

Abstract
Atypical protein kinase C (aPKC) isozymes are unique in the protein kinase C (PKC) superfamily in that they are not regulated by the lipid second messenger diacylglycerol. Whether a different second messenger acutely controls their function is unknown. Here we show that the lipid mediator, sphingosine 1-phosphate (S1P), controls the cellular activity of aPKC. Using a genetically-encoded reporter we designed, aPKC-specific C Kinase Activity Reporter (aCKAR), we demonstrate that intracellular S1P activates aPKC. Biochemical studies reveal that S1P directly binds to the kinase domain of aPKC to relieve autoinhibitory constraints. In silico studies identify potential binding sites on the kinase domain, one of which was validated biochemically. Lastly, functional studies reveal that S1P-dependent activation of aPKC suppresses apoptosis in HeLa cells. Taken together, our data reveal a previously undescribed molecular mechanism for controlling the cellular activity of atypical PKC and identify a new molecular target for S1P.
One-sentence summary:
The first genetically-encoded biosensor for atypical protein kinase C isozymes is used to identify intracellular sphingosine 1-phosphate as a novel activator of this class of protein kinase C isozymes, with computational, biochemical, and cellular studies identifying the mechanism and function of this activation.
INTRODUCTION
The atypical protein kinase C (aPKC) consists of two isozymes, PKCζ and PKCι/λ (PKCλ is mouse orthologue of human PKCι), and PKCζ gene also encodes N-terminal truncated form PKMζ, a constitutively active form of PKCζ. The aPKC isozymes are involved in diverse cellular functions, including a well-characterized role in the maintenance of cell polarity and insulin signaling (1, 2). They have also been extensively studied in cancer, where these isozymes can function as either oncogenes or tumor suppressors depending on the cellular context (3–5). In particular, the PKCι gene (PRKCI) is part of the 3q amplicon, a highly amplified region including oncogenes such as PI3 kinase (6) and its levels are thus elevated in many cancers (5). Unlike other PKC family members, aPKC isozymes do not transduce signals resulting from phospholipid hydrolysis (7). Rather, they are activated by binding protein scaffolds. Whether their activity is also acutely activated by second messengers remains to be established.
aPKC isozymes are co-translationally phosphorylated by mTORC2 on the turn motif, followed by post-translational and constitutive phosphorylation by PDK-1 on the activation loop, modifications that are required for activity (8, 9). As with other PKC family members, the phosphorylated enzymes are maintained in an autoinhibited conformation by interaction of a pseudosubstrate-C1 domain module with the kinase domain. Binding to protein scaffolds not only positions these PKC isozymes near their substrates, but also relieves autoinhibitory constraints by tethering the pseudosubstrate away from the substrate-binding cavity (1). For example, the interaction of PKCζ with the scaffold p62 results in the tethering of the basic PKCζ pseudosubstrate to an acidic surface of the PB1 (Phox-Bem1) domain of p62, stabilizing the open and active conformation of the kinase (10). Similarly, PKCζ is maintained in an open conformation when bound to Par6 (11, 12). One of the effects of insulin is to induce the interaction of aPKC isozymes with protein scaffolds such as IRS-1 (12). Colocalization of aPKC isozymes and substrates on protein scaffolds ensures efficient phosphorylation given the exceptionally slow catalytic rate of these isozymes, which is 40-fold slower compared to that of conventional PKC isozymes (9). Although aPKC isozymes are not regulated by diacylglycerol, several lines of evidence suggest that other lipid mediators may regulate the activity of aPKC isozymes. In addition to anionic lipids, the neutral lipid ceramide has been reported to directly activate aPKC in vitro (13–16). Ceramide is at the hub of sphingolipid metabolism, involving a network of bioactive lipids that are key regulators of survival and metabolic signaling (17) and pose as attractive second messenger candidates for regulation of aPKC in cells.
Sphingosine 1-phosphate (S1P) has been implicated as an important lipid mediator acting both inside and outside the cell (18–22). It is produced intracellularly by sphingosine kinase (SphK)-catalyzed phosphorylation of sphingosine and is then exported out of cells, where it binds G-protein-coupled S1P receptors (S1P1–5) to regulate a variety of cellular functions. Notably, these receptors mediate pro-inflammatory and pro-survival signaling (23–25). In addition, mounting evidence supports a role for intracellular S1P in cell signaling (17). However, less is known about the molecular mechanisms of its intracellular second messenger function. It has been reported to bind and modulate the activity of signaling molecules such as histone deacetylase (HDAC), TNF receptor-associated factor 2 (TRAF2), and human telomerase reverse transcriptase (hTERT) (26–28). Whether S1P has additional targets is unknown. Because aberrant sphingolipid signaling is associated with multiple pathologies, including cancer, metabolic disorders, and neurodegeneration (19), identifying the mechanisms and targets of S1P has important therapeutic potential.
Here we report that intracellular S1P directly binds and activates aPKC isozymes. As part of this study, we developed a genetically-encoded reporter specific for aPKC isozymes that allowed us to assess the activity of aPKC in real time in cells. Using a combination of computational, biochemical, and imaging technologies, we show that S1P binds a pocket on the surface of the kinase domain, reducing autoinhibitory contacts with the pseudosubstrate-C1 module to cause activation. Cellular studies reveal that basal S1P promotes activation of aPKC and that this basal activity inhibits apoptosis. Our results are consistent with a model in which S1P allosterically activates aPKC isozymes to suppress apoptosis.
RESULTS
Development and Validation of a Reporter for Atypical PKC
To examine the activity of aPKC in cells, we modified the phospho-acceptor sequence in our C kinase activity reporter (CKAR) (29) to be uniquely phosphorylated by aPKC. CKAR contains a phospho-Thr-binding FHA2 domain and a peptide sequence that conforms to an FHA2 binding sequence when phosphorylated (specifically having a Thr at the phospho-acceptor site and Ile at the P+3 position (30)), flanked by the fluorescence resonance energy transfer (FRET) pair CFP and YFP (Figure 1A). Phosphorylation at the phospho-acceptor site results in an intramolecular association with the FHA2 module, detected by a decrease in intramolecular FRET. In the original CKAR, this sequence is RFRRFQTLKIKAKA, where the underlined residue is the phospho-acceptor site. We previously modified this sequence to selectively monitor the activity of PKCδ by inserting a phosphorylation sequence that we optimized for this isozyme (31). Here, we took advantage of aPKC’s specific phosphorylation of insulin-regulated membrane aminopeptidase (IRAP) at Ser80 (32), but made two modifications to conform to the FHA2 consensus sequence: Ser at the phospho-acceptor site was replaced with Thr, and the residue at the P + 3 position was replaced with Ile (Figure 1A). To determine whether aCKAR is phosphorylated by endogenous or overexpressed aPKC isozymes in cells, COS7 cells were transfected with aCKAR and either mCherry or the constitutively active catalytic domain of PKCζ (PKMζ; tagged at the N-terminus with mCherry) and then treated with the aPKC-specific inhibitor, PZ09 (33). Inhibition of aPKC resulted in a small but reproducible drop in FRET (Figure 1B). The inhibitor-induced drop in FRET reflects a reduction in the basal phosphorylation of the reporter by aPKC. Thus, the reporter was sensitive to inhibition of both endogenous aPKC isozymes (blue circles) and overexpressed PKMζ (red circles). For comparison, the FRET change measured by the original CKAR was 1.7 x greater for endogenous aPKC activity (Figure 1B, blue circles, compare left and right panels) and 2.9 x greater for overexpressed PKMζ (Figure 1B, red circles, compare left and right panels). Thus, aPKC-specific phosphorylation results in a lower magnitude change of aCKAR compared to the generic CKAR. Note that both aCKAR and CKAR have a similar dynamic range, but increased phosphatase-sensitivity of aCKAR results in smaller FRET changes upon PZ09 addition; calyculin treatment to inhibit phosphatases results in a much larger change in FRET of aCKAR compared to CKAR in cells overexpressing the constitutively active PKMζ (Figure S1). Qualitatively similar results were obtained for the corresponding catalytic domain-only construct of PKCλ (Figure 1C). To determine whether aCKAR can be phosphorylated by other PKC isozymes, COS7 cells transfected with aCKAR were treated with phorbol 12,13-dibutyrate (PDBu) to induce maximal activity of all conventional and novel PKC isozymes (Figure 1D, left panel). PDBu did not cause an increase in aCKAR readout for cells co-transfected with mCherry alone (measuring endogenous PKC responses), nor did it increase significantly in cells co-transfected with PKCα (red), PKCβII (yellow), PKCδ (green), or PKCε (pink). In contrast, a robust increase in normalized FRET ratio was observed when the reporter was CKAR (Figure 1D, right panel). Thus, aCKAR specifically reads out the activity of PKCζ and PKCι/λ and is not phosphorylated following activation of phorbol ester-sensitive PKC isozymes. To further validate that the inhibitor-sensitive activity was catalyzed by aPKC, we examined PZ09-senstive activity in cells in which both PKCζ and PKCι/λ had been depleted by siRNA. The knockdown of PKCζ was >90% whereas as that of PKCι was approximately 50% (Figure 1E). Importantly, depletion of both aPKC isozymes results in a robust reduction in basal activity, as assessed by the much smaller drop in reporter read-out in cells treated with siRNA to both isozymes (red) compared to control cells (blue) (Figure 1F). Importantly, replacement of the phospho-acceptor Thr with Ala abolished the FRET change observed upon inhibition of endogenous aPKC (yellow) or overexpressed PKMζ (green) (Figure 1G). Furthermore, aCKAR showed no response to stimulation of COS7 cells with forskolin to elevate cAMP under conditions that resulted in a robust response by the A kinase activity reporter (AKAR) (34) (Figure 1H). Similarly, EGF stimulation did not cause a change in aCKAR readout under conditions where the Akt/B kinase activity reporter (BKAR) (35) gave a robust response (Figure 1I). Additionally, treatment with PDBu resulted in no change under conditions where the D kinase activity reporter (DKAR) (36) had a large response (Figure 1J). These data reveal that aCKAR reads out the basal activity of endogenous aPKC isozymes and the basal activity of the overexpressed catalytic domain of PKCζ and PKCι/λ, but does not read out the activity of conventional PKC isozymes, novel PKC isozymes, PKA, PKB, or PKD.
Fig. 1. Development of an aPKC-selective activity reporter (aCKAR).
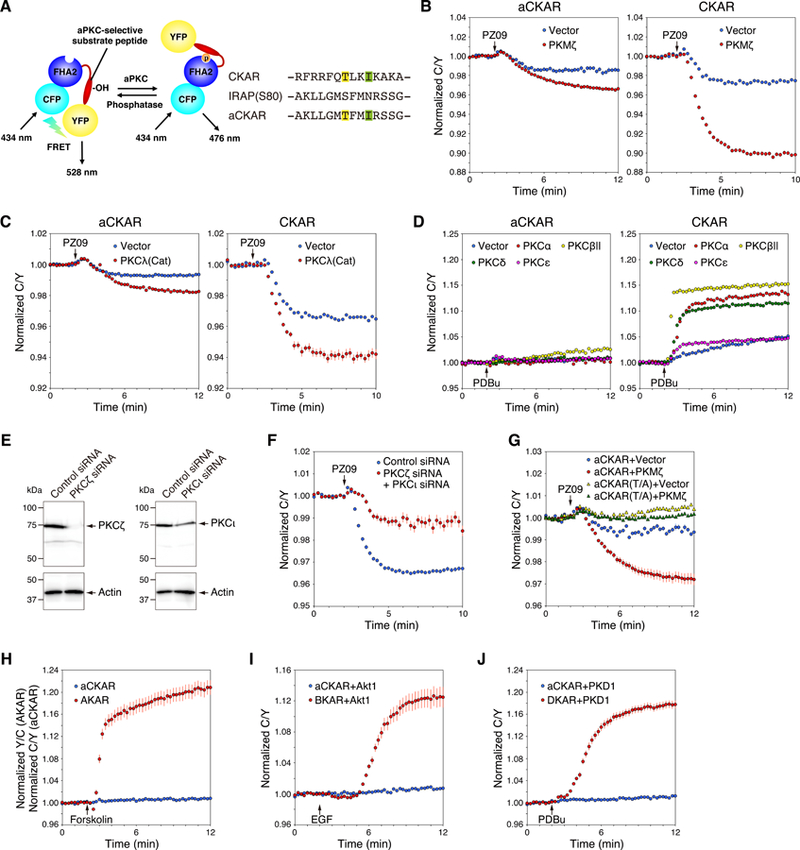
(A) The architecture of aCKAR is based on that of the pan-PKC reporter, CKAR, and consists of monomeric CFP (cyan), the FHA2 domain of Rad53p (blue), an aPKC-selective substrate peptide (red), and monomeric YFP (yellow). In the unphosphorylated state, monomeric CFP and monomeric YFP are in proximity and in an orientation resulting in FRET. Once phosphorylated by aPKC at the threonine within the substrate sequence (highlighted in yellow), the FHA2 domain binds the phosphorylated sequence, resulting in a conformational change that alters the FRET ratio. Isoleucine at the P+3 position (highlighted in green) is critical for the binding of phospho-threonine to the FHA2 domain. The substrate peptide of CKAR was replaced with an aPKC-selective substrate peptide with a sequence corresponding to residues 74 to 87 of IRAP (rat insulin-regulated membrane aminopeptidase (U76997)), except Ser80 at the phospho-acceptor was replaced with Thr (yellow) and the Asn at the P+3 position was replaced with Ile (green).
(B) COS7 cells were co-transfected with aCKAR (left panel) or CKAR (right panel) and mCherry (Vector) or mCherry-PKMζ. The CFP/YFP FRET (C/Y) emission ratio was quantified as a function of time following the addition of PZ09 (5 µM). The drop in FRET upon addition of inhibitor represents the basal (unstimulated) activity of endogenous aPKC (blue); the additional drop in cells overexpressing PKMζ (red) reflects the basal activity of PKMζ. Data represent the C/Y emission ratio normalized to the starting point (1.0) with means ± S.E. (n ≥ 25 cells).
(C) As in (B), except that cells were co-transfected with the indicated reporters and either RFP or RFP tagged to a construct of the catalytic domain of PKCλ (RFP-PKCλ(Cat)). Data represent the means ± S.E. (n ≥ 21 cells).
(D) COS7 cells were co-transfected with aCKAR (left panel) or CKAR (right panel) and mCherry, mCherry-PKCα, mCherry-PKCβII, mCherry-PKCδ, or mCherry-PKCε. The normalized C/Y emission ratio was quantified as a function of time following the addition of PDBu (200 nM). The increase in FRET represents the agonist-induced activity of these PKC isozymes. Data represent the means ± S.E. (n ≥ 16 cells for left panel, n ≥ 21 cells for right panel).
(E) Knockdown of endogenous expression of PKCζ and PKCι in HeLa cells using siRNAs for human PKCζ and PKCι. HeLa cells were transfected for 48 h with non-targeting control siRNA, human PKCζ siRNA, or human PKCι siRNA individually. Equal aliquots of lysate were blotted and probed with anti-PKCζ or anti-PKCι antibody and with anti-actin antibody as a protein loading control.
(F) HeLa cells were co-transfected with aCKAR and control siRNA or both PKCζ siRNA and PKCι siRNAs. The normalized C/Y emission ratio was quantified as a function of time following the addition of PZ09 (5 µM). Data represent the means ± S.E. (n ≥ 21 cells).
(G) COS7 cells were co-transfected with aCKAR or a construct in which the phospho-acceptor site is mutated to Ala (aCKAR (T/A)) and mCherry or mCherry-PKMζ. The normalized C/Y emission ratio was quantified as a function of time following the addition of PZ09 (5 µM). Data represent the means ± S.E. (n ≥ 28 cells).
(H) COS7 cells were transfected with aCKAR or the PKA reporter, AKAR. The normalized C/Y emission ratio was quantified as a function of time following the addition of forskolin (10 µM) to elevate cAMP. Data represent the means ± S.E. (n ≥ 28 cells).
(I) COS7 cells were co-transfected with aCKAR or the Akt/PKB reporter, BKAR, and mCherry-Akt1. The normalized C/Y emission ratio was quantified as a function of time following the addition of EGF (50 ng/ml) to activate Akt/PKB. Data represent the means ± S.E. (n ≥ 28 cells).
(J) COS7 cells were co-transfected with aCKAR or the PKD reporter, DKAR, and mCherry-PKD1. The C/Y normalized emission ratio was quantified as a function of time following the addition of PDBu (200 nM) to activate PKD (and PKC, which does not phosphorylate this reporter). Data represent the means ± S.E. (n ≥ 25 cells).
Having validated that aCKAR is appropriate to specifically monitor endogenous aPKC signaling in cells, we interrogated the endogenous signaling output of aPKC isozymes in a diverse set of cell types. Treatment with PZ09 caused a drop in aCKAR readout in all cells examined, including COS7, HEK293, HeLa, HepG2, MCF7, MDA-MB-231, and SH-SY5Y (Figure 2A). Quantification of the FRET change observed in the period 10–12 minutes after inhibitor treatment (Figure 2B) revealed that the breast cancer cell line MDA-MB-231 had the highest basal signaling output of aPKC isozymes (4.1 ± 0.2 % increase in FRET value from baseline). The lowest basal signaling was observed in the liver cells HepG2, the fibroblast cell line HEK293, and the neuroblastoma cell line SH-SY5Y, where the FRET changes were approximately 3-fold lower than that for the MDA-MB-231 cells. Western blot analysis of equal numbers for each cell type revealed that all cells expressed both PKCζ and PKCι (Figure 2C; immunoreactivity for PKCζ and PKCι quantified in Figure 2D, respectively). Cells with the highest levels of aPKC activity did not necessarily have the highest levels of PKCζ and/or PKCι protein: for example, MDA-MB-231 cells had high basal aPKC signaling but relatively low levels of aPKC protein. HeLa cells also had low protein levels of aPKC isozymes but moderate basal aPKC activity. We selected HeLa cells for subsequent experiments, reasoning that moderate basal activity might allow easier detection of both basal and agonist-stimulated activity.
Fig. 2. Basal activity of aPKC is regulated by S1P signaling.
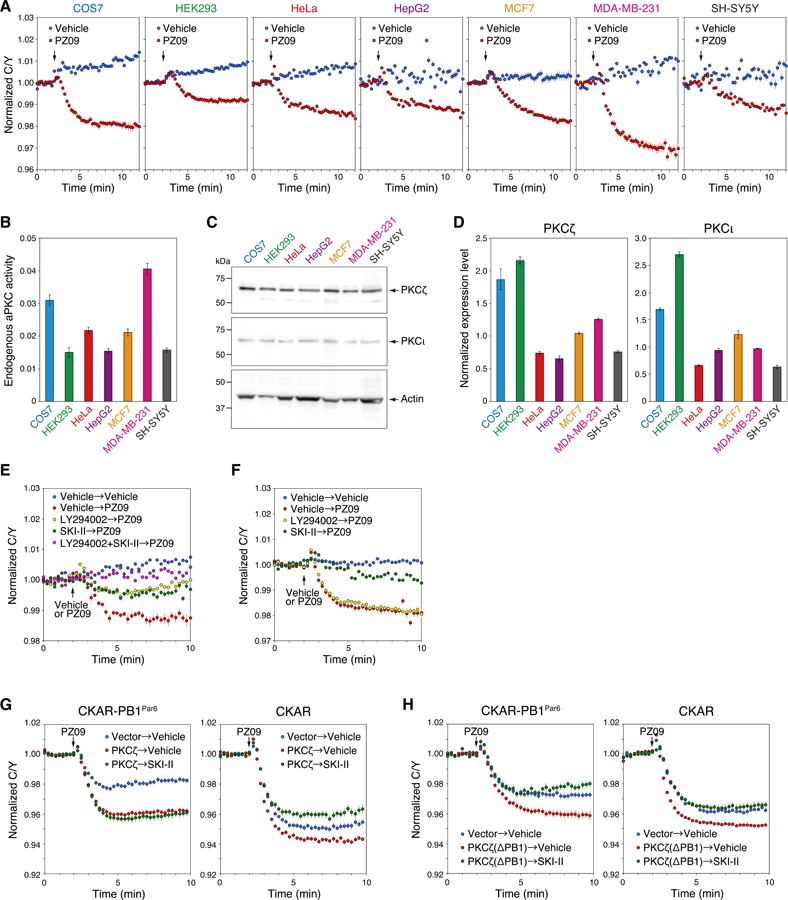
(A) COS7, HEK293, HeLa, HepG2, MCF7, MDA-MB-231, or SH-SY5Y cells were transfected with aCKAR. The basal activity of endogenous aPKC was measured following the addition of PZ09 (5 µM); DMSO vehicle was added as a control. The normalized C/Y emission ratio was quantified as a function of time following the addition of PZ09. Data represent the means ± S.E. (n ≥ 20 cells). The arrow indicates the point of DMSO vehicle or PZ09 addition.
(B) The relative basal activity of endogenous aPKC was quantified from the data in (A) and represents the difference between the C/Y emission ratios summed between minutes 10–12 for the vehicle vs PZ09 treatments.
(C) Western blot of lysates from 2.0 × 105 cells of the indicated cell lines probed with antibodies to PKCζ or PKCι. The endogenous expression level of beta-actin was also detected using an anti-β-actin antibody.
(D) Normalized expression level of PKCζ (left panel) or PKCι (right panel) was quantified from the result of (C) and represents the intensity of PKC divided by the intensity of β-actin for each cell type.
(E) HeLa cells were transfected with aCKAR and pre-treated for 16 h with DMSO vehicle, LY294002 (20 μM), SKI-II (5 μM), or LY294002 (20 μM) + SKI-II (5 μM). These cells were subsequently stimulated with DMSO vehicle or 5 μM PZ09 (addition indicated by arrow) to assess the effect of these pre-treatments on basal aPKC activity. The normalized C/Y emission ratio was quantified as a function of time. Data represent the means ± S.E. (n ≥ 22 cells). For graph legend: pre-treatment with vehicle, SKI-II, or LY294002 → treatment with vehicle or PZ09 performed at the time point indicated by the arrow.
(F) As in (E), except experiments were conducted in serum-free media. Data represent the means ± S.E. (n ≥ 22 cells). For graph legend: pre-treatment with vehicle, SKI-II, or LY294002 → treatment with vehicle or PZ09 performed at the time point indicated by the arrow.
(G) HeLa cells were co-transfected with CKAR fused to the PB1 domain of Par6 (CKAR-PB1Par6) (left panel) or CKAR (right panel) and mCherry (Vector) or mCherry-PKCζ (PKCζ). Cells were pre-treated with DMSO vehicle or SKI-II (5 μM) for 16 h, and then treated with 5 μM PZ09. The normalized C/Y emission ratio was quantified as a function of time. Data represent the means ± S.E. (n ≥ 22 cells).
(H) As in (G), except cells were transfected with mCherry-PKCζ (PB1 deletion mutant) (PKCζ(ΔPB1)) where indicated. Data represent the means ± S.E. (n ≥ 17 cells).
S1P Contributes to the Basal Activity of Non-scaffolded Atypical PKC
We used a pharmacological approach to address whether the basal activity of aPKC is regulated by S1P. As noted above, treatment of aCKAR-overexpressing HeLa cells with PZ09 in the presence of serum caused a drop in FRET ratio that represents the inhibition of basally active aPKC isozymes (Figure 2E). Pre-treatment of HeLa cells either with the SphK inhibitor SKI-II (green circles) to reduce levels of intracellular S1P (37), or the phosphatidylinositol-3 kinase (PI3K) inhibitor LY294002 (yellow circles) to reduce PIP3 levels, resulted in a 2-fold smaller drop in FRET change following PZ09 treatment. Pre-treatment with both inhibitors (pink circles) abolished all basal activity as assessed by the lack of FRET change upon addition of PZ09. In contrast, the PI3K inhibitor had no effect on the activity of endogenous aPKC in serum-starved cells (Figure 2F, yellow circles). Under these conditions, the SphK inhibitor abolished almost all endogenous aPKC activity (green circles). These data reveal that S1P stimulates the activity of endogenous aPKC isozymes in HeLa cells by a mechanism independent of serum growth conditions.
Binding to protein scaffolds such as Par6 displaces the pseudosubstrate segment of aPKC isozymes, leading us to ask whether scaffold-activated aPKC was sensitive to S1P. CKAR fused to the PB1 domain of Par6 (CKAR-PB1Par6) displayed robust sensitivity to treatment of cells with PZ09 (Figure 2G, left panel, red circles), reflecting the high basal activity of aPKC on this scaffold (10, 12). Note that the basal activity on the scaffold was two-fold greater than that read out by the cytosolic reporter (CKAR; compare blue). Importantly, aPKC activity measured on the Par6 scaffold using CKAR-PB1Par6 was insensitive to inhibition of SphK (green circles). A similar result was obtained in MCF7 and MDA-MB-231 cells (Figure S2). Importantly, deletion of the PB1 domain of PKCζ, to prevent binding to the Par6 scaffold, restored S1P sensitivity to activity readout by CKAR-PB1Par6 (Figure 2H). These data reveal that aPKC bound to Par6 is not sensitive to S1P, contrasting with the sensitivity of cytosolic aPKC to S1P.
Intracellular S1P Mediates Cellular Activation of Atypical PKC
To confirm that the SKI-II-dependent inhibition of aPKC activity was a result of depleting intracellular S1P, we tested whether this inhibition could be rescued by acute release of S1P from photolysis of caged S1P. Figure 3A shows that addition of PZ09 to HeLa cells caused the characteristic ~3% drop in FRET (red circles); this FRET change was approximately 10-fold lower when cells were pre-treated with SKI-II overnight (yellow circles). This PZ09-dependent change in FRET for the SKI-II-treated cells could be restored to control (non-SKI-II pre-treatment) levels if cells were loaded with caged S1P (38, 39) and then exposed to UV light for 5 min to uncage the lipid (pink circles). This rescue of caged S1P-loaded cells was not observed in cells loaded with caged S1P that were not exposed to UV light (green circles). To further validate that the basal activity of aPKC results from S1P generated by SphK, we knocked down Sphk1 or Sphk2 by siRNA; analysis of mRNA revealed an approximately 50% knockdown of each (Figure 3B). Figure 3C shows that knockdown of either SphK1 (red) or SphK2 (yellow) caused a modest reduction in basal activity compared to that of control cells (blue), whereas knockdown of both SphK1 and Sphk2 (green) caused a greater than 80% reduction in basal activity relative to that of control cells (blue). Lastly, addition of caged S1P to the double knockdown cells resulted in a robust increase in basal activity, similar to that for control cells (red), following photolysis to uncage S1P (pink); no effect was observed in the absence of photolysis (green) (Figure 3D). These data demonstrate that S1P is necessary and sufficient for the basal activity of aPKC observed in this study. Additionally, knockdown and inhibitor studies are consistent with basally active SphK generating S1P to basally activate aPKC.
Fig. 3. Basal activity of aPKC is regulated by intracellular S1P.
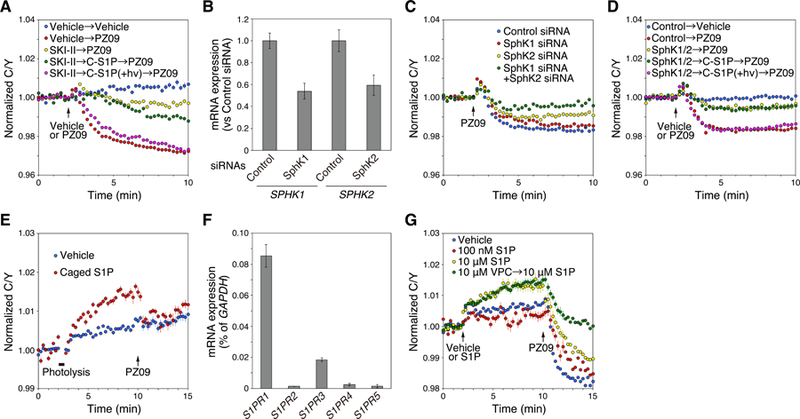
(A) HeLa cells were transfected with aCKAR and then pre-treated with DMSO vehicle or 5 µM SKI-II for 16 h; these cells were then loaded with 1 µM caged S1P (C-S1P) for 30 min, washed of extracellular caged S1P, exposed to ultraviolet light as described in methods, and incubated for another 5 min after photolysis (+ hν). Cells were subsequently treated with DMSO vehicle or 5 µM PZ09 to measure basal activity of endogenous aPKC. The normalized C/Y emission ratio was quantified as a function of time following DMSO vehicle or PZ09 treatment. Data represent the means ± S.E. (n ≥ 27 cells). The arrow indicates the point of DMSO vehicle or PZ09 addition. For graph legend: pre-treatment with vehicle, SKI-II, or caged S1P → treatment with vehicle or PZ09 performed at the time point indicated by the arrow.
(B) Knockdown of endogenous expression of SphK1 and SphK2 in HeLa cells using siRNAs for human SphK1 and SphK2. HeLa cells were transfected for 48 h with non-targeting control siRNA, human SphK1 siRNA, or human SphK2 siRNA individually. Relative mRNA expression levels of SPHK1 and SPHK2 in HeLa cells were analyzed by real-time quantitative PCR (RT-qPCR). Data represent the means ± S.E. from at least three independent experiments.
(C) HeLa cells were co-transfected with aCKAR and control siRNA, SphK1 siRNA, SphK2 siRNA, or both SphK1 siRNA and SphK2 siRNAs. The normalized C/Y emission ratio was quantified as a function of time following the addition of PZ09 (5 µM). Data represent the means ± S.E. (n ≥ 20 cells).
(D) HeLa cells were co-transfected with aCKAR and control siRNA (Control) or both SphK1 siRNA and SphK2 siRNAs (SphK1/2). They were then loaded with 1 µM caged S1P (C-S1P) for 30 min, washed of extracellular caged S1P, exposed to ultraviolet light as described in Methods, and incubated for another 5 min after photolysis (+hν). Cells were subsequently treated with DMSO vehicle or 5 µM PZ09 to measure basal activity of endogenous aPKC. The normalized C/Y emission ratio was quantified as a function of time following DMSO vehicle or PZ09 treatment. Data represent the means ± S.E. (n ≥ 57 cells). The arrow indicates the point of DMSO vehicle or PZ09 addition. For graph legend: transfection of control siRNA or SphK1 and SphK2 siRNAs → pre-treatment with caged S1P → treatment with vehicle or PZ09 performed at the time point indicated by the arrow.
(E) HeLa cells were transfected with aCKAR. Cells were then loaded with 1 µM caged S1P for 30 min, washed of extracellular caged S1P, and pre-treated with 10 µM VPC23019 for 5 min before live-cell imaging. Cells were then photolysed to detect intracellular S1P-induced activation of endogenous aPKC, and then stimulated with 5 µM PZ09. The normalized C/Y emission ratio was quantified as a function of time following photolysis. Data represent the means ± S.E. (n ≥ 37 cells).
(F) mRNA expression levels of S1PR1, S1PR2, S1PR3, S1PR4, and S1PR5 in HeLa cells were analyzed by real-time quantitative PCR (RT-qPCR). mRNA values for the S1P receptors were normalized to GAPDH. Data represent the means ± S.E. from at least three independent experiments.
(G) HeLa cells were transfected with aCKAR and then pre-treated with or without 10 µM VPC23019 (VPC) for 5 min before live-cell imaging. Cells were stimulated with DMSO vehicle, 100 nM S1P, or 10 µM S1P during live-cell imaging and then treated with 5 µM PZ09. The normalized C/Y emission ratio was quantified as a function of time following photolysis. Data represent the means ± S.E. (n ≥ 16 cells). For graph legend: pre-treatment with VPC23019 → treatment with S1P performed at the time point indicated by the first arrow.
We next asked if the basally active aPKC could be further stimulated upon addition of S1P. Figure 3E shows that uncaging of S1P in HeLa cells loaded with caged S1P resulted in an increase in aPKC activity (Figure 3E, red circles) relative to cells that had not been loaded with the caged lipid (Figure 3E, blue circles). This increase was reversed by addition of PZ09, revealing that addition of S1P induced the aPKC-dependent phosphorylation of aCKAR. To address whether extracellular S1P could also activate aPKC isozymes, we first determined which S1P receptors are expressed in HeLa cells in order to target them pharmacologically. Quantitative PCR analysis established that the major S1P receptor in HeLa cells is S1P1, with relatively low amounts of S1P3 and almost no significant amounts of S1P2, S1P4, and S1P5 (Figure 3F). Treatment with high concentration of S1P increases the total intracellular pools of S1P (26). Figure 3G shows that treatment of cells with relatively high concentrations of extracellular S1P (10 µM; yellow circles), but not low, physiologically-relevant concentrations (100 nM; red circles), caused an increase in aPKC activity. Importantly, this increase was not affected by co-treatment with a selective antagonist of the S1P1 and S1P3 receptors, VPC23019 (Figure 3G, green circles). Thus, the activation of aPKC by high concentrations of exogenously added S1P is independent of S1P receptor signaling and likely reflects accumulation of the lipid intracellularly. Taken together, these data reveal that aPKC is activated by intracellular, but not receptor-dependent extracellular, S1P.
S1P Directly Binds and Activates Atypical PKC
To gain insight into the mechanism by which intracellular S1P activates aPKC, we purified PKCζ to homogeneity and determined whether S1P stimulated its activity using an in vitro kinase assay. Addition of 30 µM S1P increased the rate of phosphorylation of myelin basic protein, a reaction that was linear up to 25 min (Figure 4A). Addition of increasing concentrations of S1P revealed dose-dependent activation, with 30 µM S1P doubling the rate of substrate phosphorylation (Figure 4B). This activation by S1P was reduced by 10 µM PZ09 (Figure 4C), was not sensitive to acyl chain unsaturation as dihydro-S1P (DH-S1P) also activated the enzyme (Figure 4D), and was specific for the phosphate group of S1P as sphingosine did not alter the activity of PKCζ (Figure 4E). To address the effect of S1P on the canonical phosphatidylserine (PS)-dependence of aPKC activation, we assessed the effect of trace S1P on the concentration of PS resulting in half-maximal activation of PKCζ using the well-characterized Triton X-100:lipid mixed micelle assay (40, 41). The presence of 5 mol% S1P reduced the concentration of PS resulting in half-maximal activity from 6.5 ± 0.8 mol% to 0.49 ± 0.01 mol% but did not affect the Vmax at saturating PS; this low concentration of S1P was also sufficient to double the basal activity in the absence of PS (Figure 4F). Consistent with the micelle data, the activation of PKCζ by PS in lipid bilayers (9, 42) was not further enhanced by S1P (Figure 4G). These results are consistent with S1P binding to aPKC and releasing autoinhibitory constraints. If this hypothesis is correct, we reasoned that the isolated kinase domain of PKCζ, PKMζ, should be insensitive to activation by S1P. Indeed, Figure 4H shows that SKI-II treatment had no effect on the PZ09-dependent inhibition of PKMζ activity (green triangles) compared to its effect on untreated cells (yellow triangles). In contrast, SKI-II reduced the PZ09-dependent inhibition of wild-type PKCζ (red circles) compared to untreated cells (blue circles). These data are consistent with S1P facilitating the activation of S1P by removing autoinhibitory constraints.
Fig. 4. Direct activation of aPKC by S1P.
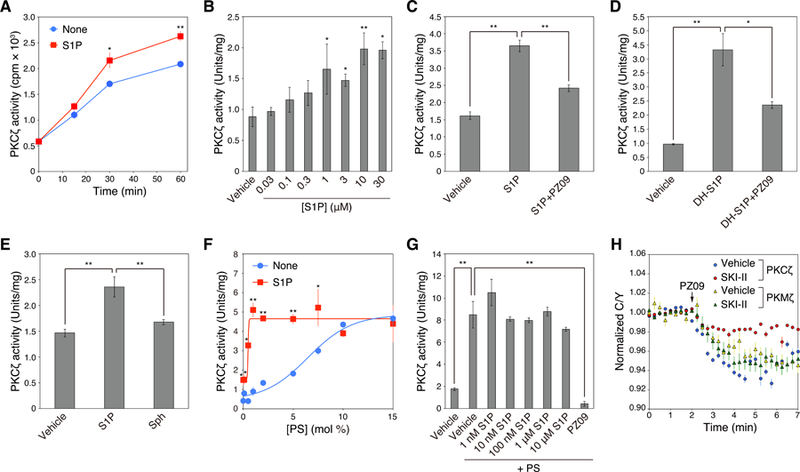
(A) Effect of S1P on activation of PKCζ assessed by an in vitro kinase assay. Kinase activity of purified GST-PKCζ was measured in the absence or presence of 30 µM S1P at the indicated time points. Data represent means ± S.E. from at least three independent experiments (*P < 0.05, **P < 0.01 for S1P versus vehicle control for each time point).
(B) Dose-dependent effects of S1P on activation of PKCζ assessed by an in vitro kinase assay. Kinase activity of purified GST-PKCζ was measured for 60 min in the absence or presence of the indicated concentrations of S1P. Data represent means ± S.E. from at least three independent experiments (*P < 0.05, **P < 0.01 for S1P versus vehicle control).
(C) Kinase activity of purified GST-PKCζ was measured in the absence or presence of 30 µM S1P or 30 µM S1P + 10 µM PZ09. Data represent means ± S.E. from at least three independent experiments (**P < 0.01).
(D) Kinase activity of purified GST-PKCζ was measured in the absence or presence of 30 µM dihydro-S1P (DH-S1P) or 30 µM DH-S1P + 10 µM PZ09. Data represent means ± S.E. from at least three independent experiments (*P < 0.05, **P < 0.01).
(E) Kinase activity of purified GST-PKCζ was measured in the absence or presence of 30 µM S1P or 30 µM sphingosine (Sph). Data represent means ± S.E. from at least three independent experiments (**P < 0.01).
(F) Kinase activity of purified GST-PKCζ was measured in the presence of Triton X-100 mixed micelles containing 0 – 15 mol% phosphatidylserine (PS) and 0 or 5 mol% S1P. Data represent mean ± S.E. from three independent experiments (*P < 0.05, **P < 0.01 for 5 mol% S1P versus 0 mol% S1P control).
(G) Kinase activity of purified GST-PKCζ was measured in the absence or presence of 140 µM phosphatidylserine (PS) with various concentrations of S1P or 10 µM PZ09. Data represent means ± S.E. from at least three independent experiments (**P < 0.01).
(H) HeLa cells were co-transfected with CKAR and mCherry-PKCζ or mCherry-PKMζ. Cells were pre-treated with DMSO vehicle or 5 µM SKI-II for 16 h, and then treated with 5 µM PZ09. The normalized C/Y emission ratio was quantified as a function of time following PZ09 treatment. Data represent the means ± S.E. (n ≥ 25 cells).
We next tested whether S1P directly binds to PKC. We developed a new assay to monitor the binding of nitrobenzoxadiazole (NBD)-labeled S1P to PKC using flow cytometry to detect protein with bound fluorescent lipid. Binding of NBD-S1P was detected on PKCδ and PKCζ but not PKCα and PKCγ (Figure 5A). We next measured the binding of (umodified) S1P to aPKC using a protein-lipid overlay (PLO) assay. The S1P-PKCζ binding was dose-dependent, with 300 pmol S1P resulting in maximal binding to 500 ng/ml PKCζ (Figure 5B). As a control, comparable S1P binding to the RING domain of TRAF2 (a known S1P-binding protein) was used (Figure 5C) (26). Binding was specific for the sphingosine backbone as sphingosine also bound, but no binding by LPA was detected (Figure 5D). Intermediate binding was observed with both ceramide and PS, known binding lipids for aPKC (Figure 5D) (13, 15, 43). Lastly, various domain deletion mutants of PKCζ were purified (Figure 5E), and their S1P binding was examined. Figure 5F reveals that S1P bound to all constructs that contained the kinase domain: wild-type (i.e. full length), one deleted in the PB1 domain (ΔPB1), one deleted in the pseudosubstrate (ΔPseudo), one deleted in the C1 domain (ΔC1), one deleted in the PDZ ligand (ΔPDZ), and one deleted in the regulatory moiety (ΔReg). However, binding of S1P to a construct in which the kinase domain was deleted (ΔCat) was not observed (Figure 5F, right panel). These data reveal that S1P binds the kinase domain of PKCζ.
Fig. 5. Direct binding of S1P to aPKC.
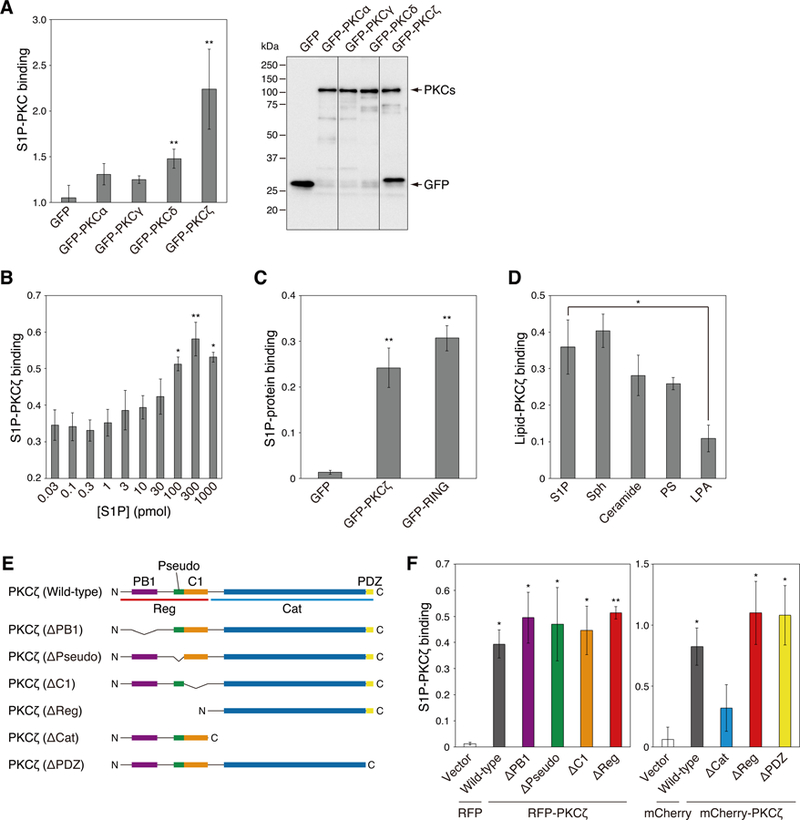
(A) COS7 cells were transfected with GFP, GFP-PKCα, GFP-PKCγ, GFP-PKCδ, or GFP-PKCζ. S1P-PKC binding ability was measured by a protein-lipid binding assay with nitrobenzoxadiazole (NBD)-labeled S1P (left panel). Data represent means ± S.E. from at least three independent experiments (*P < 0.05 versus GFP vector control). Immunoprecipitated GFP or GFP-PKC were run on SDS-PAGE (right panel). The amount of exogenously expressed GFP or GFP-PKC was detected via western blot analysis using an anti-GFP antibody.
(B) The binding affinity of PKCζ for S1P was assessed using the PLO assay. The interactions between purified GST-PKCζ and various concentrations of S1P were detected using an anti-GST antibody. Data represent means ± S.E. from at least three independent experiments (*P < 0.05, **P < 0.01 versus 0.03 pmol S1P).
(C) The binding affinity of PKCζ or the RING domain of TRAF2 for S1P was assessed by a PLO assay. The interaction between GFP, GFP-PKCζ, or GFP-tagged RING domain of TRAF2 (GFP-RING) and 30 pmol of S1P was detected using an anti-GST antibody. Data represent means ± S.E. from at least three independent experiments (**P < 0.05 versus GFP vector control).
(D) The binding affinity of PKCζ for related lipids was assessed using the PLO assay. The interactions between purified GST-PKCζ and 30 pmol S1P, sphingosine (Sph), C16-ceramide (Ceramide), phosphatidylserine (PS), or lysophosphatidic acid (LPA) were detected using an anti-GST antibody. Data represent means ± S.E. from at least three independent experiments (*P < 0.05).
(E) Domain schematic of deletion mutants of PKCζ. Structures of full-length PKCζ and six deletion mutants are shown. PB1: PB1 domain, Pseudo: pseudosubstrate domain, C1: C1 domain, PDZ: PDZ domain, Reg: regulatory domain, Cat: catalytic domain, N: N-terminus, C: C-terminus.
(F) The binding affinity of domain deletion mutants of PKCζ for S1P was assessed using the PLO assay. The interactions between RFP- or mCherry-fused full length PKCζ (RFP-PKCζ (wild-type) or mCherry-PKCζ (wild-type)), PB1 deletion mutant (ΔPB1), PS deletion mutant (ΔPseudo), C1 deletion mutant (ΔC1), regulatory domain deletion mutant (ΔReg) (PKMζ), catalytic domain deletion mutant (ΔCat), or PDZ deletion mutant (ΔPDZ) and 30 pmol of S1P were detected using an anti-GST antibody. The S1P-PKCζ binding affinity was compared to the RFP or mCherry vector control. Data represent means ± S.E. from at least three independent experiments (*P < 0.05, **P < 0.01 versus vector control).
We next set out to identify whether there is a specific binding pocket for S1P on the surface of the aPKC kinase domain by a strategy involving in silico prediction and experimental validation (Figure 6A). First, we constructed a homology model of the PKCζ kinase domain based on the crystal structure of PKCι (44) and used the Schrödinger’s SiteMap algorithm (45, 46) to screen the surface of the kinase for possible binding sites. This model identified five potential binding pockets on PKCζ (Figure 6B). Of these five potential sites, Pocket 5 was the ATP-binding site; therefore, only pockets 1, 2, 3, and 4 were selected for docking analysis. Induced fit docking (Schrödinger) (47–49), which accounts for the flexibility of the kinase, predicted the most energetically favorable docking pose for either S1P or sphingosine (Sph) in each of these four potential binding pockets. S1P and Sph did not dock to pocket 4 and thus no binding scores were generated. The five lowest binding scores for S1P or Sph are shown for pockets 1, 2, and 3 (Figure 6C). The lowest docking scores of S1P to binding site 1, 2, and 3 were −7.335 kcal/mol, −7.753 kcal/mol, and −8.583 kcal/mol, respectively. In comparison, the lowest docking scores of Sph to binding site 1, 2, and 3 were −7.066 kcal/mol, −6.586 kcal/mol, and −7.424kcal/mol, respectively. In pocket 1, only one score was lower than −6.500 kcal/mol, while in pocket 2 and 3, several scores were lower than this threshold. Additionally, in pocket 2 and 3, the docking scores for S1P were consistently better than the docking scores for Sph. These features make pocket 2 or 3 promising candidates for S1P-specific binding. The binding poses for pockets 1, 2, and 3 with the most favorable docking scores are illustrated in Figure 6D, and the two-dimensional interaction diagrams are shown in Figure 6E. A similar result was obtained for S1P-PKCι binding (Figure S3). From these poses, we identified the following basic amino acid residues, lysine (Lys: K) and arginine (Arg: R), predicted to interact with S1P: Lys265, Lys284, and Arg325 in pocket 1, Arg375 and Lys399 in pocket 2, and Lys513 in pocket 3. We then mutated these basic residues in full length PKCζ to neutral glutamine (Gln: Q) and monitored the effect of these mutations on S1P-induced activity in live cells. Figure 6F shows that the pocket 1 mutations (yellow circles) and the pocket 3 mutation (pink circles) resulted in activation similar to wild-type PKCζ (red circles). However, the mutation of the residues in pocket 2 (green circles) abolished activation above endogenous levels (blue circles). Of the two residues predicted to interact with S1P in pocket 2, Arg375 is the most critical: the single mutation R375Q (green circles), but not K399Q (pink circles), abolished the S1P-dependent activation of overexpressed PKCζ analogously to R375Q/R399Q (yellow circles) (Figure 6G). These data identify binding pocket 2 as an allosteric binding site that promotes S1P-dependent activation of PKCζ in cells. To validate that this is the site responsible for the S1P-induced activation of pure enzyme, we purified the PKCζ R375Q/R399Q mutant and examined its stimulation by S1P vs PS. Figure 6H shows that concentrations of S1P that resulted in near maximal activation of wild-type PKCζ, had no effect on the activity of the R375Q/R399Q mutant. We note that the PS-dependent activity of this mutant was approximately half that of wild-type enzyme under the conditions of the assay, likely reflecting mutation of R375 which is part of a conserved HRD motif in protein kinases (50); Q is found at this position in some kinases (51). Nonetheless, the mutant had sufficient activity to establish it was no longer sensitive to S1P. To confirm that this binding pocket is driving basal activation of aPKC by basal S1P, we examined how a mutation at this site affected SKI-II-sensitive signaling. Figure 6I shows that SKI-II pre-treatment had no effect on PZ09-mediated inhibition of PKCζ activity for the pocket 2 mutant (right panel, red circles), whereas it reduced the PZ09-mediated inhibition of wild-type PKCζ (left panel, red circles). As a control, the pocket 2 mutant had the same sensitivity to PI3K inhibition (green circles) as the wild-type enzyme. Thus, basic amino acid mutation of pocket 2 specifically prevented the activation of aPKC by basal S1P. Lastly, we show that the PKCζ R375Q/R399Q mutant has similar activity on the Par6 scaffold as wild-type enzyme (Figure 6J). Note that although the mutation decreased the Vmax 2-fold in vitro, its activity in a cellular context was similar to that of wild-type enzyme likely because the CKAR readout is saturated in overexpression studies.
Fig. 6. Identification of critical sites and amino acids for binding of aPKC to S1P.
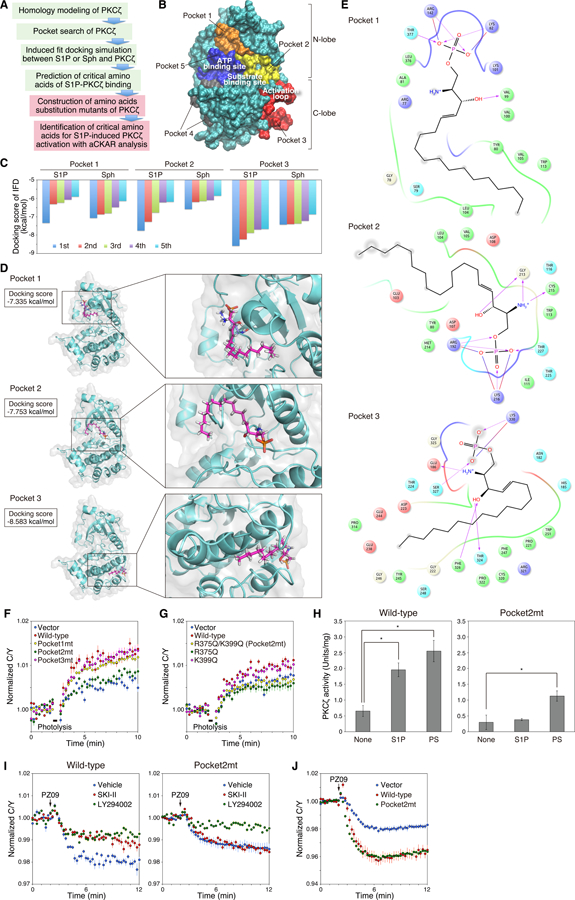
(A) Flowchart of the strategy for identifying the critical sites and amino acids of PKCζ for S1P binding and S1P-induced activation are shown. 1) Homology modeling of the catalytic domain of PKCζ from the crystal structure of PKCι, 2) A search of potential ligand-binding pockets of PKCζ, 3) Induced-fit docking simulation between S1P or sphingosine (Sph) and catalytic domain of PKCζ, 4) Identification of candidate critical pockets and amino acids for S1P-PKCζ binding, 5) Construction of single amino acid substitution mutants of candidate amino acids of S1P-PKCζ binding, 6) Identification of amino acids that are essential for S1P-PKCζ binding and S1P-induced activation of PKCζ using aCKAR-FRET analysis. The green boxes indicate in silico assay and the red boxes indicate cellular assay.
(B) Potential ligand-binding pockets on the surface of the homology model of the catalytic domain of PKCζ predicted using the Schrödinger’s SiteMap algorithm.
(C) S1P or sphingosine (Sph) was docked to the center of mass position for pocket 1, pocket 2, or pocket 3 on the catalytic domain of PKCζ using the induced-fit docking protocol in the Schrödinger package. The five lowest docking scores from the induced-fit docking for each pocket are shown in the bar graph (kcal/mol). IFD: induced-fit docking.
(D) The induced-fit docking poses of S1P (carbon atoms in pink) to pocket 1, pocket 2, and pocket 3 of the catalytic domain of human PKCζ are shown with corresponding docking scores. These docking poses have the lowest docking score (kcal/mol) in each pocket.
(E) Two-dimensional interaction diagram of the S1P-PKCζ binding as in (D). Negatively-charged, positively-charged, polar, hydrophobic, and glycine residues at the active site are represented by red, purple, cyan, green, and white spheres, respectively. Hydrogen bonds between the S1P and backbone or side chains are shown in solid pink arrows or dashed pink arrows, respectively. Salt bridges are shown in red-blue lines. Lys82, Lys101, and Arg142 in the pocket 1 correspond to Lys265, Lys284, and Arg325 of full length PKCζ. Arg192 and Lys216 in the pocket 2 correspond to Arg375 and Lys399 of full length PKCζ. Lys330 in the pocket 3 corresponds to Lys513 of full-length PKCζ.
(F) HeLa cells were co-transfected with aCKAR and mCherry (Vector), mCherry-PKCζ (wild-type), mCherry-PKCζ(K265Q/K284Q/R325Q) (mutant for pocket 1; Pocket1mt), mCherry-PKCζ(R375Q/K399Q) (mutant for pocket 2; Pocket2mt), or mCherry-PKCζ(K513Q) (mutant for pocket 3; Pocket3mt). Cells were then loaded with 1 µM caged S1P for 30 min, washed, and pre-treated with 10 µM VPC23019 for 5 min before live-cell imaging. Cells were photolysed to detect intracellular S1P-induced activation of exogenous mCherry-PKCζ. The normalized C/Y emission ratio was quantified as a function of time following photolysis. Data represent the means ± S.E. (n ≥ 27 cells).
(G) As in (F), except HeLa cells were co-transfected with aCKAR and mCherry (Vector), mCherry-PKCζ (Wild-type), mCherry-PKCζ(R375Q/K399Q) (Pocket2mt), mCherry-PKCζ(R375Q), or mCherry-PKCζ(K399Q) mutant. Data represent the means ± S.E. (n ≥ 17 cells).
(H) Kinase activity of purified GST-PKCζ (Wild-type) or GST-PKCζ(R375Q/K399Q) (Pocket2mt) was measured in the absence or presence of 30 µM S1P or 140 µM phosphatidylserine (PS). Data represent means ± S.E. from at least three independent experiments (*P < 0.05).
(I) HeLa cells were co-transfected with aCKAR and mCherry-PKCζ (Wild-type) (left panel) or mCherry-PKCζ(R375Q/K399Q) (Pocket2mt) (right panel). Cells were then pre-treated with DMSO vehicle, 20 µM LY294002, or 5 µM SKI-II for 16 h and then treated with 5 µM PZ09 during live-cell imaging to detect basal activity of exogenous mCherry-PKCζ. The normalized C/Y emission ratio was quantified as a function of time following PZ09 treatment. Data represent the means ± S.E. (n ≥ 25 cells).
(J) HeLa cells were co-transfected with CKAR-PB1Par6 and mCherry (Vector), mCherry-PKCζ (Wild-type), or mCherry-PKCζ(R375Q/K399Q) (Pocket2mt). Cells were then treated with 5 µM PZ09 during live-cell imaging to detect basal activity of exogenous mCherry-PKCζ. The normalized C/Y emission ratio was quantified as a function of time following PZ09 treatment. Data represent the mean ± S.E. (n ≥ 16 cells).
S1P Mediated Activation of Atypical PKC Protects Cells from Apoptosis
We next addressed the physiological function for S1P-mediated activation of PKCζ. Specifically, we asked whether S1P-dependent activation of PKCζ regulated apoptosis or necrosis. Treatment of HeLa cells (grown in 10% serum) with inhibitors of either SphK (SKI-II) or PI3K (LY294002) alone had no significant effect on the number of apoptotic or necrotic cells, as assessed by staining with Apopxin Green, which serves as a marker for apoptosis by detecting exposed PS, or 7-aminoactinomycin D (7-AAD, a fluorescent DNA intercalator) which serves as measure of necrosis (Figure 7A–B). However, inhibiting both SphK and PI3K caused a significant increase in the number of apoptotic cells but not necrotic cells. Inhibition of aPKC with PZ09 also increased apoptosis, suggesting that the effects of SKI-II and LY294002 could be mediated by inhibition of aPKC (Figure 7A–B). We also investigated chromatin condensation, which correlates with apoptotic morphology (52), in HeLa cells with Hoechst 33342 staining and obtained similar results using PS exposure as a measure of apoptosis (Figure 7C, Figure S4A). To examine the contribution of S1P in the absence of PI3K effects (Figure 2F), serum-starved cells were treated with SKI-II. In the absence of serum, SKI-II treatment caused a significant increase in apoptosis and cells with condensed DNA (Figure 7D–F, Figure S4B). Thus, under conditions of serum starvation, inhibition of SphK promotes apoptosis.
Fig. 7. S1P-induced basal activity of aPKC is involved in apoptosis resistance.
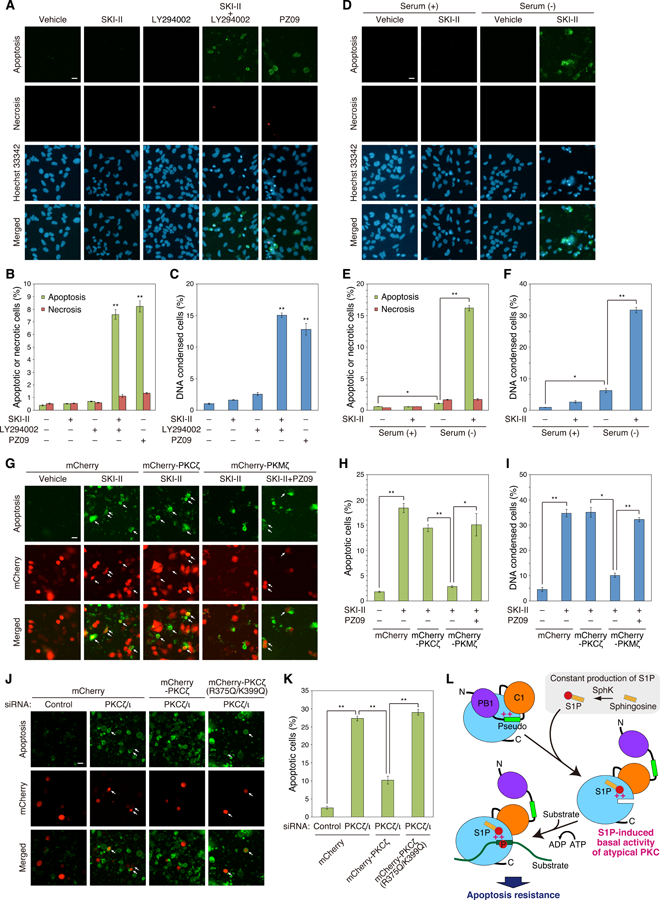
(A) HeLa cells were treated with DMSO vehicle, 5 µM SKI-II, 20 µM LY294002, 5 µM SKI-II+20 µM LY294002, or 5 µM PZ09 for 12 h. Cells were stained with Hoechst 33342, Apopxin Green (for phosphatidylserine exposure (Apoptosis)), and 7-aminoactinomycin D (for loss of plasma integrity (Necrosis)), then observed using fluorescence microscopy. Scale bar, 20 µm.
(B) Percentage of the Apopxin Green- or 7-aminoactinomycin D-positive cells was quantified respectively from the results in (A). Data represent means ± S.E. from at least three independent experiments (**P < 0.01 versus vehicle control).
(C) HeLa cells were treated with DMSO vehicle, 5 µM SKI-II, 20 µM LY294002, 5 µM SKI-II+20 µM LY294002, or 5 µM PZ09 for 24 h. DNA condensation was observed using Hoechst 33342 staining under fluorescence microscopy, and the percentage of condensed cells was quantified. Data represent means ± S.E. from at least three independent experiments (**P < 0.01 versus vehicle control).
(D) HeLa cells were treated with DMSO vehicle or 5 µM SKI-II in the presence or absence of serum for 12 h. Cells were stained with Hoechst 33342, Apopxin Green and 7-aminoactinomycin D, then observed using fluorescence microscopy. Scale bar, 20 µm.
(E) Percentage of the Apopxin Green- or 7-aminoactinomycin D-positive cells was quantified respectively from the results in (D). Data represent means ± S.E. from at least three independent experiments (*P < 0.05, **P < 0.01).
(F) HeLa cells were treated with DMSO vehicle or 5 µM SKI-II in the presence or absence of serum for 24 h. DNA condensation was observed using Hoechst 33342 staining under fluorescence microscopy, and the percentage of condensed cells was quantified. Data represent means ± S.E. from at least three independent experiments (*P < 0.05, **P < 0.01).
(G) HeLa cells were transfected with mCherry (Vector), mCherry-PKCζ, or mCherry-PKMζ. Cells were treated with DMSO vehicle, 5 µM SKI-II, or 5 µM SKI-II + 5 µM PZ09 in serum-free conditions for 12 h. Cells were stained with Apopxin Green, then observed using fluorescence microscopy. Arrows indicate both Apopxin Green- and mCherry-positive cells (apoptotic cells in mCherry-expressed cells). Scale bar, 20 µm.
(H) Percentage of the Apopxin Green-positive cells within the population of mCherry-positive cells was quantified from the results in (G). Data represent means ± S.E. from at least three independent experiments (*P < 0.05, **P < 0.01).
(I) HeLa cells were transfected with mCherry (Vector), mCherry-PKCζ, or mCherry-PKMζ. Cells were treated with DMSO vehicle, 5 µM SKI-II, or 5 µM SKI-II +5 µM PZ09 in serum-free conditions for 24 h. DNA condensation was observed using Hoechst 33342 staining under fluorescence microscopy, and the percentage of condensed cells was quantified. Data represent means ± S.E. from at least three independent experiments (*P < 0.05, **P < 0.01).
(J) HeLa cells were co-transfected with control siRNA or both PKCζ siRNA and PKCι siRNAs and mCherry (Vector), mCherry-PKCζ, or mCherry-PKCζ(R375Q/K399Q). Cells were then serum-starved with serum-free medium for 12 h. Cells were stained with Apopxin Green, then observed using fluorescence microscopy. Arrows indicate both Apopxin Green- and mCherry-positive cells (apoptotic cells in mCherry-expressing cells). Scale bar, 20 µm.
(K) Percentage of the Apopxin Green-positive cells within the population of mCherry-positive cells was quantified from the results in (J). Data represent means ± S.E. from at least three independent experiments (**P < 0.01).
(L) Model showing mechanism for S1P-mediated activation of aPKC to provide a basal signaling output that suppresses apoptosis. aPKC is autoinhibited by interaction of the pseudosubstrate (Pseudo) with the substrate-binding cavity of the kinase domain (blue circle) (upper left). S1P, constitutively produced from sphingosine by sphingosine kinase (SphK), binds to a pocket with basic amino acids (++), R375 and K399, close to the substrate-binding site in the kinase domain; this interaction displaces the pseudosubstrate to allow substrate binding and down-stream signaling. This S1P-induced basal activitation of aPKC promotes resistance to apoptosis. PB1: Phox and Bem1, P: phosphate, N: N-terminus, C: C-terminus.
To dissect out the specific contribution of aPKC activation by S1P in suppressing apoptosis, we asked whether SKI-II-induced apoptosis could be rescued by constitutively active PKMζ. SKI-II treatment induced apoptosis in cells overexpressing PKCζ but was ineffective in inducing apoptosis in cells overexpressing PKMζ (Figure 7G–I, Figure S4C). In contrast, cells overexpressing PKMζ had increased apoptosis upon treatment with SKI-II and PZ09. The sensitivity of PKCζ-overexpressing cells, but not PKMζ-overexpressing cells, to SKI-II-induced apoptosis is consistent with S1P basal activation of aPKC through relief of autoinhibition to suppress apoptosis. To validate the role of the S1P binding site in the cellular function of aPKC, we used siRNA to deplete HeLa cells of both PKCζ and PKCι. Knockdown of both aPKC isozymes caused a 10-fold increase in the number of apoptotic cells (Figure 7J), from 2.6 ± 0.7% to 27 ± 1% (Figure 7K). Overexpression of mCherry-tagged wild-type PKCζ reduced the number of apoptotic cells by 3-fold, to 10 ± 2%. In contrast, overexpression of the mCherry-tagged R375Q/R399Q mutant was unable to rescue the increased apoptosis resulting from depletion of endogenous aPKC. These results are consistent with aPKC suppressing apoptosis by a mechanism that depends on its ability to bind S1P.
DISCUSSION
We have identified intracellular S1P as a lipid second messenger that allosterically activates aPKC isozymes. Using a genetically-encoded activity reporter we developed specifically for aPKC in cells, we show that 1] intracellular S1P enables signaling of aPKC and 2] photolytic release of S1P from a caged lipid probe results in aPKC activation. This S1P-dependent activation of aPKC is not due to S1P receptor activation by extracellular S1P. Rather, it results from direct binding of intracellular S1P to the kinase domain, an event that relieves autoinhibitory constraints. Using molecular docking simulations, we identified a conserved S1P binding pocket on the kinase domains of PKCζ and PKCι/λ which we validated biochemically. Lastly, cellular studies reveal that inhibition of SphK increases apoptosis in cells overexpressing PKCζ but not PKMζ, an alternative splice variant that lacks autoinhibitory constraints. These data are consistent with a model in which S1P binds the kinase domain of aPKC isozymes to partially activate these kinases, with this enhanced activity contributing to the suppression of apoptosis. Thus, S1P is a lipid mediator that modulates aPKC function.
The lack of small molecules to modulate aPKC activity in cells has made identifying substrates, measuring cellular activity, and determining signaling pathways considerably more challenging than for the diacylglycerol-dependent isozymes. For the latter, phorbol esters, functional analogues of diacylglycerol, revolutionized the study of the conventional and novel PKC isozymes by providing a tool to acutely activate these enzymes (53). The recent development and validation of a small molecule inhibitor, PZ09, provided the first tool to successfully interrogate aPKC signaling in cells (12, 33, 54). However, the lack of specific agonists for aPKC activation made it challenging to use existing pan-PKC reporters for PKC activity, such CKAR (29), which reports on the activity of all PKC isozymes (31). Here, we engineered CKAR to specifically interrogate the spatio-temporal dynamics of aPKC signaling. This reporter, aCKAR, measures aPKC activity but not the activity of other PKC isozymes; nor does it report the activity of the related AGC kinases Akt or PKA. The combination of a reporter tailored for aPKC and an aPKC-selective inhibitor allowed us not only to assess the basal activity of aPKC isozymes in a panel of cell lines, but also to examine activation mechanisms of aPKC in cells. We found that basal aPKC activity was highest in the more aggressive breast cancer cell line MDA-MB-231 compared to the less aggressive MCF7 cell line, and lowest in the liver cell line HepG2 and the neuroblastoma cell line SH-SY5Y. In this study, we chose HeLa cells, which display readily detectable basal activity, to examine signaling mechanisms of aPKC in cells.
Live-cell imaging studies identify intracellular S1P as an activator of aPKC isozymes. This lipid mediator accounts for a significant fraction of the basal activity of aPKC isozymes, with basal signaling by PI3K accounting for the remainder. Basally active aPKC is further stimulated by an acute increase of S1P, as revealed by photolytic release of this lipid from caged S1P. Biochemical studies reveal that S1P directly binds the kinase domain of aPKC. This interaction relieves autoinhibitory constraints, as assessed by the ability of S1P to stimulate the activity of purified aPKC approximately 2-fold. Additionally, it decreases the concentration of PS necessary to half-maximally activate pure aPKC. Whereas ceramide has been previously reported to activate aPKC (15, 55, 56), the mechanism we report here is highly specific for S1P, which binds to a unique and defined pocket in the kinase domain to facilitate release of autoinhibitory constraints.
As is the case for all PKC isozymes, aPKC isozymes are maintained in an autoinhibited conformation by interaction of a pseudosubstrate segment in the substrate-binding cavity (Figure 7L, top left). Mechanisms that break intramolecular contacts of the pseudosubstrate result in substrate binding and downstream signaling. Whereas conventional and novel PKC isozymes are activated by binding the second messenger diacylglycerol, aPKC isozymes are activated by binding protein scaffolds through their PB1 domain. But whether or not there are second messengers that contribute to release of the aPKC pseudosubstrate has remained unclear. Here we show that S1P binds to a basic pocket on the surface of aPKC isozymes, disrupting autoinhibitory constraints (Figure 7L middle right) to allow substrate phosphorylation (Figure 7L bottom left). Thus, S1P is an allosteric activator of aPKC isozymes. Consistent with S1P facilitating pseudosubstrate release and accompanying activation, aPKC bound to the scaffold Par6 is insensitive to S1P; this is unsurprising as aPKC bound to this scaffold is already in an open and active conformation (11, 12). Whether scaffolded aPKC dynamically exchanges with the cytosolic pool, and whether localized pools of S1P drive localized aPKC signaling, remains to be determined.
Functional studies in serum-starved HeLa cells are consistent with S1P-dependent activation of aPKC isozymes suppressing apoptosis. In cells in which PI3K is basally active (i. e. serum conditions), inhibition of SphK alone had no effect on apoptosis, but inhibition of PI3K unmasked a robust effect of SphK inhibition in suppressing apoptosis. Under these serum growth conditions, inhibition of PI3K alone also had no effect on apoptosis, but inhibition of both SphK and PI3K resulted in a marked increase in apoptosis. The ability of the SphK inhibitor to promote apoptosis is driven in large part by preventing the S1P-dependent activation of aPKC: the SphK inhibitor effectively increased apoptosis in cells overexpressing PKCζ but had no effect on cells overexpressing the constitutively active kinase domain, PKMζ. For cells overexpressing PKMζ, treatment with the active site inhibitor, PZ09, induced a robust increase in apoptosis. Taken together, these data support a model in which basal S1P binding to aPKC activates the enzyme, with one consequence being the suppression of apoptosis.
The ability of S1P to suppress apoptosis is consistent with the mounting body of literature establishing S1P as a mediator of survival signaling and SphK as an oncogene (17). Clinical data reveal that in renal and breast cancer, high levels of SphK are associated with poor patient outcome (23, 57). Of the two aPKC isozymes, PKCι/λ serves as an oncogene in diverse cancers and diverse contexts (5). Although a number of loss-of-function mutations have been identified in cancers for both aPKC isozymes (4), our data reveal one context in which aPKC signaling is oncogenic. Importantly, our data identify an axis between oncogenic S1P signaling and oncogenic aPKC signaling.
The activation of aPKC isozymes by S1P unveils a context in which these isozymes may function as oncoproteins. Although the cancer-associated mutations in PKCζ and PKCι/λ that have been characterized to date are loss-of-function (suggesting a tumor suppressor role), considerable evidence supports an oncogenic function for these isozymes in certain contexts (58–61). Two particular cancers where PKCι/λ may be oncogenic are lung and brain cancers. In lung cancer, Fields and coworkers have identified a role for PKCι/λ in phosphorylating SOX2, a master transcriptional regulator of stemness (62, 63). This isozyme is also tumor-initiating in K-Ras-mediated lung adenocarcinomas by phosphorylating ELF-3 to control Notch expression (62). In patients with glioblastoma, high aPKC immunoreactivity (primarily for PKCι/λ) correlates with poor disease prognosis; furthermore, an aPKC inhibitor reduced tumor growth in a mouse model of glioblastoma (54). In contrast, aPKC isozymes may function as tumor suppressors in other cancers. Loss of PKCζ in intestinal cells promotes metabolic reprogramming associated with a cancer phenotype (64, 65). PKCι/λ has also been proposed to have a tumor suppressive function in the intestine: this isozyme is lost in the intestinal epithelium of patients with Crohn’s disease, a pathology associated with a high risk of cancer (66). Furthermore, mice lacking PKCι/λ in their intestinal epithelium have increased inflammation and tumorigenesis (66). Given that aPKC isozymes may function as both tumor suppressors and oncoproteins depending on the context, the link to S1P signaling suggests that patients with oncogenic SphK mutations may benefit from inhibition of aPKC signaling.
MATERIALS AND METHODS
Chemicals and antibodies
Phorbol 12, 13-dibutyrate (PDBu), forskolin, calyculin A, and LY294002 were purchased from Calbiochem. EGF was purchased from Upstate Biotechnology. PZ09 was obtained from Reagency. SKI-II, Lysophosphatidic acid (LPA), and cOmplete Mini Protease Inhibitor Cocktail were purchased from Sigma-Aldrich. Caged S1P was purchased from Santa Cruz. VPC23019 was purchased from Cayman Chemical. S1P, dihydro-S1P, Sphingosine, and C16-ceramide were purchased from Enzo Life Sciences. Nitrobenzoxadiazole (NBD)-labeled S1P and phosphatidylserine (PS) were purchased from Avanti Polar Lipids. The anti-PKCζ antibody was purchased from Santa Cruz (C-20, SC-216). The anti-PKCι antibody was purchased from BD (Clone 23, 610176). The anti-β-actin antibody was purchased from Sigma-Aldrich (clone AC-74, A2228). The anti-GFP (Clone mFX75, 012–22541) and anti-GST (Clone 5A7, 013–21851) antibodies were purchased from Wako Pure Chemical Industries. All other materials were reagent-grade.
Plasmid constructs and mutagenesis
A Kinase Activity Reporter (AKAR) was a kind gift from Dr. Roger Tsien (UCSD) (34). C kinase activity reporter (CKAR), B kinase activity reporter (BKAR), D kinase activity reporter (DKAR), and CKAR-PB1Par6 were described previously (12, 29, 35, 36). aPKC-specific CKAR (aCKAR) was generated through substitution of an aPKC substrate sequence, AKLLGMTFMIRSSG, for the substrate sequence within CKAR. The phospho-acceptor threonine was mutated to an alanine to create aCKAR-T/A using the QuikChange mutagenesis protocol (Stratagene). Human PKCζ, human PKMζ, human PKCζ(ΔPDZ) (residues 1–589 of human PKCζ), human PKCα, rat PKCβII, human PKCδ, human PKCε, mouse Akt1, and human PKD1 constructs were amplified and cloned into the pDONR221 vector; these were then recombined with various pDEST vectors constructed in-house to make fusion proteins with an N-terminal mCherry tag inus in pcDNA3 for mammalian cell expression, or an N-terminal GST-tag in a pFastBac backbone vector for insect cell expression using the Gateway cloning system (Invitrogen) (9, 12, 67, 68). mCherry-PKCζ(ΔCat) was made by removing the residues from 184 to 592 of full-length mCherry-PKCζ. Rat PKCζ, rat PKMζ, or mouse PKCλ (residues 136–595 of mouse PKCλ) were amplified and cloned into an N-terminal monomeric red fluorescent protein (mRFP)-tagged pcDNA3 vector for making RFP-PKCζ, RFP-PKMζ, or RFP-catalytic domain of PKCλ (RFP-PKCλ(Cat)). RFP-PKCζ(ΔPB1), RFP-PKCζ(ΔPseudo), or RFP-PKCζ(ΔC1) were made by removing the residues from 25 to 106 for ΔPB1, from 113 to 130 for ΔPS, or from 131 to 183 for ΔC1 of full-length RFP-rat PKCζ respectively. mCherry-PKCζ(ΔPB1) was made by removing the residues from 25 to 106 of full-length mCherry-PKCζ. For mCherry-PKCζ(K265Q/K284Q/R325Q), mCherry-PKCζ(R375Q/K399Q), mCherry-PKCζ(K513Q), mCherry-PKCζ(R375Q), and mCherry-PKCζ(K399Q), lysine or arginine were mutated to a glutamine using the QuikChange mutagenesis protocol. GFP-PKCα, GFP-PKCγ, GFP-PKCδ, and GFP-PKCζ were kindly provided by Dr. Naoaki Saito (Kobe University). The RING domain of human TRAF2 (residues 28–101) was amplified and cloned into pmCherry-N1 (Clontech) for making GFP-RING domain of TRAF2.
Short interfering RNAs (siRNAs)
For RNA interference, we used the following siRNAs: for hPKCζ, (5′-CGUGAUUGACCCUUUAACUdGdT-3′ and 5′-AGUUAAAGGGUCAAUCACGdCdG-3′; dX is deoxyribosylthymine throughout); for hPKCι, (5′-GUACUGUUGGUUCGAUUAAdAdA-3′ and 5′-UUAAUCGAACCAACAGUACdTdT-3′); for hSphK1, (5′-GGGCAAGGCCUUGCAGCUCdTdT-3′ and 5′-GAGCUGCAAGGCCUUGCCCdTdT-3′); for hSphK2, (5′-GCUGGGCUGUCCUUCAACCUdTdT-3′ and 5′-AGGUUGAAGGACAGCCCAGCdTdT-3′); and for non-targeting control, (5′-GGGCAAGGCUCUGAAGCUCdTdT-3′ and 5′-GAGCUUCAGAGCCUUGCCCdTdT-3′). The target sequence of the hPKCζ siRNA is in 3′-untranslated region of hPKCζ mRNA, and that of the other siRNAs, aside from the non-targeting control, are in the coding region of each respective mRNA. The siRNAs for hPKCζ and hPKCι were synthesized by Dharmacon, and the siRNAs for control, hSphK1, and hSphK2 were by Japan Bio Services.
Cell culture and transfection
COS7, HEK293, MCF7, MDA-MB-231, and SH-SY5Y cells were purchased from ATCC. HeLa cells were kindly gifted from Dr. Naoaki Saito (Kobe University). HepG2 cells were kindly gifted from Dr. Jerrold Olefsky (UCSD). Cells were maintained in Dulbecco’s modified Eagle’s medium (Mediatech) containing 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C in 5% CO2. Cells were plated onto sterilized plastic culture plates or glass-bottomed 35 mm culture dishes before transfection. Transient transfection was carried out using FuGENE HD (Promega), and all experiments were performed 2 to 3 days after the transfection. In case of knockdown experiments, transient transfection of siRNAs or both siRNAs and plasmids was carried out using Lipofectamine 2000 (Thermo Fisher Scientific), and all experiments were performed 48h after the transfection.
FRET imaging
Cells expressing kinase activity reporters were rinsed once with, and imaged in, Hank’s Balanced Salt Solution containing 1 mM CaCl2. Images were acquired on a Zeiss Axiovert microscope (Carl Zeiss) using a MicroMax digital camera (Roper Industries) controlled by MetaFluor software (Universal Imaging). Optical filters were obtained from Chroma Technology. Using a 10% neutral density filter, CFP and FRET images were obtained every 15 s through a 420/20-nm excitation filter, a 450-nm dichroic mirror, and a 475/40-nm emission filter (CFP) or 535/25-nm emission filter (FRET). YFP emission was also monitored as a control for photobleaching through a 495/10-nm excitation filter, a 505-nm dichroic mirror, and a 535/25-nm emission filter. Excitation and emission filters were switched in filter wheels (Lambda 10–2; Sutter Instrument). Integration times were 200 ms for CFP and FRET and 100 ms for YFP. Red fluorescent protein (RFP or mCherry) images were obtained through a 568/55-nm excitation filter, a 600-nm dichroic mirror, and a 653/95-nm emission filter. Images were reanalyzed using Metaflour Analyst (Universal Imaging). One region per cell was selected such that there was no net movement of the targeted reporter in and out of the selected region, and Metafluor Analyst was used to calculate the average FRET ratio within the selected region. The trace for each cell imaged was normalized to its t=0–2 min averaging baseline value. The normalized C/Y emission ratios were combined from three independent experiments and represented as the average of these corrected values ±S.E.
Loading of caged S1P
HeLa cells grown on glass-bottomed culture dishes were preloaded with 1 μM caged S1P for 30 min. After washing out the media, dishes were then flashed with ultraviolet light (254 nm) for 30 s from 5 cm distance for photolysis of caged S1P.
Real-time quantitative reverse transcription-PCR (RT-qPCR)
Total RNA was extracted from HeLa cells (2 × 106 cells) using NucleoSpin RNA II (Macherey-Nagel) according to the manufacturer’s instructions. Reverse transcription was carried out using 1 μg of RNA (ReverTra Ace qPCR RT kit, Toyobo). Real-time quantitative PCR (RT-q-PCR) was performed with SYBR Premix (Takara Bio) on ABI Prism 7000 (Applied Biosystems). The primer sequences (sense and antisense) were as follows: for human S1P1 (S1PR1), 5′-TTCCTGGTGTTAGCTGTGCTCAAC-3′ and 5′-TCGCTTGAATTTGCCAGCAGAGTC-3′; for human S1P2 (S1PR2), 5′-TGCGCCATTGTGGTGGAAAACC-3′ and 5′-TTGCCCAGAAACAGGTACATTGCC-3′; for human S1P3 (S1PR3), 5′-AGCAGCAACAATAGCAGCCACTC-3′ and 5′-AGTGCTGCGTTCTTGTCCATGATG-3′; for human S1P4 (S1PR4), 5′-AACTGCCTGTGCGCCTTTGAC-3′ and 5′-ATCACCAGGCAGAAGAGGATGTAGC-3′; for human S1P5 (S1PR5), 5′-TTCCTGCTGCTGTTGCTCGAC-3′ and 5′-TTCAGAAGTGAGTTGGCCATGGC-3′; for human sphingosine kinase 1 (SPHK1), 5′-AGCTTCCTTGAACCATTATGCTG-3′ and 5′-AGGTCTTCATTGGTGACCTGCT-3′; for human sphingosine kinase 2 (SPHK2), 5′-ATGAATGGACACCTTGAAGCAG-3′ and 5′-CATGGCCTTAGCCCTGACCAG-3′; for human glyceraldehyde 3-phosphate dehydrogenase (GAPDH), 5′-GCCATCAATGACCCCTTCATT-3′ and 5′-TCTCGCTCCTGGAAGATGG-3′. The expression of each mRNA was normalized to GAPDH mRNA expression.
Purification of GST-PKC
Baculoviruses were made in High Five or SF9 insect cells from pFastBac plasmids using the Bac-to-Bac expression system (Invitrogen). Batch purification using glutathione sepharose beads was used to purify the GST-tagged proteins from infected High Five or SF9 insect cell cultures. Briefly, cells were rinsed with PBS and lysed in 50 mM HEPES (pH 7.5), 100 mM NaCl, and 1 mM DTT (Buffer A) with 0.1% Triton X-100, 100 μM PMSF, 2 mM benzamidine, and 50 μg/ml leupeptin. The soluble lysate was incubated with glutathione resin beads (Novagen) for 30 min at 4°C. Protein-bound beads were washed three times in Buffer A and then eluted three times in Buffer A with 10 mM glutathione. Eluent was loaded in a 30 kDa Amicon Ultra centrifugal filter unit (Millipore) and washed/concentrated three times with Buffer B (20 mM HEPES (pH 7.5), and 1 mM DTT). Glycerol was added to 50% volume before measurement of GST-PKCζ concentration using BSA standards on a Coomassie Brilliant Blue stained gel and enzyme stocks were stored at −20°C.
In vitro kinase activity assay
PKC activity was assayed by measuring the incorporation of 32Pi from [γ−32P]ATP into substrate as described previously (9, 69). Kinase activity was measured in 80 μl reactions supplemented with PS, S1P or sphingosine at the concentrations indicated in figure legends. In some experiments, Triton X-100:lipid mixed micelles containing 0 – 15 mol% PS and 0 or 5 mol% of S1P in 1% Triton X-100 (Thermo Fisher Scientific) were made, as described previously (41), and diluted 10x in the reaction mixture. The reaction mixture contained 20 mM HEPES (pH 7.4), 5 mM MgCl2, 100 μM ATP, 10 μCi/ml [γ−32P]ATP (PerkinElmer), 50 μg/ml myelin basic protein (MBP, Sigma), 30 ng purified GST-PKCζ and the lipids indicated in the figure legends. The incubation was carried out for indicated times at 30 °C. Reactions were stopped by addition of 25 mM ATP, 25 mM EDTA (pH 8.0), and spotted on pieces of P81 Whatman cellulose phosphate filter paper. Filter papers were washed four times with 0.4% phosphoric acid before measurement using a scintillation counter (Beckman).
Western blots
Cells were harvested with lysis buffer (50 mM Na2HPO4, 1 mM sodium pyrophosphate, 20 mM NaF, 2 mM EDTA, 2 mM EGTA, 1% Triton X-100, 1 mM DTT, 200 µM benzamidine, 40 µg/ml leupeptin, and 1 mM PMSF). The detergent-solubilized cell lysate was obtained by centrifuging the whole cell lysate in a microcentrifuge at 13,000 rpm for 5 min. The detergent-solubilized lysates were separated on a 7.5% SDS-PAGE gel, blotted onto polyvinylidene difluoride membrane (Bio-Rad), and probed with an anti-PKCζ antibody (diluted 1:1000), anti-PKCι antibody (diluted 1:1,000), or anti-β-actin antibody (diluted 1:1,000), followed by incubation with HRP-conjugated secondary antibodies and further imaging using enhanced chemiluminescence with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). Images were acquired using FluorChem Q (Alpha Innotech). For the S1P-PKC binding assay, immunoprecipitated GFP or GFP-PKC bound with NBD-S1P samples were detergent-solubilized and separated on a 10% SDS–PAGE gel, blotted onto polyvinylidene difluoride membrane (Millipore), and probed with an anti-GFP antibody (diluted 1:1000). HRP-conjugated secondary antibody labeling of blots was detected using enhanced chemiluminescence with SuperSignal West Dura Extended Duration Substrate (Thermo Scientific). Images were acquired using LAS3000 (Fujifilm). Quantification of western blots was performed using ImageJ software (NIH).
In vitro PKC-S1P binding assay
COS7 cells transfected with empty vector, GFP, GFP-PKCα, GFP-PKCγ, GFP-PKCδ, or GFP-PKCζ were harvested and lysed in lysis buffer (10 mM Tris-HCl (pH 7.6), 150 mM NaCl, 0.05% Tween 20 with cOmplete, Mini Protease Inhibitor Cocktail), then sonicated on ice and centrifuged at 10,000 g for 30 min at 4°C, and the supernatant collected. GFP or GFP-PKC was immunoprecipitated using anti-GFP antibody affinity agarose beads (Nacalai Tesque) and immunoprecipitates washed with PBS. The immunoprecipitated GFP or GFP-PKC isozymes were mixed with 0.4 μg of NBD-S1P and rotated for 24 h at 4°C. After washing the beads by centrifugation, PKC bound to NBD-S1P was isolated using a chloroform/methanol method described previously (70). The aldehyde/sulfate latex beads (4 × 104) were added and the mixture was rotated overnight at 4°C. After washing the beads, NBD-S1P fluorescence on the aldehyde/sulfate latex beads was detected using FACSCalibur (BD) with an excitation wavelength of 488 nm. Total fluorescence intensity of NBD-S1P on aldehyde/sulfate latex beads was calculated and normalized with that of aldehyde/sulfate latex beads only. Finally, the S1P-PKC binding unit was calculated by ratio of none to GFP, or none to GFP-PKC isozymes.
Protein-lipid overlay (PLO) assay
Purified GST-PKCζ (500 ng/ml) was used to probe bovine serum albumin-blocked nitrocellulose filters (Echelon Research Laboratories) on which various phospholipids had been spotted. The filters were washed, and GST-PKCζ bound to the filters was immunostained with anti-GST antibody (Amersham Biosciences) and anti-mouse IgG (Alexa Fluor® 488 Conjugate) (Cell Signaling Technology) secondary antibody, then fluorescently detected using an EnSpire Multimode Plate Reader (PerkinElmer). For mammalian cell-expressed PKC, COS7 cells transfected with RFP-PKC or mCherry-PKC were harvested in lysis buffer (10 mM Tris-HCl (pH 7.6), 150 mM NaCl, 0.05% Tween 20 with cOmplete, Mini Protease Inhibitor Cocktail), sonicated on ice, and centrifuged at 10,000 x g for 30 min to collect supernatant. The cell lysates containing RFP-PKC or mCherry-PKC were applied to the S1P-spotted filters and allowed to incubate for 1 h at room temperature, filters were then washed, and the fluorescence of RFP or mCherry on the filter was detected using an EnSpire Multimode Plate Reader. The fluorescence intensity of S1P-bound RFP-PKC or mCherry-PKC on the filter was normalized to the fluorescence intensity of RFP-PKC or mCherry-PKC in the cell lysate.
Induced fit docking simulation
The crystal structure of PKCι (PDB: 4DC2) determined at a 2.40-Å resolution was used to construct a homology model of human PKMζ (NM_001033581) and 232–596 amino acids of human PKCι (NM_002740) using the SWISS-MODEL server (https://swissmodel.expasy.org) (71). The surface of the homology model of human PKCζ or human PKCι was screened for potential binding sites with Schrödinger’s SiteMap (Schrödinger Release 2016–1: SiteMap, Schrödinger, LLC) (45, 46). S1P and sphingosine were prepared by LigPrep in the Schrödinger package using default parameters (Schrödinger Release 2016–1: LigPrep, Schrödinger, LLC). These ligands were docked to the center of mass position of each identified site (1–5) using the Induced Fit Docking Protocol in Schrödinger (Schrödinger Release 2016–1: Schrödinger Suite 2016–1 Induced Fit Docking protocol; Glide, Schrödinger, LLC; Prime, Schrödinger, LLC) (47–49). The five lowest energy docking poses were further evaluated. The resulting model skeleton structures of the poses of S1P- or sphingosine-binding to PKCζ or PKCι were observed and analyzed using PyMOL software (MacPyMOL: PyMOL v1.8.6.0 Enhanced for Mac OS X, Schrödinger, LLC).
Chromatin condensation assay
After various treatments for 24 h, both attached and floating HeLa cells were collected in PBS, followed by staining for DNA using NucBlue Live ReadyProbes Reagent (Thermo Fisher Scientific); images of the fluorescence of Hoechst 33342 dye were obtained under a Zeiss Axiovert fluorescence microscope. The number of cells showing chromatin changes with two or more condensed chromatin fragments in their nuclei were manually counted using ImageJ software (NIH).
Apoptosis versus necrosis detection assay
HeLa cells were treated with various treatments for 12 h, and the cells were stained with the Apoptosis/Necrosis Detection Kit (Abcam) according to the manufacturer’s instructions. Images of Apopxin Green and 7-aminoactinomycin D (7-AAD) were obtained under a Zeiss Axiovert fluorescence microscope. The cells stained with Apopxin Green were counted as early stages of apoptotic cells, and the cells labeled with 7-AAD were counted as necrotic cells using ImageJ software (NIH).
Quantification and statistical analyses
All the statistical data are presented as mean ± S.E. The statistical significance of differences was determined with Student’s t test using GraphPad Prism 6 (GraphPad Software). p < 0.05 was considered to be statistically significant.
Supplementary Material
Fig. S1. Phosphatase sensitivity of aCKAR is greater than that of CKAR.
Fig. S2. S1P signaling does not affect the Par6-regulated basal activity of PKCζ in breast cancer cells.
Fig. S3. Identification of critical sites and amino acids for PKCι-S1P binding in silico.
Fig. S4. Apoptotic nuclear morphology images.
Acknowledgments:
We thank the late R. Y. Tsien (University of California, San Diego) for providing the AKAR, J. M. Olefsky (University of California, San Diego) for providing the HepG2 cells and N. Saito (Kobe University) for providing the constructs related to GFP-PKC isozymes and HeLa cells. We are grateful to J. E. Dixon (University of California, San Diego) and S. S. Taylor (University of California, San Diego) for sharing their equipment. We also thank all members of our laboratories for their helpful comments on this manuscript.
Funding: This work was supported by NIH GM122523 and NIH DK5444 to A.C.N., NIH GM31749 and NIH GM103426 and computing resources from SDSC to J.A.M., JSPS KAKENHI Grant Numbers JP26460341, JP15KK0307, and JP17K08594 to T.K., Kobe University Grant for International Research and Kobe University Grant for Japan-US Collaboration to T.K., Nakatani Foundation Technology Development Research Grant to T.K., JSPS KAKENHI Grant Number JP15K15066 to S.N., JSPS KAKENHI Grant Number JP15K07930 to T.O., and A.D.C., C.A.P. and A.V. were supported in part by the UCSD Graduate Training Program in Cellular and Molecular Pharmacology (T32 GM007752).
Footnotes
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Drummond ML, Prehoda KE, Molecular Control of Atypical Protein Kinase C: Tipping the Balance between Self-Renewal and Differentiation. J Mol Biol 428, 1455–1464 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suzuki A, Ohno S, The PAR-aPKC system: lessons in polarity. Journal of cell science 119, 979–987 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Diaz-Meco MT, Moscat J, The atypical PKCs in inflammation: NF-kappaB and beyond. Immunol Rev 246, 154–167 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Newton AC, Brognard J, Reversing the Paradigm: Protein Kinase C as a Tumor Suppressor. Trends in pharmacological sciences 38, 438–447 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parker PJ, Justilien V, Riou P, Linch M, Fields AP, Atypical protein kinase Ciota as a human oncogene and therapeutic target. Biochemical pharmacology 88, 1–11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qian J, Massion PP, Role of chromosome 3q amplification in lung cancer. J Thorac Oncol 3, 212–215 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Newton AC, Protein kinase C as a tumor suppressor. Semin Cancer Biol 48, 18–26 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le Good JA et al. , Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science 281, 2042–2045 (1998). [DOI] [PubMed] [Google Scholar]
- 9.Tobias IS et al. , Protein kinase Czeta exhibits constitutive phosphorylation and phosphatidylinositol-3,4,5-triphosphate-independent regulation. The Biochemical journal 473, 509–523 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsai LC et al. , Zeta Inhibitory Peptide Disrupts Electrostatic Interactions That Maintain Atypical Protein Kinase C in Its Active Conformation on the Scaffold p62. The Journal of biological chemistry 290, 21845–21856 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graybill C, Wee B, Atwood SX, Prehoda KE, Partitioning-defective protein 6 (Par-6) activates atypical protein kinase C (aPKC) by pseudosubstrate displacement. The Journal of biological chemistry 287, 21003–21011 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tobias IS, Newton AC, Protein Scaffolds Control Localized Protein Kinase Czeta Activity. The Journal of biological chemistry 291, 13809–13822 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bourbon NA, Yun J, Kester M, Ceramide directly activates protein kinase C zeta to regulate a stress-activated protein kinase signaling complex. J Biol Chem 275, 35617–35623 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Fox TE et al. , Ceramide recruits and activates protein kinase C zeta (PKC zeta) within structured membrane microdomains. J Biol Chem 282, 12450–12457 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Wang G, Krishnamurthy K, Umapathy NS, Verin AD, Bieberich E, The carboxyl-terminal domain of atypical protein kinase Czeta binds to ceramide and regulates junction formation in epithelial cells. The Journal of biological chemistry 284, 14469–14475 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang G et al. , Direct binding to ceramide activates protein kinase Czeta before the formation of a pro-apoptotic complex with PAR-4 in differentiating stem cells. J Biol Chem 280, 26415–26424 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Hannun YA, Obeid LM, Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol, 10.1038/nrm.2017.1107 (2017). [DOI] [PMC free article] [PubMed]
- 18.Le Stunff H, Milstien S, Spiegel S, Generation and metabolism of bioactive sphingosine-1-phosphate. J Cell Biochem 92, 882–899 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Maceyka M, Harikumar KB, Milstien S, Spiegel S, Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol 22, 50–60 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mendelson K, Evans T, Hla T, Sphingosine 1-phosphate signalling. Development 141, 5–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Proia RL, Hla T, Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J Clin Invest 125, 1379–1387 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strub GM, Maceyka M, Hait NC, Milstien S, Spiegel S, Extracellular and intracellular actions of sphingosine-1-phosphate. Adv Exp Med Biol 688, 141–155 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geffken K, Spiegel S, Sphingosine kinase 1 in breast cancer. Adv Biol Regul 67, 59–65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pyne NJ, Pyne S, Sphingosine 1-phosphate and cancer. Nat Rev Cancer 10, 489–503 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Xia P et al. , An oncogenic role of sphingosine kinase. Curr Biol 10, 1527–1530 (2000). [DOI] [PubMed] [Google Scholar]
- 26.Alvarez SE et al. , Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 465, 1084–1088 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hait NC et al. , Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325, 1254–1257 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Panneer Selvam S et al. , Binding of the sphingolipid S1P to hTERT stabilizes telomerase at the nuclear periphery by allosterically mimicking protein phosphorylation. Sci Signal 8, ra58 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Violin JD, Zhang J, Tsien RY, Newton AC, A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. J Cell Biol 161, 899–909 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahajan A et al. , Structure and function of the phosphothreonine-specific FHA domain. Sci Signal 1, re12 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Kajimoto T, Sawamura S, Tohyama Y, Mori Y, Newton AC, Protein kinase C {delta}-specific activity reporter reveals agonist-evoked nuclear activity controlled by Src family of kinases. J Biol Chem 285, 41896–41910 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ryu J et al. , Protein kinase C-zeta phosphorylates insulin-responsive aminopeptidase in vitro at Ser-80 and Ser-91. Arch Biochem Biophys 403, 71–82 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Trujillo JI et al. , 2-(6-Phenyl-1H-indazol-3-yl)-1H-benzo[d]imidazoles: design and synthesis of a potent and isoform selective PKC-zeta inhibitor. Bioorganic & medicinal chemistry letters 19, 908–911 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Zhang J, Hupfeld CJ, Taylor SS, Olefsky JM, Tsien RY, Insulin disrupts beta-adrenergic signalling to protein kinase A in adipocytes. Nature 437, 569–573 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Kunkel MT, Ni Q, Tsien RY, Zhang J, Newton AC, Spatio-temporal dynamics of protein kinase B/Akt signaling revealed by a genetically encoded fluorescent reporter. J Biol Chem 280, 5581–5587 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kunkel MT, Toker A, Tsien RY, Newton AC, Calcium-dependent regulation of protein kinase D revealed by a genetically encoded kinase activity reporter. The Journal of biological chemistry 282, 6733–6742 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.French KJ et al. , Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer research 63, 5962–5969 (2003). [PubMed] [Google Scholar]
- 38.Kajimoto T, Okada T, Miya S, Zhang L, Nakamura S, Ongoing activation of sphingosine 1-phosphate receptors mediates maturation of exosomal multivesicular endosomes. Nat Commun 4, 2712 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Usatyuk PV et al. , Photolysis of caged sphingosine-1-phosphate induces barrier enhancement and intracellular activation of lung endothelial cell signaling pathways. Am J Physiol Lung Cell Mol Physiol 300, L840–850 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hannun YA, Loomis CR, Bell RM, Activation of Protein Kinase C by Triton X-100 Mixed Micelles Containing Diacylglycerol and Phosphatidylserine. J. Biol. Chem 260, 10039–10043 (1985). [PubMed] [Google Scholar]
- 41.Orr JW, Newton AC, Interaction of protein kinase C with phosphatidylserine. 1. Cooperativity in lipid binding. Biochemistry 31, 4661–4667 (1992). [DOI] [PubMed] [Google Scholar]
- 42.Newton AC, Koshland DE Jr., Phosphatidylserine affects specificity of protein kinase C substrate phosphorylation and autophosphorylation. Biochemistry 29, 6656–6661 (1990). [DOI] [PubMed] [Google Scholar]
- 43.Callender JA, Newton AC, Conventional protein kinase C in the brain: 40 years later. Neuronal Signaling 1, NS20160005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang C, Shang Y, Yu J, Zhang M, Substrate recognition mechanism of atypical protein kinase Cs revealed by the structure of PKCiota in complex with a substrate peptide from Par-3. Structure 20, 791–801 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Halgren T, New method for fast and accurate binding-site identification and analysis. Chemical Biology & Drug Design 69, 146–148 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Halgren T, Identifying and Characterizing Binding Sites and Assessing Druggability. J Chem Inf Model 49, 377–389 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Farid R, Day T, Friesner RA, Pearlstein RA, New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorgan Med Chem 14, 3160–3173 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R, Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem 49, 534–553 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Sherman W, Beard HS, Farid R, Use of an induced fit receptor structure in virtual screening. Chemical Biology & Drug Design 67, 83–84 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Taylor SS, Keshwani MM, Steichen JM, Kornev AP, Evolution of the eukaryotic protein kinases as dynamic molecular switches. Philos Trans R Soc Lond B Biol Sci 367, 2517–2528 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McSkimming DI et al. , KinView: a visual comparative sequence analysis tool for integrated kinome research. Mol Biosyst 12, 3651–3665 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B, PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 7, 673–682 (2001). [DOI] [PubMed] [Google Scholar]
- 53.Newton AC, Protein kinase C: poised to signal. Am J Physiol Endocrinol Metab 298, E395–402 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kusne Y et al. , Targeting aPKC disables oncogenic signaling by both the EGFR and the proinflammatory cytokine TNFalpha in glioblastoma. Science signaling 7, ra75 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bourbon NA, Yun J, Kester M, Ceramide directly activates PKC zeta to regulate a stress-activated protein kinase signaling complex. The Journal of biological chemistry, (2000). [DOI] [PubMed]
- 56.Muller G et al. , PKC zeta is a molecular switch in signal transduction of TNF-alpha, bifunctionally regulated by ceramide and arachidonic acid. The EMBO journal 14, 1961–1969 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uhlen M et al. , A pathology atlas of the human cancer transcriptome. Science 357, eaan2507 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Butler AM et al. , Protein kinase C zeta regulates human pancreatic cancer cell transformed growth and invasion through a STAT3-dependent mechanism. PLoS One 8, e72061 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Paul A et al. , PKClambda/iota signaling promotes triple-negative breast cancer growth and metastasis. Cell Death Differ 21, 1469–1481 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Regala RP et al. , Atypical protein kinase Ciota plays a critical role in human lung cancer cell growth and tumorigenicity. J Biol Chem 280, 31109–31115 (2005). [DOI] [PubMed] [Google Scholar]
- 61.Urtreger AJ, Kazanietz MG, Bal de Kier Joffe ED, Contribution of individual PKC isoforms to breast cancer progression. IUBMB Life 64, 18–26 (2012). [DOI] [PubMed] [Google Scholar]
- 62.Ali SA, Justilien V, Jamieson L, Murray NR, Fields AP, Protein Kinase Ciota Drives a NOTCH3-dependent Stem-like Phenotype in Mutant KRAS Lung Adenocarcinoma. Cancer Cell 29, 367–378 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Justilien V et al. , The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. Cancer cell 25, 139–151 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Llado V et al. , Repression of Intestinal Stem Cell Function and Tumorigenesis through Direct Phosphorylation of beta-Catenin and Yap by PKCzeta. Cell Rep 10, 740–754 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ma L et al. , Control of nutrient stress-induced metabolic reprogramming by PKCzeta in tumorigenesis. Cell 152, 599–611 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakanishi Y et al. , Control of Paneth Cell Fate, Intestinal Inflammation, and Tumorigenesis by PKClambda/iota. Cell reports 16, 3297–3310 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Antal CE et al. , Cancer-Associated Protein Kinase C Mutations Reveal Kinase’s Role as Tumor Suppressor. Cell 160, 489–502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen D, Purohit A, Halilovic E, Doxsey SJ, Newton AC, Centrosomal anchoring of protein kinase C betaII by pericentrin controls microtubule organization, spindle function, and cytokinesis. J Biol Chem 279, 4829–4839 (2004). [DOI] [PubMed] [Google Scholar]
- 69.Kajimoto T et al. , Ceramide-induced apoptosis by translocation, phosphorylation, and activation of protein kinase Cdelta in the Golgi complex. The Journal of biological chemistry 279, 12668–12676 (2004). [DOI] [PubMed] [Google Scholar]
- 70.Fujita T et al. , Delta-catenin/NPRAP (neural plakophilin-related armadillo repeat protein) interacts with and activates sphingosine kinase 1. The Biochemical journal 382, 717–723 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bordoli L et al. , Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc 4, 1–13 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Phosphatase sensitivity of aCKAR is greater than that of CKAR.
Fig. S2. S1P signaling does not affect the Par6-regulated basal activity of PKCζ in breast cancer cells.
Fig. S3. Identification of critical sites and amino acids for PKCι-S1P binding in silico.
Fig. S4. Apoptotic nuclear morphology images.