

Abstract
Cystic fibrosis (CF) lung disease is the major cause of morbidity and mortality in people with CF. Abnormal mucociliary transport has been the leading hypothesis for the underlying pathogenesis of CF airway disease. However, this has been difficult to investigate at very early time points. A porcine CF model, which recapitulates many features of CF disease in humans, enables studies to be performed in non-CF and CF pigs on the day that they are born. In newborn CF pigs, we found that under basal conditions, mucociliary transport rates in non-CF and CF pigs are similar. However, after cholinergic stimulation, which stimulates submucosal gland secretion, particles become stuck in the CF airways owing to a failure of mucus strands to release from submucosal glands. In this review, we summarize these recent discoveries and also discuss the morphology, composition, and function of mucins in the porcine lung.
Keywords: cystic fibrosis, mucociliary transport, mucus, submucosal gland
Cystic fibrosis (CF) is a genetic disease caused by mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) anion channel (1, 2). CF-related lung disease is a major cause of morbidity and mortality in people with CF (3). Advanced CF lung disease is clinically characterized by mucus obstruction in airways, chronic bacterial airway infection, prominent neutrophilic airway inflammation, and progressive bronchiectasis. Over the last two decades, researchers have devoted efforts to understanding the pathogenesis of CF airway disease and to developing new therapeutics based on that knowledge. Newborn screening for CF has enabled the early diagnosis of CF and has revealed that even in infants with CF, airway abnormalities and host defense defects are already present (4–7). A more acidic airway surface liquid (ASL) pH is present in neonates with CF, and early bacterial infection and inflammation as well as radiographic evidence of lung disease are also present within months of birth (4–6, 8).
Mucociliary transport (MCT) helps to keep the airways sterile by clearing inhaled pathogens and particles out of the lungs. Abnormal MCT has been the leading hypothesis for the underlying pathogenesis of CF airway disease (9, 10). However, the data to support this idea have been lacking owing to difficulties with studies in infants, limitations of MCT assays, and problems with mucus sampling; this is especially true in the very early stages of CF airway disease. To circumvent these and other limitations, we developed a porcine model of CF (11, 12). On the day that CF pigs are born, their lungs lack airway infection, inflammation, or mucus accumulation, yet their airways fail to eradicate bacteria as well as non-CF control pigs (13). Within weeks to months of birth, CF pigs also develop hallmark features of CF lung disease, including airway infection, inflammation, remodeling, and mucus accumulation/obstruction (12, 14). Thus, newborn CF pigs represent a model to investigate the primary effects of loss of CFTR function on MCT in the absence of secondary manifestations of disease. In this review, we summarize recent data from our studies on MCT and mucin structure and morphology in newborn non-CF and CF pig airways. We also discuss potential clinical and therapeutic implications of these findings.
MCT Is Heterogeneous in Newborn Pigs
Most studies of mucociliary clearance have depended on using radionuclides and nuclear medicine imaging to measure tracer disappearance from the lungs (9). Although these assays can be informative, they lack the granularity needed to track individual particles as they are transported up the airway tree, and thus they do not yield insight into mechanisms of dysfunction. Therefore, to assay MCT with improved spatial and temporal resolution, we developed an X-ray computed tomography–based assay to track the movement of 350-μm diameter tantalum microdisks (Figure 1A) (15, 16). The microdisks are insufflated into the airways, and then serial computed tomographic imaging (every ∼5–15 s for 5–10 min) is performed. From these computed tomographic image datasets, x-, y-, and z-coordinates can be obtained for each microdisk as it travels up the airways toward the larynx.
Figure 1.

Three-dimensional reconstructed images created using X-ray computed tomography reveal in vivo mucociliary transport in newborn pigs. (A) Individual tantalum microdisks (diameter, ∼350 μm) travel up the airways in 3 minutes. Individual microdisks are represented by different colors spheres (enlarged for visualization). Microdisk trajectories are connected by lines. Microdisks move with substantially heterogeneous speeds at different time points. (B) Microdisks are immobile, or fail to move, a greater percentage of the time in methacholine-treated newborn cystic fibrosis (CF) pig airways. *P < 0.05. Data are from Reference 16.
In newborn non-CF pigs, we found that microdisks exhibited interesting movement behaviors. First, microdisks tended to travel toward the ventral surface of the trachea, which was related to the orientation of ciliary beating in the airways (15). Second, although on average, microdisks moved at MCT rates similar to those previously reported in the literature (17, 18), there was substantial heterogeneity in microdisk speed, both between animals and even within the same animal. Some microdisks moved fast, whereas other microdisks moved slowly. Moreover, an individual microdisk could exhibit both fast and slow transport rates at different time points during a study. The heterogeneous rates of MCT suggest that, even in healthy airways, a homogeneous blanket of mucus might not cover the airways (15, 19–22). This heterogeneity could be related to multiple factors, including variability in submucosal gland secretions, unevenness in mucus layer thickness, or regional differences in mucus biophysical properties and cell types/submucosal glands.
Cholinergic Stimulation Impairs MCT in Newborn CF Pigs
Under baseline conditions, we found that mean and maximum transport speeds of microdisks were similar in non-CF and CF newborn pigs (16). However, we hypothesized that MCT might become impaired in CF airways subjected to a stress. To test this hypothesis, we treated non-CF and CF newborn pigs with intravenous methacholine, which increases ciliary beat frequency and submucosal gland secretion. A similar cholinergic response is elicited when the airways experience an insult or injury. Methacholine stimulation increased the maximum and mean speeds of microdisks in non-CF airways. Cholinergic stimulation tended to reduce the clearance of microdisks from CF lungs and caused microdisks to become immobile or “stuck” in the CF airways. This was associated with a significant reduction in the time that microdisks spent in motion (Figure 1B) (16). Interestingly, if a “stuck” microdisk broke free in the CF airway, it typically traveled at a normal speed. These data suggest that loss of CFTR function does not induce a generalized defect in MCT, such as might be observed with uniform depletion of periciliary liquid or inhibition of ciliary beating, but instead causes more localized defects in MCT.
To further investigate the underlying mechanism(s) for impaired MCT in CF airways, we developed an ex vivo system to study submucosal gland mucus secretion and mucus transport along the airway (16). Excised tracheas were bathed in physiological solutions, and 40-nm fluorescent nanospheres were added to the solution. These fluorescent nanospheres bind to and label mucus strands as they emerge from submucosal gland duct openings, enabling real-time imaging. We developed a time-averaging procedure to preferentially visualize stationary mucus strands and provide panoramic views of entire tracheal segments. Under basal conditions, in tracheas from both non-CF and CF newborn piglets, nearly all of the mucus quickly cleared from the airway surface. However, after methacholine stimulation, we observed a markedly different appearance in CF pig tracheas. Mucus strands and globules often failed to detach from submucosal gland ducts, thus impairing mucus clearance from the airway surface. In contrast, in non-CF tracheas, mucus strands quickly cleared the airway surface. We were able to replicate the CF mucus phenotype in non-CF tracheas that were bathed in solutions lacking both Cl− and HCO3− transport. Thus, these data directly link loss of CFTR-mediated anion secretion to defective MCT.
Mucins Form Distinct Morphological Structures on the Airway Surface
In the airways, surface goblet cells and submucosal glands secrete two gel-forming mucins, MUC5AC and MUC5B (23). However, knowledge of the morphological structure of airway mucus in situ is very limited, because cultured cells lack submucosal glands; rodent species have few submucosal glands; and findings from human sputum can be impacted by isolation, processing, and storage of samples. Therefore, we investigated the morphological structure of MUC5AC and MUC5B from freshly excised non-CF and CF newborn pig tracheas (24).
To investigate the native mucin structure, we harvested freshly excised tracheas from newborn pigs and immediately fixed the airways. We visualized the gel-forming mucins with two lectins, jacalin (JAC) and wheat germ agglutinin (WGA), that preferentially bind either MUC5AC or MUC5B, respectively. In non-CF newborn pig tracheas, we observed three distinct mucin morphologies, as described below.
Mucus Strands
We observed “strands” of mucin on the airway surface that originated from submucosal gland duct openings (Figure 2). These cylindrical strands ranged in diameter from approximately 5 to 50 μm, could extend hundreds of micrometers from gland duct openings toward the larynx, and at times developed a ribbon-like shape after secretion. Strands were composed of multiple filaments that labeled with WGA but not JAC, indicating that they were composed of MUC5B. This was expected because submucosal glands predominantly secrete MUC5B (24, 25). Mucus strands sometimes attached to adjacent strands, forming a mucus network that covered the airway surface (Figure 2). Similar-appearing mucus strands have been observed by in other studies, including recent work by Hansson and colleagues (26).
Figure 2.
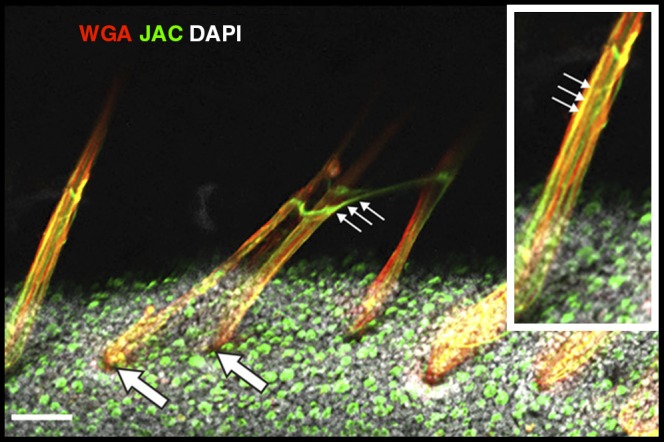
Staining of airway mucus and epithelial cells. Mucus strands, composed with MUC5B (stained red with wheat germ agglutinin [WGA]), emerge from submucosal gland duct openings (large arrows) and are partially coated with MUC5AC threads (small arrows) (stained green with jacalin [JAC]) from goblet cells. 4′,6-Diamidino-2-phenylindole (DAPI) nuclei = gray. Scale bar = 120 μm. Additional images comparing non–cystic fibrosis and cystic fibrosis can be found in Reference 24.
Mucus Threads
We also identified wispy “threads” of mucus on the tracheal surface that labeled with JAC but not WGA, suggesting MUC5AC composition (Figure 2). These threads were secreted from goblet cells and ranged in size from 1 to 4 μm in diameter. MUC5AC threads were rarely attached to goblet cells, suggesting that after secretion, they quickly break free from their site of origin. Often these MUC5AC threads coated the surface of the MUC5B strands, thus forming a complex mucus structure composed of both MUC5AC and MUC5B (Figure 2, inset) (24, 26). We rarely observed mucus threads that were composed of MUC5B.
Mucus Sheets
Sometimes we also observed thin sheets of mucus that labeled for MUC5AC. As described below, these were more common in the CF airway.
Mucus Morphology in CF Airways
We also observed mucus strands, threads, and sheets in newborn CF pig tracheas. However, we discovered that the morphologic appearance of the mucins differed from that of non-CF pig airways in several ways (24):
-
1.
In CF airways, MUC5B strands often remained attached to the submucosal glands from which they emerged. This finding is consistent with our earlier work in CF airways showing that after cholinergic stimulation, microdisks become “stuck” in the airway when they attach to mucus strands that fail to detach from submucosal gland openings (16).
-
2.
Attached mucus strands oftentimes bound to adjacent mucus strands, giving a complex, tangled, netlike appearance.
-
3.
MUC5AC sheets were more commonly observed in CF airways, and they tended to attach to MUC5B strands.
-
4.
Compared with non-CF, CF submucosal gland ducts were more often filled with mucus. This mucus labeled for MUC5B and extended from the acinus all the way to the airway surface at the duct openings.
Speculations and Perspectives for Future Research
Why Have the Airways Evolved to Have Mucus with Different Morphological Structures: Strands, Threads, and Sheets?
The airways are repeatedly exposed to inhaled particles and pathogens, which are effectively cleared out of the lungs by MCT. Impairments in MCT increase susceptibility to lung infections. For example, genetic defects in the ciliary machinery, such as primary ciliary dyskinesia (27), or targeted deletion of MUC5B (28), impairs MCT and leads to airway disease. Thus, the airways likely evolved for optimal MCT, and the presence of mucus strands, threads, and sheets probably produces the most effective MCT for various particles and pathogens (Figure 3A). Particles inhaled from the external environment have a wide range of sizes. Large particles tend to land in the larger airways, whereas small particles tend to land in the smaller-sized airways. It could be that strands, which emerge from the submucosal glands in the large airways, are required to remove large objects deposited on the surface of the large airways (Figure 3). Mucus threads and sheets, produced by surface epithelial cells in both small and large airways, might be sufficient to remove smaller-sized particles, which can land in either the large or small airways. The presence of submucosal glands only in the large airways and the lack of submucosal glands in the airways of rodents could be related, in part, to whether a mucus strand, thread, or sheet is required for MCT in a particular airway.
Figure 3.

Mucus secretion and transport in the airways. (A) Model of mucus secretion in pig large airways. Within submucosal glands (SMGs), MUC5B-predominant mucus (red lines) is secreted from mucous cells; serous cells express cystic fibrosis transmembrane conductance regulator (CFTR), determine pH, volume, and ionic composition of submucosal gland lumen and mucus. Mucus strands are coated with MUC5AC threads (green lines) or sheets. MUC5AC sheets and threads are predominantly secreted by surface goblet cells. (B) Model of mucus secretion in large and small airways.
Why Do the Airways Have Two Mucus-producing Structures: Submucosal Glands and Goblet Cells?
The answer may also be related to the production of mucus strands, threads, and sheets (Figure 3B). For example, mucus strands are present in the large airways where submucosal glands are present. We observed that mucus strands emerging from submucosal gland ducts are composed of multiple MUC5B filaments (Figure 2). It is possible that these MUC5B filaments are secreted by individual mucus-secreting cells in the gland, and their transport along the length of the ducts promotes the formation of mucus strands that emerge onto the airway surface. In contrast, mucus threads, which seem to have a much simpler structure, are simply secreted onto the airway surface from individual goblet cells. Another potential explanation for the presence of both submucosal glands and goblet cells in the airways may be related to differences in the amount of secreted mucus in the large versus small airways. The large airways require abundant amounts of mucus for effective MCT. However, the limited surface-to-volume ratio in large airways requires a structure that occupies a small area but can secrete large volumes of mucus (e.g., the submucosal gland). In contrast, in the small airways, with a high surface-to-volume ratio, goblet cell secretion of MUC5AC threads might be sufficient for effective MCT.
What Is the Role of MUC5B Strand and MUC5AC Thread Complexes in Large Airways?
The coexistence of MUC5B strands and MUC5AC threads in large airways is likely important for the clearance of both large and small particulates from the airway surface (Figure 3B). Yet, the physiological importance of the mucus strand structure consisting of an MUC5B core with partial coating of MUC5AC is less clear. Presumably, networks of MUC5B strands form a backbone structure for airway mucus. Mucus strand breakage and release from submucosal glands is likely a key determinant of MCT rates. In addition, a network of MUC5AC threads, attached to and partially covering strands, may be superior for bacterial attachment and clearance. Thus, in healthy airways, these MUC5AC–MUC5B complexes form a mucin network that is likely very important for effective MCT. However, in CF, mucus strands fail to release from submucosal glands, leading to defective MCT. Less clear is how the presence of MUC5AC sheets in the CF airway further impact airway mucus clearance.
How Does Loss of CFTR Function Cause Defective Mucus Strand Release in the CF Airway?
Although loss of CFTR function alters many factors both within the submucosal gland and on the airway surface, defective submucosal gland function is likely a key determinant of impaired MCT in CF airways. Even when the airway surface of newborn CF pig tracheas is bathed in a normal physiological solution, mucus strands fail to detach and break free. Loss of CFTR function could affect submucosal glands in multiple ways. First, lack of CFTR-mediated HCO3− secretion contributes to a more acidic pH in submucosal gland secretions (29) and ASL (30–34). In airways, CFTR mediates HCO3− secretion, and ATP12A (the α subunit of the nongastric H+/K+-ATPase) mediates H+ secretion (31). The acidic pH and/or the reduced [HCO3−] could impact the molecular conformation of mucin proteins and also stiffen mucus strands by making them more difficult to stretch and break. Similar results have been observed with other mucin-composed materials (35, 36). The role of pH at the later stages of CF disease, when secondary manifestations occur, is less clear (29–34, 37). Second, loss of CFTR function reduces Cl−- and HCO3−-dependent liquid secretion from submucosal gland serous cells (38). After exocytosis of mucin-containing vesicles to the CF submucosal gland lumen, mucus may be formed in a smaller volume, which would increase mucin concentration. Mucus strands with more condensed mucin molecules could be more difficult to break free from the duct openings. Earlier studies support these speculations. For example, Joo and colleagues reported that submucosal glands from CF pigs secrete less fluid in response to a number of secretagogues, including forskolin, substance P, carbachol, or their combinations (39). Tang and colleagues reported that the percentage of nonvolatile material is increased in CF pig ASL and that this is associated with an increased ASL viscosity (30). Although submucosal gland–related MCT defects were present in completely submerged CF tracheas, changes in ASL volume could also impact mucin secretion and concentration by surface airway epithelial cells (40, 41). Third, loss of CFTR function may disrupt the ionic environment in the submucosal gland lumen. Monovalent ions (e.g., Na+, Cl−, and K+) in solution shield electrostatic forces between mucin molecules, thus relaxing the mucin polymer networks (42). Divalent ions (e.g., Ca2+) are able to form Ca2+ bridges to cross-link mucin molecules, which could stiffen mucus strands (43, 44). Whether differences in the ionic environment impact mucin structure in the CF airway remains unknown.
Conclusions
Findings from these studies have important clinical implications as new therapeutic approaches are considered for CF airway disease. First, abnormal MCT is present early in CF, and data derived from newborn CF pigs suggest that these defects can occur in the absence of infection and inflammation (16). We suspect that similar defects are present early in infants with CF. If this is the case, then therapies targeting MCT defects at very early time points may be beneficial. However, it will also be important to perform similar studies at later time points because mechanisms regulating MCT can differ with development and age (45–47). Second, mucus strands secreted from submucosal glands are likely a key functional element for effective MCT. In CF airways, MCT is abnormal, in part owing to lack of detachment of these mucus strands from submucosal gland (16, 24). We predict that therapies designed to enhance mucus strand release would improve MCT in CF. Whether anticholinergics might play a therapeutic role requires further study (48). CFTR is also expressed in the small airways (49, 50), and thus it seems likely that small airway MCT will be impaired in CF. However, rigorous data are lacking to support this contention, and future studies are needed to address this point. Third, our data suggest that loss of CFTR function causes mucus strands to be abnormal within the submucosal gland, even before mucus emerges onto the airway surface. Thus, therapies designed to restore MCT in CF might need to be targeted at the submucosal gland rather than at the airway surface. Finally, it is possible that CF-related MCT defects and impaired detachment of mucus strands from CF submucosal glands are induced by multiple factors related to loss of CFTR function (e.g., pH, liquid secretion, ionic changes). Thus, correction of even a single factor may help restore MCT in CF. For example, normalization of submucosal gland pH, by inhibiting ATP12A, might enhance mucus strand breakage and release from CF submucosal gland duct openings.
In summary, studies of newborn non-CF and CF pigs have revealed new insights into MCT and how loss of CFTR function disrupts host defenses in early CF. By better understanding how loss of CFTR-mediated anion transport affects mucus composition, structure, and function, new therapeutic approaches may be developed that impact CF airway disease.
Supplementary Material
Acknowledgments
Acknowledgment
The authors thank Michael Welsh for valuable discussions, Shawn Roach for help with figure design, and Kortney Webber for assistance with manuscript preparation.
Footnotes
Supported in part by National Institutes of Health grants HL051670, HL091842, HL117744, HL136813, HL135433, and HL136927 and the Cystic Fibrosis Foundation (CFF Iowa Research Development Program and STOLTZ16XX0).
Author Contributions: Y.X., L.O., M.H.A.A., L.L., A.J.F., and D.A.S. contributed to the drafting and review of this manuscript.
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 2.Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. N Engl J Med. 2015;372:351–362. doi: 10.1056/NEJMra1300109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Welsh MJ, Ramsey BW, Accurso F, Cutting GR. et al. Cystic fibrosis. In: Scriver CR, Beaudet AL, Valle D, Sly WS, Childs B, Kinzler KW, editors; The metabolic and molecular basis of inherited disease, 8th ed. New York: McGraw-Hill; 2001. pp. 5121–5189. [Google Scholar]
- 4.Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW. Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med. 1995;151:1075–1082. doi: 10.1164/ajrccm/151.4.1075. [DOI] [PubMed] [Google Scholar]
- 5.Balough K, McCubbin M, Weinberger M, Smits W, Ahrens R, Fick R. The relationship between infection and inflammation in the early stages of lung disease from cystic fibrosis. Pediatr Pulmonol. 1995;20:63–70. doi: 10.1002/ppul.1950200203. [DOI] [PubMed] [Google Scholar]
- 6.Armstrong DS, Grimwood K, Carlin JB, Carzino R, Gutièrrez JP, Hull J, et al. Lower airway inflammation in infants and young children with cystic fibrosis. Am J Respir Crit Care Med. 1997;156:1197–1204. doi: 10.1164/ajrccm.156.4.96-11058. [DOI] [PubMed] [Google Scholar]
- 7.Sly PD, Brennan S, Gangell C, de Klerk N, Murray C, Mott L, et al. Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST-CF) Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am J Respir Crit Care Med. 2009;180:146–152. doi: 10.1164/rccm.200901-0069OC. [DOI] [PubMed] [Google Scholar]
- 8.Abou Alaiwa MH, Beer AM, Pezzulo AA, Launspach JL, Horan RA, Stoltz DA, et al. Neonates with cystic fibrosis have a reduced nasal liquid pH; a small pilot study. J Cyst Fibros. 2014;13:373–377. doi: 10.1016/j.jcf.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson M, Bye PT. Mucociliary clearance in cystic fibrosis. Pediatr Pulmonol. 2002;33:293–306. doi: 10.1002/ppul.10079. [DOI] [PubMed] [Google Scholar]
- 10.Boucher RC. Evidence for airway surface dehydration as the initiating event in CF airway disease. J Intern Med. 2007;261:5–16. doi: 10.1111/j.1365-2796.2006.01744.x. [DOI] [PubMed] [Google Scholar]
- 11.Rogers CS, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T, Taft PJ, et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science. 2008;321:1837–1841. doi: 10.1126/science.1163600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ostedgaard LS, Meyerholz DK, Chen JH, Pezzulo AA, Karp PH, Rokhlina T, et al. The ΔF508 mutation causes CFTR misprocessing and cystic fibrosis-like disease in pigs. Sci Transl Med. 2011;3:74ra24. doi: 10.1126/scitranslmed.3001868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stoltz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, Davis GJ, et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med. 2010;2:29ra31. doi: 10.1126/scitranslmed.3000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stoltz DA, Rokhlina T, Ernst SE, Pezzulo AA, Ostedgaard LS, Karp PH, et al. Intestinal CFTR expression alleviates meconium ileus in cystic fibrosis pigs. J Clin Invest. 2013;123:2685–2693. doi: 10.1172/JCI68867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoegger MJ, Awadalla M, Namati E, Itani OA, Fischer AJ, Tucker AJ, et al. Assessing mucociliary transport of single particles in vivo shows variable speed and preference for the ventral trachea in newborn pigs. Proc Natl Acad Sci USA. 2014;111:2355–2360. doi: 10.1073/pnas.1323633111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoegger MJ, Fischer AJ, McMenimen JD, Ostedgaard LS, Tucker AJ, Awadalla MA, et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science. 2014;345:818–822. doi: 10.1126/science.1255825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cooper JL, Quinton PM, Ballard ST. Mucociliary transport in porcine trachea: differential effects of inhibiting chloride and bicarbonate secretion. Am J Physiol Lung Cell Mol Physiol. 2013;304:L184–L190. doi: 10.1152/ajplung.00143.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu L, Chu KK, Houser GH, Diephuis BJ, Li Y, Wilsterman EJ, et al. Method for quantitative study of airway functional microanatomy using micro-optical coherence tomography. PLoS One. 2013;8:e54473. doi: 10.1371/journal.pone.0054473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van As A. Pulmonary airway clearance mechanisms: a reappraisal. Am Rev Respir Dis. 1977;115:721–726. doi: 10.1164/arrd.1977.115.5.721. [DOI] [PubMed] [Google Scholar]
- 20.Iravani J, van As A. Mucus transport in the tracheobronchial tree of normal and bronchitic rats. J Pathol. 1972;106:81–93. doi: 10.1002/path.1711060204. [DOI] [PubMed] [Google Scholar]
- 21.Van As A, Webster I. The morphology of mucus in mammalian pulmonary airways. Environ Res. 1974;7:1–12. [Google Scholar]
- 22.Sears PR, Davis CW, Chua M, Sheehan JK. Mucociliary interactions and mucus dynamics in ciliated human bronchial epithelial cell cultures. Am J Physiol Lung Cell Mol Physiol. 2011;301:L181–L186. doi: 10.1152/ajplung.00321.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med. 2010;363:2233–2247. doi: 10.1056/NEJMra0910061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ostedgaard LS, Moninger TO, McMenimen JD, Sawin NM, Parker CP, Thornell IM, et al. Gel-forming mucins form distinct morphologic structures in airways. Proc Natl Acad Sci USA. 2017;114:6842–6847. doi: 10.1073/pnas.1703228114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thornton DJ, Rousseau K, McGuckin MA. Structure and function of the polymeric mucins in airways mucus. Annu Rev Physiol. 2008;70:459–486. doi: 10.1146/annurev.physiol.70.113006.100702. [DOI] [PubMed] [Google Scholar]
- 26.Ermund A, Meiss LN, Rodriguez-Pineiro AM, Bähr A, Nilsson HE, Trillo-Muyo S, et al. The normal trachea is cleaned by MUC5B mucin bundles from the submucosal glands coated with the MUC5AC mucin. Biochem Biophys Res Commun. 2017;492:331–337. doi: 10.1016/j.bbrc.2017.08.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bush A, Chodhari R, Collins N, Copeland F, Hall P, Harcourt J, et al. Primary ciliary dyskinesia: current state of the art. Arch Dis Child. 2007;92:1136–1140. doi: 10.1136/adc.2006.096958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roy MG, Livraghi-Butrico A, Fletcher AA, McElwee MM, Evans SE, Boerner RM, et al. Muc5b is required for airway defence. Nature. 2014;505:412–416. doi: 10.1038/nature12807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song Y, Salinas D, Nielson DW, Verkman AS. Hyperacidity of secreted fluid from submucosal glands in early cystic fibrosis. Am J Physiol Cell Physiol. 2006;290:C741–C749. doi: 10.1152/ajpcell.00379.2005. [DOI] [PubMed] [Google Scholar]
- 30.Tang XX, Ostedgaard LS, Hoegger MJ, Moninger TO, Karp PH, McMenimen JD, et al. Acidic pH increases airway surface liquid viscosity in cystic fibrosis. J Clin Invest. 2016;126:879–891. doi: 10.1172/JCI83922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shah VS, Meyerholz DK, Tang XX, Reznikov L, Abou Alaiwa M, Ernst SE, et al. Airway acidification initiates host defense abnormalities in cystic fibrosis mice. Science. 2016;351:503–507. doi: 10.1126/science.aad5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pezzulo AA, Tang XX, Hoegger MJ, Abou Alaiwa MH, Ramachandran S, Moninger TO, et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature. 2012;487:109–113. doi: 10.1038/nature11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coakley RD, Grubb BR, Paradiso AM, Gatzy JT, Johnson LG, Kreda SM, et al. Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc Natl Acad Sci USA. 2003;100:16083–16088. doi: 10.1073/pnas.2634339100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garland AL, Walton WG, Coakley RD, Tan CD, Gilmore RC, Hobbs CA, et al. Molecular basis for pH-dependent mucosal dehydration in cystic fibrosis airways. Proc Natl Acad Sci USA. 2013;110:15973–15978. doi: 10.1073/pnas.1311999110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cao X, Bansil R, Bhaskar KR, Turner BS, LaMont JT, Niu N, et al. pH-dependent conformational change of gastric mucin leads to sol-gel transition. Biophys J. 1999;76:1250–1258. doi: 10.1016/S0006-3495(99)77288-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Celli JP, Turner BS, Afdhal NH, Ewoldt RH, McKinley GH, Bansil R, et al. Rheology of gastric mucin exhibits a pH-dependent sol-gel transition. Biomacromolecules. 2007;8:1580–1586. doi: 10.1021/bm0609691. [DOI] [PubMed] [Google Scholar]
- 37.Schultz A, Puvvadi R, Borisov SM, Shaw NC, Klimant I, Berry LJ, et al. Airway surface liquid pH is not acidic in children with cystic fibrosis. Nat Commun. 2017;8:1409. doi: 10.1038/s41467-017-00532-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ballard ST, Spadafora D. Fluid secretion by submucosal glands of the tracheobronchial airways. Respir Physiol Neurobiol. 2007;159:271–277. doi: 10.1016/j.resp.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joo NS, Cho HJ, Khansaheb M, Wine JJ. Hyposecretion of fluid from tracheal submucosal glands of CFTR-deficient pigs. J Clin Invest. 2010;120:3161–3166. doi: 10.1172/JCI43466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Button B, Cai LH, Ehre C, Kesimer M, Hill DB, Sheehan JK, et al. A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science. 2012;337:937–941. doi: 10.1126/science.1223012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Henderson AG, Ehre C, Button B, Abdullah LH, Cai LH, Leigh MW, et al. Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J Clin Invest. 2014;124:3047–3060. doi: 10.1172/JCI73469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robinson NP, Kyle H, Webber SE, Widdicombe JG. Electrolyte and other chemical concentrations in tracheal airway surface liquid and mucus. J Appl Physiol (1985) 1989;66:2129–2135. doi: 10.1152/jappl.1989.66.5.2129. [DOI] [PubMed] [Google Scholar]
- 43.Gibson LE, Matthews WJ, Minihan PT, Patti JA. Relating mucus, calcium, and sweat in a new concept of cystic fibrosis. Pediatrics. 1971;48:695–710. [Google Scholar]
- 44.Ambort D, Johansson ME, Gustafsson JK, Nilsson HE, Ermund A, Johansson BR, et al. Calcium and pH-dependent packing and release of the gel-forming MUC2 mucin. Proc Natl Acad Sci USA. 2012;109:5645–5650. doi: 10.1073/pnas.1120269109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seybold ZV, Mariassy AT, Stroh D, Kim CS, Gazeroglu H, Wanner A. Mucociliary interaction in vitro: effects of physiological and inflammatory stimuli. J Appl Physiol (1985) 1990;68:1421–1426. doi: 10.1152/jappl.1990.68.4.1421. [DOI] [PubMed] [Google Scholar]
- 46.Phipps RJ, Abraham WM, Mariassy AT, Torrealba PJ, Sielczak MW, Ahmed A, et al. Developmental changes in the tracheal mucociliary system in neonatal sheep. J Appl Physiol (1985) 1989;67:824–832. doi: 10.1152/jappl.1989.67.2.824. [DOI] [PubMed] [Google Scholar]
- 47.Wanner A.Autonomic control and mucociliary functions Chest 1987915, Suppl49S–51S. [PubMed] [Google Scholar]
- 48.Ermund A, Meiss LN, Dolan B, Bähr A, Klymiuk N, Hansson GC. The mucin bundles responsible for airway cleaning are retained in cystic fibrosis and by cholinergic stimulation. Eur Respir J. 2018;52:1800457. doi: 10.1183/13993003.00457-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shamsuddin AK, Quinton PM. Native small airways secrete bicarbonate. Am J Respir Cell Mol Biol. 2014;50:796–804. doi: 10.1165/rcmb.2013-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X, Lytle C, Quinton PM. Predominant constitutive CFTR conductance in small airways. Respir Res. 2005;6:7. doi: 10.1186/1465-9921-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.