

Abstract
Estrogen receptor (ER) is the most important factor in the pathophysiology of breast cancer. Consequently, modulation of ER activity has been exploited to develop drugs against ER+ breast cancer, such as tamoxifen, referred to as endocrine therapies. With deeper understanding of ER mechanism of action, posttranslational modifications (PTMs) are increasingly recognized as important in mediating ER activity. Some ER PTMs such as phosphorylation, are studied in the context of ligand-independent ER activity. However, they also play a pivotal role in defining the actions and outcome of the antiestrogen-bound ER. The complexity of these actions is increasing as new PTMs are identified, yet the functional consequences and clinical implications are not fully understood. This review will examine and summarize new emerging mechanistic knowledge and clinical data in breast cancer on how these PTMs affect antiestrogen-ER activity, with an emphasis on phosphorylation of serine 305 (S305). This phosphorylation site represents an integrated hub of oncogenic signaling to modulate ER conformation, dimerization, coregulators, and DNA binding to profoundly reduce sensitivity to endocrine therapy. Consequently, (i) S305 has the potential to become a useful marker of tamoxifen response, and (ii) blocking S305 phosphorylation defines a new therapeutic strategy to overcome tamoxifen resistance in breast cancer.
Keywords: estrogen receptor, posttranslational modification, breast cancer, tamoxifen, S305 phosphorylation, resistance
I. Estrogen receptor posttranslational modifications and endocrine therapy
Estrogen receptor (ER) remains the key factor in the development and progression of breast cancer. The two known receptors, ERα and ERβ, belong to the nuclear receptor super family of steroid ligand-activated transcription factors. ERs contain several functional domains including, an activating N-terminal AF-1 domain, and a DNA binding domain that is connected through a hinge region to a C-terminal ligand binding domain, or AF-21. In breast tumors, ERα expression dominates, hence throughout the text ER refers to ERα. The endogenous agonistic ligand, estrogen, binds to ER to induce the transcriptional regulation of genes involved in cell proliferation and growth of breast cancer cells2. Because the activity of a protein may be affected by posttranslational modifications (PTMs), significant effort on determining and quantifying such modifications in ER has been employed. The array of PTMs on ER constitutes a molecular code that dictates its conformation, dimerization, protein stability, subcellular localization, interacting partners or coregulators, and DNA binding, and therefore overall activity. In breast cancer cells, PTMs serve as triggers to mediate the coupling between the main genomic actions and extranuclear signaling cascades initiated by estrogen. PTMs including, phosphorylation, acetylation, methylation, sumoylation, palmitoylation, glycosylation, ubiquitination, nitrosylation, and thiol oxidation, have been reported and contribute to multifaceted mechanisms underlying ER action in breast cancer (Reviewed by Romancer et al.,3). By far, the most common and important PTM that modulates ER activity is phosphorylation of serine, threonine, and tyrosine residues - all implicated in endocrine therapy response.
Given ER’s critical role in breast cancer, endocrine therapy, which blocks ER action, has been the standard of care for breast cancer treatment for decades. These drugs are: Selective Estrogen Receptor Modulators (SERMs), Selective Estrogen Receptor Downregulators (SERDs), and Aromatase Inhibitors. Tamoxifen, the archetype SERMs, remains to date the most successful targeted cancer therapy4. However, tamoxifen has substantial ER agonist activity in bone and in the uterus5. Other SERMs, such as raloxifene, toremifene, lasofoxifene, bazedoxifene and arzoxifene have been developed to attenuate the agonist effects of tamoxifen. SERDs are drugs that affect ER stability (degradation) and cause ER downregulation. The main drug in this class, fulvestrant (ICI 182,780) is referred to as a “pure” antiestrogen because it is a pharmacological antagonist in all tissues. Fulvestrant is the most effective treatment for patients with metastatic ER+ breast cancer, but tolerability and side effects may be limiting6. AIs, such as anastrozole, exemestane, and letrozole, are drugs that inhibit the aromatase, the enzyme required for the conversion of androgens to estrogens, hence indirectly affect ER activity7. Although AIs have improved efficacy over tamoxifen in postmenopausal patients at prolonging relapse, the extra benefit in overall survival is unclear.
Despite the widely proven success of endocrine therapy in the clinic, unfortunately about 40-50% of patients treated endocrine therapy either fail to respond (de novo resistance) or become resistant (acquired resistance) to the treatment8. Interestingly, most of endocrine therapy resistance occurs despite continued expression of ER9, suggesting that ER PTMs may play a role. Therefore, understanding how these PTMs modulate endocrine therapy-ER actions is important in breast cancer. In general, phosphorylation is thought to be the main cause driving ligand-independent ER activity. In fact, most phosphorylation target residues are found within the AF-1 domain, which results in ligand-independent ER transactivation10. If ER is able to act on its own, employing drugs that lead to estrogen ablation, such as AIs, may not be sufficient to inhibit ER-driven activities. Three well-documented phosphorylation sites of ER (S118, S167, and S305) have been implicated in both ligand-independent activation of ER and resistance to antiestrogen therapies, particularly within the context of crosstalk with other growth factor and survival pathways. While S118 and S167 have generally been associated with good outcome, S305 is consistently associated with poor outcome3,11. The aim of this review is to examine and summarize the mechanistic evidence and clinical data on how S305 impacts endocrine therapy-ER actions and its consequences in breast cancer.
II. Molecular events controlling phosphorylation of S305 and clinical implications
Several kinases have been reported to phosphorylate S305 leading to ER transcriptional activation. P21-activated kinase 1 (Pak1) was the first kinase shown to phosphorylate S305 in a murine model of mammary gland hyperplasia by Kumar lab12. Pak1 is a protein involved in a number of cellular functions, including cell morphogenesis, motility, survival, angiogenesis, and mitosis, hence is implicated in both oncogenesis and cancer progression13. Pak1 directly phosphorylated S305, and mammary tissues with active Pak1 shows ER transactivation and elevated expression of endogenous ER target genes12. Since then, additional in vivo and clinical data have emerged supporting the role of Pak1 and S305 in tamoxifen resistance14,15. Michalides and colleagues showed that cAMP-dependent protein kinase A (PKA) can phosphorylate S305 in vitro16. They examined two consensus PKA phosphorylation sites in ER, S236 and S305. Only S305 was a bona fide site and the main target of PKA activity16. In the presence of tamoxifen, PKA led to ER transcriptional activity via S305 phosphorylation. This modification converted tamoxifen from an antagonist to an agonist, but did not change the actions of a SERD like fulvestrant. More importantly, in clinical samples, they found that downregulation of a negative regulator of PKA, PKA-RIα, was associated with tamoxifen resistance prior to treatment16. Later, Zwart and colleagues showed that the PKA-anchoring protein, AKAP13, mediates the interaction between ER and PKA and is essential for the phosphorylation of S305 upon PKA activation17. In addition, AKAP13 mRNA expression levels correlated with poor outcome in patients who received tamoxifen treatment in the metastatic setting17.
A well-established ER crosstalk with the inflammatory NFκB pathway has been shown to promote aggressiveness in breast cancer18,19. Not surprisingly, at least two NFκB pathway-related kinases have been shown to phosphorylate S305. Wei et al. reported that TANK binding kinase 1 (TBK1) regulates phosphorylation of S305 independent of its role in innate immune response20. This is important because patients with tumors highly expressing TBK1 respond poorly to tamoxifen treatment and show high risk for relapse20. Stender et al. demonstrated that inflammatory cytokine-induced phosphorylation of S305 is mediated by the inhibitor of NFκB kinase 2 (IKKβ)21. Frasor lab has shown that IKKβ promotes the expansion of stem/basal-like cells and a dormant, metastatic phenotype in ER+ breast cancer22, suggesting that phosphorylation of S305 by IKKβ may play a role.
Bostner et al. observed that in primary tumors from postmenopausal patients, an overactive PI3K / Akt / mTOR pathway together with nuclear phosphorylated ER at S167 and S305 sites, was significantly associated with reduced response to tamoxifen23. More recently, nuclear expression of raptor, which is also part of PI3K / mTOR signaling, was coupled to elevated ER phosphorylation at S167 and S30524. The increase of nuclear raptor was inversely correlated with tamoxifen benefit in clinical samples, and indicated worse prognosis on long-term follow-up24. While mTOR signaling cascade is shown to phosphorylate ER at S104/106 and S167 residues, there is yet no evidence on direct phosphorylation of S305.
Additional molecular events that may impact S305 phosphorylation status include ER mutations and crosstalk with other PTM sites. Generally, these modifications can act sequentially and/or in concert to fine-tune the outcome of ER actions. ER mutations, although rare, have been documented to affect PTM target sites of the receptor. Fuqua lab identified an ER mutation that results in a lysine to arginine substitution at residue 303 (K303R) that promotes enhanced breast tumor cell growth under estrogen deprivation and resistance to both anastrozole25 and tamoxifen26. This mutation through enhanced crosstalk to growth factor pathways such as HER2 / IGF1R / IRS1 / PI3K / Akt results in S305 phosphorylation, which is then key in mediating the mutant actions, and failure to antiestrogen drugs25–27.
While individual PTMs have been well-documented, interactions between two or more PTMs remain to be fully elucidated. An example of positive phosphorylation crosstalk has been reported for S305 and S11815. Using a S305E mutant, which mimics constitutive phosphorylation, Kumar group found that it triggers subsequent phosphorylation of S118 that is further potentiated by tamoxifen15. Furthermore, S305-induced ER transactivation requires a functional S118 and is mediated by Pak115. This indicates a cooperative interaction between these two phosphorylation sites although located in different ER domains, yet are capable of coordinating overall transactivation activity of ER. More recently, Diakonova and colleagues have reported a positive feedback loop where estrogen activation of Pak1 requires tyrosyl phosphorylation of a complex that includes PKA28. In turn, this complex potentiates PKA activity for maximal ER S305 phosphorylation28. Once activated, S305 led to enhanced phosphorylation of S118 to promote cell proliferation and tumor growth28.
An example of negative crosstalk was reported between S305 phosphorylation and K303 acetylation29. Acetylation of K303 is constitutive and was shown to inhibit ER transcriptional activity. However, K303 resides adjacent to S305, and phosphorylation and acetylation in this region are inversely correlated. Fuqua and colleagues showed that phosphorylation of S305 by PKA blocks acetylation of K30329. In addition, a S305D mutant that mimics S305 phosphorylation, also blocks acetylation of the K303 and enhances transcriptional response to that seen with the naturally occurring K303R mutant receptor29. Hence, this is an example where upstream PKA signaling coordinates negative crosstalk between PTMs by coupling high S305 phosphorylation to low K303 acetylation to enhance ER hyperactivity29. The full extent of crosstalk between phosphorylated S305 and additional ER mutations and/or other PTMs remains to be determined in clinical samples.
III. S305 phosphorylation modulates ER activity through multiple mechanisms
S305 phosphorylation has been demonstrated to affect ER conformation, dimerization, interacting partners or coregulators, and DNA binding, thus overall ER function. Such perturbation results in ligand-independent constitutive ER activity and failure to be inhibited by tamoxifen, both of which have major implications in breast cancer. S305 residue is located in the hinge region of ER, which is known to coordinate and determine the functional synergy between AF-1 and AF-2 in response to estrogen and tamoxifen30. Mechanistically, Stender et al. showed that phosphorylated S305 forms a charge-linked bridge with the AF-2 domain of ER that enables inter-domain communication and constitutive activity from the coactivator-binding site21. Stable expression of wild type ER or S305E mutant in cervical cancer HeLa cells revealed that although a ligand was required for dimerization in both, the capability for dimerization was significantly higher in S305E mutant cells31. Furthermore, S305E mutants exhibited enhanced binding to target gene promotors, which may in part explain the increased ligand-independent cell growth observed in S305E mutant HeLa cells31. Kumar lab also reported that overexpression of S305E mutant in ER-negative MDA-MB-231 cells was sufficient to upregulate the expression of a few but not all ER-regulated genes, including zinc finger protein 147 and cyclin D1, a gene implicated in breast cancer progression32. These phenotypes of ligand-independent cell growth and regulation of bona fide ER-target genes should be validated in additional breast cancer cell lines and in clinically relevant animal models.
Using fluorescence resonance energy transfer (FRET), a technique that allows monitoring of conformational changes, Michalides and colleagues showed that upon S305 phosphorylation, tamoxifen bound to ER, but could not induce the inactive conformation, instead it led to ER-dependent transcriptional transactivation16. PKA activity thus converts tamoxifen from an antagonist to an agonist16. Interestingly, this activation does not modify the response to SERDs such as fulvestrant16. In a comprehensive in vitro study using the same FRET technique, various SERMs and SERDs were screened for inducing rapid ER conformational changes upon binding in the context of S305, S236, and S118 phosphorylation33. Tamoxifen and EM-652 were the most susceptible to kinase activities, whereas fulvestrant and ICI-164,384 were the most stringent33. The different responses of antiestrogens to the various combinations of phospho-modifications in ER elucidate as to why certain antiestrogens are more inclined than others to develop resistance.
Phosphorylation of S305 is reported to modulate the binding of coactivators to ER. Zwart et al. showed that S305 phosphorylation did not influence overall binding of coactivator SRC-1, but rather changed the orientation between ER and SRC-134. This altered orientation renders ER transcriptionally active in the presence of tamoxifen, also corroborated in additional biological assays34. This finding was extended beyond just SRC-1 in the study by Houtman et al35. A high throughput peptide array, which allowed assessment of multiple coregulators at once, revealed that in the presence of tamoxifen, wild type ER adopted a confirmation that reduced binding of coactivators35. However, phosphorylated S305 had enhanced binding to coregulators in a ligand-independent manner, further providing evidence of ER’s ability to circumvent the suppressive effects of tamoxifen. This was tested also in clinical samples where two ER+ tumors were selected, one of which was phosphorylated at S305. In the patient with phosphorylated S305, the ER lost substantial binding capacity when the sample was dephosphorylated, suggesting phosphorylation was essential for ER activation. This patient also experienced recurrence after tamoxifen treatment, further providing a correlation between S305 phosphorylation of ER and tamoxifen resistance35.
Zwart and colleagues were the first to characterize the phosphorylated S305-ER transcriptome in breast cancer cells36. They found that S305 modification upon activation of PKA signaling redirects ER to new transcriptional start sites. By altering the chromatin-binding pattern, S305 phosphorylation of ER translates into a 26-gene expression classifier that identifies breast cancer patients with a poor disease outcome after tamoxifen treatment36. Stender et al., showed that inflammatory cytokine-induced S305 phosphorylation establishes an ER cistrome that substantially overlaps with the estrogen-dependent ER cistrome, supporting a cytokine-induced constitutive activation of ER21. This activation requires and is mediated by IKKβ21. Cytokine treatment prevented the effects of tamoxifen in MCF-7 breast cancer cells with wild-type ER, while not affecting the anti-proliferative effects of tamoxifen in MCF7 cells expressing the S305A ER mutant unable to be phosphorylayed21. Cytokine treatment of MCF-7 cells also increased extravasation and invasion. Most importantly, inhibition of IKKβ restores tamoxifen sensitivity in the presence of inflammatory cytokines21.
Overall these findings demonstrate how, upon binding of tamoxifen, subtle changes in ER caused by phosphorylation of S305 confer resistance to tamoxifen in this context. Additional cellular consequences S305 phosphorylation include ligand-independent cell growth, regulation of some bona-fide ER-target genes and others, and invasiveness as depicted in Figure 1.
Figure 1.
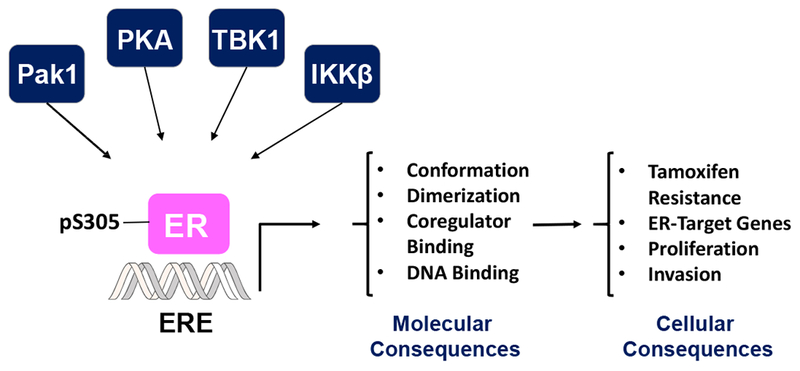
A schematic representation of ER S305 phosphorylation and its molecular and cellular consequences in breast cancer.
IV. The relevance of S305 phosphorylation in a clinical setting
The experimental data summarized above suggests that phosphorylation of S305 has an important role in tamoxifen resistance in breast cancer. However, there is no evidence linking S305 to SERD resistance16, and only limited in vitro data connecting S305 to AI sensitivity27. In clinical studies, inconsistent results are sometimes observed possibly due to small numbers of cases, different patient characteristics, type of antibodies used, differences in scoring and quantification, as well as varying definitions of positivity and negativity. However, the common theme that has emerged is that S305 phosphorylation is associated with poor response to tamoxifen. This is important because tamoxifen for decades was the first-in-line drug to treat breast cancer, and is expected to continue to be in wide use for some time. As more specific antibodies become available, this will enable better determination the S305 relevance in breast cancer and in predicting response to tamoxifen. Validation of such antibodies for immunohistochemistry (IHC) is extremely important, and more effort is required to generate reliable, high-quality antibodies suitable for IHC in larger patient cohorts. Additionally, incorporating other techniques to detect S305 phosphorylation that are amendable to clinical samples, such as mass spectrometry, may prove useful in the future.
Numerous clinical studies show that tamoxifen resistance occurred in endocrine-treated patients with detectable phosphorylated S30514,23,37,38. S305 phosphorylation has no effect on patients that received no endocrine treatment, but was a predictive marker for treatment outcome in patients that received it38. This suggest that S305 is a marker of therapy benefit, but does not predict general ER+ tumor progression. In the study by Holm et al. S305 phosphorylation alone was negatively correlated with tamoxifen response38. However, in two later studies, phosphorylated S305 failed to predict tamoxifen sensitivity by itself, but rather in combination with other markers 14,23,37 . These findings strongly imply that S305 phosphorylation should be examined in the context of other ER phosphorylation PTMs and overactivated oncogenic signaling pathways that may control S305 phosphorylation status.
As previously noted, in contrast to S305, phosphorylation of S118 and S167 are associated with good prognosis3,11. Bostner et al. reported that coexpression of S305 with S167 showed borderline statistical significance in terms of tamoxifen resistance23. Detection of multiple phosphorylation sites and well-documented crosstalk between them supports the notion that phospho-profiling of ER in breast tumors to establish an ER phosphorylation score may be a more precise marker of prognosis and/or response to endocrine therapy. Numerous studies emphasize the interplay between a kinase activity or an oncogenic pathway with S305 phosphorylation status in predicting tamoxifen effectiveness. Therefore, therapeutic interventions aimed at inhibiting these kinases or pathways would be desirable, as they would simultaneously achieve blocking of the target and S305 phosphorylation. Bostner et al. showed that in samples from postmenopausal patients nuclear localization of Pak1 together with phosphorylated S305 decrease benefit from tamoxifen treatment14. Kok et al. also confirmed the correlation between Pak1 and tamoxifen resistance37. Additionally, they showed that coexpression of phosphorylated PKA and S305 predicts tamoxifen response37. A later study by Bostner et al. showed that a combination score of phosphorylated ER at S167 and S305 together with phosphorylated Akt and mTOR predicts further reduced benefit form tamoxifen treatment23. Hence, inhibiting these kinases or pathways may provide a personalized breast cancer treatment for patients unlikely to respond to tamoxifen alone.
In conclusion, phosphorylation of ER at S305 site represents an integrated hub of oncogenic signaling to modulate ER conformation, dimerization, coregulatory binding, and DNA binding to profoundly reduce sensitivity to endocrine therapy. Consequently, (i) S305 has the potential to become a useful marker of tamoxifen therapy benefit, and (ii) blocking S305 phosphorylation defines a new therapeutic strategy to overcome tamoxifen resistance.
Highlights:
PTMs and especially phosphorylation are critical to modulate ER activity in breast cancer
Numerous kinases and crosstalk to other PTMs control phosphorylation of S305
S305 phosphorylation of ER affects its conformation, dimerization, coregulator and DNA binding
Important cellular consequences of phosphorylated S305 include, tamoxifen resistance, ligand-independent cell growth, gene regulation and invasion
Phosphorylated S305 predicts poor outcome to tamoxifen therapy
Acknowledgements
This work was supported by a grant from the National Institutes of Health (NIH), R01 CA200669 to Jonna Frasor. We thank our mentor Dr. Jonna Frasor for her support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kumar V, et al. Functional domains of the human estrogen receptor. Cell 51, 941–951 (1987). [DOI] [PubMed] [Google Scholar]
- 2.Ciocca DR & Fanelli MA Estrogen receptors and cell proliferation in breast cancer. Trends in endocrinology and metabolism: TEM 8, 313–321 (1997). [DOI] [PubMed] [Google Scholar]
- 3.Le Romancer M , et al. Cracking the estrogen receptor’s posttranslational code in breast tumors. Endocr Rev 32, 597–622 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Jordan VC Tamoxifen as the first targeted long-term adjuvant therapy for breast cancer. Endocrine-related cancer 21, R235–246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wardell SE, Nelson ER & McDonnell DP From empirical to mechanism-based discovery of clinically useful Selective Estrogen Receptor Modulators (SERMs). Steroids 90, 30–38 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nathan MR & Schmid P A Review of Fulvestrant in Breast Cancer. Oncology and therapy 5, 17–29 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brueggemeier RW Update on the use of aromatase inhibitors in breast cancer. Expert Opin Pharmacother 7, 1919–1930 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Ali S & Coombes RC Endocrine-responsive breast cancer and strategies for combating resistance. Nature reviews. Cancer 2, 101–112 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Robertson JF Oestrogen receptor: a stable phenotype in breast cancer. British journal of cancer 73, 5–12 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lannigan DA Estrogen receptor phosphorylation. Steroids 68, 1–9 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Murphy LC, Seekallu SV & Watson PH Clinical significance of estrogen receptor phosphorylation. Endocrine-related cancer 18, R1–14 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Wang RA, Mazumdar A, Vadlamudi RK & Kumar R P21-activated kinase-1 phosphorylates and transactivates estrogen receptor-alpha and promotes hyperplasia in mammary epithelium. The EMBO journal 21, 5437–5447 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rane CK & Minden A P21 activated kinase signaling in cancer. Seminars in cancer biology (2018). [DOI] [PubMed] [Google Scholar]
- 14.Bostner J, Skoog L, Fornander T, Nordenskjold B & Stal O Estrogen receptor-alpha phosphorylation at serine 305, nuclear p21-activated kinase 1 expression, and response to tamoxifen in postmenopausal breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 16, 1624–1633 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Rayala SK, et al. P21-activated kinase 1 regulation of estrogen receptor-alpha activation involves serine 305 activation linked with serine 118 phosphorylation. Cancer research 66, 1694–1701 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Michalides R, et al. Tamoxifen resistance by a conformational arrest of the estrogen receptor alpha after PKA activation in breast cancer. Cancer cell 5, 597–605 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Bentin Toaldo C, et al. Protein Kinase A-induced tamoxifen resistance is mediated by anchoring protein AKAP13. BMC cancer 15, 588 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frasor J, El-Shennawy L, Stender JD & Kastrati I NFkappaB affects estrogen receptor expression and activity in breast cancer through multiple mechanisms. Molecular and cellular endocrinology 418 Pt 3, 235–239 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baumgarten SC & Frasor J Inflammation: an instigator of more aggressive estrogen receptor (ER) positive breast cancers. Molecular endocrinology 26, 360–371 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei C, et al. Elevated expression of TANK-binding kinase 1 enhances tamoxifen resistance in breast cancer. Proceedings of the National Academy of Sciences of the United States of America 111, E601–610 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stender JD, et al. Structural and Molecular Mechanisms of Cytokine-Mediated Endocrine Resistance in Human Breast Cancer Cells. Molecular cell 65, 1122–1135 e1125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El-Shennawy L, et al. Coactivation of Estrogen Receptor and IKKbeta Induces a Dormant Metastatic Phenotype in ER-Positive Breast Cancer. Cancer research 78, 974–984 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bostner J, et al. Activation of Akt, mTOR, and the estrogen receptor as a signature to predict tamoxifen treatment benefit. Breast cancer research and treatment 137, 397–406 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bostner J, et al. Raptor localization predicts prognosis and tamoxifen response in estrogen receptor-positive breast cancer. Breast cancer research and treatment 168, 17–27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barone I, et al. Expression of the K303R estrogen receptor-alpha breast cancer mutation induces resistance to an aromatase inhibitor via addiction to the PI3K/Akt kinase pathway. Cancer research 69, 4724–4732 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giordano C, et al. Growth factor-induced resistance to tamoxifen is associated with a mutation of estrogen receptor alpha and its phosphorylation at serine 305. Breast cancer research and treatment 119, 71–85 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barone I, et al. Phosphorylation of the mutant K303R estrogen receptor alpha at serine 305 affects aromatase inhibitor sensitivity. Oncogene 29, 2404–2414 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oladimeji P, Skerl R, Rusch C & Diakonova M Synergistic Activation of ERalpha by Estrogen and Prolactin in Breast Cancer Cells Requires Tyrosyl Phosphorylation of PAK1. Cancer research 76, 2600–2611 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui Y , et al. Phosphorylation of estrogen receptor alpha blocks its acetylation and regulates estrogen sensitivity. Cancer research 64, 9199–9208 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Zwart W, et al. The hinge region of the human estrogen receptor determines functional synergy between AF-1 and AF-2 in the quantitative response to estradiol and tamoxifen. Journal of cell science 123, 1253–1261 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Tharakan R, Lepont P, Singleton D, Kumar R & Khan S Phosphorylation of estrogen receptor alpha, serine residue 305 enhances activity. Molecular and cellular endocrinology 295, 70–78 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Balasenthil S, Barnes CJ, Rayala SK & Kumar R Estrogen receptor activation at serine 305 is sufficient to upregulate cyclin D1 in breast cancer cells. FEBS letters 567, 243–247 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Zwart W , et al. Classification of anti-estrogens according to intramolecular FRET effects on phospho-mutants of estrogen receptor alpha. Molecular cancer therapeutics 6, 1526–1533 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Zwart W , et al. PKA-induced resistance to tamoxifen is associated with an altered orientation of ERalpha towards co-activator SRC-1. The EMBO journal 26, 3534–3544 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Houtman R, et al. Serine-305 phosphorylation modulates estrogen receptor alpha binding to a coregulator peptide array, with potential application in predicting responses to tamoxifen. Molecular cancer therapeutics 11, 805–816 (2012). [DOI] [PubMed] [Google Scholar]
- 36.de Leeuw R, et al. PKA phosphorylation redirects ERalpha to promoters of a unique gene set to induce tamoxifen resistance. Oncogene 32, 3543–3551 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Kok M, et al. PKA-induced phosphorylation of ERalpha at serine 305 and high PAK1 levels is associated with sensitivity to tamoxifen in ER-positive breast cancer. Breast Cancer Res Treat 125, 1–12 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Holm C, et al. Phosphorylation of the oestrogen receptor alpha at serine 305 and prediction of tamoxifen resistance in breast cancer. J Pathol 217, 372–379 (2009). [DOI] [PubMed] [Google Scholar]