

Abstract
Inactivation of the TP53 tumor suppressor gene is essential during cancer development and progression. Mutations of TP53 are often missense and occur in various human cancers. In some fraction of wild‐type (wt) TP53 tumors, p53 is inactivated by upregulated murine double minute homolog 2 (MDM2) and MDM4. We previously reported that simultaneous knockdown of MDM4 and MDM2 using synthetic DNA‐modified siRNAs revived p53 activity and synergistically inhibited in vitro cell growth in cancer cells with wt TP53 and high MDM4 expression (wtTP53/highMDM4). In the present study, MDM4/MDM2 double knockdown with the siRNAs enhanced 5‐fluorouracil (5‐FU)‐induced p53 activation, arrested the cell cycle at G1 phase, and potentiated the antitumor effect of 5‐FU in wtTP53/highMDM4 human colon (HCT116 and LoVo) and gastric (SNU‐1 and NUGC‐4) cancer cells. Exposure to 5‐FU alone induced MDM2 as well as p21 and PUMA by p53 activation. As p53‐MDM2 forms a negative feedback loop, enhancement of the antitumor effect of 5‐FU by MDM4/MDM2 double knockdown could be attributed to blocking of the feedback mechanism in addition to direct suppression of these p53 antagonists. Intratumor injection of the MDM4/MDM2 siRNAs suppressed in vivo tumor growth and boosted the antitumor effect of 5‐FU in an athymic mouse xenograft model using HCT116 cells. These results suggest that a combination of MDM4/MDM2 knockdown and conventional cytotoxic drugs could be a promising treatment strategy for wtTP53/highMDM4 gastrointestinal cancers.
Keywords: 5‐fluorouracil, colon cancer, gastric cancer, MDM4, p53
1. INTRODUCTION
5‐Fluorouracil (5‐FU) is a key drug in the treatment of colon and gastric cancers. Various combination therapies have been developed and improved survival. However, survival duration remains short, and cure with chemotherapy is rarely expected.1, 2, 3, 4, 5 To circumvent such problems, new therapeutic strategies are needed.
In human cancers, the tumor protein (TP)53 tumor suppressor gene is often inactivated by missense mutation, or its function is suppressed by enhanced expression of oncogenes such as murine double minute 2 (MDM2) and MDM4.6, 7, 8 MDM2 and its complex with MDM4 destabilizes p53 through binding and ubiquitin‐dependent protein degradation.9, 10 MDM4 can also repress p53 transcriptional activity by directly binding to the transactivating domain.11, 12
Studies have shown that reactivation of wild‐type (wt) TP53 by inhibiting MDM2‐p53 interaction or knockdown of MDM2 and MDM4 induces cell cycle arrest and apoptotic cell death, inhibiting tumor growth in tumors carrying wtTP53.6, 13, 14, 15, 16, 17, 18, 19 Thus, MDM2 and MDM4 are ideal targets for cancer therapy in such tumors. Various kinds of small molecular compounds and peptides inhibiting MDM2 function have been developed.6, 18, 20, 21 Among them, idasanutlin has been shown to be an effective treatment in some clinical studies of patients with malignant lymphomas and acute myeloblastic leukemias.22, 23, 24
A previous study reported that cultured tumor cells with wtTP53 can be divided into 2 types: high MDM2 expressers and high MDM4 expressers.16 The former expresses a high level of MDM2 and a very low level of MDM4, whereas the latter expresses a high level of MDM4 and an intermediate level of MDM2. Knockdown of either MDM4 or MDM2 alone using synthetic siRNAs with DNA‐substituted seed arms (chiMDM4, chiMDM2) specifically suppressed the growth of high MDM4 expresser cancer cells, whereas only MDM2 knockdown but not MDM4 knockdown suppressed that of high MDM2 expresser cancer cells. Simultaneous knockdown of MDM4 and MDM2 synergistically inhibited the growth of high MDM4 expresser cancer cells.
Overexpression or amplification of MDM4 has been found in 19%‐49% and 43% of colon and gastric cancers, respectively, whereas those of MDM2 have been reported in 17.3% and 32.7%‐41.8% of colon and gastric cancers, respectively.25, 26, 27, 28, 29 Therefore, reactivation of wtTP53 by chiMDM4 and chiMDM2 could be used for the treatment of these cancers. In the present study, the effects of double knockdown of MDM4 and MDM2 using chiMDM4 and chiMDM2 on the antitumor activity of 5‐FU in colon and gastric cancer cells with wtTP53 and high MDM4 (wtTP53/highMDM4) were investigated. In vivo antitumor activity of chiMDM4 plus chiMDM2 (chiMDM4/chiMDM2) and a combination of chiMDM4/chiMDM2 with 5‐FU were also explored.
2. MATERIALS AND METHODS
2.1. Cell culture
Four tumor cell lines with wtTP53 were used: HCT116 colon cancer, LoVo colon cancer, SNU‐1 gastric cancer, and NUGC‐4 gastric cancer. The HCT116 cell line was purchased from Horizon Discovery (Cambridge, UK). LoVo and SNU‐1 cell lines were purchased from ATCC (Rockville, MD, USA). The NUGC‐4 cell line was obtained from the Riken BioResource Center Cell Bank (Tsukuba, Japan). HCT116, SNU‐1, and NUGC‐4 cells were cultured in RPMI‐1640 medium (Sigma‐Aldrich, St. Louis, MO, USA) supplemented with 10% FBS (Nichirei Biosciences, Tokyo, Japan). LoVo cells were cultured in Ham's F‐12 nutrient mixture medium (Sigma‐Aldrich) with 10% FBS. 5‐Fluorouracil was purchased from Kyowa Hakko Kirin (Tokyo, Japan). Nutlin‐3 was purchased from Calbiochem (San Diego, CA, USA).
2.2. Small interfering RNAs and transfection
Sequences of DNA‐modified siRNAs used in this study were: chimera Control (chiControl, chiCtrl) sense strand, 5′‐GUACCGCACGUCAttcgtatc‐3′; chiCtrl antisense strand, 5′‐tacgaaUGACGUGCGGUACGU‐3′; chiMDM2 sense strand, 5′‐CAGCCAUCAACUUctagtagc‐3′; chiMDM2 antisense strand, 5′‐tactagAAGUUGAUGGCUGAG‐3′; chiMDM4 sense strand, 5′‐CCCUCUCUAUGAUatgctaag‐3′; chiMDM4 antisense strand, 5′‐tagcatAUCAUAGAGAGGGCU‐3′; chiCtrl (in vivo) sense strand, 5′‐gtaGUACCGCACGUCAttctc‐3′; and chiCtrl (in vivo) antisense strand, 5′‐gaaUGACGUGCGGUACtacGU‐3′ (capital letters, ribonucleotides; small letters, deoxynucleotides). The control DNA‐modified siRNA was designed to have the least homology to human and mouse genes. For the in vitro experiments, DNA‐modified siRNAs were synthesized, cartridge‐purified, and annealed (Sigma‐Aldrich). For the in vivo experiments, DNA‐modified siRNAs were synthesized, annealed, and purified using HPLC (ST Pharm., Seoul, Korea). The siRNA transfection in vitro experiment was carried out using Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, CA, USA) as reported previously,30 except for SNU‐1 cells. Because Lipofectamine RNAiMAX was toxic to SNU‐1 cells, the cells were exposed to siRNA‐Lipofectamine RNAiMAX complex for 4 hours, then centrifuged, resuspended in a complete medium, and cultivated. The siRNA transfection in vivo experiment was undertaken using AteloGene Local Use (Koken, Tokyo, Japan).
2.3. Cell viability
Water‐soluble tetrazolium salt (WST‐8) colorimetric assays were carried out using a CCK‐8 (Dojin Laboratories, Kumamoto, Japan) according to the manufacturer's protocol. Because the maximum knockdown effects of siRNAs were usually observed 2‐3 days after transfection, cells were incubated for 5 days after transfection with siRNAs (4 days after treatment with 5‐FU), which was longer than the period described in the manufacturer's protocol (1‐3 days), then analyzed using an iMark microplate reader (Bio‐Rad, Hercules, CA, USA). The absorbance of the plates was read at wavelengths of 450 and 620 nm.
2.4. Combination index
Quantification of the mixture of chiMDM4/chiMDM2 and 5‐FU synergy was determined by the Chou‐Talalay method for drug combination using CalcuSyn software (Biosoft, Cambridge, UK).31
2.5. Immunoblot analysis
Both SDS‐PAGE and immunoblot analysis were carried out as previously described.16 The primary and secondary Abs used in this study were: mouse mAb against MDM2 (2A10) (Abcam, Cambridge, UK); goat polyclonal Ab against MDMX (D‐19) (Santa Cruz Biotechnology, Dallas, TX, USA); anti‐TP53 mouse mAb (BP53‐12) (Cell Sciences, Canton, MA, USA); mouse mAbs against p21Wafl/Cip1 (DCS60), and rabbit polyclonal Ab against p53 upregulated modulator of apoptosis (PUMA) (Cell Signaling Technology, Danvers, MA, USA); and rabbit polyclonal Ab against β‐actin (Medical & Biological Laboratories, Nagoya, Japan). Both HRP‐conjugated sheep anti‐mouse IgG and donkey anti‐rabbit IgG sera were purchased from GE Healthcare (Chicago, IL, USA). The HRP‐conjugated rabbit anti‐goat IgG was purchased from Sigma‐Aldrich. Chemiluminescent detection was carried out using ECL Select Western Blotting Detection Reagent (GE Healthcare) and the Ez‐Capture Imaging System (Atto, Tokyo, Japan).
2.6. Quantitative RT‐PCR
RNA samples were extracted from cell lysate using 40 μL RealTime Ready Cell Lysis reagent (Roche Diagnostics, Mannheim, Germany) per well of a 96‐well culture plate, according to the manufacturer's instructions. Complementary DNA was synthesized using 2 μL RNA and 8 μL Transcriptor Universal cDNA Master (Roche Diagnostics) in 20‐μL reactions. Quantitative RT‐PCR assays were carried out using the Applied Biosystems 7500 Fast Real‐Time PCR system (Applied Biosystems, Foster City, CA, USA) in 96‐well plates. Primer and TaqMan probe for CDKN1A (p21Cip1) and ACTB (β‐actin) were obtained from Applied Biosystems (Assay ID: Hs00355782_m1 and Hs99999903_m1, respectively). Reactions were carried out in triplicate under standard thermocycling conditions in a 20 μL volume containing 5 μL cDNA, 900 nmol/L primers, 250 nmol/L probe, and 10 μL TaqMan Gene Expression Master Mix (Applied Biosystems), according to the manufacturer's protocol. The amount of target mRNA was examined and normalized to that of β‐actin.
2.7. Cell cycle assay
Cells were seeded into 60‐mm dishes at 1 × 105/dish. After overnight cultivation, cells were transfected with DNA‐modified siRNAs (0.5‐2 nmol/L) for 24 hours, then cultured in the presence of 5‐FU (4 μmol/L). After 2 days of cultivation, cells were gently lifted with Accutase (US Biotechnologies, Parker Ford, PA, USA) at room temperature for 10 minutes. The cells were then washed once with PBS and stained with a Cycletest Plus DNA reagent kit (BD Biosciences, Franklin Lakes, NJ, USA), according to the manufacturer's protocol. Flow cytometry was carried out using a FACSCalibur flow cytometer and CellQuest software (both BD Biosciences). The percentage of cells in different cell cycle phases was calculated using ModFit LT software (Verity Software House, Topsham, ME, USA).
2.8. In vivo antitumor effect of 5‐FU plus knockdown of MDM4 and MDM2
All animal experiments were undertaken according to procedures approved by the Institutional Animal Care and Use Committee of the University of Tsukuba (Tsukuba, Japan). Female BALB/c nude mice (5 weeks old) were obtained from Charles River Japan (Kanagawa, Japan) and maintained under specific pathogen‐free conditions in a temperature and humidity‐controlled environment. HCT116 cells were suspended in saline solution (Otsuka Pharmaceutical, Tokyo, Japan) at a concentration of 5 × 104/μL. One hundred microliters of the adjusted cell suspension of HCT116 was s.c. injected into the right flank of mice under anesthesia. Ten days after inoculation, the s.c. xenografted tumors grew to approximately 50 mm3 in size. The mice were randomly assigned to 4 groups (n = 5 per group) as follows: chiCtrl alone (1 mg/kg), mixture of chiMDM4 and chiMDM2 (0.5 mg/kg, each), chiCtrl (1 mg/kg) plus 5‐FU (30 mg/kg), and a mixture of chiMDM4 and chiMDM2 (0.5 mg/kg, each) plus 5‐FU (30 mg/kg). DNA‐modified siRNA was directly injected into tumor once a week (days 0, 7, and 14) using AteloGene as described in the manufacturer's instruction. 5‐FU was i.p. injected 3 times a week for 3 weeks (days 1, 4, 6, 8, 11, 13, 15, 18, and 20). Tumor volume was measured with a caliper 3 times a week and calculated using a formula of V = (length × width2)/2. To monitor health, the mice were weighed 3 times a week, and their general physical status was recorded daily. Experiments were terminated before the largest size of tumor reached 2000 mm3.
2.9. Statistical analysis
Statistical significance of differences between various groups was evaluated using Dunnett's or Tukey's test (in vitro assay). A repeated‐measures ANOVA was used to evaluate the in vivo antitumor effects of the drugs. A difference between the experimental groups was considered statistically significant at a P‐value of <.05. All statistical analyses were undertaken using SPSS version 25.0 (SPSS, Chicago, IL, USA).
3. RESULTS
3.1. Cell growth inhibition of MDM4/MDM2 double knockdown and 5‐FU in wt TP53 colon and gastric cancer cells
To test if double knockdown of MDM4 and MDM2 could enhance the antitumor activity of 5‐FU in colon and gastric cancers with wtTP53/highMDM4, the effect of chiMDM4/chiMDM2 on the growth inhibitory activity of 5‐FU was examined by WST‐8 assay using 2 colon cancer (HCT116 and LoVo) and 2 gastric cancer (SNU‐1 and NUGC‐4) cell lines. As shown in Figure 1A, a mixture of chiMDM4 and chiMDM2 in equimolar amounts and 5‐FU alone suppressed the growth of HCT116 cells in a dose‐dependent manner as compared with control DNA‐modified siRNA (chiCtrl). Combination of chiMDM4/chiMDM2 and 5‐FU suppressed the growth more than each alone. Similar enhancement of 5‐FU‐mediated growth suppression by chiMDM4/chiMDM2 was observed in LoVo (Figure 1B), SNU‐1 (Figure 1C), and NUGC‐4 cells (Figure 1D).
Figure 1.
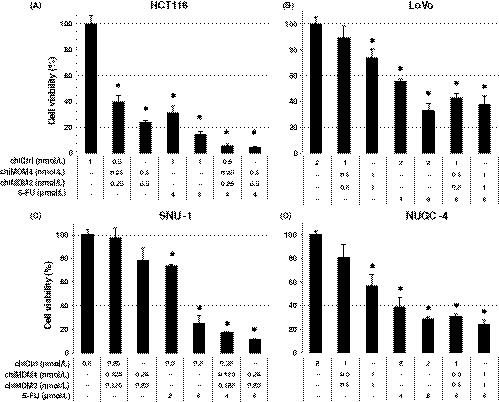
Effects of double knockdown of MDM4 and MDM2 and 5‐fluorouracil (5‐FU) on the growth of colon (HCT116 and LoVo) and gastric cancer cell lines (SNU‐1 and NUGC‐4) with wtTP53/high MDM4. HCT116 (A), LoVo (B), SNU‐1 (C), and NUGC‐4 cells (D) were transfected with either DNA‐modified control siRNA (chiCtrl) or mixture of DNA‐modified siRNA targeting MDM4 (chiMDM4) and MDM2 (chiMDM2). After 4‐16 hours of incubation, cells were exposed to 5‐FU at the indicated concentrations. Five days after transfection, cell viability was determined using the WST‐8 assay. Cell viability relative to those transfected with chiCtrl are shown (mean ± SD; n = 3) . *P < .05, compared with the chiCtrl
Combination index (CI) values were calculated, and they are summarized in Table 1. The CI value of HCT116 cells was lowest (<0.3), followed by NUGC‐4 cells (0.83, 0.77), LoVo cells (0.90, 0.97), and SNU‐1 cells (0.95, 0.97), showing that MDM4/MDM2 double knockdown enhanced the antitumor activity of 5‐FU synergistically in HCT116 cells and NUGC‐4 cells and additively in SNU‐1 cells and LoVo cells.
Table 1.
Combination index of mixture of chiMDM4/chiMDM2 and 5‐fluorouracil (5‐FU)
| Cell line | chiMDM4 (nmol/L) | chiMDM2 (nmol/L) | 5‐FU (4 μmol/L) | Combination index |
|---|---|---|---|---|
| HCT116 | 0.250 | 0.250 | + | 0.28 |
| 0.500 | 0.500 | + | 0.29 | |
| LoVo | 0.500 | 0.500 | + | 0.90 |
| 1.000 | 1.000 | + | 0.97 | |
| SNU‐1 | 0.125 | 0.125 | + | 0.95 |
| 0.250 | 0.250 | + | 0.97 | |
| NUGC‐4 | 0.500 | 0.500 | + | 0.83 |
| 1.000 | 1.000 | + | 0.77 |
We tested whether nutlin‐3, an inhibitor of MDM2‐p53 interaction, could serve as a substitute for chiMDM2 to enhance the antitumor effect of chiMDM4 in 4 cell lines (NUGC‐4, SNU‐1, HCT116, and LoVo). As shown in Figure 2A and Table 2, a synergistic antitumor effect of chiMDM4 and nutlin‐3 was observed in 3 cell lines (NUGC‐4, HCT116, and LoVo), whereas a mostly additive effect was seen in the SNU‐1 cell line. Next, we examined whether the combination of chiMDM4/nutlin‐3 could enhance the antitumor effect of 5‐FU in HCT116 and NUGC‐4 cells (Figure 2B, Table 3). We found that chiMDM4/nutlin3 synergistically enhanced the 5‐FU effect in HCT116 (CI, 0.55‐0.93), whereas its effect was additive or even antagonistic to 5‐FU in NUGC‐4 (CI, 0.82‐1.42).
Figure 2.
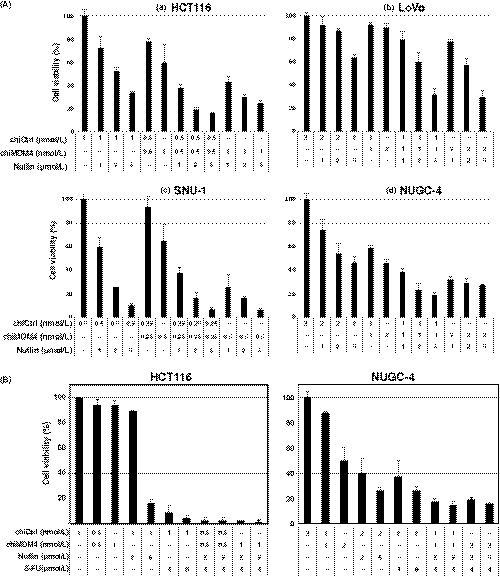
Effects of MDM4 knockdown and nutlin‐3 on tumor cell growth and antitumor activity of 5‐fluorouracil (5‐FU) in colon and gastric cancer cells. A, Growth inhibitory effect of MDM4 knockdown and nutlin‐3 in HCT116 (a), LoVo (b), SNU‐1 (c), and NUGC‐4 cells (d). Cells were transfected with either control siRNA (chiCtrl) or DNA‐modified siRNA targeting MDM4 (chiMDM4). After 4‐16 hours of incubation, cells were exposed to nutlin‐3 at the indicated concentrations. Five days after transfection, cell viability was determined using the WST‐8 assay. Cell viability relative to those transfected with chiCtrl are shown (mean ± SD; n = 3). B, Enhancement of MDM4 knockdown/nutlin‐3 on antitumor activity of 5‐FU in colon (HCT116) and gastric cancer cells (NUGC‐4). HCT116 (left) and NUGC‐4 cells (right) were transfected with either chiCtrl or chiMDM4. After 16 hours of incubation, cells were exposed to nutlin‐3 and 5‐FU at the indicated concentrations. Five days after transfection, cell viability was determined using the WST‐8 assay. Cell viability relative to those transfected with chiCtrl are shown (mean ± SD; n = 3)
Table 2.
Combination index of chiMDM4 and nutlin‐3
| Cell line | Nutlin‐3 (μmol/L) | chiMDM4 (nmol/L) | Combination index |
|---|---|---|---|
| HCT116 | 1.00 | 0.50 | 0.49 |
| 2.00 | 0.50 | 0.31 | |
| 5.00 | 0.50 | 0.49 | |
| 1.00 | 1.00 | 0.88 | |
| 2.00 | 1.00 | 0.72 | |
| 5.00 | 1.00 | 0.97 | |
| LoVo | 1.00 | 1.00 | 0.48 |
| 2.00 | 1.00 | 0.35 | |
| 5.00 | 1.00 | 0.30 | |
| 1.00 | 2.00 | 0.51 | |
| 2.00 | 2.00 | 0.33 | |
| 5.00 | 2.00 | 0.28 | |
| SNU‐1 | 1.00 | 0.25 | 0.96 |
| 2.00 | 0.25 | 0.82 | |
| 5.00 | 0.25 | 0.94 | |
| 1.00 | 0.50 | 0.98 | |
| 2.00 | 0.50 | 1.04 | |
| 5.00 | 0.50 | 1.07 | |
| NUGC‐4 | 1.00 | 1.00 | 0.47 |
| 2.00 | 1.00 | 0.23 | |
| 5.00 | 1.00 | 0.26 | |
| 1.00 | 2.00 | 0.54 | |
| 2.00 | 2.00 | 0.53 | |
| 5.00 | 2.00 | 0.68 |
Table 3.
Combination index of chiMDM4/nutlin‐3 and 5‐fluorouracil (5‐FU)
| Cell line | chiMDM4 (nmol/L) | Nutlin‐3 (μmol/L) | 5‐FU (4 μmol/L) | Combination index |
|---|---|---|---|---|
| HCT116 | 0.5 | 2.0 | + | 0.55 |
| 0.5 | 5.0 | + | 0.93 | |
| 1.0 | 2.0 | + | 0.48 | |
| 1.0 | 5.0 | + | 0.66 | |
| NUGC‐4 | 1.0 | 2.0 | + | 0.82 |
| 1.0 | 5.0 | + | 1.06 | |
| 2.0 | 2.0 | + | 1.19 | |
| 2.0 | 5.0 | + | 1.42 |
3.2. Expression of MDM2, MDM4, p53, and their downstream molecules by MDM4/MDM2 double knockdown
To explore the mechanisms by which chiMDM4/chiMDM2 enhanced 5‐FU‐mediated growth suppression in colon and gastric cancer cells, expression changes of MDM2, MDM4, p53, p21, and PUMA were examined in 2 colon cancer (HCT116 and LoVo) and 2 gastric cancer cells (SNU‐1 and NUGC‐4) by immunoblotting (Figure 3). The results of quantification of immunoblotting bands are shown in Table S1.
Figure 3.
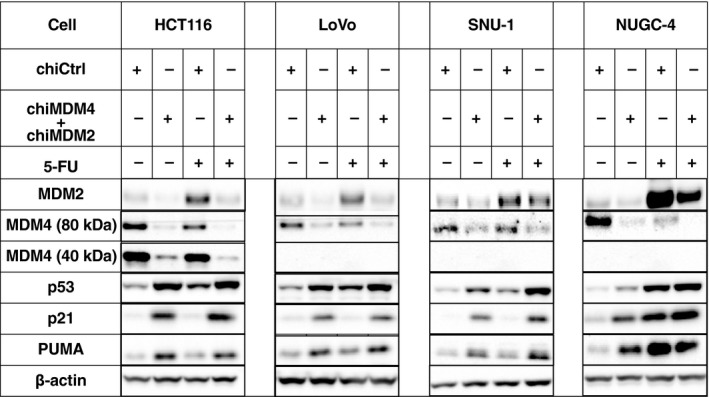
Effects of double knockdown of MDM2 and MDM4 and 5‐fluorouracil (5‐FU) on levels of p53, p21, and p53 upregulated modulator of apoptosis (PUMA) in colon (HCT116 and LoVo) and gastric cancer cells (SNU‐1 and NUGC‐4). HCT116, LoVo, SNU‐1, and NUGC‐4 cells were transfected with either control siRNA (chiCtrl) or a mixture of DNA‐modified siRNA targeting MDM4 (chiMDM4) and MDM2 (chiMDM2). After 4‐16 hours of incubation, cells were exposed to 5‐FU at the indicated concentrations. Twenty‐four hours after exposure to 5‐FU, cells were analyzed for levels of MDM2, MDM4, p53, p21, and PUMA using immunoblotting. β‐actin was used as an internal control
HCT116 cells are known to have wild and mutant alleles of MDM4. The mutant allele contained 1 base deletion of the third nucleotide of codon 279 and resulted in frameshift and premature termination. This gave rise to a smaller protein of 289 amino acids,32 which retained a p53‐binding region and could exert an inhibitory effect toward p53. chiControl‐transfected HCT116 cells expressed two bands of 80 and 40 kDa (Figure 4), representing wild and mutant MDM4, respectively.
Figure 4.
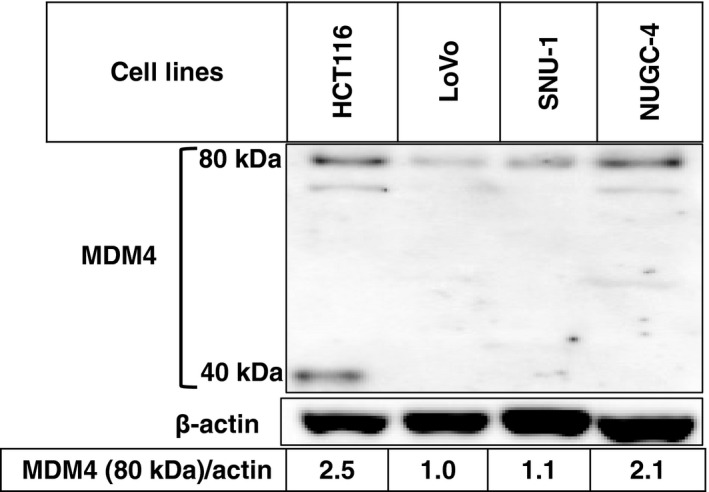
Expression level of MDM4 in wtTP53 colon (HCT116 and LoVo) and gastric cancer cell lines (SNU‐1 and NUGC‐4). Expression levels of MDM4 were analyzed by immunoblotting
chiMDM4/chiMDM2 suppressed both wild and mutant MDM4 in HCT116 and NUGC‐4 cells by 11‐ and 10‐fold, respectively, which was more efficient than in LoVo (3.8‐fold) and SNU‐1 cells (1.8‐fold). chiMDM4/chiMDM2 also decreased the levels of MDM2 in all cell lines by 1.3 to 3.7‐fold. Knockdown of MDM4/MDM2 concomitantly induced the accumulation of p53 and its downstream gene products of p21 and PUMA. The enhancing effects on p53 expression were almost equivalent among the 4 cell lines (2.2 to 3.0‐fold).
5‐FU increased p53 and its responsive gene products, MDM2, p21, and PUMA to various degrees in cell lines tested here. 5‐FU accumulated MDM2 more in NUGC‐4 (8.8‐fold) and HCT116 cells (4.2‐fold) than in LoVo (3.3‐fold) and SNU‐1 cells (1.4‐fold). In contrast, the level of MDM4 was inversely related to that of MDM2, suggesting that MDM4 might be destabilized by induced MDM2 in these cells.
Treatment with chiMDM4/chiMDM2 plus 5‐FU accumulated a lower level of MDM2 than 5‐FU alone. Furthermore, chiMDM4/chiMDM2 plus 5‐FU most potently suppressed the level of MDM4 than either alone in all tested cell lines. As a result, induction of p53 and p21 was highest in these cells treated with chiMDM4/chiMDM2 plus 5‐FU compared with cells treated with either alone.
Although both MDM2 and p21 were products of p53‐responsive genes, 5‐FU increased the p21 level less than the MDM2 level in HCT116 and LoVo cells. Therefore, we analyzed p21 mRNA levels in HCT116 cells treated with chiMDM4/chiMDM2, 5‐FU alone, and chiMDM4/chiMDM2 plus 5‐FU using quantitative RT‐PCR (Figure 5). Compared with p21 mRNA levels in chiCtrl‐treated cells, 5‐FU alone, chiMDM4/chiMDM2 alone, and chiMDM4/chiMDM2 plus 5‐FU increased the level of p21 mRNA by 2.4‐, 4.1‐, and 5.1‐fold, respectively. These results suggested that p53 activity was highest in cells treated with chiMDM4/chiMDM2 plus 5‐FU, and p21 might be destabilized by increased MDM2 in cells treated with 5‐FU.
Figure 5.
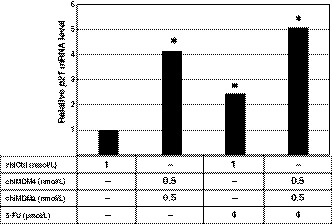
Effects of double knockdown of MDM4 and MDM2 and 5‐fluorouracil (5‐FU) on the level of p21 mRNA in HCT116 cells. HCT116 cells were transfected with either control siRNA (chiCtrl) or a mixture of DNA‐modified siRNA targeting MDM4 (chiMDM4) and MDM2 (chiMDM2) for 16 hours and then cultured in the presence of 5‐FU. Forty‐eight hours after transfection, the cells were analyzed for their p21 mRNA level using quantitative RT‐PCR. p21 mRNA levels relative to those transfected with chiCtrl are shown. *P < .05, compared with the chiCtrl
3.3. Cell cycle distribution and apoptosis
Effects of chiMDM4/chiMDM2, 5‐FU alone, and chiMDM4/chiMDM2 plus 5‐FU on the cell cycle distribution and apoptosis of colon cancer and gastric cancer cells were examined by flow cytometry (Figure 6).
Figure 6.
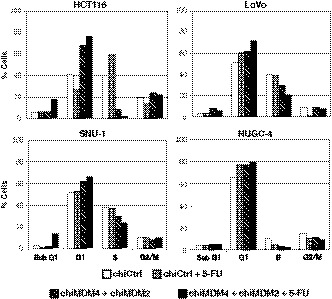
Effects of double knockdown of MDM4 and MDM2 and 5‐fluorouracil (5‐FU) on cell cycle distribution of colon (HCT116 and LoVo) and gastric cancer cells (SNU‐1 and NUGC‐4). HCT116 (top left), LoVo (top right), SNU‐1 (bottom left), and NUGC‐4 cells (bottom right) were transfected with control siRNA (chiCtrl) or a mixture of DNA‐modified siRNA targeting MDM4 (chiMDM4) and MDM2 (chiMDM2) overnight, then exposed to 5‐FU. After overnight cultivation, the cells were analyzed for cell cycle distribution by flow cytometry
In HCT116 cells, chiMDM4/chiMDM2 increased the fraction of the G1 phase and decreased that of the S phase, showing that MDM4/MDM2 double knockdown caused G1 arrest. 5‐FU decreased the G1 phase fraction and increased the S phase fraction, showing that 5‐FU caused early S phase arrest. The combination of these enhanced chiMDM4/chiMDM2‐induced G1 arrest, as well as apoptotic cell death detected as sub‐G1 fraction.
In SNU‐1 cells and LoVo cells, chiMDM4/chiMDM2 caused G1 arrest. 5‐FU alone caused weak G1 arrest in LoVo cells but had an undetectable effect on the cell cycle distribution in SNU‐1 cells. Simultaneous treatment with chiMDM4/chiMDM2 and 5‐FU alone enhanced G1 arrest. chiMDM4/chiMDM2 plus 5‐FU increased the population of apoptotic cells in SNU‐1 cells but not in LoVo cells.
In NUGC‐4 cells, 5‐FU alone as well as chiMDM4/chiMDM2 induced G1 arrest but not apoptosis. 5‐FU and chiMDM4/chiMDM2 decreased the S phase fraction, whereas a combination of these two had a faint change in the S phase fraction. Furthermore, a small increase in the G1 phase fraction was observed with a combination of 5‐FU and chiMDM2/chiMDM4 (5‐FU alone, 77%; chiMDM2/chiMDM4 alone, 76%; 5‐FU plus chiMDM4/chiMDM2, 80%), suggesting that chiMDM4/chiMDM2 marginally enhanced 5‐FU‐induced G1 arrest.
3.4. In vivo antitumor activity
To test whether chiMDM4/chiMDM2 could inhibit in vivo tumor growth and enhance the antitumor activity of 5‐FU, we examined the effects of chiCtrl, chiCtrl plus 5‐FU, chiMDM4/chiMDM2, and chiMDM4/chiMDM2 plus 5‐FU on the growth of HCT116 xenograft tumors in mice. chiMDM4/chiMDM2 alone and chiCtrl plus 5‐FU slowed the tumor growth rate compared with chiCtrl alone (Figure 7), showing that double knockdown suppressed in vivo tumor growth of wtTP53/highMDM4 colon cancer. 5‐FU plus chiMDM4/chiMDM2 most potently inhibited the tumor growth compared with chiCtrl/5‐FU and chiMDM4/chiMDM2.
Figure 7.
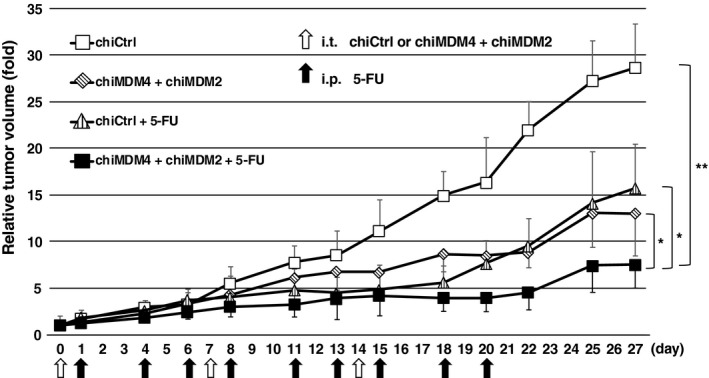
In vivo antitumor activity of a mixture of DNA‐modified siRNA targeting MDM4 (chiMDM4) and MDM2 (chiMDM2), 5‐fluorouracil (5‐FU), and combinations of these two in a xenograft model of HCT116 colon cancer cells. Results are expressed as means ± SE. *P < .05, **P < .001, compared with the control group (those injected with control siRNA [chiCtrl])
4. DISCUSSION
5‐FU is widely used for chemotherapy in various cancers including colon, stomach, and breast cancer. In our previous study, simultaneous knockdown of MDM4 and MDM2 using synthetic DNA‐substituted siRNAs (chiMDM4 and chiMDM2) was shown to synergistically suppress the growth of cancer cells with wtTP53/highMDM4.16 In this study, we showed that double knockdown of MDM4 and MDM2 enhanced the antitumor activity of 5‐FU in colon and gastric cancer cells with wtTP53/highMDM4.
In all cell lines used in this study, 5‐FU induces p53 expression and concomitant MDM2. Accumulated MDM2, functioning as a negative feedback regulator, can compromise p53‐mediated antitumor activity in 5‐FU‐treated cancer cells. In HCT116 cells, 5‐FU increased p21 mRNA and MDM2 but failed to accumulate p21, suggesting that MDM2 might antagonize p53‐mediated growth inhibition through ubiquitination and destabilization of p21.33 In this context, MDM2 knockdown using chiMDM4/chiMDM2 might disrupt these negative effects of MDM2 on p53‐ and p21‐mediated growth inhibition and potentiate the antitumor activity of 5‐FU.
We revealed that chiMDM4‐mediated growth inhibition could be synergistically enhanced by nutlin‐3, as was observed by chiMDM2.34 However, enhancement of 5‐FU antitumor activity by chiMDM4/nutlin‐3 was less efficient than that by chiMDM4/chiMDM2. Particularly in NUGC‐4 cells, chiMDM4/nutlin‐3 was even antagonistic to 5‐FU at high concentrations. 5‐FU caused MDM2 accumulation in NUGC‐4 more intensely than in HCT116 cells. Addition of nutlin‐3 to 5‐FU‐exposed cells may further the increase MDM2 level, which partially inactivates negative growth signals by direct interactions with p53, p21, RB, and E2F1 in these cells.33, 35 Small molecules and peptides targeting MDM2 and MDM4 have been developed,18 most of which disrupt MDM2‐p53 interactions and increase MDM2 expression, similar to nutlin‐3.13, 36 Thus, MDM2 knockdown might have some advantages over MDM2‐p53 inhibitors in the treatment of cancers carrying wtTP53.
The magnitude of enhancement of chiMDM4/chiMDM2 on 5‐FU‐mediated antitumor activity appears to be related to the magnitude of MDM4 suppression. 5‐FU suppresses MDM4 in synergistic responders (HCT116 and NUGC‐4) more strongly than in additive responders (SNU‐1 and LoVo). It remains unknown how 5‐FU decreases MDM4 levels in these cells. 5‐FU might destabilize MDM4 through the increase of MDM2 induced by p53 activation because MDM2 can ubiquitinate and destabilize MDM4.37 Furthermore, MDM4 knockdown by chiMDM4/chMDM2 is more efficient in synergistic responders (HCT116 and NUGC‐4) than additive responders (LoVo and SNU‐1). The expression level of MDM4 differs among cell lines (Figure 4). Synergistic responders (HCT116 and NUGC‐4) express higher levels of MDM4 than additive responders (SNU‐1 and LoVo). Synergistic responders could be more dependent on MDM4 expression for their growth and survival than additive responders.38, 39 Knockdown efficiency of synthetic siRNA might depend on various factors, including transfection efficiencies of siRNA and RNA‐induced silencing complex formation. RNA‐induced silencing complex formation could be determined by the abundance of AGO2 protein, Hsc70/Hsp90 chaperone, and endoribonuclease complex, consisting of Trax and Translin.40 Factors regulating MDM4 knockdown efficiency are now being analyzed.
This study shows that 5‐FU causes similar p53 accumulation in all colon and gastric cancer cell lines carrying wtTP53/highMDM4. However, the effect of 5‐FU on the cell cycle distribution differs among them: 5‐FU causes cell cycle arrest in the early S phase in HCT116 cells, whereas it induces G1 arrest in LoVo cells and NUGC‐4 cells. 5‐FU has two independent mechanisms of action.41, 42, 43 It blocks DNA replication by inhibiting thymidine synthesis, resulting in cell cycle arrest in the early S phase. It also causes nucleolar stress by being incorporated into ribosomal RNA and interfering in subsequent ribosomal RNA processing. This leads to p53 activation, which induces G1 arrest and apoptosis. 5‐FU might activate p53 more potently in LoVo and NUGC‐4 cells than in HCT116 cells. Although inhibition of DNA replication has been reported as the major mechanism of action of 5‐FU in most cancer cells, the magnitude of nucleolar stress could vary among cell lines, dependent on the efficiency of fluorouridine incorporation into ribosomal RNA. Although 5‐FU causes early S‐phase arrest in HCT116 cells, its combination with chiMDM4/chiMDM2 dramatically shifts the effect on cell cycle distribution from early S‐phase arrest to G1 arrest with strong p53 activation. This result suggests that the addition of chiMDM4/chiMDM2 might change the main action mechanism of 5‐FU from inhibition of DNA replication to augmented activation of p53.
chiMDM4/chiMDM2 plus 5‐FU induced larger populations of apoptotic cells in SNU‐1 cells than in LoVo cells. Activated p53 can trigger apoptosis by modulating the expression of genes involved in intrinsic (eg, PUMA, BAX, and BCL2) and extrinsic apoptosis pathways (FAS‐L and FAS).44 Inducibility of p53‐mediated apoptosis depends on the expression and structures of these genes in individual cell lines. LoVo cells but not SNU‐1 cells carry BAX mutations,45, 46 which could be one of the mechanisms by which some cancer cells acquire resistance to p53‐triggered apoptosis.
In the present study, we show that MDM4/MDM2 double knockdown inhibits in vivo tumor growth and enhances the antitumor effect of 5‐FU without any intolerable toxicity. Recent advances in the delivery system for oligonucleotides47 could enable chiMDM4/chiMDM2 to be applied to the treatment of wtTP53/highMDM4 tumors. Approximately 50% of human cancers express wtTP53,48 in some fraction of which wtTP53 is directly suppressed by MDM2 alone or MDM2 combined with MDM4. Thus, MDM2 and MDM4 are ideal targets of therapy for these tumors. In addition, MDM2 knockout was shown to suppress the growth of tumors lacking TP53 alleles by inducing p53‐responsive gene through TP73‐mediated transactivation.49 MDM4/MDM2 knockdown can be potentially used for the treatment of cancers lacking p53 expression.
In conclusion, the double knockdown of MDM4 and MDM2 enhances in vitro and in vivo antitumor activity of 5‐FU toward gastrointestinal cancer with wtTP53/highMDM4. Combination of siRNAs targeting MDM4 and MDM2 and cytotoxic anticancer drugs including 5‐FU could be a novel therapeutic strategy for such cancers.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
We thank Ms. J. Yamaguchi and Ms. M. Aida for their administrative work.
Imanishi M, Yamamoto Y, Wang X, et al. Augmented antitumor activity of 5‐fluorouracil by double knockdown of MDM4 and MDM2 in colon and gastric cancer cells. Cancer Sci. 2019;110:639–649. 10.1111/cas.13893
Funding information
Grant‐in‐Aid for Scientific Research (C) from the Ministry of Education, Culture, Sports, Science and Technology of Japan to H.I. (grant number 26460624).
REFERENCES
- 1. Douillard JY, Cunningham D, Roth AD, et al. Irinotecan combined with fluorouracil compared with fluorouracil alone as first‐line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet. 2000;355:1041‐1047. [DOI] [PubMed] [Google Scholar]
- 2. Saltz LB, Cox JV, Blanke C, et al. Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. Irinotecan Study Group. N Engl J Med. 2000;343:905‐914. [DOI] [PubMed] [Google Scholar]
- 3. de Gramont A, Figer A, Seymour M, et al. Leucovorin and fluorouracil with or without oxaliplatin as first‐line treatment in advanced colorectal cancer. J Clin Oncol. 2000;18:2938‐2947. [DOI] [PubMed] [Google Scholar]
- 4. Goldberg RM, Sargent DJ, Morton RF, et al. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J Clin Oncol. 2004;22:23‐30. [DOI] [PubMed] [Google Scholar]
- 5. Falcone A, Ricci S, Brunetti I, et al. Phase III trial of infusional fluorouracil, leucovorin, oxaliplatin, and irinotecan (FOLFOXIRI) compared with infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) as first‐line treatment for metastatic colorectal cancer: the Gruppo Oncologico Nord Ovest. J Clin Oncol. 2007;25:1670‐1676. [DOI] [PubMed] [Google Scholar]
- 6. Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009;9:862‐873. [DOI] [PubMed] [Google Scholar]
- 7. Wade M, Wang YV, Wahl GM. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 2010;20:299‐309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Merkel O, Taylor N, Prutsch N, et al. When the guardian sleeps: reactivation of the p53 pathway in cancer. Mutat Res. 2017;773:1‐13. [DOI] [PubMed] [Google Scholar]
- 9. Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell. 2006;21:307‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gu J, Kawai H, Nie L, et al. Mutual dependence of MDM2 and MDMX in their functional inactivation of p53. J Biol Chem. 2002;277:19251‐19254. [DOI] [PubMed] [Google Scholar]
- 11. Shvarts A, Steegenga WT, Riteco N, et al. MDMX: a novel p53‐binding protein with some functional properties of MDM2. EMBO J. 1996;15:5349‐5357. [PMC free article] [PubMed] [Google Scholar]
- 12. Linares LK, Hengstermann A, Ciechanover A, Muller S, Scheffner M. HdmX stimulates Hdm2‐mediated ubiquitination and degradation of p53. Proc Natl Acad Sci USA. 2003;100:12009‐12014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Endo S, Yamato K, Hirai S, et al. Potent in vitro and in vivo antitumor effects of MDM2 inhibitor nutlin‐3 in gastric cancer cells. Cancer Sci. 2011;102:605‐613. [DOI] [PubMed] [Google Scholar]
- 14. Li Q, Lozano G. Molecular pathways: targeting Mdm2 and Mdm4 in cancer therapy. Clin Cancer Res. 2012;19:34‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Duffy MJ, Synnott NC, McGowan PM, Crown J, O'Connor D, Gallagher WM. p53 as a target for the treatment of cancer. Cancer Treat Rev. 2014;40:1153‐1160. [DOI] [PubMed] [Google Scholar]
- 16. Hirose M, Yamato K, Endo S, et al. MDM4 expression as an indicator of TP53 reactivation by combined targeting of MDM2 and MDM4 in cancer cells without TP53 mutation. Oncoscience. 2014;1:830‐843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pellegrino M, Mancini F, Luca R, et al. Targeting the MDM2/MDM4 interaction interface as a promising approach for p53 reactivation therapy. Cancer Res. 2015;75:4560‐4572. [DOI] [PubMed] [Google Scholar]
- 18. Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E. Clinical overview of MDM2/X‐targeted therapies. Front Oncol. 2016;6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tisato V, Voltan R, Gonelli A, Secchiero P, Zauli G. MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. J Hematol Oncol. 2017;10:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Q, Zeng SX, Lu H. Targeting p53‐MDM2‐MDMX loop for cancer therapy. Subcell Biochem. 2014;85:281‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wachter F, Morgan AM, Godes M, Mourtada R, Bird GH, Walensky LD. Mechanistic validation of a clinical lead stapled peptide that reactivates p53 by dual HDM2 and HDMX targeting. Oncogene. 2017;36:2184‐2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Blotner S, Chen LC, Ferlini C, Zhi J. Phase 1 summary of plasma concentration‐QTc analysis for idasanutlin, an MDM2 antagonist, in patients with advanced solid tumors and AML. Cancer Chemother Pharmacol. 2018;81:597‐607. [DOI] [PubMed] [Google Scholar]
- 23. Herting F, Herter S, Friess T, et al. Antitumour activity of the glycoengineered type II anti‐CD20 antibody obinutuzumab (GA101) in combination with the MDM2‐selective antagonist idasanutlin (RG7388). Eur J Haematol. 2016;97:461‐470. [DOI] [PubMed] [Google Scholar]
- 24. Lehmann C, Friess T, Birzele F, Kiialainen A, Dangl M. Superior anti‐tumor activity of the MDM2 antagonist idasanutlin and the Bcl‐2 inhibitor venetoclax in p53 wild‐type acute myeloid leukemia models. J Hematol Oncol. 2016;9:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Valassiadou KE, Stefanaki K, Tzardi M, et al. Immunohistochemical expression of p53, bcl‐2, mdm2 and waf1/p21 proteins in colorectal adenocarcinomas. Anticancer Res. 1997;17:2571‐2576. [PubMed] [Google Scholar]
- 26. Danovi D, Meulmeester E, Pasini D, et al. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol Cell Biol. 2004;24:5835‐5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gilkes DM, Pan Y, Coppola D, Yeatman T, Reuther GW, Chen J. Regulation of MDMX expression by mitogenic signaling. Mol Cell Biol. 2008;28:1999‐2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gunther T, Schneider‐Stock R, Hackel C, et al. Mdm2 gene amplification in gastric cancer correlation with expression of Mdm2 protein and p53 alterations. Mod Pathol. 2000;13:621‐626. [DOI] [PubMed] [Google Scholar]
- 29. Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6:909‐923. [DOI] [PubMed] [Google Scholar]
- 30. Yamato K, Egawa N, Endo S, et al. Enhanced specificity of HPV16 E6E7 siRNA by RNA‐DNA chimera modification. Cancer Gene Ther. 2011;18:587‐597. [DOI] [PubMed] [Google Scholar]
- 31. Chou TC, Talalay P. Quantitative analysis of dose‐effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27‐55. [DOI] [PubMed] [Google Scholar]
- 32. Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Z, Wang H, Li M, Agrawal S, Chen X, Zhang R. MDM2 is a negative regulator of p21WAF1/CIP1, independent of p53. J Biol Chem. 2004;279:16000‐16006. [DOI] [PubMed] [Google Scholar]
- 34. Hu B, Gilkes DM, Farooqi B, Sebti SM, Chen J. MDMX overexpression prevents p53 activation by the MDM2 inhibitor Nutlin. J Biol Chem. 2006;281:33030‐33035. [DOI] [PubMed] [Google Scholar]
- 35. Ganguli G, Wasylyk B. p53‐independent functions of MDM2. Mol Cancer Res. 2003;1:1027‐1035. [PubMed] [Google Scholar]
- 36. Kitagawa M, Aonuma M, Lee SH, Fukutake S, McCormick F. E2F‐1 transcriptional activity is a critical determinant of Mdm2 antagonist‐induced apoptosis in human tumor cell lines. Oncogene. 2008;27:5303‐5314. [DOI] [PubMed] [Google Scholar]
- 37. Pan Y, Chen J. MDM2 promotes ubiquitination and degradation of MDMX. Mol Cell Biol. 2003;23:5113‐5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gembarska A, Luciani F, Fedele C, et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat Med. 2012;18:1239‐1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Patton JT, Mayo LD, Singhi AD, Gudkov AV, Stark GR, Jackson MW. Levels of HdmX expression dictate the sensitivity of normal and transformed cells to Nutlin‐3. Cancer Res. 2006;66:3169‐3176. [DOI] [PubMed] [Google Scholar]
- 40. Kobayashi H, Tomari Y. RISC assembly: coordination between small RNAs and Argonaute proteins. Biochim Biophys Acta. 2016;1859:71‐81. [DOI] [PubMed] [Google Scholar]
- 41. Akpinar B, Bracht EV, Reijnders D, et al. 5‐Fluorouracil‐induced RNA stress engages a TRAIL‐DISC‐dependent apoptosis axis facilitated by p53. Oncotarget. 2015;6:43679‐43697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fang F, Hoskins J, Butler JS. 5‐Fluorouracil enhances exosome‐dependent accumulation of polyadenylated rRNAs. Mol Cell Biol. 2004;24:10766‐10776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Longley DB, Harkin DP, Johnston PG. 5‐fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330‐338. [DOI] [PubMed] [Google Scholar]
- 44. Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis – the p53 network. J Cell Sci. 2003;116:4077‐4085. [DOI] [PubMed] [Google Scholar]
- 45. Ku JL, Park JG. Biology of SNU cell lines. Cancer Res Treat. 2005;37:1‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rampino N, Yamamoto H, Ionov Y, et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science. 1997;275:967‐969. [DOI] [PubMed] [Google Scholar]
- 47. Kim HJ, Kim A, Miyata K, Kataoka K. Recent progress in development of siRNA delivery vehicles for cancer therapy. Adv Drug Deliv Rev. 2016;104:61‐77. [DOI] [PubMed] [Google Scholar]
- 48. Hainaut P, Hernandez T, Robinson A, et al. IARC database of p53 gene mutations in human tumors and cell lines: updated compilation, revised formats and new visualisation tools. Nucleic Acids Res. 1998;26:205‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Feeley KP, Adams CM, Mitra R, Eischen CM. Mdm2 is required for survival and growth of p53‐deficient cancer cells. Cancer Res. 2017;77:3823‐3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials