

Abstract
The association of immunodeficiency-related vaccine-derived rubella virus (iVDRV) with cutaneous and visceral granulomatous disease has been reported in patients with primary immunodeficiency disorders (PIDs). The majority of these PID patients with rubella-positive granulomas had DNA repair disorders. To support this line of inquiry, we provide additional descriptive data on seven previously reported patients with Nijmegen breakage syndrome (NBS) (n = 3) and ataxia telangiectasia (AT) (n = 4) as well as eight previously unreported patients with iVDRV-induced cutaneous granulomas and DNA repair disorders including NBS (n = 1), AT (n = 5), DNA ligase 4 deficiency (n = 1), and Artemis deficiency (n = 1). We also provide descriptive data on several previously unreported PID patients with iVDRV-induced cutaneous granulomas including cartilage hair hypoplasia (n = 1), warts, hypogammaglobulinemia, immunodeficiency, myelokathexis (WHIM) syndrome (n = 1), MHC class II deficiency (n = 1), Coronin-1A deficiency (n = 1), X-linked severe combined immunodeficiency (X-SCID) (n = 1), and combined immunodeficiency without a molecular diagnosis (n = 1). At the time of this report, the median age of the patients with skin granulomas and DNA repair disorders was 9 years (range 3–18). Cutaneous granulomas have been documented in all, while visceral granulomas were observed in six cases (40%). All patients had received rubella virus vaccine. The median duration of time elapsed from vaccination to the development of cutaneous granulomas was 48 months (range 2–152). Hematopoietic cell transplantation was reported to result in scarring resolution of cutaneous granulomas in two patients with NBS, one patient with AT, one patient with Artemis deficiency, one patient with DNA Ligase 4 deficiency, one patient with MHC class II deficiency, and one patient with combined immunodeficiency without a known molecular etiology. Of the previously reported and unreported cases, the majority share the diagnosis of a DNA repair disorder. Analysis of additional patients with this complication may clarify determinants of rubella pathogenesis, identify specific immune defects resulting in chronic infection, and may lead to defect-specific therapies.
Keywords: DNA ligase 4 deficiency, ataxia telangiectasia, Nijmegen breakage syndrome, Artemis deficiency, combined immunodeficiency, chronic rubella infection resulting in cutaneous granuloma formation
Introduction
The long-term presence of immunodeficiency-related vaccine-derived rubella virus (iVDRV) in association with cutaneous and sometimes visceral granuloma formation was recently identified using deep sequencing of lesions [1] and then extended by rubella-targeted testing in a total of 19 patients with various primary immunodeficiency disorders (PIDs) [1–4]. Based on the clinical and laboratory features, it has been hypothesized that underlying immunodeficiency allowed for persistence of the attenuated rubella virus vaccine strain. Polarization of the macrophages to a M2 phenotype appears to be associated with an inability to clear the virus [2]. Aberrant cytotoxic CD8+ T cell responses also appear to be important in the persistence of rubella virus in association with cutaneous granuloma formation [2]. Interestingly, the majority of PID patients with rubella-positive granulomas had DNA repair disorders such as ataxia telangiectasia (AT) (n = 9), Nijmegen breakage syndrome (NBS) (n = 3), or V(D)J recombination defects such as RAG1 (n =1) and RAG2 (n = 1) deficiency [1–4].
DNA repair disorders encompass a spectrum of PIDs including AT, NBS, XRCC4-like factor (XLF)/Cernunnos, Bloom’s syndrome (BS), DNA ligase 4 deficiency, and Artemis deficiency. The importance of DNA repair by homologous recombination, non-homologous end joining (NHEJ), and cell cycle checkpoint control is underscored by its crucial role in the development of the adaptive immune system. Processes involving NHEJ such as V(D)J recombination and associated development of T and B cell receptor diversity is one such example. It is unknown, however, how DNA repair disorders are permissive for the salient features that have been hypothesized to permit the development of iVDRV infection often presenting as chronic cutaneous granulomas (see Fig. 1). The potential impact of DNA repair disorders on cytotoxic CD8+ T cell responses and macrophage polarity are intriguing areas that warrant more detailed evaluation.
Fig. 1.
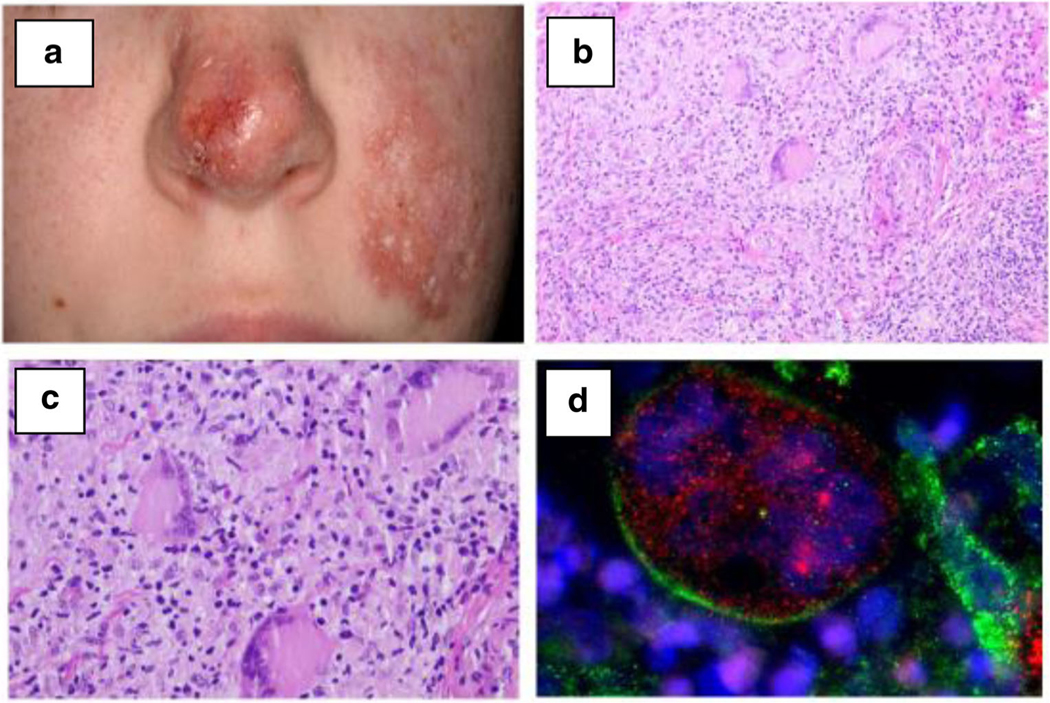
a Macroscopic features in patient no. 21 with acute and chronic ulcerations of the cheek and destruction of the nasal cartilage. b, c Histopathology of lesional skin in patient no. 21 with granulomatous dermatitis—H&E staining. Nodular, superficial, and deep dermal lymphohistiocytic infiltrate. d Histological immunofluorescent staining demonstrating the presence of rubella virus capsid (red) in Lanrgerhans’ giant cells (green) in patient no. 21
Methods
To support the importance of this line of inquiry, we provide additional information on previously reported patients (three patients with NBS and four patients with AT) and iVDRV-associated cutaneous granulomatous disease as well as eight not yet reported patients with DNA repair disorders and iVDRV-associated cutaneous granulomatous disease (one patient with NBS, five patients with AT, one patient with DNA ligase 4 deficiency, and one patient with Artemis deficiency). To our knowledge, the latter two disorders have not yet been associated with this complication. We are also including six not yet reported patients with other combined immunodeficiencies and iVDRV cutaneous granuloma formation (one patient each with cartilage hair hypoplasia (CHH), warts, hypogammaglobulinemia, immunodeficiency, myelokathexis (WHIM) syndrome, MHC class II deficiency, Coronin-1A deficiency, X-linked severe combined immunodeficiency (X-SCID), and combined immunodeficiency without a known molecular cause).
Patients with a PID diagnosis with cutaneous granulomas that were rubella virus-positive were identified though the Clinical Immunology Society Listserve which is a tool utilized by the physician and scientist members of this professional society to enhance collaboration and exchange information focused on PID patients. The authors also identified relevant cases following a literature search or personal communication. Physicians were individually contacted. A structured datasheet was utilized to collect clinical information from treating physicians.
Results
Table 1 provides a description of the clinical and immunological characteristics of iVDRV-associated cutaneous granulomatous disease patients with DNA repair disorders (n =15) and other PIDs (n = 6).
Table 1.
Clinical and immunological characteristics of new and previously reported cases of immunodeficiency-related vaccine-derived rubella virus-associated cutaneous granulomatous disease in patients with DNA repair disorders and other primary immunodeficiency disorders
| Patient no./Ref. | Age (yrs) | Sex | DNA breakage syndrome | Mutation | Clinical features | Immunophenotype | Age at vaccination with rubella and start of cutaneous granulomas (months) |
|---|---|---|---|---|---|---|---|
| 1 Deripapa 2017 | 8 | M | Nijmegen breakage syndrome | c. 657_661_del5 and c. 657_661 del5 | Granulomatous dermatitis | T and B cell lymphopenia, hypogammaglobulinemia | 13 months/24 months |
| 2 Deripapa 2017 | 15 | F | Nijmegen breakage syndrome | c. 657_661_del5 and c. 657_661_del5 | Diffuse large B cell lymphoma, granulomatous dermatitis | T and B cell lymphopenia, hypogammaglobulinemia | 16 months/168 months |
| 3 Deripapa 2017 | 15 | F | Nijmegen breakage syndrome | c. 657_661_del5 and c. 657_661_del5 | Diffuse large B cell lymphoma, ganglioglioma, granulomatous dermatitis | T and B cell lymphopenia, hypogammaglobulinemia | 14 months/120 months |
| 4 New Case | 10 | M | Nijmegen breakage syndrome | c. 657_661_del5 and c. 657_661_del5 | Lymphoma treated with chemotherapy and rituximab, granulomatous dermatitis of cheek | T and B cell lymphopenia, hypogammaglobulinemia | 13 months/NA |
| 5 New Case | 11 | F | AT | NA | ILLD, EBV- lymphoproliferative disease, granulomatous dermatitis | T and B cell lymphopenia, hypogammaglobulinemia | 12 months/60 months |
| 6 New Case | 10 | F | AT | NA | ILLD, granulomatous dermatitis, infections | T and B cell lymphopenia, hypogammaglobulinemia hyper M | 16 months/108 months |
| 7 New Case | 7 | F | AT | NA | ILLD, granulomatous dermatitis, granulomatous spleen and liver lesions | T and B cell lymphopenia, hypogammaglobulinemia hyper IgM | 13 months/24 months |
| 8 New Case | 7 | F | AT | c. 8781_8786 + 2del and c. 8781 8786 + 2del | Pancytopenia, transaminitis, granulomatous dermatitis / spleen lesions, chronic lung disease, probably partial thymectomy after heart surgery for Tetralogy of Fallot | T and B cell lymphopenia. Poor mitogen responses, hypogammaglobulinemia poor antibody responses. Constantly positive rubella IgM | 13 months/39 months |
| 9 Perelygina 2016 “ | 9 | F | AT | NA | Granuloma annulare, lymphoproliferative disorder, non-Hodgkin lymphoma, immune cytopenias | T and B cell lymphopenia, hypogammaglobulinemia hyper IgM | NA/NA |
| 10 Perelygina 2016 | 3 | F | AT | IVS32 + 1 and 5726 T>Gpl909 Meth>Arg | Failure to thrive, developmental delay, granulomatous skin lesions, developed B cell lymphoma | T and B cell lymphopenia. Poor mitogen responses, hypogammaglobulinemia-poor antibody responses | 12 months/NA |
| 11 Perelygina 2016 | 5 | F | AT | NA | Proteinuria, granulomatous dermatitis, trunk, pelvic lesion on x-ray | Hyper IgM | NA/NA |
| 12 New Case | 18 | M | AT | NA | Cutaneous/hepatic granulomatosis, diabetes, polyarthritis, squamous cell carcinoma | T and B cell lymphopenia, hypogammaglobulinemia-poor antibody responses. Hyper IgM | NA/48 months |
| 13 Perelygina 2016 | 7 | F | AT | Homozygous deletion with frameshift, absent protein | Diffuse granulomas, skin, liver, lungs, skeleton after chemotherapy for Hodgkin lymphoma | Hyper IgM | NA/NA |
| 14 New Case | 13 | M | Artemis deficiency | c. 194C > T and c. 194C > T | Granulomatous dermatitis | Poor antibody responses | 12 month/86 month |
| 15 New Case | 4 | F | Ligase 4 deficiency | c.1341G>T and c.1103A>T | Bronchiectasis, villous atrophy, marrow failure, granulomatous dermatitis and liver lesions | T and B cell lymphopenia, hypogammaglobulinemia-poor antibody responses | 12 months/14 months |
| 16 New Case | 33 | F | Cartilage hair hypoplasia | Compound heterozygous | Diffuse large B cell lymphoma (20 years); autoimmune hemolytic anemia (27 years), hepatomegaly splenomegaly | T cell lymphopenia, naive T cell and memory B cell lymphopenia | 216 months/324 months |
| 17 New Case | 3 | F | MHC class II Deficiency | RFXANK c.338–25_338del homozygous | Severe chicken pox, cerebral vasculitis, hemiparesis, skin granuloma | T cell lymphopenia | 9 months/22 months |
| 18 New Case | 18 | F | WHIM syndrome | NA skin granuloma (cheeks) | T and B cell lymphopenia, neutropenia | NA/192 months | |
| 19 New Case | 18 | M | Coronin 1A Deficiency | Homozygous c.717G > A in CORO 1A exon 4 resulting in VI34 M | EBV-induced B cell lymphoproliferation (1 year), recurrent infections, bronchiectasis, reported in Moshous et al. JACI 2013. | T cell lymphopenia, naive T cell and memory B cell lymphopenia, antibody deficiency | NA/156 months |
| 20 New Case | 16 | M | X-SCID | hemizygous c.209 T > C | Failure to thrive, multiple viral and bacterial infections, bronchiectasis, skin granulomas (face shoulder, knee) | T and NK lymphopenia | 24 months/120 months |
| 21 New Case | 14 | F | Combined immune deficiency | No mutation identified with whole exome sequencing | Facial dermatitis with destruction of nasal cartilage | T and B cell lymphopenia, moderate B lymphopenia | 12 month/147 month |
| Patient no./Ref. | Biopsy Findings | Treatment of cutaneous granuloma and outcomes | Effect | Diagnosis of rubella Follow-up virus | |
|---|---|---|---|---|---|
| 1 Deripapa 2017 | Superficial and dermal epithelioid granuloma with lymphocytic infiltration | Multiple courses of antibiotics and topical steroids, HSCT conditioning (busulfan, fludarabin, ATG, cyclophosfamide, rituximab) | None. Effect post-conditioning | PCR positive | Alive, after HSCT |
| 2 Deripapa 2017 | Superficial and dermal granulomas with necrosis, perivascular infiltration with lymphocytes, plasma cells and histiocytes | HSCT conditioning (busulfan, fludarabin, ATG, cyclophosfamide, rituximab) | None. Effect post-conditioning | PCR positive | Alive, after HSCT |
| 3 Deripapa 2017 | Epithelial ulcers. Dermal granulomas with central necrosis. Mixed cell infiltration (CD8 lymphocytes, plasmacytes, neutrophils) | Multiple courses of antibiotics and topical steroids | None | PCR positive | Died from ganglioglioma progression |
| 4 New Case | Sarcoidal granulomatous dermatitis and perifoliculitis | Permethrin ivermectin keflex minocycline metronidazole gel | None | Positive | Alive |
| 5 New Case | Dermal epitheliod granulomas with central necrosis. Mixed cells perivascular | Multiple courses of antibiotics and topical steroids | None | PCR positive | Died from severe infection |
| infiltration (CD4, CD8, CD20 | |||||
| 6 New Case | Intraderamal mixed cells infiltration (CD8, eosinophils, histiocytes) | Nitazoxanide | None | PCR positive | Alive, on regular IVIG, antifungal and anti-bacterial prophylaxis |
| 7 New Case | Dermal epitheliod granulomas with necrosis, giant cells formation. Mixed cells perivascular infiltration | Interferon-alfa | None | NA | Alive, on regular IVIG, antifungal and anti-bacterial prophylaxis |
| 8 New Case | Dermal lymphohistiocytic infiltration with granuloma-like macrophage aggregations. Infiltrate predominantly positive for CD68 and CD163 | Nitazoxanide | Progressive dermatitis | PCR positive (RA27/3), IHC positive (skin and spleen) | Alive after HSCT |
| 9 Perelygina 2016 | Necrotizing granulomatous inflammation | Oral/topical antibiotics, topical steroids | None | IHC positive | Deceased |
| 10 Perelygina 2016 | Dermal chronic inflammation. Deep dermis with large and small necrotizing granulomas | Topical steroids and nutrition | Treatment effective | IHC positive | Deceased |
| 11 Perelygina 2016 | Necrotic vasculitis with neutrophilic infiltrates and necrotizing granulomatous inflammation of dermis | Oral/topical steroids | None | IHC positive | Deceased |
| 12 New Case | Granulomatous inflammation with necrosis | Surgery, emollients, topical steroid, nitizoxanide, etanercept | NA | IHC positive skin and liver | Alive, squamous cell carcinoma |
| 13 Perelygina 2016 | Non-infectious granulomatous process | Oral and topical steroids | Partial | IHC positive | Alive |
| 14 New Case | Dermal epitheloid granulomas with necrosis, giant cells formation | Topical steroids, IVIG, cotrimoxazole, conditioning with alemtuzumab, fludarabine, treosulfan | Dermatitis resolved after HSCT | IHC positive (skin) | Alive after HSCT |
| 15 New Case | Nodular, superficial and deep dermal lymphohistiocytic infiltrate with scattered lymphohistiocytic cells | Topical steroids, IVIG (for immune deficiency), HSCT conditioning (fludarabine, cyclophosfamide, alemtuzumab), nitazoxanide | None. Effect post-conditioning. | IHC positive (skin and liver) | Deceased, after HSCT (respiratory failure) |
| 16 New Case | Granuloma skin biopsy | Cyclosporine, corticosteroids | Progressive disease | PCR positive | Progressive disease |
| 17 New Case | Granuloma skin biopsy | None | Stable disease | PCR positive | Alive and well after HSCT |
| 18 New Case | Granuloma skin biopsy | Topical steroids, anti-TNF therapy | Improved with anti-TNF therapy | PCR positive | Stable |
| 19 New Case | Granuloma skin biopsy | Antibiotics, topical steroids, surgical excision | Stable disease | PCR positive | Stable |
| 20 New Case | Granulomatous dermaitis with epitheliod granulomas | Subcutaneous immunoglobulin | Stable disease | IHC positive | Stable disease |
| 21 New Case | Granulomatous dermaitis with non-necrotizing epitheliod granulomas, giant cell formation, and dermal microcysts | Topical: steroids, tacrolimus; systemic: steroids, chloroquin, methotrexate, clarithromycin, doxycyclin, ciprofloxacin; nitazoxanide | Dermatitis resolved after HSCT, but plastic surgery will be needed | PCR positive (RA27/3) | Alive and well after HSCT |
AT ataxia telangiectasia, HSCT hematopoietic stem cell transplantation, IHC immunohistochemistry, ILLD interstitial lymphocytic lung disease, IVIG intravenous immunoglobulin, NA not available, PCR polymerase chain reaction, X-SCID X-linked severe combined immunodeficiency
Demographic Features
The median age at the time of report of the patients with DNA repair disorders (n =15) was 9 years (range 3–18). The majority were females (73%). For patients with other PIDs (n = 6), the median age at the time of report was 17 years (range 3–33). The majority were also females (67%).
Clinical and Immunologic Features
Cutaneous granuloma formation has been documented in all 15 DNA repair disorder cases while visceral granuloma formation impacting the spleen, liver, bone, and lungs were observed in six cases (40%). The severity of the cutaneous granulomas was variable including superficial lesions as well as deep ulcerating lesions and destruction of soft tissues (Fig. 1). Among patients with other PIDs, only cutaneous granuloma formation has been documented. T cell and B cell lymphopenia as well as hypogammaglobulinemia or impaired antibody formation was present in the majority of patients with DNA repair disorders consistent with a combined immunodeficiency phenotype. Four AT patients demonstrated a hyper IgM phenotype. Moreover, T cell and B cell lymphopenia was also present in the majority of patients with other PIDs. Among DNA repair disorder patients and other PIDs, the median age at onset of cutaneous granuloma formation was 54 months (range 14–168) and 151 months (range 22–324), respectively. The median duration of time elapsed from vaccination to the development of cutaneous granuloma formation in these two groups was 48 months (range 2–152) and 102 months (range 13–135), respectively. Two patients with DNA repair disorders developed cutaneous granulomas following chemotherapy for lymphoma (both AT patients). The presence of rubella was confirmed in all skin and visceral granulomas. The diagnosis of rubella was made by PCR (47%) or immunohistochemistry (53%) in patients with DNA repair disorders. For patients with other PIDs, the diagnosis of rubella was made by PCR in the majority of cases (83%).
Treatments and Outcomes
Among DNA repair disorder patients, treatments included topical corticosteroids (53%), hematopoietic stem cell transplantation (33%), and nitazoxanide (27%). Similarly, among patients with other PIDs, treatments included topical (n = 1) and systemic corticosteroids (n = 1) or both (n =1). One patient demonstrated clinical improvement following anti-tumor necrosis factor therapy. Hematopoietic stem cell transplantation was reported to result in scarring resolution of granulomata including two patients with NBS, one patient with AT, one patient with Artemis deficiency, one patient with DNA Ligase 4 deficiency, one patient with MHC class II deficiency, and one patient with combined immunodeficiency without a known molecular cause [4]. Rubella-associated complications did not contribute to death among those patients who died (40%).
Discussion
The majority of the previously reported (14 of 19) and not yet reported (8 of 14) cases listed here share the clinical diagnosis of a DNA repair disorder and the presence of rubella virus, strongly suggesting the association between iVDRV and cutaneous granuloma formation. The addition of patients with DNA ligase 4 and Artemis deficiency to the existing literature of PIDs (i.e., AT, NBS, RAG1/RAG2 deficiency) with cutaneous granuloma formation in association with chronic rubella virus infection [1–4] suggests that aberrations in the genes involved in DNA repair result in a specific deficient immune response that places these patients at risk for the development of this complication. However, the presence of iVDRV- associated granulomatous disease in other PIDs may indicate a general predisposition of patients with combined immunodeficiencies who are not severe enough to be diagnosed before the initiation of live viral vaccines.
Common to all of these patients is the presence of a significant T cell deficiency. For many, there is also a concomitant antibody deficiency. Although not much is known about rubella virus-specific T cell responses, we hypothesize that adequate rubella virus-specific T cell responses are vital to the control of rubella virus. Likely common to many of these patients is the presence of rubella virus-specific antibodies. Since many of these patients were recipients of immunoglobulin prophylaxis with concomitant levels of rubella virus-specific antibodies reflective of the immunoglobulin donor pool, it appears that rubella virus-specific antibody is not able to eliminate persistent infection. Moreover, no cases of rubella virus-associated granulomas have been described in patients with agammaglobulinemia, and intravenous immunoglobulin does not appear to be an effective treatment of cutaneous granulomas.
Although a prominent T cell deficiency and concomitant antibody deficiency are common to the majority of these patients, the significant variability that exists even among specific diagnoses supports the importance of these aforementioned immunological lacunae in the pathogenesis of iVDRV-associated cutaneous granulomatous disease. For example, among patients with AT, there is a wide spectrum of symptoms, but granulomatous disease is not a frequent clinical finding. Moreover, the vast majority of the AT patients represented here have documented clinical and laboratory evidence of significant and profound immunodeficiency and immune dysregulation. This includes end organ autoimmunity and lymphoproliferation as well as interesting features such as abnormal isotype switching resulting in a hyper IgM phenotype and lack of anti-rubella IgG antibody formation [5]. Additional data focusing on detailed immunological evaluation among unbiased cohorts of patients (e.g., AT patients) without iVDRV-associated granulomatous disease would be a useful comparison.
The period of latency between vaccination and the evolution of iVDRV-associated cutaneous granulomatous disease was wide. Notably, the period of latency was shorter among patients with DNA repair disorders when compared with patients suffering from other PIDs. We hypothesize that the immunologic status of the patient plays an important role in shaping this period of latency. For example, diminution of CD8+ T cell cytotoxicity or the T cell repertoire over time may be forces favoring the persistence of vaccine-associated rubella virus and the subsequent development of cutaneous granulomatous disease. The presence of additional host factors (e.g., the receipt of cytotoxic chemotherapeutic agents in some of the reported patients), environmental factors, and pathogen-specific factors (e.g., viral escape mechanisms to include the accumulation of mutations by the virus) may also influence the risk for the development of this complication and the timing of its development in individual patients.
We acknowledge the limitations imposed by our study design and the possibility that there may be a selection bias such that patients with DNA repair disorders and cutaneous granuloma formation may be over-represented. We can only speculate as to the risk for iVDRV-associated cutaneous granuloma formation in DNA repair disorders and other combined immunodeficiencies. We acknowledge that no exact evidence exists that proves that rubella virus causes cutaneous granulomatous disease. It is clear, however, that rubella virus can be demonstrated in granulomas of patients with PIDs including patients with DNA repair disorders. For these PID patients, vaccination exposure preceded the development of cutaneous granulomatous disease. Indeed, there is a body of literature describing the presence of cutaneous granulomatous disease of unknown etiology in the context of disorders such as Artemis, DNA-dependent protein kinase (DNA-PK), and RAG deficiencies [1, 6, 7]. Careful evaluation of these cases for the presence of chronic rubella virus infection may be a productive clinical exploration. Moreover, confirmation of the presence of rubella virus strain RA27/3 is necessary. Many of these analyses can be achieved through the application of immunohistochemical staining and more sensitive techniques including RT-PCR. Analysis on fresh tissue is desirable as extraction of intact RNA from formalin-fixed paraffin-embedded is limited.
This relatively common association underscores the risk of live viral vaccination in patients with these disorders. If the diagnosis of a primary immunodeficiency disorder is known at the time of vaccination (e.g., AT), rubella virus vaccine should only be given in full awareness of this potential complication. Moreover, the presence of chronic granulomatous lesions should prompt aggressive pathogen identification to include rubella virus. Consideration should be given to use of curative treatment with hematopoietic stem cell transplantation using modified reduced intensity conditioning in selected patients with DNA repair disorders [8]. Other areas of active exploration include the use of agents such as nitazoxanide that may possess broad antiviral properties [9]. Interestingly, the responses reported post-conditioning among several of the NBS patients, one AT patient, one DNA ligase 4-deficient patient, and one Artemis-deficient patient suggests that the elimination of rubella virus-specific (functionally impaired) effector T cells may have resulted in the disappearance of the cutaneous granulomatous disease. We anticipate that the rubella virus itself remains; however, the engrafted donor immune system is then able to control the rubella virus as would be anticipated in the normal host.
Additional basic and clinical research analyses of confirmed patients with DNA repair disorders and other PIDs with iVDRV as well as cutaneous and visceral granuloma formation are planned. A focus of these analyses will include the evaluation of the cellular and humoral immune response to rubella vaccination. These efforts will help clarify those determinants that influence rubella pathogenesis, as new patients continue to be identified retrospectively and prospectively.
Acknowledgements
We thank the patients and their families for participating in our research studies. We would also like to acknowledge the support of the U.S. Centers for Disease Control and Prevention in Atlanta, GA. Patient blood samples and biopsy material were obtained after provision of informed consent.
Footnotes
Compliance with Ethical Standards
Conflicts of Interest The authors declare that they have no conflict of interest.
Disclaimer The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention, US Department of Health and Human Services.
Ethics Statements 1. CDC, VVPDB personnel were not involved in any clinical decisions regarding these patients. All decisions were made by physicians treating these cases. Patient blood samples and biopsy material were obtained by physicians after provision of informed consent. CDC’s direct role involving the patients was only laboratory testing of specimens.
2. Archived FFPE specimens from patients with granuloma were tested with non-disclosure of patient information, which was determined to be ethically acceptable by the Internal Review Board at the CDC. This work was determined to be non-applicable for human subject regulations (ID number 2014 6417).
3. In addition, RV RT-PCR detection and sequencing analysis in the biopsy material for possible rubella virus in these tissue specimens was done for the purpose of possible public health response as a part of ongoing CDC surveillance for rubella virus. Note that rubella virus-positive granulomas have been shown to shed divergent infectious rubella virus, which is the public health concern. In addition, CDC, VVPDB, rubella team personnel have international public health responsibilities as part of the CDC Global Specialized Laboratory in the WHO Measles and Rubella Laboratory Network.
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bodemer C, Sauvage V, Mahlaoui N, Cheval J, Couderc T, Leclerc-Mercier S, et al. Live rubella virus vaccine long-term persistence as an antigenic trigger of cutaneous granulomas in patients with primary immunodeficiency Clin Microbiol Infect. 2014;20(10):0656–63. [DOI] [PubMed] [Google Scholar]
- 2.Perelygina L, Plotkin S, Russo P, et al. Rubella persistence in epidermal keratinocytes and granuloma M2 macrophages in patients with primary immunodeficiencies. J Allergy Clin Immunol. 2016;138(5): 1436–1439.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neven B, Pérot P, Bruneau J, Pasquet M, Ramirez M, Diana JS, et al. Cutaneous and visceral chronic granulomatous disease triggered by a rubella virus vaccine strain in children with primary immunodeficiencies. Clin Infect Dis. 2017;64(1):83–6. [DOI] [PubMed] [Google Scholar]
- 4.Deripapa E, Balashov D, Rodina Y, Laberko A, Myakova N, Davydova NV, et al. Prospective study of a cohort of Russian Nijmegen breakage syndrome patients demonstrating predictive value of low kappa-deleting recombination excision circle (KREC) numbers and beneficial effect of hematopoietic stem cell transplantation (HSCT). Front Immunol. 2017;8:807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noordzij JG, Wulffraat NM, Haraldsson A, Meyts I, Veer LJ, Hogervorst FBL, et al. Ataxia-telangiectasia patients presenting with hyper-IgM syndrome. Arch Dis Child. 2009;94(6):448–9. [DOI] [PubMed] [Google Scholar]
- 6.Felgentreff K, Lee YN, Frugoni F, du L, van der Burg M, Giliani S, et al. Functional analysis of naturally occurring DCLRE1C mutations and correlation with the clinical phenotype of ARTEMIS deficiency. J Allergy Clin Immunol. 2015;136(1):140–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mathieu AL, Verronese E, Rice GI, et al. PRKDC mutations associated with immunodeficiency, granuloma, and autoimmune regulator- dependent autoimmunity. J Allergy Clin Immunol. 2015;135(6): 1578–88.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slack J, Albert MH, Balashov D, Belohradsky BH, Bertaina A, Bleesing J, et al. Outcome of hematopoietic cell transplantation for DNA double-strand break repair disorders. J Allergy Clin Immunol. 2018;141(1):322–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perelygina L, Hautala T, Seppanen M, Adebayo A, Sullivan KE, Icenogle J. Inhibition of rubella virus replication by the broad-spectrum drug nitazoxanide in cell culture and in a patient with a primary immune deficiency. Antivir Res. 2017;147:58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]