EP2 in modulation of inflammatory response has been documented.13, 15 Consistent with previous findings, results from gene expression analysis suggested that EP2 functions as an immune modulator in the heart after injury. In the absence of EP2, a significant reduction in the expression level of proinflammatory response‐associated genes, including inducible NO synthase, tumor necrosis factor‐α, cyclooxygenase 2, and interleukin‐1β, was observed (Figure S2). The effect of EP2 on modulating proinflammatory response was more prominent at the early time point, 3 days, after MI. To further confirm the role of EP2 in modulating inflammatory response, the levels of proinflammatory and anti‐inflammatory cytokines were examined in the infarcted myocardium at day 3 after injury. Consistent with the gene expression analysis, the levels of most proinflammatory cytokines were found to be reduced on loss of EP2 (Figure 2A). The results indicate that loss of EP2 affects the inflammatory microenvironment after infarction. In response to tissue injury, the major immune cell populations that are recruited shortly after injury and participate in orchestrating the microenvironment during the acute inflammatory stage are neutrophils and macrophages. Because accumulation of neutrophils and macrophages dominates at the early time point after injury,20 the effect of EP2 deletion on cell infiltration to the injured myocardium was evaluated 3 days after MI. In sham‐operated animals, the numbers of both cell populations were not significantly different (Figure S3A and S3B). After myocardial injury, flow cytometric analysis suggested that there was a significant reduction in the number of macrophages in the injured heart of EP2−/− mice (Figure 2B and 2C). However, the number of neutrophils remained unchanged (Figure 2B and 2D). Quantitative polymerase chain reaction analysis of the pan‐macrophage marker CD11b showed that the expression peaked at day 3 after MI (Figure S4) and was undetectable at day 14 after injury (data not shown). Histological analysis also consistently demonstrated that there were fewer macrophages in the injured myocardium of EP2−/− mice (Figure 2E and 2F). It has been reported that PGE2 can act through the EP2 and EP4 receptors to augment production of anti‐inflammatory cytokine interleukin‐10 from macrophages.4 We, therefore, assessed whether macrophage polarity was altered on loss of EP2. The results revealed that the ratio of M1/M2 macrophage subtypes was not significantly changed on loss of EP2 expression (Figure S5). Furthermore, cytokine analysis revealed that the levels of anti‐inflammatory cytokines, including interleukin‐4, interleukin‐10, interleukin‐13, and interleukin‐22, were not altered on loss of EP2 at day 3 after MI (Figure 2G). This observation indicates that change in macrophage polarity does not contribute to attenuated proinflammatory response after loss of EP2. Further examination indicated that the macrophages expressed a higher level of EP2 in response to heart injury (Figure S6), suggesting that EP2 potentially contributes to the regulation of macrophage activities after injury.
Figure 2.
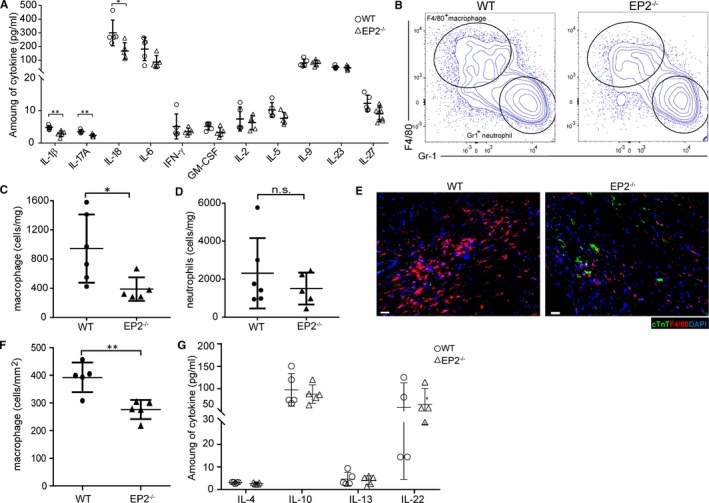
Prostaglandin E2 receptor 2 (EP2) knockout mice show less inflammation response and decreased macrophage recruitment at the infarcted myocardium. A, At day 3 after myocardial infarction (MI), the levels of a panel of proinflammatory cytokines in the infarct tissue of EP2‐null (EP2−/−) mice were compared with those in the wild‐type (WT) animals. Quantification was performed with 5 animals for each genotype. Statistical analysis was performed using an unpaired t test or a nonparametric test. The levels of the rest of cytokines are not statistically different. *P=0.0159 for IL‐18; **P=0.0051 and **P=0.0072 for interleukin‐1β and interleukin‐17A, respectively. B, At day 3 after MI, the infarcted region of injured heart was excised and enzymically digested for quantification of immune cells. The representative flow cytometric results of F4/80+ macrophages and Gr1+ neutrophils from the infarcted hearts of EP2−/− and WT mice are shown. C and D, Quantification of F4/80+ macrophages (C) and Gr1+ neutrophils (D) relative to the weight of injured tissue from EP2−/− (n=5) and WT (n=6) mice at day 3 after MI. Statistical analysis was performed using an unpaired t test or a nonparametric test. *P=0.0173. E and F, The distribution of F4/80+ macrophages in the injured hearts of EP2−/− and WT mice was analyzed. Quantification was performed with 5 animals for each genotype. Statistical analysis was performed using an unpaired t test. Bar=20 µm. **P=0.0036. G, Levels of anti‐inflammatory cytokines in the injured regions of EP2−/− and WT animals were compared. Quantification was performed with 5 animals for each genotype. Statistical analysis was performed using an unpaired t test, and no significant difference is observed. GM‐CSF indicates granulocyte‐macrophage colony‐stimulating factor; IFN‐γ, interferon‐γ; n.s., not significant.
EP2 Deficiency Abolishes Macrophage Migration Ability
The role of EP2 in regulating cell apoptosis has been documented.21 We, therefore, wondered whether the change in macrophage number in the EP2‐deficient mice resulted from increased cell apoptosis. The ischemic heart is under hypoxic conditions22; therefore, the apoptotic rate of EP2‐deficient macrophages was examined in hypoxic culture conditions. The cells were cultured in 1% oxygen for 24 and 72 hours, followed by examination of the degree of apoptotic rate. Results from a 3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5 diphenyl tetrazolium bromide assay indicated that loss of EP2 in the macrophages did not significantly alter cell viability (Figure S7). In response to tissue injury, monocytes migrate through the bloodstream to the injured tissue, where they mature into macrophages. To examine whether changes in the level of EP2 expression affected production of monocytes, the numbers of Ly6c+/CD115+ monocytes from the bone marrow, the spleen, and circulation were quantified in sham‐operated animals as well as before and after heart injury. Flow cytometric analysis showed that there was no significant difference in monocyte numbers between the WT and EP2−/− mice, regardless of the presence or absence of heart injury (Figure 3A and 3B) or the sham operation (Figure S3C and S3D). This finding implies that EP2 does not play any role in monocyte generation.
Figure 3.

Macrophages isolated from prostaglandin E2 receptor 2 (EP2) knockout mice show attenuated migration ability. A, The effect of EP2 deficiency on the number of monocytes was examined in various tissues. Representative flow cytometric analyses of CD115+/Ly6c+ monocytes, as indicated in the red boxes, in the bone marrow, spleen, and peripheral blood before and after myocardial infarction (MI) are shown. B, The number of monocytes examined in different tissues of wild‐type (WT) and EP2‐null (EP2−/−) mice before and after MI was quantified and statistically analyzed. Before MI, n=5 to 6 animals per group. After MI, n=5 to 9 animals per group. Statistical analysis was performed using an unpaired t test or a nonparametric test. C and D, The mobilization ability of macrophages isolated from WT and EP2−/− mice was examined. C, Representative images showing the macrophages, as presented by 4,6‐diamidino‐2‐phenylindole staining, which migrated in response to different levels of monocyte chemoattractant protein 1 (MCP‐1). The experiment was repeated 4 times. D, The mobilization ability of macrophages was quantified as cell migration index. Statistical analysis was performed using 2‐way ANOVA with Sidak's test. Bar=50 µm. n.s. indicates not significant. **P=0.0040 and ****P<0.0001 at MCP‐1 concentrations of 3 and 30 ng/mL, respectively.
In response to tissue injury, macrophages must migrate to a designated location to exert their effects. Therefore, we investigated whether EP2 signaling affected the ability of monocytes to migrate to the injury site, explaining the reduced number of cells found in the injured myocardium. Results of a migration assay showed that the migration ability of macrophages was impaired after loss of EP2. A dose‐dependent effect was observed in the WT macrophages in response to monocyte chemoattractant protein 1. However, such an effect was attenuated in EP2−/− macrophages (Figure 3C and 3D). This result suggests that loss of EP2 affects the mobilization ability of the macrophages, thereby contributing to a reduced number of cells being recruited in response to heart injury.
Erdr1 Acts Downstream of the EP2 Pathway to Negatively Regulate Macrophage Migration
To uncover the molecular mechanism involved in EP2‐dependent regulation of macrophage migration, microarray analysis was performed to examine the differences in WT and EP2−/− macrophages. Consistent with the literature and our current findings, gene ontology analysis revealed that loss of EP2 had a profound effect on immune response and cell mobilization (Figure 4A). Microarray data showed that expression of a panel of genes associated with cell migration was changed in the EP2−/− macrophages, which included P‐selectin glycoprotein ligand‐1 (Selp), Gata2, thrombospontin 1 (Thbs1), IL‐1α, Tgf‐β2, and Erythroid differentiation regulator 1 (Erdr‐1). Quantitative polymerase chain reaction analysis showed that the most strongly induced gene in EP2−/− macrophages was Erdr1 (Figure 4B), suggesting that Erdr1 may be a critical factor acting downstream of the EP2 signaling pathway to negatively regulate cell mobilization. To test this hypothesis, the Erdr1‐specific shRNA was designed to knock down the gene expression in EP2−/− macrophages. The quantitative polymerase chain reaction result showed that the expression of Erdr1 was dramatically reduced in the EP2−/− macrophages after introduction of shRNAs (Figure S8). The macrophages transduced with shRNA were subjected to in vitro migration assay to examine whether knockdown of Erdr1 expression rescued the migration defect observed in EP2−/− macrophages. In comparison with the EP2 macrophages, which were transduced with empty vector alone, the migration ability of EP2−/− cells expressing Erdr1 shRNA was effectively improved (Figure 4C). In addition to in vitro examination, the effect of knocking down Erdr1 shRNA in rescuing EP2−/− macrophage migration was further tested in vivo. Macrophages transduced with vector alone or Erdr1 shRNA were systemically injected into the WT mice at day 2 after MI. The number of injected cells that migrated to the injured myocardium was then analyzed the next day (ie, day 3 after MI). Because the transduced cells were positive for GFP, the infused cells that migrated to the injured myocardium could be distinguished from the endogenous cells. Flow cytometric analysis revealed that ≈5% of infused GFP+ macrophages could be detected in the injured myocardium of the mice receiving WT macrophages. The percentage of GFP+ cells in the injured heart of the mice receiving EP2−/− macrophages transduced with vector alone decreased significantly (Figure 4D and 4E). However, the number of GFP+ EP2−/− macrophages significantly increased when the mice were infused with cells expressing Erdr1 shRNA (Figure 4D and 4E). Together, these findings showed that Erdr1 acts downstream of EP2 to negatively regulate macrophage mobilization, and lowering the expression level of the gene could rescue the migration defect of EP2−/− macrophages.
Figure 4.

Erythroid differentiation regulator 1 (Erdr1) acts downstream of prostaglandin E2 receptor 2 (EP2) signaling to regulate macrophage mobilization. A, Transcriptome heat map and gene ontology analysis (right panel) show the pathways that were most affected by loss of EP2. B, Comparison of candidate gene expression in macrophages isolated from EP2‐null (EP2−/−) and wild‐type (WT) mice; n=6 for each genotype. Statistical analysis was performed using an unpaired t test or a nonparametric test. n.s. indicates not significant. *P=0.0228 for Selp; *P=0.037 for Gata2; *P=0.0169 for Thbs1; *P=0.0343 for Tgf‐β2; and **P=0.0022 for Erdr1. C, In vitro migration assay was performed to examine the mobilization ability of the EP2−/− macrophages on small hairpin RNA (shRNA)–mediated knockdown of Erdr1. Cells transduced with vector alone were used as the control group. The mobilization ability of cells was evaluated by the migration index. Statistical analysis was performed using 2‐way ANOVA with Dunnett's test. MCP‐1 indicates monocyte chemoattractant protein 1; and n.s., not significant vs vector alone control. ***P=0.0004; ****P=0.0001. D and E, At day 3 after myocardial infarction (MI), cardiomyocyte‐depleted small cells were isolated for quantification of infused F4/80+/green fluorescent protein–positive (GFP +) macrophages. Control groups were the mice injected with wild type (WT; WT control) or EP2−/− (EP2−/− control) macrophages transduced with vector alone. D, Representative flow cytometric examination of the GFP + cells in the injured hearts of WT mice with or without cell infusion is shown. E, The number of GFP + cells in the injured myocardium among groups was quantified. MI alone, n=5; WT control, n=5; EP2−/− control, n=5; EP2−/− Erdr1 shRNA, n=5. Statistical analysis was performed using 1‐way ANOVA with Dunnett's test. n.s. indicates not significant vs EP2−/− control. *P=0.0383; ****P=0.0001.
EP2 Signaling Regulates Cardiomyocyte Regeneration
In response to external stimuli (eg, tissue injury), the stem cells need to be activated for effective tissue repair. After heart injury, cardiac stem cells can be observed at both infarct and peri‐infarct regions of the injured myocardium (Figure S9). A recent study suggests that macrophages are necessary for activation of resident stem cells on muscle damage.23 In addition, it has also been reported that inflammatory macrophages are the key activator of hair follicle stem cells for neogenesis after injury.24 Consistent with these studies, it is known that PGE2 improves cardiac regeneration by acting through EP2 to regulate macrophage polarization and endogenous stem cell activities.12 We showed herein that EP2‐dependent signaling is involved in regulating the mobilization ability of macrophages. To more directly assess the importance of EP2 on stem cell–dependent cardiomyocyte replenishment, trigenic EP2−/− MZ mice were generated. In the absence of heart injury, ≈80% of cells expressed GFP (Figure 5A), suggesting the labelling efficiency in the trigenic MZ mice was not affected by EP2 ablation. After MI, an increase in the replenished cardiomyocytes was observed at the infarct border zone of both WT and EP2+/− MZ mice. At the remote area, however, the regeneration efficiency was less profound (Figure 5B). Nevertheless, we did not observe a significant change in the number of cells being repopulated at the infarct region of the EP2−/− MZ mice (Figure 5A). This finding confirms that loss of EP2 abolished stem cell–dependent cardiomyocyte regeneration.
Figure 5.
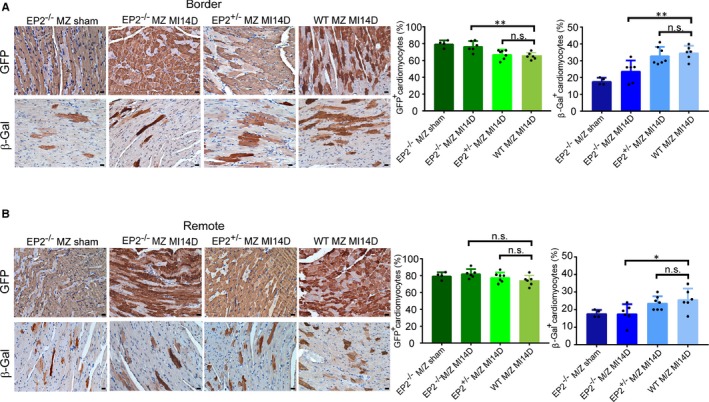
Cardiomyocyte replenishment is abolished on loss of prostaglandin E2 receptor 2 (EP2) expression after myocardial infarction (MI). A and B, At day 14 after MI (MI14D), the hearts of MCM/ZEG (MZ) mice with homozygous (EP2−/−/MZ) or heterozygous (EP2+/−/MZ) deletion of EP2 expression were harvested for evaluation of adult cardiomyocyte replenishment after MI. Shown are representative images of green fluorescent protein–positive (GFP +) and β‐Gal+ cardiomyocytes at the border zone (A, left panels) and the remote area (B, left panels) of the injured hearts from different groups. The percentages of resident GFP + cardiomyocytes and regenerated β‐Gal+ cardiomyocytes were quantified and statistically analyzed at the border zone (A, right panels) and remote region (B, right panels). Wild‐type MZ MI14D, n=7; EP2−/−/MZ sham, n=4; EP2−/−/MZ MI14D, n=6; EP2+/−/MZ MI14D, n=7. Statistical analysis was performed using 1‐way ANOVA with Dunnett's test. Bar=20 µm. n.s. indicates not significant. At the border zone, **P=0.0031 and **P=0.0032 for GFP + and β‐Gal+ cells, respectively. At the remote area, *P=0.0240.
Discussion
In this study, we used EP2‐deficient transgenic mice and demonstrated that the EP2 signaling pathway is involved in cardiac repair by modulating macrophage activities. We further showed that depletion of EP2 significantly impairs macrophage recruitment to the injured myocardium, thereby attenuating tissue repair.
EP2 is one of the receptors on which PGE2 acts to exert an immune modulatory effect. In some tissues, the EP2‐dependent signaling pathway has been shown to be biphasic. For example, in the lungs, activation of EP2 has an anti‐inflammatory effect by inhibiting recruitment of granulocytes and promoting release of anti‐inflammatory cytokine interleukin‐10.25 In the brain, loss of EP2 leads to enlarged infarct volume after induction of cerebral ischemia, suggesting that the PGE2‐EP2 signaling axis plays a neuroprotective role.26 However, in the presence of intracerebral hemorrhage, activation of EP2 is neurotoxic and deletion of EP2 has been shown to improve functional outcome after brain injury.27 In the present study, we provide evidence that activation of EP2 plays a positive role in the heart by modulating macrophage mobilization. We showed that deletion of EP2 resulted in worse cardiac performance after injury. Together, these findings provide evidence that the effect of the EP2 signaling pathway is tissue and damage dependent.
In agreement with our current findings, EP2‐dependent regulation of cell mobilization has also been shown in cancer cells.28 We used transcriptomic analysis to find the downstream target of EP2 in macrophages, revealing that the expression of Erdr1 was significantly induced in EP2‐deficient macrophages. The role of Erdr1 in regulating cell migration has been documented. The gene has been shown to exert an antimetastatic effect by inhibiting cancer cell migration and proliferation.29, 30 Furthermore, it has been demonstrated that different pathways act downstream of Erdr1 to negatively regulate the mobilization of different types of cancer cells. For example, in melanoma cells, the heat shock protein 90 expression is significantly reduced on Erdr1 overexpression.30 In gastric cancer cells, on the other hand, Erdr1 inhibits cell mobilization via activation of the JNK pathway. Collectively, these studies suggest that EP2 and Erdr1 are involved in regulation of cell mobilization; however, further study is required to examine precisely how Erdr1 expression is regulated in macrophages. Furthermore, detailed examination is also necessary to delineate the pathway acting downstream of the EP2‐Erdr1 axis for regulation of macrophage mobilization in response to tissue damage. In the present study, the monocyte/macrophage population examined is primarily from the bone marrow and in the circulation. It has been reported that the heart possesses tissue‐resident macrophage populations, which are distinct from the bone marrow– or circulation‐derived macrophages and can be renewed via in situ proliferation.31 Thus, whether EP2 signaling has an effect on the myocardium‐resident macrophages will require further investigation.
It has been shown that PGE2 can directly promote stem cell–driven cardiomyocyte replenishment through EP2 after MI.12 Using a trigenic EP2‐deficient MZ mouse model, we found that depletion of EP2 effectively impairs cardiomyocyte regeneration. The resident cardiac stem cells are shown to express EP2, and attenuated stem cell activity is observed on loss of EP2.12 In addition to direct modulation of stem cell function, EP2 may indirectly regulate stem cells via macrophages. Macrophages have been shown to be necessary for activation of stem cells in several tissues for regeneration.23, 24 Considering the direct and indirect effects of EP2 on stem cells, further experiments will be necessary to delineate its role in stem cell–dependent tissue repair after heart injury. Immunomodulation for tissue repair is an important research field that holds great potential for therapeutic application.32, 33 In addition, more and more new therapies target monocyte and macrophage subsets to condition the inflammatory microenvironment, thereby favoring and stimulating myocardial repair. It has been shown that mesenchymal stem cell therapy improves cardiac repair by elevating the number of reparative subtype M2 macrophages in the injured myocardium.34 Furthermore, cardiosphere‐derived cells have been shown to exert a cardioprotective effect by promoting polarization of macrophages into a distinct phenotype via the paracrine effect.35 Our findings taken together with these studies suggest that a macrophage‐conditioned inflammatory microenvironment is critical for effective tissue regeneration.
In conclusion, our findings provide evidence that EP2 plays an important role in modulating macrophage mobilization and that such an effect involves downregulation of Erdr1 expression in cells. In EP2‐deficient animals, cardiac performance is attenuated after injury, suggesting the importance of EP2 in cardiac repair. In the current study, the examinations were performed in EP2−/− mice, in which the receptor is knocked out in all cell types. Therefore, a future study examining the effect of macrophage‐specific knockout of EP2 on the injured myocardium will be important. The involvement of PGE2 in cardiac repair is clear.7, 12 However, PGE2 is an immune modulator regulating a wide range of inflammatory responses. A better understanding of the mechanisms underlying EP2‐dependent regulation of macrophage functions at various stages of myocardial injury would be valuable for development of the therapy for treating ischemic heart diseases.
Sources of Funding
This work was supported by the Ministry of Science and Technology (102‐2321‐B‐001‐069‐MY3, 104‐2325‐B‐001‐010, and 105‐2325‐B‐001‐009), the National Health Research Institutes (EX105‐10512SI), and the Academia Sinica Translational Medicine Program. The National RNAi Core Facility at the Institute of Molecular Biology/Genomics Research Center, Academia Sinica, is supported by the National Core Facility Program for Biotechnology Grants from the National Science Council (100‐2319‐B‐001‐002).
Disclosures
Hsieh receives research grants from AstraZeneca and Takeda, neither of which participated in funding this work. The remaining authors have no disclosures to report.
Supporting information
Data S1. Supplemental methods.
Figure S1. Loss of EP2 receptor did not alter cardiac function in sham‐operated animals but worsened cardiac function after injury. A, Cardiac function of EP2 null (EP2−/−) mice was not significantly different from their WT and heterozygous (EP2+/−) knockout littermates when subjected to sham surgery. B, Cardiac function of EP2−/− mice and their heterozygous and WT littermates was examined at 1 month post‐MI. The experiments were performed with n≧14 animals per genotype. Statistical analyses were performed using one‐way ANOVA with Dunnette's test. *P=0.0376, ns=not significant.
Figure S2. Loss of EP2 resulted in reduced expression of inflammation modulators. At day 3 and day 7 post‐MI, the expression of genes of interest in the injured hearts of EP2−/− mice was compared to that in the WT animals. Analysis was performed with 3 animals per time point for each genotype. Statistical analysis was performed using one‐way ANOVA with Tukey's test. *P=0.0466 and *P=0.0292 for TNF‐α and IL‐1β, respectively; **P=0.0017, ***P=0.0007, ****P<0.0001 and ns, not significant vs WT control at the same time point.
Figure S3. Immune cell number was not significantly altered in sham‐operated animals. Following sham operation, the number of immune cells was examined in excised heart tissue and the number of monocytes was analysed in various tissues. Representative flow cytometric results of F4/80+ macrophages and Gr1+ neutrophils are shown in (A). The numbers of F4/80+ macrophages (B, left panel) and Gr1+ neutrophils (B, right panel) relative to the weight of excised tissue from EP2−/− and WT mice were compared. Statistical analysis was performed using unpaired t test. ns, not significant. C, Representative flow cytometric analyses of CD115+/Ly6c+ monocytes, as indicated in the red boxes, are shown in various tissues from sham‐operated animals. D, The number of monocytes examined in different tissues of sham‐operated WT and EP2−/− mice were quantified and statistically analysed using unpaired t test. ns, not significant.
Figure S4. Macrophage marker expression peaked at day 3 post‐injury. The expression of pan‐macrophage marker CD11b in the injured hearts of WT mice was examined at different time points after myocardial injury. Analysis was performed on 4 animals per time point. The gene expression was undetectable in 2 samples at day 7 post‐injury. Statistical analyses were performed using one‐way ANOVA with Dunnette's test. ***P=0.0001 and **P=0.0072 and ns, not significant vs sham control.
Figure S5. EP2 signaling had little effect on polarization of M1 and M2 macrophage subpopulations. The cardiomyocyte‐depleted small cell population was analyzed for the F4/80+/CD206− M1 and F4/80+/C206+ M2 macrophage subpopulations at day 3 post‐myocardial injury. Shown is a representative image of the gating of the M1 and M2 macrophage subpopulations (Left). Statistical analysis revealed that the M1 and M2 macrophage ratio was not significantly changed in WT (n=5) and EP2 null (EP2−/−, n=5) mice after injury (Right). Statistical analysis was performed using unpaired t test. ns indicates not significant.
Figure S6. Cardiac injury induced EP2 expression in macrophages. In the injured myocardium, the macrophages in the cardiomyocyte‐depleted small cell fraction were analyzed for expression of EP2 receptor. The results showed that EP2 expression was induced in response to heart injury.
Figure S7. EP2 deficiency had negligible effect on cell viability. Macrophages isolated from WT and EP2 null (EP2−/−) mice were exposed to hypoxic conditions for induction of apoptosis. Cell viability was then analyzed at different time points following culturing under hypoxic conditions. The experiment was repeated 4 and 5 times for the 24 and 72 hours time point, respectively. Statistical analysis was performed using unpaired t test. ns indicates not significant.
Figure S8. The shRNA‐dependent gene knockdown effectively lowered expression of Erdr1 gene in EP2 deficient macrophages. Following viral transduction of Erdr1 shRNA in the EP2 null (EP2−/−) macrophages, quantitative PCR was conducted to examine the expression level of the target gene. The cells transduced with vector alone were the control. Macrophages were isolated from 3 animals for viral transduction. Statistical analysis was performed using one‐way ANOVA with Tukey's test. ****P<0.0001.
Figure S9. Sca‐1+ stem cells were observed at in the injured myocardium. At day 3 post‐infarction, the injured myocardium were stained for the stem cell marker Sca‐1. Shown are representative images of Sca‐1+ cells observed at the infarct region (A) and peri‐infarct area (B) in the injured myocardium. Scale bars, 20 μm.
Acknowledgments
We thank Dr Shu‐Jen Chou of the Institute of Plant and Microbial Biology, Academia Sinica, for assistance in microarray analysis. We thank Dr Ying‐Chang Hsueh at the National Health Research Institute for providing insightful comments on the manuscript. The fluorescence‐activated cell sorting, high‐content imaging, and animal studies were supported by the Core Facility at the Institute of Biomedical Sciences, Academia Sinica. RNA interference reagents were obtained from the National RNAi Core Facility at the Institute of Molecular Biology/Genomics Research Center, Academia Sinica.
(J Am Heart Assoc. 2018;7:e009216 DOI: 10.1161/JAHA.118.009216.)
References
- 1. Rieser C, Bock G, Klocker H, Bartsch G, Thurnher M. Prostaglandin E2 and tumor necrosis factor alpha cooperate to activate human dendritic cells: synergistic activation of interleukin 12 production. J Exp Med. 1997;186:1603–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sheibanie AF, Tadmori I, Jing H, Vassiliou E, Ganea D. Prostaglandin E2 induces IL‐23 production in bone marrow‐derived dendritic cells. FASEB J. 2004;18:1318–1320. [DOI] [PubMed] [Google Scholar]
- 3. Jing H, Vassiliou E, Ganea D. Prostaglandin E2 inhibits production of the inflammatory chemokines CCL3 and CCL4 in dendritic cells. J Leukoc Biol. 2003;74:868–879. [DOI] [PubMed] [Google Scholar]
- 4. Nemeth K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, Robey PG, Leelahavanichkul K, Koller BH, Brown JM, Hu X, Jelinek I, Star RA, Mezey E. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)‐dependent reprogramming of host macrophages to increase their interleukin‐10 production. Nat Med. 2009;15:42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Murakami M, Naraba H, Tanioka T, Semmyo N, Nakatani Y, Kojima F, Ikeda T, Fueki M, Ueno A, Oh S, Kudo I. Regulation of prostaglandin E2 biosynthesis by inducible membrane‐associated prostaglandin E2 synthase that acts in concert with cyclooxygenase‐2. J Biol Chem. 2000;275:32783–32792. [DOI] [PubMed] [Google Scholar]
- 6. Antman EM, Bennett JS, Daugherty A, Furberg C, Roberts H, Taubert KA; American Heart Association . Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation. 2007;115:1634–1642. [DOI] [PubMed] [Google Scholar]
- 7. Degousee N, Fazel S, Angoulvant D, Stefanski E, Pawelzik SC, Korotkova M, Arab S, Liu P, Lindsay TF, Zhuo S, Butany J, Li RK, Audoly L, Schmidt R, Angioni C, Geisslinger G, Jakobsson PJ, Rubin BB. Microsomal prostaglandin E2 synthase‐1 deletion leads to adverse left ventricular remodeling after myocardial infarction. Circulation. 2008;117:1701–1710. [DOI] [PubMed] [Google Scholar]
- 8. Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282:11613–11617. [DOI] [PubMed] [Google Scholar]
- 9. Hishikari K, Suzuki J, Ogawa M, Isobe K, Takahashi T, Onishi M, Takayama K, Isobe M. Pharmacological activation of the prostaglandin E2 receptor EP4 improves cardiac function after myocardial ischaemia/reperfusion injury. Cardiovasc Res. 2009;81:123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Martin M, Meyer‐Kirchrath J, Kaber G, Jacoby C, Flogel U, Schrader J, Ruther U, Schror K, Hohlfeld T. Cardiospecific overexpression of the prostaglandin EP3 receptor attenuates ischemia‐induced myocardial injury. Circulation. 2005;112:400–406. [DOI] [PubMed] [Google Scholar]
- 11. Qian JY, Harding P, Liu Y, Shesely E, Yang XP, LaPointe MC. Reduced cardiac remodeling and function in cardiac‐specific EP4 receptor knockout mice with myocardial infarction. Hypertension. 2008;51:560–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hsueh YC, Wu JM, Yu CK, Wu KK, Hsieh PC. Prostaglandin E(2) promotes post‐infarction cardiomyocyte replenishment by endogenous stem cells. EMBO Mol Med. 2014;6:496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Montine TJ, Milatovic D, Gupta RC, Valyi‐Nagy T, Morrow JD, Breyer RM. Neuronal oxidative damage from activated innate immunity is EP2 receptor‐dependent. J Neurochem. 2002;83:463–470. [DOI] [PubMed] [Google Scholar]
- 14. Quan Y, Jiang J, Dingledine R. EP2 receptor signaling pathways regulate classical activation of microglia. J Biol Chem. 2013;288:9293–9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johansson JU, Pradhan S, Lokteva LA, Woodling NS, Ko N, Brown HD, Wang Q, Loh C, Cekanaviciute E, Buckwalter M, Manning‐Bog AB, Andreasson KI. Suppression of inflammation with conditional deletion of the prostaglandin E2 EP2 receptor in macrophages and brain microglia. J Neurosci. 2013;33:16016–16032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Amemiya H, Kono H, Fujii H. Liver regeneration is impaired in macrophage colony stimulating factor deficient mice after partial hepatectomy: the role of M‐CSF‐induced macrophages. J Surg Res. 2011;165:59–67. [DOI] [PubMed] [Google Scholar]
- 17. Mounier R, Theret M, Arnold L, Cuvellier S, Bultot L, Goransson O, Sanz N, Ferry A, Sakamoto K, Foretz M, Viollet B, Chazaud B. AMPKα1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab. 2013;18:251–264. [DOI] [PubMed] [Google Scholar]
- 18. Shireman PK, Contreras‐Shannon V, Ochoa O, Karia BP, Michalek JE, McManus LM. MCP‐1 deficiency causes altered inflammation with impaired skeletal muscle regeneration. J Leukoc Biol. 2007;81:775–785. [DOI] [PubMed] [Google Scholar]
- 19. Takagawa J, Zhang Y, Wong ML, Sievers RE, Kapasi NK, Wang Y, Yeghiazarians Y, Lee RJ, Grossman W, Springer ML. Myocardial infarct size measurement in the mouse chronic infarction model: comparison of area‐ and length‐based approaches. J Appl Physiol (1985). 2007;102:2104–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J, Yamamoto T, Takeshima A, Shinmura K, Shen W, Fukuda K, Sano M. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol. 2013;62:24–35. [DOI] [PubMed] [Google Scholar]
- 21. Takadera T, Shiraishi Y, Ohyashiki T. Prostaglandin E2 induced caspase‐dependent apoptosis possibly through activation of EP2 receptors in cultured hippocampal neurons. Neurochem Int. 2004;45:713–719. [DOI] [PubMed] [Google Scholar]
- 22. Kim HA, Rhim T, Lee M. Regulatory systems for hypoxia‐inducible gene expression in ischemic heart disease gene therapy. Adv Drug Deliv Rev. 2011;63:678–687. [DOI] [PubMed] [Google Scholar]
- 23. Du H, Shih CH, Wosczyna MN, Mueller AA, Cho J, Aggarwal A, Rando TA, Feldman BJ. Macrophage‐released ADAMTS1 promotes muscle stem cell activation. Nat Commun. 2017;8:669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang X, Chen H, Tian R, Zhang Y, Drutskaya MS, Wang C, Ge J, Fan Z, Kong D, Wang X, Cai T, Zhou Y, Wang J, Wang J, Wang S, Qin Z, Jia H, Wu Y, Liu J, Nedospasov SA, Tredget EE, Lin M, Liu J, Jiang Y, Wu Y. Macrophages induce AKT/beta‐catenin‐dependent Lgr5(+) stem cell activation and hair follicle regeneration through TNF. Nat Commun. 2017;8:14091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Medeiros AI, Serezani CH, Lee SP, Peters‐Golden M. Efferocytosis impairs pulmonary macrophage and lung antibacterial function via PGE2/EP2 signaling. J Exp Med. 2009;206:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McCullough L, Wu L, Haughey N, Liang X, Hand T, Wang Q, Breyer RM, Andreasson K. Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. J Neurosci. 2004;24:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Leclerc JL, Lampert AS, Diller MA, Immergluck JB, Dore S. Prostaglandin E2 EP2 receptor deletion attenuates intracerebral hemorrhage‐induced brain injury and improves functional recovery. ASN Neuro. 2015;7:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pan MR, Hou MF, Chang HC, Hung WC. Cyclooxygenase‐2 up‐regulates CCR7 via EP2/EP4 receptor signaling pathways to enhance lymphatic invasion of breast cancer cells. J Biol Chem. 2008;283:11155–11163. [DOI] [PubMed] [Google Scholar]
- 29. Jung MK, Houh YK, Ha S, Yang Y, Kim D, Kim TS, Yoon SR, Bang SI, Cho BJ, Lee WJ, Park H, Cho D. Recombinant Erdr1 suppresses the migration and invasion ability of human gastric cancer cells, SNU‐216, through the JNK pathway. Immunol Lett. 2013;150:145–151. [DOI] [PubMed] [Google Scholar]
- 30. Jung MK, Park Y, Song SB, Cheon SY, Park S, Houh Y, Ha S, Kim HJ, Park JM, Kim TS, Lee WJ, Cho BJ, Bang SI, Park H, Cho D. Erythroid differentiation regulator 1, an interleukin 18‐regulated gene, acts as a metastasis suppressor in melanoma. J Invest Dermatol. 2011;131:2096–2104. [DOI] [PubMed] [Google Scholar]
- 31. Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, Schilling JD, Schwendener R, Sergin I, Razani B, Forsberg EC, Yokoyama WM, Unanue ER, Colonna M, Randolph GJ, Mann DL. Embryonic and adult‐derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40:91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shang F, Liu S, Ming L, Tian R, Jin F, Ding Y, Zhang Y, Zhang H, Deng Z, Jin Y. Human umbilical cord MSCs as new cell sources for promoting periodontal regeneration in inflammatory periodontal defect. Theranostics. 2017;7:4370–4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mayfield AE, Kanda P, Nantsios A, Parent S, Mount S, Dixit S, Ye B, Seymour R, Stewart DJ, Davis DR. Interleukin‐6 mediates post‐infarct repair by cardiac explant‐derived stem cells. Theranostics. 2017;7:4850–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ben‐Mordechai T, Holbova R, Landa‐Rouben N, Harel‐Adar T, Feinberg MS, Abd Elrahman I, Blum G, Epstein FH, Silman Z, Cohen S, Leor J. Macrophage subpopulations are essential for infarct repair with and without stem cell therapy. J Am Coll Cardiol. 2013;62:1890–1901. [DOI] [PubMed] [Google Scholar]
- 35. Hasan AS, Luo L, Yan C, Zhang TX, Urata Y, Goto S, Mangoura SA, Abdel‐Raheem MH, Zhang S, Li TS. Cardiosphere‐derived cells facilitate heart repair by modulating M1/M2 macrophage polarization and neutrophil recruitment. PLoS One. 2016;11:e0165255. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental methods.
Figure S1. Loss of EP2 receptor did not alter cardiac function in sham‐operated animals but worsened cardiac function after injury. A, Cardiac function of EP2 null (EP2−/−) mice was not significantly different from their WT and heterozygous (EP2+/−) knockout littermates when subjected to sham surgery. B, Cardiac function of EP2−/− mice and their heterozygous and WT littermates was examined at 1 month post‐MI. The experiments were performed with n≧14 animals per genotype. Statistical analyses were performed using one‐way ANOVA with Dunnette's test. *P=0.0376, ns=not significant.
Figure S2. Loss of EP2 resulted in reduced expression of inflammation modulators. At day 3 and day 7 post‐MI, the expression of genes of interest in the injured hearts of EP2−/− mice was compared to that in the WT animals. Analysis was performed with 3 animals per time point for each genotype. Statistical analysis was performed using one‐way ANOVA with Tukey's test. *P=0.0466 and *P=0.0292 for TNF‐α and IL‐1β, respectively; **P=0.0017, ***P=0.0007, ****P<0.0001 and ns, not significant vs WT control at the same time point.
Figure S3. Immune cell number was not significantly altered in sham‐operated animals. Following sham operation, the number of immune cells was examined in excised heart tissue and the number of monocytes was analysed in various tissues. Representative flow cytometric results of F4/80+ macrophages and Gr1+ neutrophils are shown in (A). The numbers of F4/80+ macrophages (B, left panel) and Gr1+ neutrophils (B, right panel) relative to the weight of excised tissue from EP2−/− and WT mice were compared. Statistical analysis was performed using unpaired t test. ns, not significant. C, Representative flow cytometric analyses of CD115+/Ly6c+ monocytes, as indicated in the red boxes, are shown in various tissues from sham‐operated animals. D, The number of monocytes examined in different tissues of sham‐operated WT and EP2−/− mice were quantified and statistically analysed using unpaired t test. ns, not significant.
Figure S4. Macrophage marker expression peaked at day 3 post‐injury. The expression of pan‐macrophage marker CD11b in the injured hearts of WT mice was examined at different time points after myocardial injury. Analysis was performed on 4 animals per time point. The gene expression was undetectable in 2 samples at day 7 post‐injury. Statistical analyses were performed using one‐way ANOVA with Dunnette's test. ***P=0.0001 and **P=0.0072 and ns, not significant vs sham control.
Figure S5. EP2 signaling had little effect on polarization of M1 and M2 macrophage subpopulations. The cardiomyocyte‐depleted small cell population was analyzed for the F4/80+/CD206− M1 and F4/80+/C206+ M2 macrophage subpopulations at day 3 post‐myocardial injury. Shown is a representative image of the gating of the M1 and M2 macrophage subpopulations (Left). Statistical analysis revealed that the M1 and M2 macrophage ratio was not significantly changed in WT (n=5) and EP2 null (EP2−/−, n=5) mice after injury (Right). Statistical analysis was performed using unpaired t test. ns indicates not significant.
Figure S6. Cardiac injury induced EP2 expression in macrophages. In the injured myocardium, the macrophages in the cardiomyocyte‐depleted small cell fraction were analyzed for expression of EP2 receptor. The results showed that EP2 expression was induced in response to heart injury.
Figure S7. EP2 deficiency had negligible effect on cell viability. Macrophages isolated from WT and EP2 null (EP2−/−) mice were exposed to hypoxic conditions for induction of apoptosis. Cell viability was then analyzed at different time points following culturing under hypoxic conditions. The experiment was repeated 4 and 5 times for the 24 and 72 hours time point, respectively. Statistical analysis was performed using unpaired t test. ns indicates not significant.
Figure S8. The shRNA‐dependent gene knockdown effectively lowered expression of Erdr1 gene in EP2 deficient macrophages. Following viral transduction of Erdr1 shRNA in the EP2 null (EP2−/−) macrophages, quantitative PCR was conducted to examine the expression level of the target gene. The cells transduced with vector alone were the control. Macrophages were isolated from 3 animals for viral transduction. Statistical analysis was performed using one‐way ANOVA with Tukey's test. ****P<0.0001.
Figure S9. Sca‐1+ stem cells were observed at in the injured myocardium. At day 3 post‐infarction, the injured myocardium were stained for the stem cell marker Sca‐1. Shown are representative images of Sca‐1+ cells observed at the infarct region (A) and peri‐infarct area (B) in the injured myocardium. Scale bars, 20 μm.