

Abstract
Membrane proteins are crucial for cellular sensory cascades and metabolite transport, and hence are key pharmacological targets. Structural studies by traditional high-resolution techniques are limited by the requirements for high purity and stability when handled in high concentration and nonnative buffers. Hence, there is a growing requirement for the use of alternate methods in a complementary but orthogonal approach to study the dynamic and functional aspects of membrane proteins in physiologically relevant conditions. In recent years, significant progress has been made in the field of X-ray radiolytic labeling in combination with mass spectroscopy, commonly known as X-ray Footprinting and Mass Spectrometry (XFMS), which provide residue-specific information on the solvent accessibility of proteins. In combination with both low-resolution bio-physical methods and high-resolution structural data, XFMS is capable of providing valuable in-sights into structure and dynamics of membrane proteins, which have been difficult to obtain by standalone high-resolution structural techniques. The XFMS method has also demonstrated a unique capability for identification of structural waters and their dynamics in protein cavities at both a high degree of spatial and temporal resolution, and thus capable of identifying conformational hot-spots in transmembrane proteins. Here, we provide a perspective on the place of XFMS amongst other structural biology methods and showcase some of the latest developments in its usage for studying conformational changes in membrane proteins.
Keywords: Hydroxyl-radical footprinting, oxidative labeling, mass spectrometry, ion channels, transporters, radiolysis
1. INTRODUCTION
Membrane proteins are crucial for gating key metabolites across membranes and for cellular signaling, and hence play a critical role in the cellular energy production and initiation of numerous cellular signaling cascades. Membrane proteins often act in concert with diverse accessory proteins, together which form an intricate and highly organized biomolecular network underlying the complex function of living cells. This makes membrane proteins prime pharmacological targets, and elucidation of their functional mechanisms is important for therapeutic usages. While classical structure determination methods such as crystallography and NMR provide high-resolution information, they are of limited usage for membrane proteins because of sample preparation requirements, which require either crystallization, preparation of stable protein at high concentrations, and/or use of non-physiological buffer conditions [1–3]. Cryo-electron microscopy [4–6] has recently grown into a tool capable of producing structural information with a resolution ≥ 3.5 Å. However, this technique requires stable and symmetrical complexes to produce higher resolution structural data. Other structural techniques such small-angle scattering [7], which is difficult to carry out in the presence of detergents or liposomes, and DEER spectroscopy [8], which require cysteine point mutation followed by labeling with a sterically resistant paramagnetic group, can provide novel structural information but are limited in resolution and usages. Thus, membrane proteins have earned a deserved reputation of being difficult targets, and their structural investigation requires complementary and orthogonal approaches, and hence is still a relatively new area of research. Recent advances in the development of radiolytic labeling and mass-spectrometry (known as X-ray Footprinting and Mass Spectrometry, XFMS, and also referred to a Hydroxyl Radical Footprinting, HRF) [9], an in situ hydroxyl radical labeling method coupled with extensive bottom-up mass spectrometry, provide an alternative and complementary approach to these more common structural tools. The method has the advantage of providing single residue resolution and experiments can be carried out under near-physiological conditions with minimum sample manipulation, giving many options in sample preparation under various functional states [10]. In this review, we describe the basic principles of XFMS, its place relative to other mass-spectrometry-based methods, and its advantages in comparison to other available methods in application to membrane proteins, as well as highlighting recent examples of its applications. We also outline the latest developments and future promises of this technology to study membrane protein structure and dynamics.
2. STRUCTURAL MASS SPECTROMETRY FOR STUDYING THE GLOBAL AND LOCAL STRUCTURE OF MEMBRANE PROTEINS
In contrast to the characterization of membrane proteins by quantitative proteomics approaches, such as membrane protein expression analysis, post-translational modification analysis and estimation of stoichiometry and molecular size, and imaging mass spectrometry of membrane protein in tissues, studies to determine structure, dynamics and interactions of membrane proteins by MS are rather limited because of their low abundance, the complexity of membrane protein sample handling conditions, and the challenges in obtaining a high degree of sequence coverage of the transmembrane domains [11–13]. Broadly, structural mass spectrometry on membrane proteins can be divided into two categories. In the first category, covalent labeling methods, such hydroxyl radical labeling [14, 15], Hydrogen-Deuterium Exchange MS (HDX-MS) [16, 17] and cross-linking MS [13, 18] are carried out in solution conditions to label accessible side chains and modify proteins, which in turn are analyzed by bottom-up or top-down mass spectrometry methods. In the bottom-up method the known peptide fragment– generated by enzymatic digestion and liquid chromatography coupled to mass spectrometry – are used to separate the peptides and identify their sequence and position of modification, and to quantify levels of modification at the peptide or residue level. Alternatively, identification of sites of covalent labeling can be done by the top-down method, where the intact labeled protein is introduced in the mass spectrometer and fragmented in the gas phase to identify the site of covalent labeling [19]. Top-down methods provide a major advantage by bypassing the protein digestion step; however, such fragmentation methods need specific instrument configurations and sample solubilizing conditions and are often limited by the usability of covalent labeling to analyze membrane protein structure with high sequence coverage. Among the various covalent labeling approaches, HDX-MS is a powerful method to study structure and dynamics of backbone amide bonds as well as to provide information of thermodynamics and kinetics of membrane protein interactions. In this method the backbone hydrogen is exchanged with deuterium from D2O and the rate of exchange measures the flexibility and accessibility of the exchanged peptide bond of the target sequence of the protein. The post H/D exchange steps, which need low pH and low temperature condition to quench the exchange reaction, limits the choice of protease fragmentation and often results in limited coverage of the hydrophobic regions of transmembrane domains. In addition, scrambling of H/D by collision-induced dissociation, a method which is used in bottom-up MS, poses a major challenge in data analysis and interpretation. HDX-MS with top-down MS is very useful for obtaining information on the stability of local the secondary structure, folding and interactions with other proteins. Top-down approaches using electron capture/transfer dissociation fragmentation are under development to reduce the error generated by back exchange. The application of HDX-MS to the investigation of transmembrane proteins is a growing area of application [17]. The HDX method is combined with other solution state methods to study structure and dynamics of membrane transporters and receptors [20, 21]. The chemical- or photo-crosslinking and mass spectrometry is another solution based approach, which covalently modifies selective sites to study spatial relationship between strongly interacting domains and subunits of membrane proteins and their complexes. Conventional crosslinkers with a nonreactive spacer domain between two reactive terminal groups generally targets amino, thiol, or carboxyl groups. The location of the crosslinked residues can be directly identified by bottom-up mass spectrometry and provide information about tertiary contacts [13, 18]. Guided by available crystal structures of proteins in isolation, this is a powerful technique which can be used to map the interactions in large complexes and membrane proteins [22]. This method is restricted by both the accessibly of probe residues in transmembrane domains and availability of cross-linkable residues, typically Lys or Cys, depending on the type of the cross-linker and the cross-linking efficiency, which may vary significantly. It is also rather difficult to quantify the effects and provide time-resolved studies by chemical cross-linking, although some progress has recently been achieved using isotope-labels [23]. Another covalent labeling method, known as GEE labeling, has been used to study soluble protein though has limited usage in membrane protein system as this method can only probe solvent accessible Glu residues in the periplasmic domain [24]. Hydroxyl radical labeling followed by mass spectrometry is well suited to the study of structure and dynamics of versatile classes of protein systems, including membrane proteins. The relative ease of generation of the small, highly diffusible, and highly reactive hydroxyl radical probe enables structural studies with a high degree of spatial and temporal resolution both for transmembrane and periplasmic domains [14, 25, 26]. Each of the solution based methods has its own advantages and limitations. In this review, we focus on X-ray mediated hydroxyl radial labeling also known as X-ray Footprinting and Mass Spectrometry (XFMS), which is discussed in the next section with recent examples on the studies of membrane protein complexes. In the second category of structural mass spectrometry, studies are done with intact protein system in semi-gaseous or partially dehydrated phase using native MS. Under careful electro-spray instrumental conditions, the detergent-solubilized membrane protein complexes can be ionized and detected as an intact molecule by native-MS to determine size and stoichiometry, and ion mobility MS can further provide information about global structural dynamics and interactions [27, 28]. Native MS is only suitable for analysis of membrane complexes, which are either very stable or can be sampled using mild conditions. Native MS in conjunction with the top-down approach can reveal oligomeric state as well as sequence information in a single experiment. The laser-induced liquid bead ion desorption technique is another approach, which does not require membrane protein solubilization and can be used to analyze subunit stoichiometry and quaternary structure of large membrane protein complexes [29]. Finally, integration of these MS based data with X-ray or NMR structures and / or EM density maps provides detailed models of previously intractable membrane protein structure and dynamics.
3. X-RAY RADIOLYTIC LABELING AND MASS SPECTROMETRY (XFMS)
Synchrotron X-rays in the energy range of 5 – 12 keV interact with water almost exclusively by the photoelectric effect, ionizing water molecules and forming highly reactive hydroxyl radicals (•OH) [30]. The •OH radical, which is generated wherever water is present, diffuses and reacts with side chains in the vicinity of the site of origin, and under aerobic conditions, resulting in covalent labeling such as hydroxylation, carbonylation, and di-oxidation, with specific signature mass adducts (e.g., + 16, +14 and +32 Da). The reaction mechanisms for each peptide are known, and details are reported elsewhere [10]. The site and extent of labeling is identified and quantified by reverse phase liquid chromatography coupled with high-resolution mass spectrometry in a standard protocol for bottom-up proteomics analysis (Figure 1) [31]. Increasing the •OH dose in steps results in a dose-response plot for each residue that provides the side chain-specific hydroxyl radical reactivity rate. This rate depends on both the intrinsic reactivity and the solvent accessibility of the residues; however, since XFMS studies compare two or more states of proteins, the ratio of reactivity rates of the same residue from one state to another depends solely on the solvent accessibility difference between the two states. The information obtained can be used to enhance understanding of existing structures or incorporated into molecular modeling strategies that provide information about protein or ligand docking and conformational changes [26, 32]. When sample is exposed to X-rays a steady-state concentration of •OH is achieved as a result of many primary and secondary free radical reactions in solution. The counterproductive reactions, which limit the sensitivity of the method, consist of •OH •OH recombination and reaction of •OH with buffer constituents in samples. In general, unlike globular proteins, membrane protein samples are surrounded by a high concentration of extrinsic •OH scavengers like detergents and phospholipids, which reduce the effective dose to the protein. A low flux X-ray beamline necessitates long irradiation times, resulting in secondary damage, which results in specific structural perturbations. In contrast, a short pulse of high flux density photons produces an adequate •OH concentration to overcome counterproductive reactions and scavenging reactions while preserving the structural integrity of the samples, which is particularly important for studying membrane proteins and their complexes. The XFMS method was first developed at the bending magnet beamline X28C at NSLS and initial experiments required long irradiation times to yield detectable labeling on globular protein samples [33–35]. The use of a focusing mirror revolutionized the XFMS approach at X28C by delivering a high flux density in milliseconds, leading to successful studies of megaDalton and membrane proteins complexes [14, 36, 37]. Currently, a new XFMS facility is under operation at the Advanced Light Source (ALS) at beamline 3.2.1, and at the National Synchrotron Light Source II (NSLS II) Beamline 17BM [9, 30]. Both the beamlines, ALS 5.3.1 and NSLS II 17BM deliver a broadband X-ray beam with an average flux of ~1×1016 photons/sec. These two beamlines, and the ALS beamline 3.3.1 under construction, are described in other articles in this issue. A microfocus beam and microfluidic sample handling address the challenge of inadequate flux density and extend the time scale of the method to the microsecond range [30]. However, the challenge of obtaining sufficient hydroxyl radical dose without excessive exposure time remain, despite improvements in mass spectrometry resolution, sensitivity, and new analysis approaches targeting low yield modification products. The use of lowest possible concentration of non-scavenging buffer constituents still limits the sample that can be brought to the XFMS facility, particularly in the case of membrane protein samples reconstituted in liposome, and further increases in achieved dose are currently being pursued using various sample exposure methods, such as drop on demand, as described in another article in this issue.
Figure 1. Schematic of the X-ray footprinting method using synchrotron X-ray radiolysis and mass spectrometry.
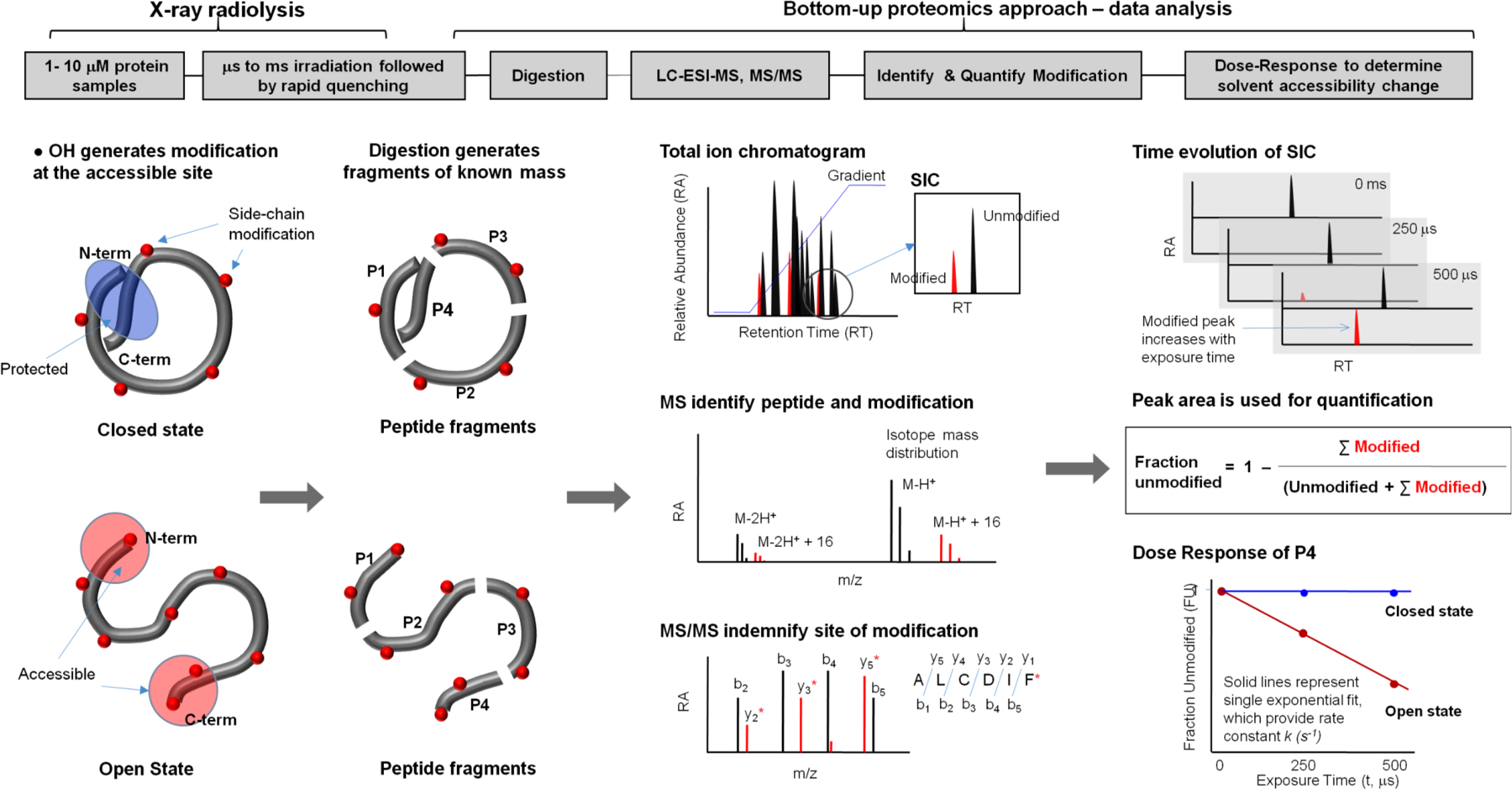
The top panel shows the overall method, and the bottom panel illustrates a case study for a protein that undergoes a conformational change from the closed to the open state. The protein is covalently modified after a series of X-ray irradiations on the order of microseconds followed by rapid quenching by methionine amide. Irradiated protein is digested with proteases to generate peptide fragments of known mass. Digested protein is analyzed by reverse phase Liquid Chromatography coupled with Electrospray Ionization Mass Spectrometry (LC-ESI-MS), in which peptides are separated in the Total Ion Chromatogram (TIC) or mass chromatogram. For any peptide fragment the unmodified and modified m/z is extracted and visualized by Selected Ion Chromatogram (SIC). High resolution mass spectrometry is used to identify the unmodified and modified peptide fragments by their accurate m/z and isotope mass distribution. The site of modification is identified from the mass assignment of the y and b fragment ions from the tandem mass spectrometry (MS/MS) of the corresponding peptide. The extent of modification for the series of irradiation points are quantified for the SICs of unmodified and modified peptide fragments. The fraction of unmodified peptide vs. exposure time (dose-response or DR-plot) provides site-specific modification rate constants (k s−1). The rates are compared among different sample conditions, and their ratios, which are independent of intrinsic reactivity, account for the degree in solvent accessibility changes due to any conformational transition/interactions. The final results are mapped onto available structures or used as constraints for structural modeling. Reproduced in part with the permission from Gupta et al. J Sync. Rad. 2016.
4. OTHER METHODS TO GENERATE •OH RADICALS
An X-ray source is not strictly necessary for protein foot-printing. For instance, chemical generation of •OH by oxidative Fenton chemistry is a simple lab-based method [38]. With this method, the radicals are produced using reagents such as Fe(II)-EDTA and H2O2 which can affect conformation or damage/unfold proteins, as well as remove essential metal ions necessary for protein function. Another lab-based technique for generating radicals is the Fast Photochemical Oxidation of Proteins (FPOP) which uses dissociation of H2O2 by UV-laser to generate a high concentration of •OH on the microsecond timescale [39]. However, H2O2 concentrations at the millimolar level are necessary with this method. Electrospray-Ionization (ESI) can also be used to generate •OH using the electric discharge in the ESI component of the mass spectrometer, in which the protein is labeled in its semi-hydrated state in the presence of ammonium carbonate [40, 41]. Electron pulses via a Van de Graaff generator can be used to radiolyze water and generate hydroxyl radicals in situ. Oxidative modification obtained in sub-microsecond timescale on Cyt c and rhodopsin using high energy electron is comparable to that of microsecond – millisecond labeling by X-ray radiolysis (Gupta et al. unpublished). However, the energy deposition by high energy electrons can cause significant protein damage [30]. The various •OH-based footprinting techniques offer distinct advantages and disadvantages and therefore can be selected to suit the system under study. One of the unique advantages of synchrotron-based XFMS is that varying the flow rate of the sample across a continuous x-ray beam provides a technically simple way to vary the •OH dose by an order of magnitude [30], allowing precise hydroxyl radical reactivity rates for residues within complex biological samples [25, 26], while in situ •OH generation allows a very large range of sample conditions without the necessity of adding deleterious external reagents such as H2O2. Also, the method is straightforward enough to allow control of additional parameters, such as varying temperature and utilizing H2 18O, which can be used to distinguish between bulk and protein-bound water, and to give information on water dynamics [42].
5. XFMS IN THE STUDY OF MEMBRANE PROTEINS
5.1. Detection of Internal Water Interactions
In dilute solutions, amino acids or small polypeptides, •OH undergoes diffusion-controlled recombination reactions with other •OH and side chain residue by a homogeneous kinetics process. The intrinsic reactivities of free amino acids towards •OH in aqueous aerobic solution vary widely; their order of reactivity as measured in aerobic aqueous solution is Cys > Met, Trp > Tyr > Phe > His > Iie > Leu > Arg which react with •OH rapidly (rate constants 109 to 1010 M−1s−1) in contrast to Lys ~ Val > Ser ~ Thr ~ Pro> Gln~ Glu>Asp~ ASN > Ala > Gly, which react 10 to 1000 fold slower. However, •OH induced modifications in large protein systems follow a complex kinetic pathway, and in many instances do not follow the order of amino acid reactivity listed above. The protein molecules create distinct hydration environments which are different from that of bulk water (Figure 2). The cavities, grooves and active sites on a protein surface contain H-bonding networks and ionic interlocks with amino acid side chains that can bind three times as many water molecules as the solvent-exposed surface. Inherently, the time scale for the sequence of events in the radiolysis of pure water leading to the generation of the highly reactive, but short-lived •OH is on the order of 10−14 sec, which is extremely fast. Low linear energy transfer ionizing radiation, such as broadband X-ray photon beams, can generate local population of •OH (spurs) inside such cavities and those •OH can result in selective modification of adjacent side chains. To date there is no report of side chain reactivity order within a protein, however XFMS results directly show that the reactivity order is governed by the proximity of the side chain to •OH, which originates from the ionization of a local water in an H-bonding network with the reactive side chain. A fully exposed side chain in the bulk water environment has to compete with equally or more reactive •OH and/or additives (buffer constituents) in order to yield detectable amounts of modification. Hence, in analyzing XFMS results it is often not surprising to see limited modification of a fully surface exposed residue as compared to considerable modification of a similar side chain inside a cavity. Temperature-dependent XFMS has shown to selectively label amino acid residues adjacent to bound water in globular proteins [43]. In general, the bound or structural waters, which form polar contacts with backbone, side chains and other internal waters, are required for folding, stability, enzymatic activity and protein-protein interactions [44]. Internal water or bound water in TM domains is often important for structural stabilization and receptor and transport activity in membrane proteins. It has been reported that the number of internal waters in TM helices is higher than that of globular proteins [45]. Study have shown the contact between the internal water and residues are mostly conserved among membrane protein families and the disease-causing mutations occur at buried water contact site at higher than random frequency [45]. Thus, XFMS is highly advantageous for the study of active sites and cavities inside TM domains. The recent development of time-resolved radiolytic labeling coupled to H2 18O exchange demonstrates that footprinting can probe the dynamics of residues adjacent to bound waters [42]. This is based on the fact that the 18O from the attacking hydroxyl radical remains attached to the Met, Phe, Tyr, Trp, Cys residues, while in other side chains the covalently attached oxygen is derived from a dissolved molecular oxygen [42]. In membrane proteins such as ion channels, receptors, and transporters this method not only has the potential to detect specific modifications of specific residues inside the transmembrane domain where bound waters are located, but also can shed light on water and conformational dynamics using time-resolved XFMS [14, 43].
Figure 2. Major reactions scenarios for X-ray radiolysis in dilute protein samples.
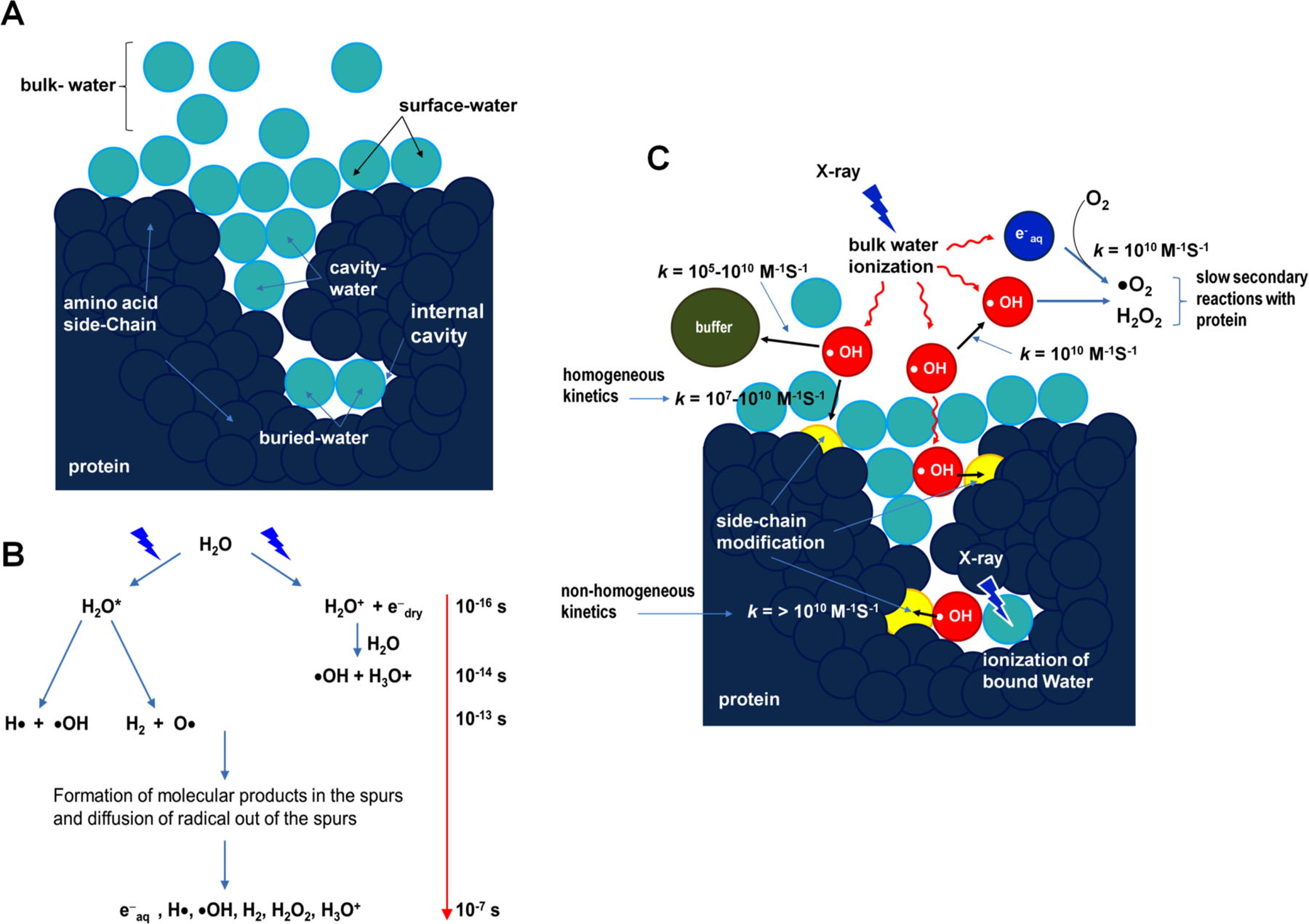
(A) Schematic showing the position of bulk- , surface- , and bound-water (cavity- and buried-water) (light blue) in a protein molecule (dark blue). (B) Radiolysis of water and the timescale of sequence of events reproduced from Gupta et al. J. Sync. Rad. 2014 and Liljenzin, J., Radiation Effects on Matter, in Radiochemistry and Nuclear Chemistry, 2002, Butterworth-Heinemann. (C) The location of hydroxyl radical (red) generated in situ from ionization or activation of water by X-ray irradiation. The •OH radicals react with nearby side chains in close proximity and yield covalent modifications on the protein side chains (yellow). Radiolysis of bulk water starts with the ionization of water on the time scale of 10−16 s . The key product, •OH, diffuses (10−7 s) out in the bulk (red arrows) and undergoes reactions with other •OH, buffer molecules, and protein side chains (bimolecular reactions are indicated by black arrows, and approximate values of the rate constants for different reactions are shown). The rapid counterproductive reactions, such as •OH - •OH recombination, as well as various other reactions scavenge •OH and reduce the concentration of •OH in the bulk. Thus, a sufficient X-ray dose or high flux density beam is needed to maintain a steady concentration of •OH that will lead to a detectable yield of side chain modification on the protein in solution. In contrast, •OH radicals formed from activation of a bound water can react faster with side chains in proximity because of the translational and rotational ordering of water and because fewer scavenging reactions by other •OH or highly reactive buffer constituents are available. Radiolysis of water also produces electrons, which rapidly become solvated and react with O2 to produce superoxide radicals. In general, the reactivity of side-chains to solvated electrons is lower than to hydroxyl radicals (Xu & Chance 2007). Peroxides and superoxides undergo slow reactions with protein side chains and are quenched as described in sections 1.1 and 1.3. Solvated electrons consume O2, which is also required for side chain labeling by •OH radicals; thus short irradiation time as well as high flux density are the key factors for success of the XF-MS experiment. Reproduced in part with the permission from Gupta et al. J. Sync. Rad. 2014 and Gupta et al. J Sync. Rad. 2016.
5.2. XFMS in the Study of GPCRs
Comparison of known GPCR structures has revealed that conserved water molecules in the TM domains generally interact with conserved residues, implying that these water networks in the transmembrane domains are probably as important for functions as the conserved residues of GPCRs, and that they participate in the transfer of signal from the chromophore or the agonist-binding site to allosteric sites ultimately regulating the G-protein activation for signal transduction [46]. In particular, in the photo-activation processes, which are well studied in the example of mammalian rhodopsin, studies demonstrated the involvement of transient intermediates leading to the so-called Meta II state that is competent for G-protein binding [20]. The X-ray crystal structures of rhodopsin and of its photo-intermediates have dramatically increased our understanding of structural rearrangements upon the activation of GPCRs [20]. However, it is also increasingly clear that static structures alone are not sufficient to provide a complete understanding of GPCR function, especially given the prominent role that is played by structural waters, which are only visible in very high-resolution crystal structures, and are difficult to obtain for a number of membrane receptors. XF, on the other hand, has emerged as a novel approach to study GPCRs by in situ labeling of transmembrane residues located in proximity to bound water. The first molecular details of the photoactivation process came from the comparative XF studies of the dark state, meta II and opsin states from detergent-solubilized samples [14]. These results are summarized in Figure 3. Results indicated an increase in labeling efficiency near the retinal-binding region, conserved TM domains and in a few residues of cytoplasmic and extracellular loops upon activation. The local conformational changes arising from the isomerization of the covalently bound retinal appear to be propagated to the cytoplasmic surface by means of water reorganization, and rearrangement of the H-bonding network between bound water and amino-acid side chains in the TM domain. The combined XFMS and hydrogen-deuterium exchange mass spectrometry (HDX-MS) studies on rhodopsin, activated rhodopsin and rhodopsin-G-protein complex have elucidated the finer details of the dynamics of the internal water rearrangements that accompany G-protein binding, as well as revealing the location of the protein-protein interactions in both rhodopsin and G protein [20]. These studies allowed XFMS to be used to structurally validate a homology model for the 5-HT4R receptor, for which no high resolution structure was available [47]. The method predicted the sites for internal waterside chain interaction in the 5-HT4R receptor necessary for its activation process, highlighting another important application of the XFMS method. In another more recent study, the method identified individual residues that are part of the interaction interface between a chemokine receptor (ACKR3) and chemokines ligands, CXCL12 and CCX777, which form fully and partially activated receptor-ligand complexes respectively [48]. The results, in combination with disulfide trapping and mutagenesis studies, were used to generate experimentally guided 3D modeling of the ACKR3 complex. While the free ACKR3 was highly unstable, rendering many other types of experiments practically impossible, comparison of XFMS labeling data between fully activated and partially activated complex showed recruitment and ordering of water molecules in the TM domains similar to a structural mechanism for agonist-induced activation of canonical GPCR.
Figure 3. XF-MS probes bound water mediated signal transfer pathway in bovine rhodopsin.
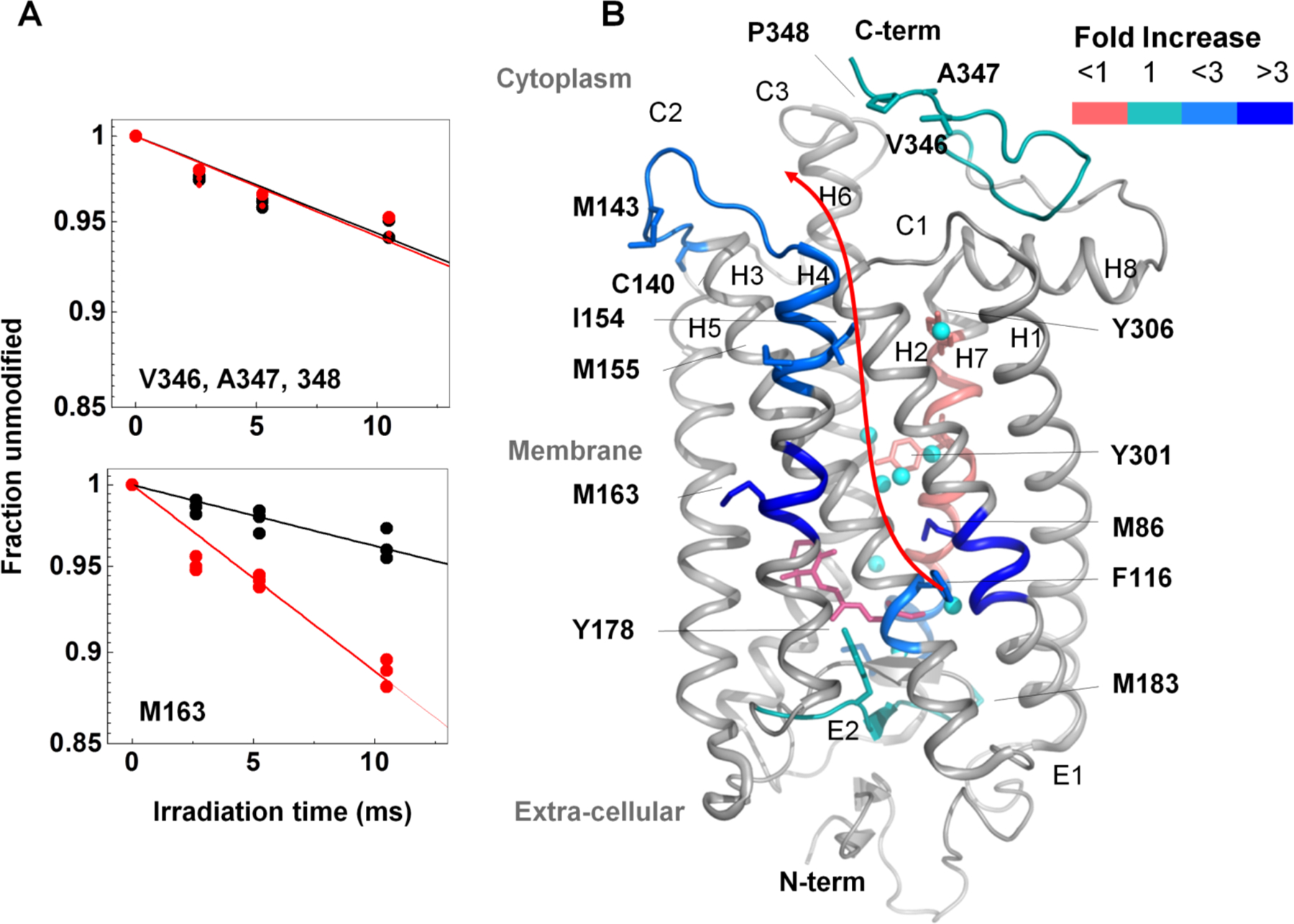
(A) DR plots of dark (black) vs. meta II (red) for the peptide p333–348 (at the solvent exposed cytoplasmic side) and p160–164 (at the TM region) for modified residues indicated. (B) Pictorial summary of relative solvent accessibility changes for the photoactivation of dark to meta II state. Residues with rate constants > 0.1 s−1 are shown as sticks. The color coding represents the ratio of rate constants between meta-II and rhodopsin. Conserved transmembrane waters are shown as cyan spheres. The changes in rates of modification reflect local structural changes inside the TM domain upon formation of meta II. The results demonstrate disruption and reorganization of multiple close-packing interactions, mediated by both side chains and bound waters. The information is transmitted from the chromophore (ligand-binding site) to the cytoplasmic surface for G-protein activation. Results from Angel et al. PNAS 2009 and reproduced from Gupta et al. J. Sync. Rad. 2016.
5.3. FMS Applied to the Study of K+ Ion Channels
Ion channels present exceptionally good candidates for study by XFMS, as their activation and the associated channel “gating” events usually result in dramatic changes in water permeability of the central pore of the channel which is, in the resting state, devoid of water, presenting a major energetic barrier to ion conduction [49]. In the case of potassium channels, these gating events are connected to a variety of regulatory stimuli and include several critical conformational transitions that are suggested to propagate from the inner side of the membrane, including the so-called bundle crossing [50], which is thought to be the principle gating mechanism for the majority of the K+ channels, and is absolutely required to be in an open conformation in order for the channel to be in a conductive state. The study of the bundle crossing gating using crystallography proved to be challenging as the closed state appears to be energetically favorable, resulting in channels preferentially crystallizing in the closed state [51, 52], while their size precluded NMR analysis, leaving the question of structural transitions during gating unanswered for a number of years. In potassium channels, bound water was also shown to play a major role in their slow inactivation, while cavities inside transmembrane domains are formed by the interaction between bound water and amino acid residues [53]. The first K-channel to be studied by XFMS was KirBac 3.1 [25], and the comparison of the closed state of the channel with an EDTA-induced opening revealed a dramatic increase in the solvent accessibility along the central cavity of the channel, as well as along the interface between the transmembrane (gating) domain and the cytoplasmic regulatory domain of the channel, and conformational transitions that were suggested to induce opening of the channel (Figure 4). In addition, this study [25] highlighted the possibility of the presence of a hydrophobic gate in this inward rectifying potassium channel, namely the residue L124 which showed the highest level of change in accessibility between the two open and closed conformations of the channel. These predictions have been confirmed in full by the crystal structure of the open state of the channel [54]. Notably, although the existence of such hydrophobic gating for K-channels had been suggested much earlier [55], the XFMS directly visualized the role of the L124 during the transition [43]. As hydrophobic gating is suggested to play a major role in a number of channels lacking the traditional “bundle crossing gate”, such as K2P channels and a number of other channels including pentameric ligand-gated and the bacterial mechanoselective channels [49], XFMS appears an ideally suited tool to interrogate the gating transitions in these channels in the future. Similarly, during activation the pH-dependent bacterial KcsA channel undergoes gating at the bundle crossing with several charged residues suggested to play the role of proton sensors [56]. KcsA also possesses a unique C-terminal domain forming a cytoplasmic protrusion, which imposes steric limits on the bundle crossing gate and the contraction of which appears to control the opening of the channel and its inactivation [57]. The comparison of water accessibility between the wildtype and non-inactivatable mutant E71A of KcsA enabled not only confirmation of the significant changes of the water accessibility of the C-terminal domain (Gupta, unpublished data) but also demonstrated radical changes in the solvent profiles of residues behind the selectivity filter of the channel which harbors the E71A mutation. These findings are consistent with the recent suggestion of the important role played by structured/bound water in stabilizing the conductive selectivity filter conformation [58] and highlights the potential of XFMS for the study of these systems.
Figure 4. XF-MS identifies conformation changes during gating of a K+ ion channel, KirBac3.1.
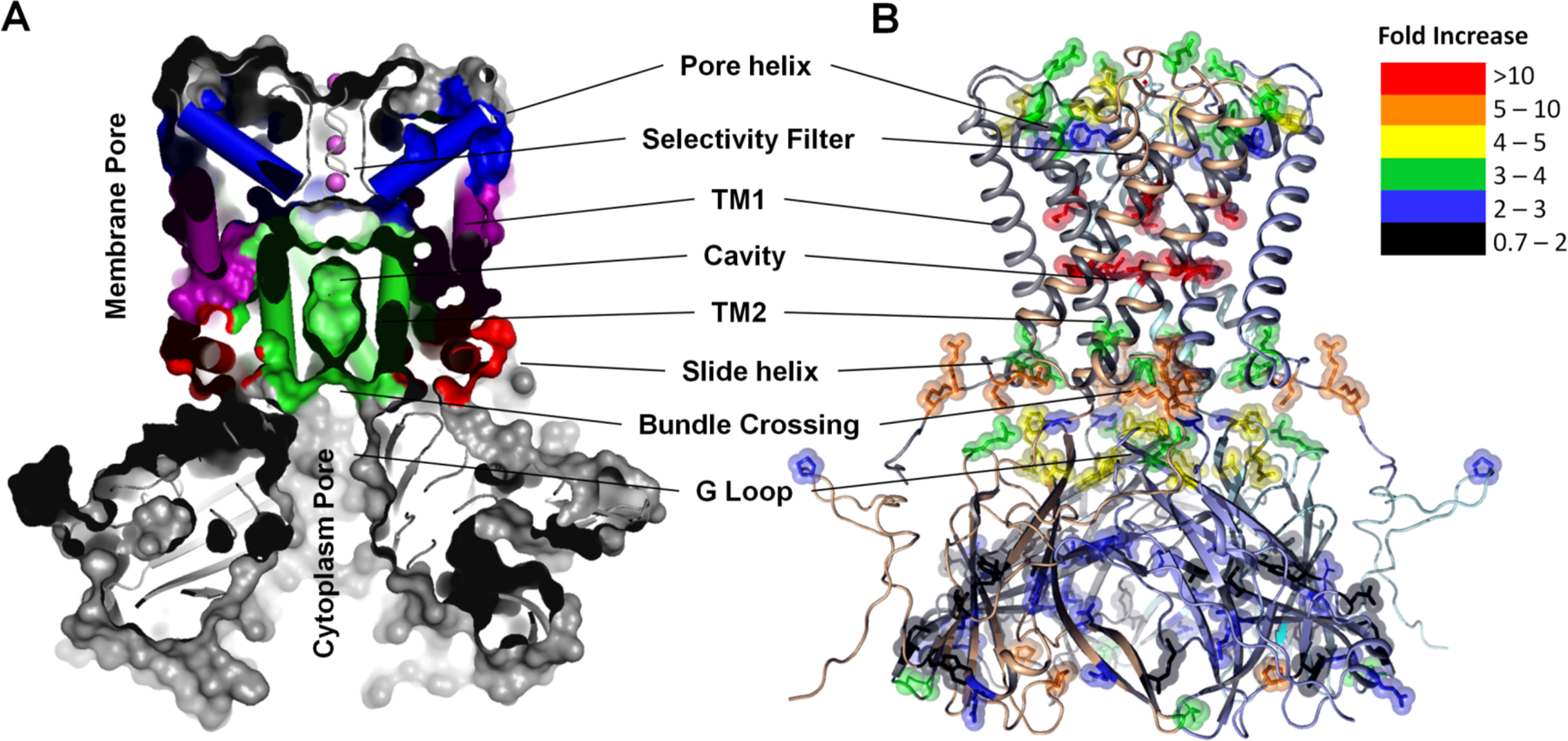
(A) Cross sectional view showing multiple surface exposed and buried cavities in close KirBac3.1 (PDB-1XL4). The TM1 (purple) and TM2 (green) denote transmembrane helix-1 (Outer), and transmembrane helix-2 (Inner) respectively. Pore helix and side helix are colored as blue and red respectively. (B) Solvent accessibility changes from the closed to the open conformation in KirBac3.1 are visualized on the structure of closed KirBac3.1, where the subunits are represented by different colors. The modified residues are shown by sticks. Color coding indicates the changes in the modification rates or solvent accessibilities upon transition from the closed to the open state. Residues that undergo increased interactions with water due to changes in the structure of the channel in the open state show dramatic increase in labeling efficiency. The results support the proposed existence of three potential gates within the channel. Results from Gupta et al. Structure. 2010 and reproduced in part with the permission from Gupta et al. J Sync. Rad. 2016.
5.4. XFMS in the Study of Ion Transporters
The unique capabilities of XFMS to label side chains inside TM domains close to structural water was used to investigate the mechanism of a prototypical proton-coupled Zn2+ transporter, YiiP, from the cytoplasmic membrane of E. coli. [26]. This transporter transports Zn2+ against the concentration gradient with the exchange of protons [59]. The crystal structure of Zn-YiiP is available and showed the absence of any polar residues that could carry protons to the Zn binding or transport site, and showed the presence of a hydrophobic barrier that divides the transport pathway between the intra- and extra-cellular cavity [60, 61]. A comparative XFMS analysis of Zn2+-YiiP and Apo-YiiP identified specific and reciprocal solvent accessibility changes in the residues adjacent to these cavities and within the hydrophobic barrier (Figure 5). At the extracellular side, Zn2+ binding residue D49 (TM2, at the active site) and L152 (TM5, at the hydrophobic gate) showed a significant decrease in water accessibility upon zinc binding which is consistent with the available crystal structure of Zn-YiiP. Residue M197, which is at the entrance of the intracellular surface near L152, also showed a decrease in water accessibility. L152 and M197 interact with residues from TM3 and TM6, and this cluster of residues forms a highly conserved TM5→TM3-TM6 packing core that stabilizes the TM helical arrangement and hydrophilic barrier. Solvent accessibility data show that Zn2+ access to the transport-site shuts off water access to L152, suggesting that L152 on TM5 may function as an inter-cavity gate that controls alternating access of zinc ions and water molecules to the transport-site. In contrast, increased water accessibility was observed in several methionine residues located on the TM5 but facing opposite to D49, L152, M197. This reciprocal change in water accessibility on two opposite TM5 faces is consistent with a TM5 re-orientation in response to zinc binding. This result suggested that the transport-site in apo-YiiP was constitutively open to the intracellular cavity, in agreement with a low-resolution cryo-EM structure of an apo-YiiP homolog. Millisecond time-resolved XFMS was also used to monitor the time course of closing of the inter-cavity water pathway in response to rapid zinc binding to the detergent solubilized YiiP. The analysis showed the reciprocal pattern of steady-state responses for residues on opposite faces of TM5; the exponential rise in the accessibility of M197 and D49 mirrored the exponential decrease of the M151 and M159/M160 accessibilities at an average rate of 1.8 sec−1. The rates of water accessibility changes for these four positions along TM5 were identical within experimental errors, suggesting that TM5 underwent a rigid-body re-orientation upon zinc binding. XF-MS data suggested that the zinc-binding energy in the hydrophobic core at the intra-cellular interface might get transformed into mechanical energy to reorient TM5 and close the L152 gate. This conformational change can alternatively expose the transport site to the intracellular and extracellular cavity. The difference in pH at the extra- and intra-cellular cavities can modulate the protonation state of His153, a critical residue for Zn2+ binding in the transport site, which in turn can regulate Zn2+ binding. XFMS thus provide a valuable understanding of structural and dynamic elements of the coupling mechanism by which the proton gradient is utilized to pump substrates against their concentration gradient the transporters.
Figure 5. XF-MS probes proton-coupled Zn2+ transfer mechanism in Zn transporter YiiP.
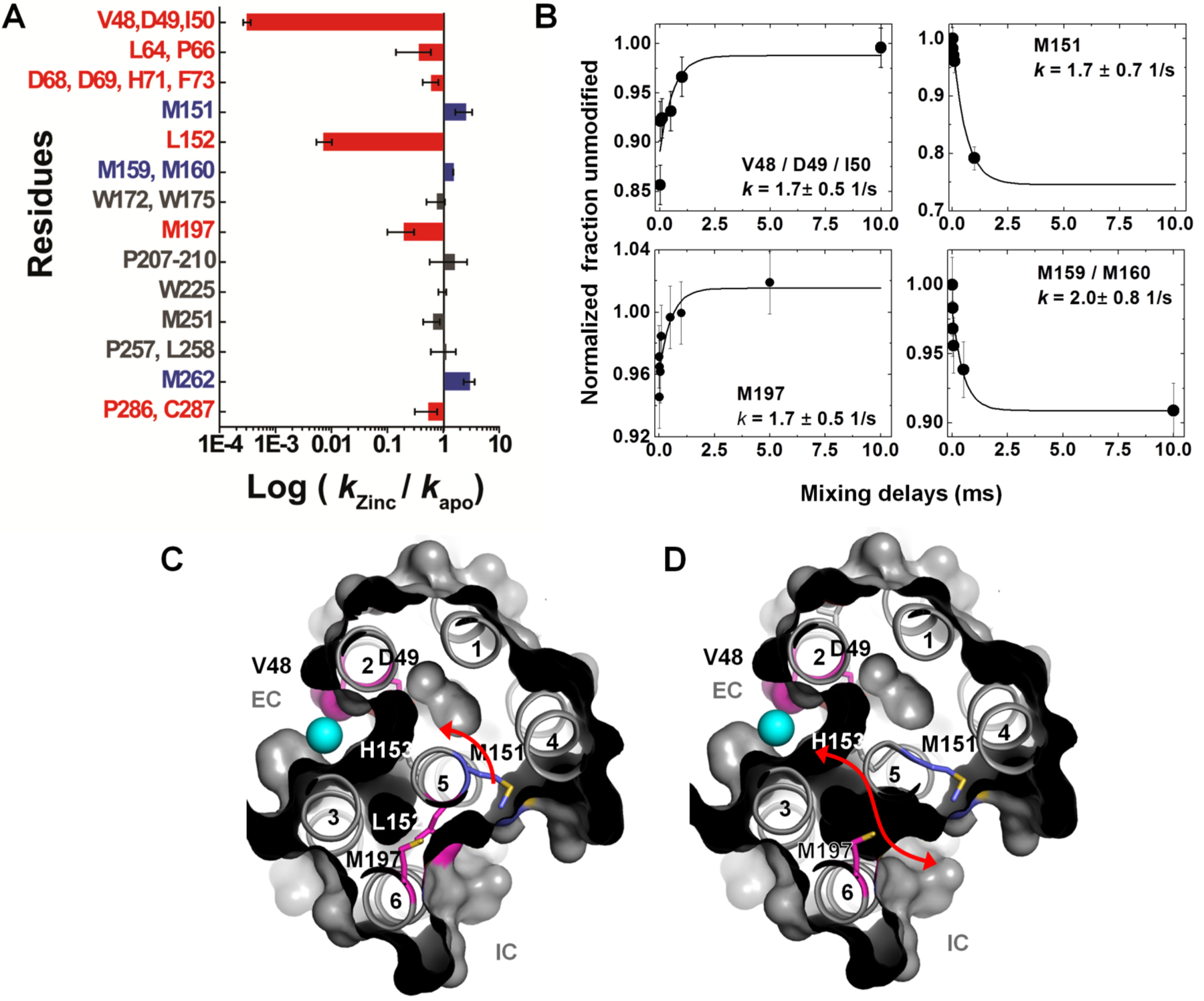
(A) Bar plot showing radiometric water accessibility changes in response to Zn2+ binding measured by ratio (R) of •OH labeling rates for residues with increase (blue), decrease (red) and no change (grey) in accessibility after rapid Zn2+ exposure. (B) Time courses of water accessibility changes for the indicated residues. Solid lines represent single exponential fits, which provide rate constants of conformational changes associated with Zn2+ binding and translocation. (C) X-ray footprinting reveals a hydrophobic gate at residue L152, which controls the opening of the inner cavity water pathway for zinc-proton exchange in the YiiP transporter. The cross-sectional view shows the position of TM helices, which separate two cavities at the intra-cellular (IC) and extra-cellular (EC) sides. Residues are color coded as in (A). XF-MS results suggest the protein conformational change alternates the membrane-facing on–off mode of zinc coordination (in D49) and protonation–deprotonation (H153) of the transport site in a coordinated fashion. (D) Red arrow indicates the proposed water pathway, which connects EC with IC after excluding the residue L152 from the surface drawing of the TM helices. Results from Gupta et al. Nature. 2014 and reproduced in part with the permission from Gupta et al. J Sync. Rad. 2016.
CONCLUSION
Synchrotron-based XFMS has been used to elucidate structural changes within several membrane proteins, including G-protein coupled receptors [14, 20, 47], potassium channels [43], and ion transporters [26] associated with their activated or functional states. Availability of high-resolution X-ray crystal structures of membrane proteins in their ground state has validated the ability of the XFMS approach to determine plausible roles of transmembrane structural waters in the regulation of their functional states. Moreover, recent advancements in experimental strategies with high flux-density X-ray beams at synchrotron facilities [30] together with significant improvement in mass spectrometry-based data analysis methodologies [62, 63], serve as a promising platform for the development of a unique, integrative structural method universally suited to study any membrane protein system. XFMS is an ideal complement to both high- and low-resolution structural studies, e.g. allowing the accurate placing of components within low-to-medium resolution maps from cryo-EM or solution scattering studies. The method is also highly complementary with HDX-MS for comparing backbone stability data obtained by HDX-MS with that of the side chain solvent accessibility by XFMS. The advantages of XFMS, which can be used for various solutions conditions, is that it requires very low protein sample concentrations, yields residue-specific information, detects locations of structural or bound water, and provides both structural and protein-dynamics data with a high degree of temporal resolution, and this makes XFMS one of the most important tools in solving structural problems related membrane proteins.
ACKNOWLEDGEMENTS
The authors thank Dr. Vass Bavily and Dr. Dax Fu for helpful discussions regarding the application of XFMS to membrane proteins.
CONFLICT OF INTEREST
SG and CR are supported by NIH-NIGMS R01 GM126218.
Footnotes
CONSENT FOR PUBLICATION
Not applicable.
REFERENCES
- [1].Carpenter EP; Beis K; Cameron AD; Iwata S Overcoming the challenges of membrane protein crystallography. Curr. Opin. Struct. Biol, 2008, 18(5), 581–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hong M; Zhang Y; Hu F Membrane protein structure and dynamics from NMR spectroscopy. Annu. Rev. Phys. Chem, 2012, 63, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ubarretxena-Belandia I; Stokes DL Present and future of membrane protein structure determination by electron crystallography. Adv. Protein Chem. Struct. Biol, 2010, 81, 33–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Goldie KN; Abeyrathne P; Kebbel F; Chami M; Ringler P; Stahlberg H Cryo-electron microscopy of membrane proteins. Methods Mol. Biol, 2014, 1117, 325–341. [DOI] [PubMed] [Google Scholar]
- [5].Bartesaghi A; Subramaniam S Membrane protein structure determination using cryo-electron tomography and 3D image averaging. Curr. Opin. Struct. Biol, 2009, 19(4), 402–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Schmidt-Krey I Electron crystallography of membrane proteins: Two-dimensional crystallization and screening by electron microscopy. Methods, 2007, 41(4), 417–426. [DOI] [PubMed] [Google Scholar]
- [7].Breyton C; Gabel F; Lethier M; Flayhan A; Durand G; Jault JM; Juillan-Binard C; Imbert L; Moulin M; Ravaud S; Härtlein M; Ebel C Small angle neutron scattering for the study of solubilised membrane proteins. Eur. Phys. J.E. Soft Matter, 2013, 36(7), 71. [DOI] [PubMed] [Google Scholar]
- [8].Jeschke G DEER distance measurements on proteins. Annu. Rev. Phys. Chem, 2012, 63, 419–446. [DOI] [PubMed] [Google Scholar]
- [9].Gupta S; Feng J; Chan LJ; Petzold CJ; Ralston CY Synchrotron X-ray footprinting as a method to visualize water in proteins. J. Synchrotron. Radiat, 2016, 23(Pt 5), 1056–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Xu G; Chance MR Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem. Rev, 2007, 107(8), 3514–343. [DOI] [PubMed] [Google Scholar]
- [11].Gilmore JM; Washburn MP Advances in shotgun proteomics and the analysis of membrane proteomes. J. Proteomics, 2010, 73(11), 2078–2091. [DOI] [PubMed] [Google Scholar]
- [12].Savas JN; Stein BD; Wu CC; Yates JR 3rd. Mass spectrometry accelerates membrane protein analysis. Trends Biochem. Sci, 2011, 36(7), 388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Schey KL; Grey AC; Nicklay JJ Mass spectrometry of membrane proteins: A focus on aquaporins. Biochemistry, 2013, 52(22), 3807–3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Angel TE; Gupta S; Jastrzebska B; Palczewski K; Chance MR Structural waters define a functional channel mediating activation of the GPCR, rhodopsin. Proc. Natl. Acad. Sci. USA, 2009, 106(34), 14367–14372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lu Y; Zhang H; Niedzwiedzki DM; Jiang J; Blankenship RE; Gross ML Fast photochemical oxidation of proteins maps the topology of intrinsic membrane proteins: Light-harvesting complex 2 in a nanodisc. Anal. Chem, 2016, 88(17), 8827–8834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pirrone GF; Iacob RE; Engen JR Applications of hydrogen/deuterium exchange MS from 2012 to 2014. Anal. Chem, 2015, 87(1), 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hebling CM; Morgan CR; Stafford DW; Jorgenson JW; Rand KD; Engen JR Conformational analysis of membrane proteins in phospholipid bilayer nanodiscs by hydrogen exchange mass spectrometry. Anal. Chem, 2010, 82(13), 5415–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Weerasekera R; Schmitt-Ulms G Crosslinking strategies for the study of membrane protein complexes and protein interaction interfaces. Biotechnol. Genet. Eng. Rev, 2006, 23, 41–62. [DOI] [PubMed] [Google Scholar]
- [19].Pan J; Borchers CH Top-down mass spectrometry and hydrogen/deuterium exchange for comprehensive structural characterization of interferons: Implications for biosimilars. Proteomics, 2014, 14(10), 1249–1258. [DOI] [PubMed] [Google Scholar]
- [20].Orban T; Jastrzebska B; Gupta S; Wang B; Miyagi M; Chance MR; Palczewski K Conformational dynamics of activation for the pentameric complex of dimeric G protein-coupled receptor and heterotrimeric G protein. Structure, 2012, 20(5), 826–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pan Y; Piyadasa H; O’Neil JD; Konermann L Conformational dynamics of a membrane transport protein probed by H/D exchange and covalent labeling: The glycerol facilitator. J. Mol. Biol, 2012, 416(3), 400–413. [DOI] [PubMed] [Google Scholar]
- [22].Debelyy MO; Waridel P; Quadroni M; Schneiter R; Conzelmann A Chemical crosslinking and mass spectrometry to elucidate the topology of integral membrane proteins. PloS One, 2017, 12(10), e0186840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fischer L; Chen ZA; Rappsilber J Quantitative cross-linking/mass spectrometry using isotope-labelled cross-linkers. J. Proteom, 2013, 88, 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wecksler AT; Kalo MS; Deperalta G Mapping of Fab-1:VEGF interface using carboxyl group footprinting mass spectrometry. J. Am. Soc. Mass. Spectrom, 2015, 26(12), 2077–2080. [DOI] [PubMed] [Google Scholar]
- [25].Gupta S; Bavro VN; D’Mello R; Tucker SJ; Vénien-Bryan C; Chance MR Conformational changes during the gating of a potassium channel revealed by structural mass spectrometry. Structure, 2010, 18(7), 839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gupta S; Chai J; Cheng J; D’Mello R; Chance MR; Fu D Visualizing the kinetic power stroke that drives proton-coupled zinc (II) transport. Nature, 2014, 512(7512), 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Marcoux J; Wang SC; Politis A; Reading E; Ma J; Biggin PC; Zhou M; Tao H; Zhang Q; Chang G; Morgner N Mass spectrometry reveals synergistic effects of nucleotides, lipids, and drugs binding to a multidrug resistance efflux pump. Proc. Nat. Acad. Sci. USA, 2013, 110(24), 9704–9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Laganowsky A; Reading E; Hopper JT; Robinson CV Mass spectrometry of intact membrane protein complexes. Nat. Protoc, 2013, 8(4), 639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Morgner N; Kleinschroth T; Barth HD; Ludwig B; Brutschy B A novel approach to analyze membrane proteins by laser mass spectrometry: From protein subunits to the integral complex. J. Am. Soc. Mass Spectrom, 2007, 18(8), 1429–1438. [DOI] [PubMed] [Google Scholar]
- [30].Gupta S; Celestre R; Petzold CJ; Chance MR; Ralston C Development of a microsecond X-ray protein footprinting facility at the Advanced Light Source. J. Synchrotron Radiat, 2014, 21(4), 690–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Takamoto K; Chance MR Radiolytic protein footprinting with mass spectrometry to probe the structure of macromolecular complexes. Annu. Rev. Biophys. Biomol. Struct, 2006, 35, 251–276. [DOI] [PubMed] [Google Scholar]
- [32].Kamal JK; Chance MR Modeling of protein binary complexes using structural mass spectrometry data. Protein Sci, 2008, 17(1), 79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Guan JQ; Almo SC; Chance MR Synchrotron radiolysis and mass spectrometry: A new approach to research on the actin cytoskeleton. Acc. Chem. Res, 2004, 37(4), p. 221–9. [DOI] [PubMed] [Google Scholar]
- [34].Kiselar JG; Mahaffy R; Pollard TD; Almo SC; Chance MR Visualizing Arp2/3 complex activation mediated by binding of ATP and WASp using structural mass spectrometry. Proc. Natl. Acad. Sci. USA, 2007, 104(5), 1552–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gupta S; Sullivan M; Toomey J; Kiselar J; Chance MR The Beamline X28C of the center for synchrotron biosciences: A national resource for biomolecular structure and dynamics experiments using synchrotron footprinting. J. Synchrotron. Radiat, 2007, 14(Pt 3), 233–243. [DOI] [PubMed] [Google Scholar]
- [36].Sullivan MR; Rekhi S; Bohon J; Gupta S; Abel D; Toomey J; Chance MR Installation and testing of a focusing mirror at beamline X28C for high flux X-ray radiolysis of biological macromolecules. Rev. Sci. Instrum, 2008, 79(2 Pt 1), 025101. [DOI] [PubMed] [Google Scholar]
- [37].Bohon J; Jennings LD; Phillips CM; Licht S; Chance MR Synchrotron protein footprinting supports substrate translocation by ClpA via ATP-induced movements of the D2 loop. Structure, 2008, 16(8), 1157–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tullius TD; Dombroski BA Iron(II) EDTA used to measure the helical twist along any DNA molecule. Science, 1985, 230(4726), 679–681. [DOI] [PubMed] [Google Scholar]
- [39].Hambly DM; Gross ML Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J. Am. Soc. Mass Spectrom, 2005, 16(12), 2057–2063. [DOI] [PubMed] [Google Scholar]
- [40].Maleknia SD; Downard KM On-plate deposition of oxidized proteins to facilitate protein footprinting studies by radical probe mass spectrometry. Rapid Commun. Mass Spectrom, 2012, 26(19), 2311–2318. [DOI] [PubMed] [Google Scholar]
- [41].Maleknia SD; Chance MR; Downard KM Electrospray-assisted modification of proteins: A radical probe of protein structure. Rapid Commun. Mass Spectrom, 1999, 13(23), 2352–2358. [DOI] [PubMed] [Google Scholar]
- [42].Gupta S; D’Mello R; Chance MR Structure and dynamics of protein waters revealed by radiolysis and mass spectrometry. Proc. Natl. Acad. Sci. USA, 2012, 109(37), 14882–14887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Gupta S; Bavro VN; D’Mello R; Tucker SJ; Vénien-Bryan C; Chance MR Conformational changes during the gating of a potassium channel revealed by structural mass spectrometry. Structure, 2010, 18(7), 839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ball P Water as an active constituent in cell biology. Chem. Rev, 2008, 108(1), 74–108. [DOI] [PubMed] [Google Scholar]
- [45].Renthal R Buried water molecules in helical transmembrane proteins. Protein Sci, 2008, 17(2), 293–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Angel TE; Chance MR; Palczewski K Conserved waters mediate structural and functional activation of family A (rhodopsin-like) G protein-coupled receptors. Proc. Natl. Acad. Sci. USA, 2009, 106(21), 8555–8560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Padayatti PS; Wang L; Gupta S; Orban T; Sun W; Salom D; Jordan SR; Palczewski K; Chance MR A hybrid structural approach to analyze ligand binding by the serotonin type 4 receptor (5-HT4). Mol. Cell Proteomics, 2013, 12(5), 1259–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gustavsson M; Wang L; van Gils N; Stephens BS; Zhang P.; Schall TJ; Yang S; Abagyan R; Chance MR; Kufareva I; Handel TM Structural basis of ligand interaction with atypical chemokine receptor 3. Nat. Commun, 2017, 8, 14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Aryal P; Sansom MS; Tucker SJ Hydrophobic gating in ion channels. J. Mol. Biol, 2015, 427(1), 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Swartz KJ Towards a structural view of gating in potassium channels. Nat. Rev. Neurosci, 2004, 5(12), 905–916. [DOI] [PubMed] [Google Scholar]
- [51].Tao X; Avalos JL; Chen J; MacKinnon R Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 A resolution. Science, 2009, 326(5960), 1668–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Uysal S; Vásquez V; Tereshko V; Esaki K; Fellouse FA; Sidhu SS; Koide S; Perozo E; Kossiakoff A Crystal structure of full-length KcsA in its closed conformation. Proc. Natl. Acad. Sci. USA, 2009, 106(16), 6644–6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ostmeyer J; Chakrapani S; Pan AC; Perozo E; Roux B Recovery from slow inactivation in K+ channels is controlled by water molecules. Nature, 2013, 501(7465), 121–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Bavro VN; De Zorzi R; Schmidt MR; Muniz JR; Zubcevic L; Sansom MS; Vénien-Bryan C; Tucker SJ Structure of a KirBac potassium channel with an open bundle crossing indicates a mechanism of channel gating. Nat. Struct. Mol. Biol, 2012, 19(2), 158–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zimmerberg J; Parsegian VA Polymer inaccessible volume changes during opening and closing of a voltage-dependent ionic channel. Nature, 1986, 323(6083), 36–39. [DOI] [PubMed] [Google Scholar]
- [56].Thompson AN; Posson DJ; Parsa PV; Nimigean CM Molecular mechanism of pH sensing in KcsA potassium channels. Proc. Natl. Acad. Sci. USA, 2008, 105(19), 6900–6905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Uysal S; Cuello LG; Cortes DM; Koide S; Kossiakoff AA; Perozo E Mechanism of activation gating in the full-length KcsA K+ channel. Proc. Natl. Acad. Sci. USA, 2011, 108(29), 11896–11899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Raghuraman H; Islam SM; Mukherjee S; Roux B; Perozo E Dynamics transitions at the outer vestibule of the KcsA potassium channel during gating. Proc. Natl. Acad. Sci. USA, 2014, 111(5), 1831–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Chao Y; Fu D Kinetic study of the antiport mechanism of an Escherichia coli zinc transporter, ZitB. J Biol. Chem, 2004, 279(13), 12043–12050. [DOI] [PubMed] [Google Scholar]
- [60].Lu M; Fu D Structure of the zinc transporter YiiP. Science, 2007, 317(5845), 1746–1748. [DOI] [PubMed] [Google Scholar]
- [61].Lu M; Chai J; Fu D Structural basis for autoregulation of the zinc transporter YiiP. Nat. Struct. Mol. Biol, 2009, 16(10), 1063–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kaur P; Kiselar J; Yang S; Chance MR Quantitative protein topography analysis and high-resolution structure prediction using hydroxyl radical labeling and tandem-ion mass spectrometry (MS). Mol. Cell. Proteomics, 2015, 14(4), 1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kaur P; Kiselar JG; Chance MR Integrated algorithms for high-throughput examination of covalently labeled biomolecules by structural mass spectrometry. Anal. Chem, 2009, 81(19), 8141–8149. [DOI] [PMC free article] [PubMed] [Google Scholar]