

Abstract
Nuclear localization of androgen receptor (AR) directs transcriptional regulation of a host of genes, referred to as genomic signaling. Additionally, nonnuclear or nongenomic activities of the AR have long been described, but understanding of these activities remains elusive. Here, we report that AR is imported into and localizes to mitochondria and has a novel role in regulating multiple mitochondrial processes. Employing complementary experimental approaches of AR knockdown in AR-expressing cells and ectopic AR expression in AR-deficient cells, we demonstrate an inverse relationship between AR expression and mitochondrial DNA (mtDNA) content and transcription factor A, mitochondrial (TFAM), a regulator of mtDNA content. We show that AR localizes to mitochondria in prostate tissues and cell lines and is imported into mitochondria in vitro. We also found that AR contains a 36-amino-acid-long mitochondrial localization sequence (MLS) capable of targeting a passenger protein (GFP) to the mitochondria and that deletion of the MLS abolishes the import of AR into the mitochondria. Ectopic AR expression reduced the expression of oxidative phosphorylation (OXPHOS) subunits. Interestingly, AR also controlled translation of mtDNA-encoded genes by regulating expression of multiple nuclear DNA-encoded mitochondrial ribosomal proteins. Consistent with these observations, OXPHOS supercomplexes were destabilized, and OXPHOS enzymatic activities were reduced in AR-expressing cells and restored upon AR knockdown. Moreover, mitochondrial impairment induced AR expression and increased its translocation into mitochondria. We conclude that AR localizes to mitochondria, where it controls multiple mitochondrial functions and mitonuclear communication. Our studies also suggest that mitochondria are novel players in nongenomic activities of AR.
Keywords: androgen receptor, mitochondria, prostate cancer, cell signaling, gene transcription, genomic signaling, mitochondrial localization sequence, nongenomic signaling, oxidative phosphorylation, retrograde signaling, castration-resistant, nuclear receptor
Introduction
Prostate cancer is the second leading cause of cancer deaths in men in the United States (1–3). Despite recent advances in prostate cancer diagnosis and treatment, mortality from prostate cancer remains very high. Androgen deprivation therapy is the standard treatment regimen, but patients initially responding well to this treatment develop resistance to it, and progression to androgen-independent or castration-resistant prostate cancer (CRPC)3 occurs. Metastatic CRPC remains the biggest challenge to treat, as androgen independence leaves no straightforward way to its management (4). Numerous hypotheses have been put forward to explain the development of CRPC; overexpression of AR and constitutive AR activity as a result of AR amplification and/or mutations remain the most acceptable (5). Recent studies have identified AR mutations in prostate cancer that lead to ligand-free translocation of the receptor into the nucleus, resulting in constitutively active AR (6). A number of studies have also reported cytoplasmic functions of AR not only in prostate cancer cells (7–11) but also in nonprostate cells, where androgen-dependent AR translocation in the nucleus is not observed (12). Another study demonstrated that the role of AR in CRPC is not to direct the androgen-dependent gene expression, but to execute a different program, resulting in androgen-independent growth (13). Likewise, the ability of AR to enhance the invasion of prostate cancer cell lines independent of its nuclear localization has also been shown (14). These studies suggest that besides nuclear genomic signaling, AR also participates in nongenomic (outside the nucleus) signaling that may play an important role in prostate carcinogenesis (15). Unfortunately, the origin and nature of nongenomic AR signaling remain elusive.
Mitochondria regulate cell growth, survival, and death by an intricate and as yet poorly understood mitochondrial-nuclear cross-talk. This cross-talk is mediated by anterograde (nucleus to mitochondria) and retrograde (mitochondria to the nucleus) signaling, which continuously monitor and fine-tune signaling and cellular metabolism as a part of adaptation to a changing cellular environment. mtDNA depletion has been associated with multiple cancers (16–18), including prostate cancer (19, 20). It has been demonstrated that apoptosis is inhibited in mtDNA-depleted cells, favoring cancer progression (21–23). mtDNA-depleted cells also show Warburg effect, metabolic reprogramming, and cancer stem cell properties (24, 25). We have provided evidence that, in African-Americans, a low mtDNA content is a risk factor for poor prognosis and aggressive prostate cancer (26). An interesting study demonstrated that the depletion of mtDNA from androgen-dependent LNCaP results in the loss of androgen dependence, and restoration of mtDNA restores androgen dependence (27), suggesting that mitochondria are central to advanced stages of prostate cancer. These results indicate that mtDNA may play an important role in androgen dependence of prostate cancer cells. These studies establish an undisputed role of mitochondria in prostate carcinogenesis and in cancer disparities in ethnically diverse populations.
The above pieces of evidence suggest that androgen receptor and mitochondrial functions are interlinked and are two important determinants of prostate cancer, which strongly affect risk and may play critical roles in the progression from androgen dependence to independence. However, very little is known about the cellular significance of the connection between these two. In the present study, we demonstrate that AR, besides being nuclear, also localizes into mitochondria, thereby regulating the mitochondrial function and retrograde signaling. We demonstrate the existence of an authentic mitochondrial localization sequence in the N terminus of AR protein. We established a distinct AR function regulating mitochondrial processes directly and indirectly. Our study provides insights into a previously unrecognized nongenomic role of AR in modulating cellular functions.
Results
We utilized two complementary approaches in our studies. First, a CRISPR-Cas9 approach was employed to knock out AR in LNCaP cells. Western blotting analyses showed about 80% decrease in AR expression in AR knockout cells compared with mock control (Fig. 1A). We also used harmalol hydrochloride (HH) in a pharmacological approach to inhibit AR expression (Fig. 1B). In a complementary approach, we ectopically expressed AR in PC-3 cells lacking AR expression. Expression of AR in PC-3 cells was confirmed by Western blotting (Fig. 1C). It is noteworthy that AR re-expression in PC3 cells has been shown to result in constitutively active nuclear AR, which does not require ligand for activation and transcription of its target genes (10, 28, 29).
Figure 1.
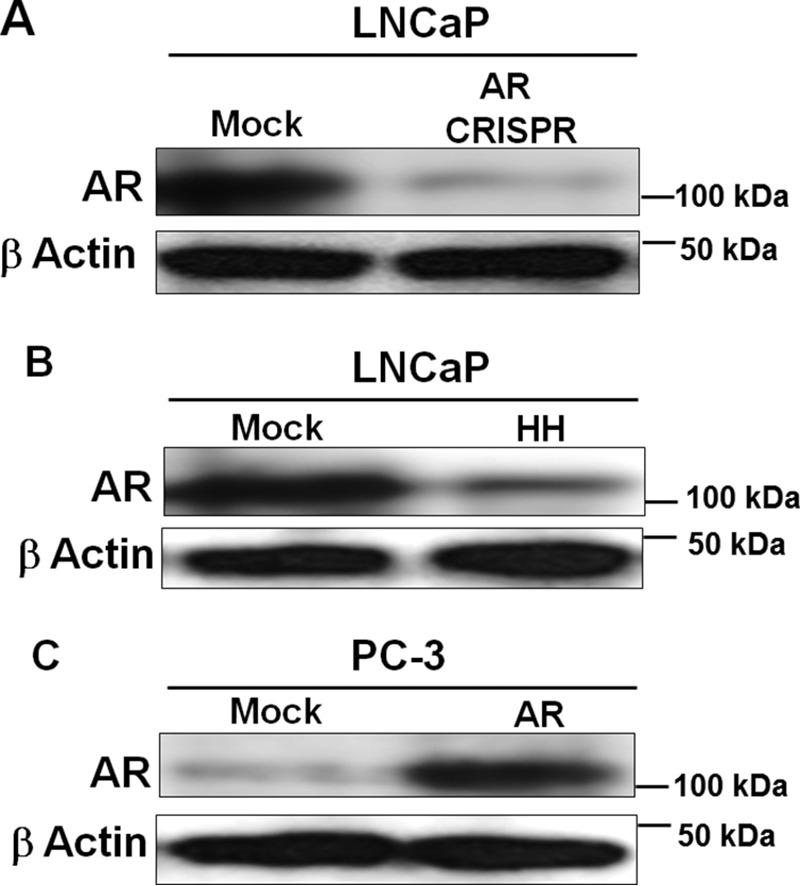
Cellular models employed in this study. A and B, genetic knockout of AR in LNCaP cells was achieved using CRISPR-Cas9 (A), and pharmacological inhibition was achieved by HH treatment for 48 h (B). C, AR overexpression in PC-3 cells was achieved by transfecting these cells with AR cDNA. Western blots show AR down-regulation in LNCaP cells (A and B) and up-regulation in PC3 cells (C).
AR maintains mtDNA homeostasis
To elucidate an association between AR and mtDNA, we measured mtDNA content in AR knockdown LNCaP cells and in PC-3 cells expressing AR ectopically. An 18% increase in mtDNA content was observed after AR knockdown in LNCaP, whereas about 25% decrease was observed in PC-3 upon AR expression (Fig. 2A). These studies inversely correlate mtDNA content with AR expression. The changes in mtDNA content led us to investigate whether AR modulates mtDNA via regulation of TFAM (transcription factor A, mitochondrial), a protein known to regulate mtDNA content. It has been previously shown that fibroblasts derived from TFAM+/− mice contain reduced mtDNA content (30). Interestingly, AR knockdown in LNCaP cells increased TFAM expression (Fig. 2B-i), whereas ectopic AR expression in PC3 cells led to a decrease in TFAM expression (Fig. 2B-ii). To validate the semiquantitative changes in TFAM expression, we performed real-time quantitative PCR analysis and observed a 2.3-fold increase and 73% decrease in TFAM expression in AR knockout LNCaP and AR-expressing PC-3 cells, respectively (Fig. 2, C-i and C-ii). These results suggest that AR maintains mtDNA homeostasis via regulation of TFAM.
Figure 2.

Androgen receptor expression regulates mtDNA content and TFAM level. A, AR inactivation in LNCaP cells increases mtDNA content, whereas ectopic AR expression in PC3 cells reduces mtDNA content. The ratio of mtDNA to nuclear DNA was used as an index for measuring the mtDNA content. B-i, AR knockout in LNCaP cells increased TFAM expression; B-ii, AR expression in PC-3 cells decreased TFAM expression. TFAM expression was analyzed by semiquantitative PCR. C-i and -ii, real-time quantitative PCR analysis of TFAM expression: validation of semiquantitative PCR results showing increased TFAM expression in AR knockout LNCaP cells (C-i) and decreased TFAM expression in PC-3 cells ectopically expressing AR (C-ii). Statistical significance was calculated by Student's t test, and significant differences (p < 0.05) are marked with asterisks. All experiments were done in triplicates. Error bars, S.D.
Mitochondrial stress induces expression of AR
To determine whether mitochondrial stress impacts the expression of AR, we employed two different approaches. First, we used rho0 cells that were devoid of mtDNA. Second, we utilized rotenone to inhibit OXPHOS complex I. Interestingly, we observed about a 2-fold increase in AR expression in rho0 cells (Fig. 3A). An alternative approach was also used to introduce mitochondrial stress by inactivating mtDNA polymerase γ (POLG1) by CRISPR-Cas9 (Fig. 3B) and inhibiting complex I by rotenone (Fig. 3C). POLG1 inactivation is known to deplete mtDNA and hence reduce mitochondrial OXPHOS function (31, 32). Our experiments show increased AR expression with a parallel increase in prostate specific antigen expression due to defects in mitochondrial function (Fig. 3, B and C). These studies suggest that AR is a retrograde responsive protein regulating nuclear genes.
Figure 3.
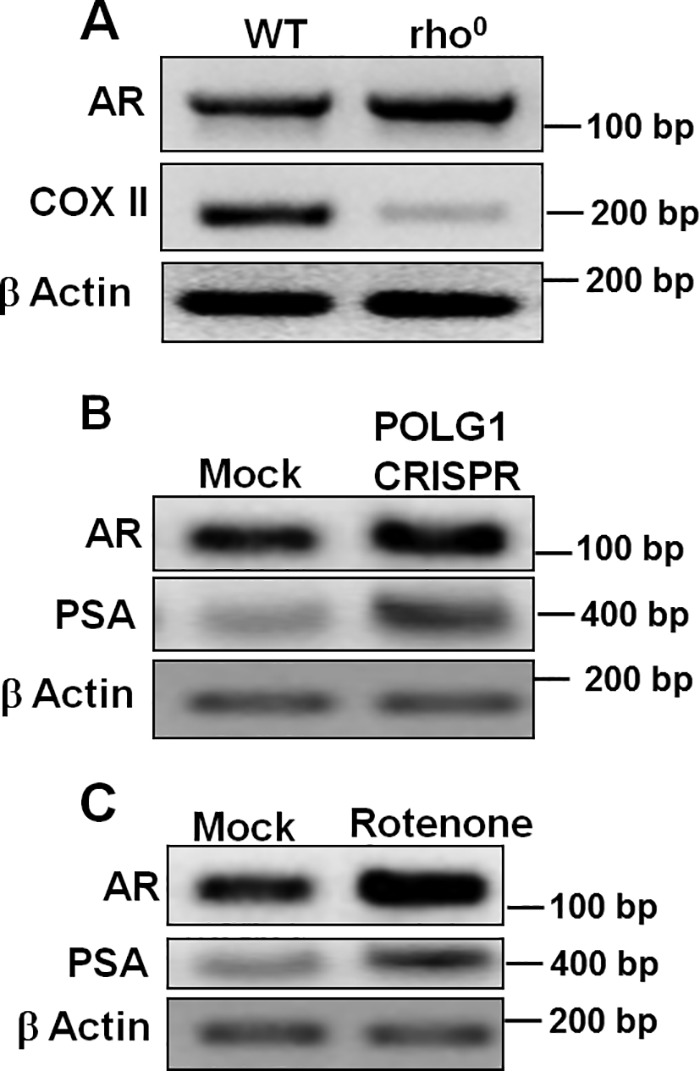
Mitochondrial dysfunction increases androgen receptor expression. A, rho0 (mtDNA-depleted) cell line showed increased AR expression when compared with parental cells. CRISPR targeting POLG1 (B) and rotenone treatment (25 nm for 24 h) (C) were used to induce mitochondrial stress in LNCaP cells, both of which showed increased expression of AR and prostate specific antigen.
AR localizes into mitochondria
To analyze the subcellular localization of AR, we isolated nuclear, mitochondrial, and cytosolic fractions from prostate PC-3 cells expressing AR (Fig. 4A-i), LNCaP endogenously expressing AR (mock), CRISPR-Cas9 AR knockout LNCaP cells (Fig. 4A-ii), and normal mouse prostate tissues (Fig. 4A-iii). In addition to its expected nuclear and cytoplasmic localizations, in each case, we observed a strong AR immunoreactive in the mitochondrial fraction (Fig. 4A, i–iii). Lamin A/C (nuclear), COXII (mitochondrial), and tubulin (cytoplasmic) antibodies were used as controls. Because these experiments suggest the presence of AR in the mitochondria, we investigated whether AR contained mitochondrial localization signal (MLS). To predict the probability of AR import, we analyzed AR protein sequence by using MitoProt database. The MitoProt software predicted MLS and a cleavage site after the first 14 amino acids at the N terminus with a probability score of 0.25 (Fig. 4B-ii). We cloned WT-AR in pHTC-Halo-Tag CMV-neo vector, and using this template, we generated an AR mutant (Δ36n-MLS-AR) lacking the first 36-amino-acid-long putative MLS present in the N terminus of AR. We used the CRISPR-Cas9 system to knock out AR in LNCaP cells (Fig. 4Aii, right). LNCaP cells lacking AR expression were transfected with WT (Fig. 4B-i) and Δ36n-MLS-AR (Fig. 4B-iii). We then isolated nuclear, mitochondrial, and cytoplasmic fractions and conducted Western blotting analyses. As expected, WT-AR (containing the MLS) localized into the mitochondrial compartment within the cell (Fig. 4B-i) (top), whereas Δ36n-MLS-AR (without MLS) lacked mitochondrial localization (Fig. 4B-iii) (top). A very faint immunoreactive band was detected in mitochondrial fractions of Δ36n-MLS-AR, which was sensitive to trypsin treatment (Fig. 4B-iii) (bottom), indicating that AR lacking MLS was bound to the outer membrane and not targeted inside mitochondria. (Fig. 4B-iii). Interestingly, the mitochondrial fraction of WT-AR was resistant to trypsin treatment, further confirming the intramitochondrial localization of WT-AR (Fig. 4B-i (bottom)). Additionally, mitochondrial fractions were lysed by treatment with 1% Triton X-100 (v/v) before trypsin treatment. The WT-AR mitochondrial protein became sensitive to trypsin following disruption of the mitochondrial membrane by treatment with Triton X-100 (TT), further confirming that it is localized in the mitochondrial matrix compartment (Fig. 4B-i (bottom)). Consistent with earlier observations, WT (and mutant) AR localized to nuclear and cytosolic fractions within the cell (Fig. 4, B-i and B-iii (top)).
Figure 4.

Mitochondrial Localization of the androgen receptor. A-i, exogenous expression of WT-AR in PC-3 cells. PC-3 cells transiently transfected with AR construct showed a prominent presence of AR in the nuclear and mitochondrial fractions in addition to its presence in the cytosolic fraction. A-ii, endogenous expression of AR in LNCaP cells and LNCaP CRISPR-Cas9 AR knockout (KO) cells. Shown is Western blot analysis of nuclear, mitochondrial, and cytosolic fractions to analyze endogenous AR expression in LNCaP cells (mock) and AR knockout by CRISPR-Cas9 in LNCaP cells (AR-KO). 80 μg of protein was resolved on 10% SDS gel. A-iii, endogenous expression of AR in mitochondrial fractions of mouse prostate tissue was analyzed by fractionation followed by Western blotting. Blots were probed with antibodies against AR, lamin A/C (nuclear control), tubulin (cytosolic control), and COXII (mitochondrial control) to analyze cross-contamination with nuclear, cytosolic, and mitochondrial fractions, respectively. B-i, WT-AR expression in AR knockout cells. LNCaP CRISPR-Cas9 AR knockout cells (KO) cells were transfected with WT-AR cloned in pHTC-Halo-Tag vector. Western blotting with subcellular nuclear, cytosolic, and mitochondrial fractions was done to analyze WT-AR expression (top). To confirm intramitochondrial localization of WT-AR, mitochondrial fractions of WT-AR were treated with trypsin (T). Additionally, one set was treated with 1% Triton X-100 (v/v) (bottom) before trypsin treatment. WT-AR containing MLS translocates to mitochondria and is resistant to trypsin (T). The WT-AR mitochondrial protein became sensitive to trypsin following disruption of the mitochondrial membrane by treatment with Triton X-100 (TT), further confirming that it is localized in the mitochondrial matrix compartment. B-ii, schematic of predicted MLS in the AR protein. B-iii, mutant Δ36n-MLS-AR expression in AR knockout cells. LNCaP CRISPR-Cas9 AR knockout cells (KO) cells were transfected with Δ36n-MLS-AR cloned in pHTC-Halo-Tag vector. Western blotting demonstrates that AR expression in mitochondrial fractions was drastically reduced upon truncating 36 amino acids from the N terminus (Δ36n-MLS-AR). This Δ36n-MLS-AR mutant lacked mitochondrial localization and was detected in nuclear and cytosolic fractions (top). The blot was probed with lamin A/C (nuclear control), COXII (mitochondrial control), and tubulin (cytosolic control). Further, trypsin treatment in mitochondrial fractions of Δ36n-MLS-AR, lacking MLS, was sensitive to trypsin, indicating that it was membrane-bound and did not translocate to mitochondria. NT, no trypsin; T, trypsin treatment; TT, trypsin treatment with 1% Triton X-100 (v/v). B-iv, schematic showing cDNA constructs cloned in pHTC-Halo-Tag vector used for in vitro transcription and translation and Western blot analysis as presented in B-i and B-ii. B-v, in vitro mitochondrial import of WT and Δ36n-MLS-AR mutant AR. We carried out an in vitro import experiment in a mouse brain mitochondrial system after translating AR cDNA in rabbit reticulocyte lysate (RRL). Limited trypsin treatment showed protection to trypsin in WT-AR, accounting for the intramitochondrial localization of AR. 36n-MLS-AR mutant was sensitive to trypsin treatment, indicating that AR protein lacking 36 amino acids of MLS did not translocate to mitochondria. Su9-DHFR and DHFR were used as positive and negative controls, respectively. Su9-DHFR contains a classic MTS, a presequence of subunit 9 of N. crassa F0F1-ATPase that has been fused to DHFR. Upon successful import, this MTS is cleaved after entry into mitochondria; thus, only the cleaved protein is present inside mitochondria after import and protected from trypsin treatment. Because DHFR is a cytosolic protein, it was used as a negative control in the experiment. Western blots were probed with anti-Halo-Tag antibody. C-i, mitochondrial localization of GFP by AR-MLS. pEGFP-N2 containing MLS derived from AR shows strong GFP localization in mitochondria (bottom). pEGFP-N2 lacking MLS was used as a negative control (top) and lacked co-localization with Mitotracker signal. D, increased translocation of AR into mitochondria under stress conditions generated by POLG1 CRISPR knockout and rotenone treatment (a complex I inhibitor) was observed. Western blotting showed nuclear and mitochondrial fractions isolated from LNCaP exposed to mitochondrial stress. The mitochondrial stress was induced by (i) POLG1 CRISPR and (ii) rotenone (25 nm for 24 h) inhibition of OXPHOS complex I. The blot was probed with lamin A/C (nuclear control) and TOM20 (mitochondrial control) to analyze cross-contamination. COXII was used as a marker of mitochondrial dysfunction.
We also performed in vitro mitochondrial import experiments (Fig. 4, B-iv and B-v). For these experiments (Fig. 4, B-iv and B-v), WT-AR, Δ36n-MLS-AR, Su-9 dihydrofolate reductase (Su9-DHFR) (positive control), and DHFR (negative control) pHTC-Halo-Tag constructs were used. 35S-Radiolabeled methionine was used label the translated protein, (Fig. S1). The constructs used in in vitro experiments were transcribed and translated using rabbit reticulocyte lysate. The translated protein was used for import into isolated mouse brain mitochondria. Our results show that whereas WT-AR containing the MLS is imported into mitochondria (Fig. 4B-v) (Fig. S1i), Δ36n-MLS-AR lacking the MLS is not imported into the isolated mitochondria (Fig. 4B-v). Su9-DHFR and DHFR were used as positive and negative controls, respectively. Su9-DHFR contains a classic mitochondrial targeting signal (MTS), a presequence of subunit 9 of Neurospora crassa F0F1-ATPase that has been fused to DHFR. Notably, the Su9-DHFR trypsin-untreated lane shows both MLS-processed and unprocessed forms. Upon successful import, the MTS is cleaved. Thus, only the cleaved protein is present and protected in the mitochondria after trypsin treatment (Fig. 4B-v) (Fig. S1ii). Because DHFR is a cytosolic protein, it was used as a negative control in the experiment (Fig. 4B-v and Fig. 1iii). Whereas Δ36n-MLS-AR was not imported in the mitochondria, WT-AR was imported and protected from digestion by trypsin treatment, indicating that WT-AR is localized inside the mitochondria (Fig. 4B-v). These studies suggest that the first 36 N-terminal amino acids in AR serve as MLS for import into the mitochondria.
Additionally, we took a complementary third approach utilizing the confocal microscopy. We cloned cDNA sequences containing MLS amino acids in frame with GFP. Cells expressing AR-MLS cloned in frame with GFP showed co-localization of GFP with mitotracker, which stains mitochondria in red (Fig. 4C, bottom). We additionally transfected cells with GFP alone (lacking AR-MLS). These cells lack colocalization with mitotracker (Fig. 4C, top). These studies suggest that AR contains authentic MLS capable of transporting a passenger protein such as GFP into mitochondria.
We also asked whether mitochondrial stress (induced by depletion of mtDNA-encoded COXII by POLG1 knockout and by inhibition of OXPHOS by rotenone (33)) increased AR translocation into mitochondria. No change in AR translocation was observed in nuclear fractions after cellular stress. However, we found increased AR translocation in mitochondrial fractions after mitochondrial stress. There was a concomitant decrease in COX-II expression (mtDNA-encoded) upon POLG1-induced mtDNA depletion and after rotenone-induced mitostress. These results demonstrate that mitochondrial stress either by genetic or metabolic stress induces increased translocation of AR into mitochondria (Fig. 4, D-i and D-ii).
Together, our studies suggest that AR (i) localizes into the mitochondria of prostate tissue and prostate cell lines, (ii) contains an authentic MLS capable of transporting a passenger protein into mitochondria, and (iii) is transported into mitochondria in vitro; also, (iv) translocation is increased upon mitochondrial genetic and metabolic stresses, and (v) AR is an authentic mitochondria-localized protein.
AR regulates expression of mtDNA-encoded OXPHOS subunit
AR is a well-known transcriptional regulator of a large number of nuclear genes (34). To analyze the effect of AR on mtDNA-encoded OXPHOS subunits, we conducted OXPHOS blot analyses of proteins isolated from AR CRISPR-Cas9 knockdown LNCaP cells. We observed increased expression of NDUFB8 (complex I), SDHB (complex II), and UQCRC2 (complex III) subunits (Fig. 5A). In PC3 cells ectopically expressing AR, mtDNA-encoded OXPHOS subunit COXII (complex IV) was markedly reduced. Interestingly, expression of nuclear DNA-encoded OXPHOS subunits, ATP5A (complex V), UQCRC2 (complex III), SDHB (complex II), and NDUFB8 (complex I), was also significantly down-regulated (Fig. 5B). These studies suggest that AR controls the expression of mitochondrial genes as well as nuclear DNA-encoded subunits involved in OXPHOS function.
Figure 5.
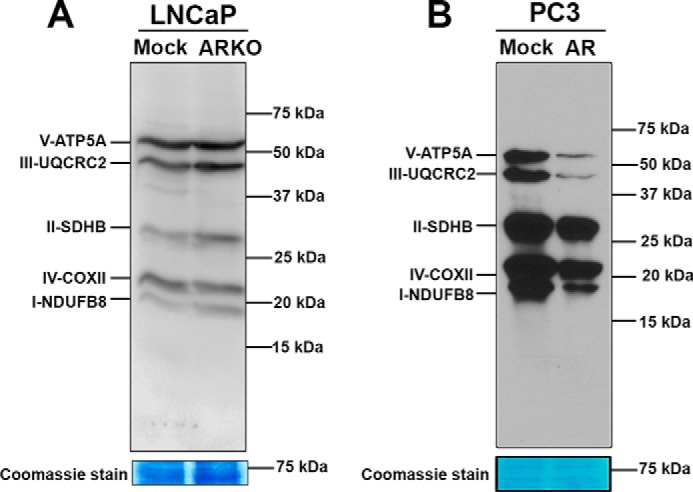
Androgen receptor regulation of OXPHOS subunits. Western blotting showing the effect of AR on expression of OXPHOS subunits. AR knockdown LNCaP cells showed an increase in OXPHOS subunit expression (A), and AR expression in PC3 cells led to a decrease in expression (B). Blots were probed with OXPHOS antibody mixture. Coomassie staining shows equal loading of proteins.
AR regulates expression of nuclear DNA-encoded OXPHOS subunits
Based on the above observations, we expanded our studies to undertake detailed expression analyses of other nuclear DNA-encoded OXPHOS subunits. Increased expression of all four complex II subunits was observed upon AR knockdown in LNCaP (Fig. 6A-i); conversely, all four subunits of complex II were down-regulated in AR-expressing PC-3 cells (Fig. 6A-ii). Likewise, subunits of complex III (Fig. 6B-i) and complex IV (Fig. 6B-ii) were up-regulated after AR knockdown. complex IV subunits were down-regulated upon AR expression in PC-3 cells (Fig. 6B-iii). Together, these studies suggest that AR regulates a large number of nuclear genes encoding mitochondrial OXPHOS subunits.
Figure 6.

Expression of nucleus-encoded OXPHOS subunits modulated by AR. Expression analyses of OXPHOS subunits by PCR are shown. Complex II subunits were up-regulated in AR knockdown LNCaP cells and in HH-treated LNCaP cells (A-i) and down-regulated in AR-expressing PC-3 cells (A-ii). B-i and -ii, expression of complex III and IV subunits was up-regulated in AR knockdown LNCaP cells; B-iii, complex IV subunit expression was down-regulated in AR-expressing PC-3 cells.
AR regulates expression of OXPHOS assembly factors
Various OXPHOS subunits encoded by nuclear DNA are synthesized in the cytoplasm and transported into mitochondria, which are then assembled into functional units by OXPHOS assembly factors (35, 36). In contrast to SDHAF1, expressions of SDHAF2 and SDHAF3 (complex II) were increased upon AR knockdown (Fig. 7A). Interestingly, a marked increase in the expression of other known complex III assembly factors, UQCC1, UQCC2, UQCC3, and TTC19 (Fig. 7A), and complex IV assembly factors SCO1, COX10, and COX15 (Fig. 7A) was observed. The mRNA quantification in PC-3 cells expressing AR showed a robust decrease in the expression of complex II assembly factors (Fig. 7B), complex III assembly factors (Fig. 7B), and complex IV assembly factors (Fig. 7B). These results further confirm that the androgen receptor is a negative regulator of OXPHOS assembly factors.
Figure 7.

Regulation of OXPHOS assembly factors by AR. PCR was done for expression analysis. Various OXPHOS assembly factors were up-regulated in AR knockout LNCaP cells (A) and down-regulated in AR-expressing PC-3 cells (B).
AR impacts the stability of OXPHOS supercomplexes
OXPHOS complexes are organized as supercomplexes (37, 38). Because our studies suggest that AR regulates nuclear DNA- and mtDNA-encoded OXPHOS subunits and OXPHOS assembly factors, we further analyzed the impact of AR on the stability of OXPHOS supercomplexes. A unique pattern for OXPHOS supercomplexes was revealed by blue-native PAGE (BN-PAGE) analysis in LNCaP mitochondrial fractions of both CRISPR knockdown and HH-treated samples. The OXPHOS supercomplex I was more stabilized in AR knockdown cells as compared with the parental counterparts (Fig. 8A-i). Minor changes were observed in the stability of complex III2/complex IV and complex III2 supercomplexes. Interestingly, mitochondrial fractions from HH-treated LNCaP cells showed more stabilized supercomplex IV and supercomplex II (Fig. 8A-ii). Furthermore, to validate the impact of AR on OXPHOS supercomplexes, we conducted BN-PAGE analysis in mitochondrial fractions of PC-3 cells ectopically expressing AR. We found destabilized supercomplex I, supercomplex II, and supercomplex IV. Supercomplex comprising complex III2/complex IV and supercomplex III2 also appeared destabilized in these cells (Fig. 8B). We conclude that AR negatively impacts the stability of OXPHOS supercomplexes.
Figure 8.

Androgen receptor affects stability of OXPHOS supercomplexes. BN-PAGE was performed with mitochondrial fractions from AR knockdown, HH-treated LNCaP cells, and PC3 cells expressing AR ectopically were probed with OXPHOS antibody mixture. A-i and -ii, more stabilized supercomplexes were observed upon AR knockdown in LNCaP and after pharmacological inhibition of AR by HH in treated LNCaP cells. B, destabilized supercomplexes were observed in AR-expressing PC3 cells.
AR regulates nucleus-encoded mitochondrial ribosomal genes and mitochondrial translational machinery
Our study suggested that AR regulates the expression of nuclear genes controlling mtDNA content (Fig. 2) and COXII gene expression (Fig. 5, A and B). We analyzed selective AR gene targets involved in mtDNA metabolism (GFM1 and GFM2) and mtDNA translation (MRPL-14, MRPL-27, MRPL-33, MRPL-39, MRPS-6, MRPS-29, and MRPS-33). The expressions of most of these gene targets were increased in AR knockdown LNCaP cells (Fig. 9, A and C). Interestingly, increased expressions of mitoribosomal genes were observed after AR knockdown (Fig. 9C). The expressions of these nucleus-encoded mitochondrial protein synthesis genes decreased upon ectopic expression of AR in PC-3 cells (Fig. 9, B and D). Using 35S labeling, we carried out mtDNA-encoded protein translation in PC-3 cells expressing AR and observed a decrease in all 13 mtDNA-encoded proteins, compared with its mock control, with a pronounced difference in ND5 expression (Fig. 9E). These results suggest that AR controls mitochondrial protein translation by controlling the expression of several nucleus-encoded mitochondrial ribosomal genes.
Figure 9.

Androgen receptor regulates mitochondrial translation. Gene expression was analyzed by RT-PCR. Expression of GFM1 and GFM2 genes was up-regulated after AR knockout in LNCaP cells (A), whereas in PC3 cells expressing AR it was down-regulated (B). Expression of mitoribosomal genes was up-regulated in AR knockout in LNCaP cells (C), while AR ectopic expression in PC-3 cells negatively regulates the expression of mitoribosomal genes (D). E, a pulse-chase labeling experiment showed the electrophoretic pattern of the de novo synthesized translational products of complex I (ND1, ND2, ND3, ND4, ND4L, ND5, and ND6 subunits); complex III (cytochrome b subunit), complex IV (COI, COII, and COIII subunits), and complex V (ATP6 and ATP8 subunits). Coomassie Blue staining of the same gel (total protein) shows equal loading of protein.
AR regulates OXPHOS enzymatic activity
Because the expression of OXPHOS subunits and the stability of OXPHOS supercomplexes were altered, we analyzed the effect of AR on mitochondrial OXPHOS complex activities. Results indicate that AR knockdown affects complex I activity. A statistically significant increase of 16 and 6% was observed by genetic knockdown and pharmacological inhibition of AR, respectively (Fig 10A-i). Similarly, a statistically significant increase in complex II activity of 28 and 30% by genetic knockdown and pharmacological inhibition, respectively, was observed (Fig. 10A-ii). AR knockdown by genetic and pharmacological means showed an increase of 22 and 10% of complex III activity, respectively (Fig. 10A-iii). We also observed a statistically significant increase in complex IV activity by 27 and 13% upon genetic knockdown and pharmacological inhibition, respectively. (Fig. 10A-iv). Conversely, PC-3 cells expressing AR consistently showed a decrease in complex I, complex II, and complex III activities (Fig. 10B, i–iii). However, complex IV activity in PC-3 cells expressing AR remained unaltered (Fig. 10B-iv). Together, these studies show that AR modulates mitochondrial OXPHOS complex activities.
Figure 10.

Androgen receptor regulates mitochondrial respiratory complex activities. A-i to -iv, increased complex I to IV activities upon AR knockdown; B-i to -iv, decreased activities upon AR expression. The statistical significance (p < 0.05) is marked with asterisks. Error bars, S.D.
Discussion
Mitochondria communicate with the nucleus to appraise the metabolic health of the cell. This involves signaling that alters expressions of many nuclear genes that are brought into action to modulate mitochondrial functions. Androgen receptor and mitochondria have been independently linked to prostate cancer risk, aggressiveness, and outcome (26, 39). We discovered that AR knockdown increases mtDNA content, and ectopic AR expression decreases the same, establishing an inverse correlation between AR expression and mtDNA content. In a complementary approach, we used rho0 (devoid of mtDNA) cells, inactivated POLG1 by CRISPR-Cas9, and inhibited OXPHOS complex I by rotenone to induce mitochondrial stress and found increased AR expression. Interestingly, mitochondrial stress has been strongly correlated with prostate cancer (40, 41). TFAM encodes a protein that participates in mtDNA replication and transcription and is thus crucial for the maintenance of mtDNA content and mitochondrial function (42). We found that ectopic AR expression decreased TFAM expression and AR down-regulation increased TFAM expression. A TFAM-dependent mechanism appears to underlie the inverse correlation between AR expression and mtDNA level.
It is well-established that mitochondria import and export a number of proteins and metabolites to communicate with the nucleus in anterograde and retrograde manners to modulate metabolism and signaling, depending upon mitochondrial health (43, 44). We provide compelling evidence for AR localization into mitochondria. With ectopic AR expression in PC3 cells, we demonstrate a strong immunoreactive band in the mitochondrial fraction in addition to its presence in the cytoplasmic and nuclear fractions. To further substantiate these findings, we identified a putative MLS and confirmed by GFP tagging that AR indeed contains a genuine 36-amino-acid-long MLS at the N terminus, which helps localize AR into mitochondria. Subcellular fractions, as analyzed by Western blotting and in vitro import results with WT-AR and Δ36n-MLS-AR, confirm that the MLS resides in the first 36 amino acids from the N terminus of AR protein and is a prerequisite for AR to be imported into mitochondria. Consistent with our studies, a previous study has reported the presence of AR in human sperm mitochondria (45). We found that AR impedes the expression of a number of mtDNA-encoded proteins along with many nDNA-encoded mitochondrial proteins. This suggests that AR is a retrograde protein that plays a crucial role in mitochondrial–nuclear cross-talk, which may play a significant role in carcinogenesis. The list of nuclear proteins participating in such noncanonical signaling is rapidly growing. Other nuclear hormone receptors, such as estrogen receptor (46), glucocorticoid receptor (47), and thyroid hormone receptor (48), have also been reported to migrate to mitochondria to fine-tune mitochondrial metabolism (49–51).
We have convincingly demonstrated that AR reduces the mtDNA content and the expression, assembly, stability, and activity of OXPHOS, resulting in the loss of mitochondrial function. A strong negative impact on mitochondrial ribosomal genes and translational machinery highlights diminished protein synthesis activity in the mitochondria upon AR expression. Mitochondrial function has been described as a tumor suppressor (16, 18, 41). Notably, the mitochondrial dysfunction has been shown to result in a pro-oncogenic state and shift to glycolysis (41). An energy-demanding process, such as cell division, is intricately wired to mitochondrial health and cellular signaling (52). The mitochondrial translocation of AR suggests a novel function impacting mitochondrial function, probably generating a pro-oncogenic environment. The rewired metabolism as a result of altered mitochondrial function may confer aggressive traits of metastatic competency and drug resistance, resulting in worse outcome in prostate cancer (41).
Nuclear localization of AR is well-known to direct transcriptional regulation of a host of genes, which is referred to as genomic signaling. Nongenomic signaling or cytoplasmic functions of AR have long been emphasized (7–11), but the mechanisms remain elusive. AR and other steroid receptors activate many signaling molecules, such as Src family kinases, Ras, MAPK, Akt, PKC, PLC, EGFR, and other secondary messengers. An interesting recent study demonstrated the ability of AR to enhance the invasion of prostate cancer cell lines via Src that was independent of its nuclear localization (10). Wang et al. (13) in an extensive study demonstrated that AR in androgen-independent prostate cancer regulates an entirely different signaling program than it does in an androgen-dependent manner (13). Signaling via cell membrane localization is the only mode of nonnuclear signaling in which steroid receptors, including AR, are known to participate in prostate cancer (53). Our discovery of AR localization into mitochondria may be central to nongenomic signaling in prostate cancer as the depletion of mtDNA has been shown to induce epigenetic modulation of miRNAs (54) and the activation of CREB (55), NF-κB (56), and other pathways (57) affecting cell survival and proliferation (11). This nongenomic mode may also explain the androgen-independent nature of prostate cancer. Because AR has both NLS and MLS, it is plausible that the shunting of AR between nucleus and mitochondria plays a major role in prostate tumorigenesis. Interestingly, a number of mutations in prostate cancer are reported within the NLS (58, 59). Notably, mutations in the NLS have been shown to result in the retention of AR in the cytoplasm, but localization of the mutant protein in mitochondria has not been explored (60). Consistent with AR dual localization, other proteins containing dual localization signals have been reported. Interestingly, defects in one localization signal are known to result in preferential localization of the protein at an alternate location (61). It is likely that the NLS mutations in AR propel mutant protein into the mitochondria, invoking distinct retrograde signaling when compared with the AR in the nucleus (Fig. 11). However, further investigation is needed to establish a role for such signaling in prostate cancer.
Figure 11.

Schematic showing a novel role of mitochondria in nongenomic AR signaling. In the traditional genomic pathway, AR undergoes conformational change and dimerization upon ligand binding, followed by its migration to the nucleus. Subsequent binding to the androgen response elements induces target gene transcription. In the nongenomic pathway, membrane-bound AR signals in the cytoplasm, which via second messengers activates other transcription factors. Our studies suggest that mitochondria are novel players in the nongenomic action of AR. Signaling cascades triggered upon translocation of AR into mitochondria may, in a retrograde manner, affect nuclear gene transcription that may contribute to aggressive prostate cancer.
Ligand-free translocation of AR into the nucleus has been reported and suggested to occur as a result of constitutive AR expression in PC3 cells and due to alternative splicing, mutations, and/or truncated versions of the protein. A previous study demonstrated that mitochondrial function played a crucial role in androgen dependence, the loss of which resulted in androgen independence (27). Our findings of AR localization into mitochondria suggest that mitochondrial dual localization may underlie androgen responsiveness. We found that AR translocation into mitochondria was independent of ligand presence. AR mutations have been reported in 30% of advanced prostate cancer patients; these mutations produce various truncated, alternate, and mutated forms of AR that are more promiscuous concerning localization (5, 6, 62, 63). Under normal conditions, nuclear transcription is the primary activity of AR; however, as suggested earlier (13), it appears that in advanced stages of prostate cancer, AR drives different programming in which mitochondria may play a key role in conferring aggressiveness to prostate cancer. Because androgen biosynthesis and translocation of AR to the nucleus are prime targets of prostate cancer drugs, ligand-independent constitutive activation and transportation to nucleus and mitochondria pose a significant challenge. Identification of signaling originating from mitochondrial localization of AR adds another dimension that needs attention to prostate cancer treatment. The mitochondrial–AR axis to prostate cancer not only contributes to the complex biology of androgen signaling in prostate cancer but also brings new avenues for improving the success of treatment.
In conclusion, we have demonstrated that AR in addition to nuclear localization is imported into the mitochondria. The identification of MLS along with a proof of mitochondrial import is an interesting finding of this study. We found that mitochondrial stress increases not only the expression of AR but also its translocation to the mitochondria, suggesting an intricate relationship between the two. The localization of AR in mitochondria makes mitochondrion a significant contributor to nongenomic signaling described so often in the literature (7–11). AR negatively regulates the expression, assembly, integrity, and functions of the mitochondrial complexes, thus impacting mitochondrial overall functions. The regulation and translocation of AR into mitochondria suggests that AR is a retrograde signaling protein. The establishment of mitochondrial AR suggests complex mitonuclear signaling in prostate cancer, which may offer new avenues for PC treatment.
Experimental procedures
Cell culture
LNCaP cells were grown in RPMI 1640 medium containing penicillin (100 units/ml), streptomycin (100 μg/ml), and 10% fetal bovine serum at 37 °C in 5% CO2. PC-3 cells were grown in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum (v/v), penicillin (100 units/ml), and streptomycin (100 μg/ml).
Transfection of AR cDNA and generation of stable cell lines
Transfection of PC-3 cells with WT-AR cDNA construct (a gift from Dr. Koocheckpour, Roswell Park Cancer Institute, Buffalo, NY), was carried out using FuGENE HD transfection reagent (Promega, Madison, WI). Cells were harvested 48 h post-transfection and used for preparing subcellular fractions. Cell lysates and mitochondria were prepared to analyze AR expression. For a generation of stable cell lines, cells were selected with G418 (100 μg/ml) antibiotic, and single colonies were selected and propagated.
Preparation of cell lysate and subcellular fractions and immunoblotting
LNCaP and PC-3 cell lysates were made in M-PER mammalian protein extraction reagent (Pierce) to analyze androgen receptor expression. For isolation of mitochondria, cells were trypsinized, and the cell pellet was collected by centrifugation at 800 × g for 5 min at 4 °C. The cell pellets were suspended in H medium (20 mm potassium HEPES buffer (pH 7.5), 70 mm sucrose, 220 mm mannitol, 2 mm EDTA), and mitochondria were prepared as described previously (64). For isolation of nuclear and cytosolic fractions, cells were washed twice with PBS and suspended in 0.5 ml of Buffer A (10 mm HEPES, pH 7.9, 10 mm KCl, 0.1 mm EDTA, 10% IGEPAL with protease inhibitor mixture) and incubated at room temperature for 10 min. Cells were collected in Eppendorf tubes and placed on ice and later centrifuged at 4 °C at 15,000 × g for 5 min. The cytosolic supernatant was saved separately, and to the pellet 150 μl of Buffer B (20 mm HEPES, pH 7.9, 0.4 mm NaCl, 1 mm EDTA, 10% glycerol with protease inhibitor mixture) was added. This was vortexed vigorously for 15 s and returned to the ice for 15 s every 10 min, for a total of 40 min, and later centrifuged at 4 °C at 15,000 × g for 5 min. Protein was measured using a PierceTM 660-nm protein assay reagent (Thermo Scientific, Waltham, MA). Proteins were solubilized in Laemmli sample buffer and incubated at 95 °C for 5 min. Protein preparation was resolved by electrophoresis on SDS-polyacrylamide (10%, w/v) for analyzing AR expression and on 14% (w/v) for analyzing OXPHOS subunit expression. SDS gels were subjected to immunoblot analysis using primary antibodies for AR (1:500 dilution), lamin A/C, β-actin, and tubulin (1:1500 dilution) (Santa Cruz Biotechnology, Inc., Dallas, TX). For OXPHOS subunits (MitoProfile Total OXPHOS Rodent WB Antibody Mixture, MitoScience, Eugene, OR) and COX-II (Life Technologies, Inc.) 1:2,000, dilutions of primary antibody and 1:50,000 dilutions of IR dye-conjugated secondary antibodies were used. Blots were imaged using an Odyssey scanner (LI-COR Biosciences, Bad Homburg, Germany) and Image Studio software.
Genetic knockout and pharmacological inhibition of androgen receptor
AR knockdown was achieved by using gene-specific CRISPR. The CRISPRs were designed using an online tool, generated from the Zhang lab at Massachusetts Institute of Technology (MIT). Primers for gene-specific guide RNAs were designed and annealed into the pSpCas9(BB)-2A-GFP vector. The transformation was performed in DH5-α Escherichia coli, and transformants were screened by PCR. The plasmid was isolated and transfected into cells using the Fugene HD according to the manufacturer's specifications. After 24 h of transfection, cells were FACS-sorted for GFP. Single colonies were selected and grown for screening. Pharmacological inhibition was achieved using 300 nm harmalol hydrochloride for 24 h (65).
Mitochondrial OXPHOS complex activities
Mitochondrial OXPHOS complex activities were assayed with minor modifications as described previously (66), using a Genesys 10S UV-visible spectrophotometer (Thermo Scientific, Waltham, MA). Briefly, 30–50 μg of mitochondrial samples were used to measure complex I activity by monitoring the oxidation of NADH at 340 nm for 3 min, indicating the extent of NADH oxidation (slope 1). Rotenone (5 μg/ml) was added to each reaction, and the absorbance was measured at 340 nm for another 2 min (slope 2). The rotenone-sensitive activity (slope 1–slope 2) was used to calculate the final complex I activity for each sample. Complex II activity was measured by analyzing the reduction of 2′,6-dichlorophenolindophenol (DCPIP). 30–50 μg of mitochondria were preincubated with 20 mm succinate at 30 °C for 10 min in assay medium (25 mm potassium phosphate, pH 7.2, 5 mm magnesium chloride). After incubation, 2 μg/ml antimycin A, 2 mm KCN, 2 μg/ml rotenone, and 50 μm DCPIP were added, and a baseline absorbance was recorded for 2 min at 600 nm. Subsequently, 65 μm ubiquinone was added, and the absorbance was recorded for 3 min. The baseline slope was subtracted from the ubiquinone slope, and the activity was calculated. Mitochondrial complex III activity was measured by monitoring the reduction of cytochrome c by ubiquinol at 550 nm. 10–30 μg of mitochondria was added in assay medium (25 mm potassium phosphate, pH 7.2, 5 mm magnesium chloride, 2.5 mg/ml BSA, 2 mm potassium cyanide, 15 μm cytochrome c, 0.6 mm lauryl maltoside, 1 μg/ml rotenone, and 35 μm ubiquinol), and the increase in absorbance was measured at 550 nm for 1 min. Further, complex IV activity was assessed by measuring the oxidation of cytochrome c at 550 nm for 1 min after the addition of 10 μg of mitochondria to complex IV assay buffer (20 mm potassium phosphate, pH 7.0, 15 μm reduced cytochrome c, and 0.45 lauryl maltoside).
Blue-native PAGE
Impact of AR knockdown (in LNCaP cells) and its expression (in PC-3 cells) on mitochondrial supercomplexes was analyzed by BN-PAGE as described previously with minor modifications (67). Gradient gel of 4–13% (w/v) was used for the analysis. Mitochondrial samples were solubilized with 1% Triton (v/v) and with 1% (w/v) digitonin in sample loading buffer (10% (v/v) glycerol, 50 mm NaCl, and 20 mm BisTris, pH 7.4). After a brief incubation on ice for 15 min, insoluble material was removed by spinning the samples at 16,000 × g for 5 min at 4 °C. Electrophoresis was done for 4 h at 600 V with anode buffer (50 mm BisTris, pH 7.0) and cathode buffer (50 mm Tricine, 15 mm BisTris, pH 7.0, 0.02% (w/v) Coomassie blue G-250). After the samples had moved from stacking gel, the cathode buffer was replaced with a fresh cathode buffer lacking dye.
[35S]Methionine pulse labeling of mitochondrial translation products in vivo
[35S]Methionine pulse labeling of mitochondrial translation products in vivo pulse labeling experiments were performed in minimum essential Dulbecco's modified Eagle's medium, without methionine, glutamine, or cysteine, and dialyzed serum (25 mm Tris-HCl, pH 7.4, 137 mm NaCl, and 10 mm KCl) following a previous protocol (68, 69). 0.2 mCi/ml [35S]methionine-containing medium (PerkinElmer Life Sciences) was added to the cells to label the mitochondrially encoded proteins. 2 h postincubation, cells were lysed in lysis buffer (50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 0.1% SDS, and 0.5% Nonidet P-40) supplemented with 1 mm phenylmethylsulfonyl fluoride and protease inhibitor mixture (Sigma-Aldrich). To separate the products formed, electrophoresis of whole-cell lysates (40 μg) was performed through 12% SDS-PAGE. The gels were dried on 3-mm chromatography paper, and the total intensities of the signals were quantified by phosphorimaging analysis.
Cloning in pHTC-Halo-Tag vector
To conduct another set of in vitro mitochondrial import assays, we cloned WT-AR (with MLS), Δ36n-MLS-AR (lacking 36 amino acids from the N terminus containing the MLS), Su9-DHFR (positive control), and DHFR (negative control) in pHTC-Halo-Tag vector (Promega) between NheI and XhoI restriction sites.
Mitochondrial targeting sequence cloning in GFP expression vector
Androgen receptor protein sequence was analyzed by using MitoProt, which predicts the probability of protein to be imported into mitochondria. A probability score of 0.25 was noted, and the software predicted a cleavage site after the first 14 amino acids. To validate the prediction that mitochondrial localization sequence resides in 1–36 amino acids from N-terminal, this stretch of sequence (containing 1–36 amino acids) was cloned in the pEGFP-N2 vector by PCR amplification within the EcoRI site and BamHI site. Cells were transfected using Fugene (Promega) according to the manufacturer's protocol. Immunocytochemical analysis was done to analyze the co-localization of GFP with MitoTracker dye signal and mounted with Prolong Gold antifade containing DAPI (Thermo Fisher Scientific, Waltham, MA) 48 h post-transfection. Images were obtained using fluorescence microscopy.
In vitro mitochondrial import
To conduct new in vitro mitochondrial import, WT-AR (with MLS), Δ36n-MLS-AR Su9-DHFR, and DHFR were cloned in pHTC-Halo-Tag vector (Promega) between NheI and XhoI restriction sites. Using T7 polymerase–coupled rabbit reticulocyte lysate transcription-translation systems (Promega), all of the above-mentioned pHTC-Halo-Tag constructs were transcribed and translated and modified later (70, 71). 500 μg of freshly isolated mouse brain mitochondria were used for import of translated proteins. The import assays were carried out in a 200-μl final volume and contained 500 μg of mitochondria (from a 10 mg/ml suspension in sucrose mannitol buffer), 60 μl of energy mixture (10 mm ATP, 10 mm GTP, 2.5 mm CDP, 2.5 mm UDP, 50 mm malate, 20 mm isocitrate), 70 μl of transport buffer (0.6 m mannitol, 20 mm Hepes, pH 7.4, 1 mm MgCl2). The import reactions were incubated at 28 °C for 60 min. After import, the reaction mixtures were cooled on ice for 5 min, and each mixture was divided into two equal portions (no trypsin and trypsin treatment). The trypsin digestion of mitochondria was performed for 20 min on ice (150 μg of trypsin/mg of mitochondrial protein). Control mitochondria were incubated similarly without adding trypsin. Soybean trypsin inhibitor (1.5 mg/mg of protein) was added to all samples to terminate the reactions. Mitochondria from both trypsin-treated and untreated samples were re-isolated by pelleting through 0.8 m sucrose, and the proteins were subjected to SDS-PAGE followed by Western blotting and analyzed by the Odyssey imaging system.
Initially, the androgen receptor cDNA construct (in pCDNA3.1 vector) was used as template in T7 polymerase–coupled rabbit reticulocyte lysate transcription-translation systems (Promega) (Fig. S1) in the presence of [35S]Met, as described previously (70), and modified later (70, 71). Su9-DHFR and DHFR cDNA construct were used as template in Sp6 polymerase-coupled rabbit reticulocyte lysate transcription-translation systems (Promega) in the presence of [35S]Met as described previously (70). Freshly isolated rat liver mitochondria were used for import of 35S-labeled translation products. Control experiments were carried out by preincubating mitochondria with carbonyl cyanide m-chlorophenylhydrazone (50 μm; Sigma-Aldrich) or oligomycin (50 μm; Sigma-Aldrich) at 25 °C for 20 min before initiation of the import reaction. After import, trypsin digestion of mitochondria was performed as the above-mentioned procedure. Mitochondria from both trypsin-treated and untreated samples were re-isolated by pelleting through 0.8 m sucrose, and the proteins were subjected to SDS-PAGE followed by fluorography.
RNA extraction and PCR amplification
For mRNA quantification, total RNA was isolated from cells using TRI reagent per the manufacturer's instructions (Molecular Research Center, Cincinnati, OH). RNA was digested with RQ1 RNase-free DNase (Promega). Total RNA (2 μg) was reverse-transcribed using the iScript cDNA synthesis kit (Bio-Rad). 25 ng of cDNA was used for quantification of mtDNA content with iQ SYBR Green Super Mix (Bio-Rad) on an ABI 7300 real-time PCR machine and analyzed using Primer Express version 3.0 (Applied Biosystems). Other gene targets were PCR-amplified using a Bio-Rad T100 thermal cycler and resolved on ethidium bromide–stained 1.5% agarose gel. A list of primer sequences is given in Table S1.
Statistical analysis
The means ± S.D. were calculated from 3–5 experimental values. p values were calculated using a one-tailed distribution and unequal variance to calculate significance (compared with vector control).
Author contributions
K. K. S. conceived the project. K. K. S. and G. S. designed the project. P. B. performed the experiments described in Figs. 1–8 and 10. E. K. conducted the mitochondrial translation studies (Fig. 9). P. B., R. S., and K. K. S. wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Ved Mooga for help with the BN-PAGE, Bhupendra Singh for help with prostate tissues, and Dr. Narayan Avadhani (University of Pennsylvania) for assistance with the initial import assay.
This study was supported by National Institutes of Health Grant R01 CA204430 and a grant from the Mike Slive Foundation for Prostate Cancer Research. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1 and Fig. S1.
- CRPC
- castration-resistant prostate cancer
- mtDNA
- mitochondrial DNA
- AR
- androgen receptor
- POLG1
- mtDNA polymerase γ
- MLS
- mitochondrial localization sequence
- DHFR
- dihydrofolate reductase
- MTS
- mitochondrial targeting signal
- OXPHOS
- oxidative phosphorylation
- BN-PAGE
- blue-native PAGE
- TFAM
- transcription factor A, mitochondrial
- DCPIP
- 2′,6-dichlorophenolindophenol
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- Tricine
- N,N-bis(2-hydroxyethyl)glycine.
References
- 1. Siegel R. L., Miller K. D., and Jemal A. (2015) Cancer statistics, 2015. CA Cancer J. Clin. 65, 5–29 10.3322/caac.21254 [DOI] [PubMed] [Google Scholar]
- 2. Siegel R. L., Miller K. D., and Jemal A. (2016) Cancer statistics, 2016. CA Cancer J. Clin. 66, 7–30 10.3322/caac.21332 [DOI] [PubMed] [Google Scholar]
- 3. Torre L. A., Siegel R. L., Ward E. M., and Jemal A. (2016) Global cancer incidence and mortality rates and trends–an update. Cancer Epidemiol. Biomarkers Prev. 25, 16–27 10.1158/1055-9965.EPI-15-0578 [DOI] [PubMed] [Google Scholar]
- 4. Ritch C. R., and Cookson M. S. (2016) Advances in the management of castration resistant prostate cancer. BMJ 355, i4405 10.1136/bmj.i4405 [DOI] [PubMed] [Google Scholar]
- 5. Jernberg E., Bergh A., and Wikström P. (2017) Clinical relevance of androgen receptor alterations in prostate cancer. Endocr. Connect. 6, R146–R161 10.1530/EC-17-0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ni L., Llewellyn R., Kesler C. T., Kelley J. B., Spencer A., Snow C. J., Shank L., and Paschal B. M. (2013) Androgen induces a switch from cytoplasmic retention to nuclear import of the androgen receptor. Mol. Cell Biol. 33, 4766–4778 10.1128/MCB.00647-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jagla M., Fève M., Kessler P., Lapouge G., Erdmann E., Serra S., Bergerat J. P., and Céraline J. (2007) A splicing variant of the androgen receptor detected in a metastatic prostate cancer exhibits exclusively cytoplasmic actions. Endocrinology 148, 4334–4343 10.1210/en.2007-0446 [DOI] [PubMed] [Google Scholar]
- 8. Liao R. S., Ma S., Miao L., Li R., Yin Y., and Raj G. V. (2013) Androgen receptor-mediated non-genomic regulation of prostate cancer cell proliferation. Transl. Androl. Urol. 2, 187–196 10.3978/j.issn.2223-4683.2013.09.07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lorin T., Salzburger W., and Böhne A. (2015) Evolutionary fate of the androgen receptor-signaling pathway in ray-finned fishes with a special focus on cichlids. G3 (Bethesda) 5, 2275–2283 10.1534/g3.115.020685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zarif J. C., Lamb L. E., Schulz V. V., Nollet E. A., and Miranti C. K. (2015) Androgen receptor non-nuclear regulation of prostate cancer cell invasion mediated by Src and matriptase. Oncotarget 6, 6862–6876 10.18632/oncotarget.3119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leung J. K., and Sadar M. D. (2017) Non-genomic actions of the androgen receptor in prostate cancer. Front. Endocrinol. (Lausanne) 8, 2 10.3389/fendo.2017.00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Castoria G., Lombardi M., Barone M. V., Bilancio A., Di Domenico M., Bottero D., Vitale F., Migliaccio A., and Auricchio F. (2003) Androgen-stimulated DNA synthesis and cytoskeletal changes in fibroblasts by a nontranscriptional receptor action. J. Cell Biol. 161, 547–556 10.1083/jcb.200211099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Q., Li W., Zhang Y., Yuan X., Xu K., Yu J., Chen Z., Beroukhim R., Wang H., Lupien M., Wu T., Regan M. M., Meyer C. A., Carroll J. S., Manrai A. K., et al. (2009) Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 138, 245–256 10.1016/j.cell.2009.04.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Efstathiou E., Titus M., Wen S., Hoang A., Karlou M., Ashe R., Tu S. M., Aparicio A., Troncoso P., Mohler J., and Logothetis C. J. (2015) Molecular characterization of enzalutamide-treated bone metastatic castration-resistant prostate cancer. Eur. Urol. 67, 53–60 10.1016/j.eururo.2014.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zarif J. C., and Miranti C. K. (2016) The importance of non-nuclear AR signaling in prostate cancer progression and therapeutic resistance. Cell. Signal. 28, 348–356 10.1016/j.cellsig.2016.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Modica-Napolitano J. S., Kulawiec M., and Singh K. K. (2007) Mitochondria and human cancer. Curr. Mol. Med. 7, 121–131 10.2174/156652407779940495 [DOI] [PubMed] [Google Scholar]
- 17. Higuchi M. (2007) Regulation of mitochondrial DNA content and cancer. Mitochondrion 7, 53–57 10.1016/j.mito.2006.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Singh K. K., and Modica-Napolitano J. S. (2017) Special issue: mitochondria in cancer. Semin. Cancer Biol. 47, iv–vi 10.1016/j.semcancer.2017.10.013 [DOI] [PubMed] [Google Scholar]
- 19. Moro L., Arbini A. A., Marra E., and Greco M. (2008) Mitochondrial DNA depletion reduces PARP-1 levels and promotes progression of the neoplastic phenotype in prostate carcinoma. Cell Oncol. 30, 307–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van Gisbergen M. W., Voets A. M., Starmans M. H., de Coo I. F., Yadak R., Hoffmann R. F., Boutros P. C., Smeets H. J., Dubois L., and Lambin P. (2015) How do changes in the mtDNA and mitochondrial dysfunction influence cancer and cancer therapy? Challenges, opportunities, and models. Mutat. Res. Rev. Mutat. Res. 764, 16–30 10.1016/j.mrrev.2015.01.001 [DOI] [PubMed] [Google Scholar]
- 21. Amuthan G., Biswas G., Ananadatheerthavarada H. K., Vijayasarathy C., Shephard H. M., and Avadhani N. G. (2002) Mitochondrial stress-induced calcium signaling, phenotypic changes and invasive behavior in human lung carcinoma A549 cells. Oncogene 21, 7839–7849 10.1038/sj.onc.1205983 [DOI] [PubMed] [Google Scholar]
- 22. Carew J. S., and Huang P. (2002) Mitochondrial defects in cancer. Mol. Cancer 1, 9 10.1186/1476-4598-1-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pelicano H., Xu R. H., Du M., Feng L., Sasaki R., Carew J. S., Hu Y., Ramdas L., Hu L., Keating M. J., Zhang W., Plunkett W., and Huang P. (2006) Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J. Cell Biol. 175, 913–923 10.1083/jcb.200512100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ayyasamy V., Owens K. M., Desouki M. M., Liang P., Bakin A., Thangaraj K., Buchsbaum D. J., LoBuglio A. F., and Singh K. K. (2011) Cellular model of Warburg effect identifies tumor-promoting function of UCP2 in breast cancer and its suppression by genipin. PLoS One 6, e24792 10.1371/journal.pone.0024792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li J., Ren S., Piao H. L., Wang F., Yin P., Xu C., Lu X., Ye G., Shao Y., Yan M., Zhao X., Sun Y., and Xu G. (2016) Integration of lipidomics and transcriptomics unravels aberrant lipid metabolism and defines cholesteryl oleate as potential biomarker of prostate cancer. Sci. Rep. 6, 20984 10.1038/srep20984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Koochekpour S., Marlowe T., Singh K. K., Attwood K., and Chandra D. (2013) Reduced mitochondrial DNA content associates with poor prognosis of prostate cancer in African American men. PLoS One 8, e74688 10.1371/journal.pone.0074688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Higuchi M., Kudo T., Suzuki S., Evans T. T., Sasaki R., Wada Y., Shirakawa T., Sawyer J. R., and Gotoh A. (2006) Mitochondrial DNA determines androgen dependence in prostate cancer cell lines. Oncogene 25, 1437–1445 10.1038/sj.onc.1209190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rimler A., Lupowitz Z., and Zisapel N. (2002) Differential regulation by melatonin of cell growth and androgen receptor binding to the androgen response element in prostate cancer cells. Neuro Endocrinol. Lett. 23, 45–49 [PubMed] [Google Scholar]
- 29. Lamb L. E., Zarif J. C., and Miranti C. K. (2011) The androgen receptor induces integrin α6β1 to promote prostate tumor cell survival via NF-κB and Bcl-xL independently of PI3K signaling. Cancer Res. 71, 2739–2749 10.1158/0008-5472.CAN-10-2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ekstrand M. I., Falkenberg M., Rantanen A., Park C. B., Gaspari M., Hultenby K., Rustin P., Gustafsson C. M., and Larsson N. G. (2004) Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet. 13, 935–944 10.1093/hmg/ddh109 [DOI] [PubMed] [Google Scholar]
- 31. Singh K. K., Ayyasamy V., Owens K. M., Koul M. S., and Vujcic M. (2009) Mutations in mitochondrial DNA polymerase-γ promote breast tumorigenesis. J. Hum. Genet. 54, 516–524 10.1038/jhg.2009.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Singh B., Owens K. M., Bajpai P., Desouki M. M., Srinivasasainagendra V., Tiwari H. K., and Singh K. K. (2015) Mitochondrial DNA polymerase POLG1 disease mutations and germline variants promote tumorigenic properties. PLoS One 10, e0139846 10.1371/journal.pone.0139846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rubio M. A., Rinehart J. J., Krett B., Duvezin-Caubet S., Reichert A. S., Söll D., and Alfonzo J. D. (2008) Mammalian mitochondria have the innate ability to import tRNAs by a mechanism distinct from protein import. Proc. Natl. Acad. Sci. U.S.A. 105, 9186–9191 10.1073/pnas.0804283105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bolton E. C., So A. Y., Chaivorapol C., Haqq C. M., Li H., and Yamamoto K. R. (2007) Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes Dev. 21, 2005–2017 10.1101/gad.1564207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fernández-Vizarra E., Tiranti V., and Zeviani M. (2009) Assembly of the oxidative phosphorylation system in humans: what we have learned by studying its defects. Biochim. Biophys. Acta 1793, 200–211 10.1016/j.bbamcr.2008.05.028 [DOI] [PubMed] [Google Scholar]
- 36. Ghezzi D., and Zeviani M. (2012) Assembly factors of human mitochondrial respiratory chain complexes: physiology and pathophysiology. Adv. Exp. Med. Biol. 748, 65–106 10.1007/978-1-4614-3573-0_4 [DOI] [PubMed] [Google Scholar]
- 37. Dudkina N. V., Sunderhaus S., Boekema E. J., and Braun H. P. (2008) The higher level of organization of the oxidative phosphorylation system: mitochondrial supercomplexes. J. Bioenerg. Biomembr. 40, 419–424 10.1007/s10863-008-9167-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dudkina N. V., Kouril R., Peters K., Braun H. P., and Boekema E. J. (2010) Structure and function of mitochondrial supercomplexes. Biochim. Biophys. Acta 1797, 664–670 10.1016/j.bbabio.2009.12.013 [DOI] [PubMed] [Google Scholar]
- 39. Zhou Y., Bolton E. C., and Jones J. O. (2015) Androgens and androgen receptor signaling in prostate tumorigenesis. J. Mol. Endocrinol. 54, R15–R29 10.1530/JME-14-0203 [DOI] [PubMed] [Google Scholar]
- 40. Khandrika L., Kumar B., Koul S., Maroni P., and Koul H. K. (2009) Oxidative stress in prostate cancer. Cancer Lett. 282, 125–136 10.1016/j.canlet.2008.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Caino M. C., and Altieri D. C. (2016) Molecular pathways: mitochondrial reprogramming in tumor progression and therapy. Clin. Cancer Res. 22, 540–545 10.1158/1078-0432.CCR-15-0460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kang D., Kim S. H., and Hamasaki N. (2007) Mitochondrial transcription factor A (TFAM): roles in maintenance of mtDNA and cellular functions. Mitochondrion 7, 39–44 10.1016/j.mito.2006.11.017 [DOI] [PubMed] [Google Scholar]
- 43. Barbour J. A., and Turner N. (2014) Mitochondrial stress signaling promotes cellular adaptations. Int. J. Cell Biol. 2014, 156020 10.1155/2014/156020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Harbauer A. B., Zahedi R. P., Sickmann A., Pfanner N., and Meisinger C. (2014) The protein import machinery of mitochondria-a regulatory hub in metabolism, stress, and disease. Cell Metab. 19, 357–372 10.1016/j.cmet.2014.01.010 [DOI] [PubMed] [Google Scholar]
- 45. Solakidi S., Psarra A. M., Nikolaropoulos S., and Sekeris C. E. (2005) Estrogen receptors α and β (ERα and ERβ) and androgen receptor (AR) in human sperm: localization of ERβ and AR in mitochondria of the midpiece. Hum. Reprod. 20, 3481–3487 10.1093/humrep/dei267 [DOI] [PubMed] [Google Scholar]
- 46. Yang S. H., Liu R., Perez E. J., Wen Y., Stevens S. M. Jr., Valencia T., Brun-Zinkernagel A. M., Prokai L., Will Y., Dykens J., Koulen P., and Simpkins J. W. (2004) Mitochondrial localization of estrogen receptor β. Proc. Natl. Acad. Sci. U.S.A. 101, 4130–4135 10.1073/pnas.0306948101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Scheller K., Sekeris C. E., Krohne G., Hock R., Hansen I. A., and Scheer U. (2000) Localization of glucocorticoid hormone receptors in mitochondria of human cells. Eur. J. Cell Biol. 79, 299–307 10.1078/S0171-9335(04)70033-3 [DOI] [PubMed] [Google Scholar]
- 48. Morrish F., Buroker N. E., Ge M., Ning X. H., Lopez-Guisa J., Hockenbery D., and Portman M. A. (2006) Thyroid hormone receptor isoforms localize to cardiac mitochondrial matrix with potential for binding to receptor elements on mtDNA. Mitochondrion 6, 143–148 10.1016/j.mito.2006.04.002 [DOI] [PubMed] [Google Scholar]
- 49. Psarra A. M., and Sekeris C. E. (2008) Steroid and thyroid hormone receptors in mitochondria. IUBMB Life 60, 210–223 10.1002/iub.37 [DOI] [PubMed] [Google Scholar]
- 50. Sepuri N. B. V., Tammineni P., Mohammed F., and Paripati A. (2017) Nuclear transcription factors in the mitochondria: a new paradigm in fine-tuning mitochondrial metabolism. Handb. Exp. Pharmacol. 240, 3–20 10.1007/164_2016_3 [DOI] [PubMed] [Google Scholar]
- 51. Choudhury A. R., and Singh K. K. (2017) Mitochondrial determinants of cancer health disparities. Semin. Cancer Biol. 47, 125–146 10.1016/j.semcancer.2017.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Antico Arciuch V. G., Elguero M. E., Poderoso J. J., and Carreras M. C. (2012) Mitochondrial regulation of cell cycle and proliferation. Antioxid. Redox Signal. 16, 1150–1180 10.1089/ars.2011.4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Levin E. R., and Hammes S. R. (2016) Nuclear receptors outside the nucleus: extranuclear signalling by steroid receptors. Nat. Rev. Mol. Cell Biol. 17, 783–797 10.1038/nrm.2016.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Carden T., Singh B., Mooga V., Bajpai P., and Singh K. K. (2017) Epigenetic modification of miR-663 controls mitochondria-to-nucleus retrograde signaling and tumor progression. J. Biol. Chem. 292, 20694–20706 10.1074/jbc.M117.797001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Arnould T., Vankoningsloo S., Renard P., Houbion A., Ninane N., Demazy C., Remacle J., and Raes M. (2002) CREB activation induced by mitochondrial dysfunction is a new signaling pathway that impairs cell proliferation. EMBO J. 21, 53–63 10.1093/emboj/21.1.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Higuchi M., Manna S. K., Sasaki R., and Aggarwal B. B. (2002) Regulation of the activation of nuclear factor kappaB by mitochondrial respiratory function: evidence for the reactive oxygen species-dependent and -independent pathways. Antioxid. Redox Signal. 4, 945–955 10.1089/152308602762197489 [DOI] [PubMed] [Google Scholar]
- 57. Biswas G., Adebanjo O. A., Freedman B. D., Anandatheerthavarada H. K., Vijayasarathy C., Zaidi M., Kotlikoff M., and Avadhani N. G. (1999) Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: a novel mode of inter-organelle crosstalk. EMBO J. 18, 522–533 10.1093/emboj/18.3.522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cutress M. L., Whitaker H. C., Mills I. G., Stewart M., and Neal D. E. (2008) Structural basis for the nuclear import of the human androgen receptor. J. Cell Sci. 121, 957–968 10.1242/jcs.022103 [DOI] [PubMed] [Google Scholar]
- 59. Gottlieb B., Beitel L. K., Nadarajah A., Paliouras M., and Trifiro M. (2012) The androgen receptor gene mutations database: 2012 update. Hum. Mutat. 33, 887–894 10.1002/humu.22046 [DOI] [PubMed] [Google Scholar]
- 60. Chan S. C., Li Y., and Dehm S. M. (2012) Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J. Biol. Chem. 287, 19736–19749 10.1074/jbc.M112.352930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yogev O., and Pines O. (2011) Dual targeting of mitochondrial proteins: mechanism, regulation and function. Biochim. Biophys. Acta 1808, 1012–1020 10.1016/j.bbamem.2010.07.004 [DOI] [PubMed] [Google Scholar]
- 62. Xu J., and Qiu Y. (2016) Role of androgen receptor splice variants in prostate cancer metastasis. Asian J. Urol. 3, 177–184 10.1016/j.ajur.2016.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chan S. C., and Dehm S. M. (2014) Constitutive activity of the androgen receptor. Adv. Pharmacol. 70, 327–366 10.1016/B978-0-12-417197-8.00011-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bajpai P., Srinivasan S., Ghosh J., Nagy L. D., Wei S., Guengerich F. P., and Avadhani N. G. (2014) Targeting of splice variants of human cytochrome P450 2C8 (CYP2C8) to mitochondria and their role in arachidonic acid metabolism and respiratory dysfunction. J. Biol. Chem. 289, 29614–29630 10.1074/jbc.M114.583062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jones J. O., Bolton E. C., Huang Y., Feau C., Guy R. K., Yamamoto K. R., Hann B., and Diamond M. I. (2009) Non-competitive androgen receptor inhibition in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 106, 7233–7238 10.1073/pnas.0807282106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Birch-Machin M. A., and Turnbull D. M. (2001) Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 65, 97–117 10.1016/S0091-679X(01)65006-4 [DOI] [PubMed] [Google Scholar]
- 67. Schägger H., and von Jagow G. (1991) Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 199, 223–231 10.1016/0003-2697(91)90094-A [DOI] [PubMed] [Google Scholar]
- 68. Yang Y., Cimen H., Han M. J., Shi T., Deng J. H., Koc H., Palacios O. M., Montier L., Bai Y., Tong Q., and Koc E. C. (2010) NAD+-dependent deacetylase SIRT3 regulates mitochondrial protein synthesis by deacetylation of the ribosomal protein MRPL10. J. Biol. Chem. 285, 7417–7429 10.1074/jbc.M109.053421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Koc E. C., Cimen H., Kumcuoglu B., Abu N., Akpinar G., Haque M. E., Spremulli L. L., and Koc H. (2013) Identification and characterization of CHCHD1, AURKAIP1, and CRIF1 as new members of the mammalian mitochondrial ribosome. Front. Physiol. 4, 183 10.3389/fphys.2013.00183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gasser S. M., Daum G., and Schatz G. (1982) Import of proteins into mitochondria: energy-dependent uptake of precursors by isolated mitochondria. J. Biol. Chem. 257, 13034–13041 [PubMed] [Google Scholar]
- 71. Bhat N. K., and Avadhani N. G. (1985) Transport of proteins into hepatic and nonhepatic mitochondria: specificity of uptake and processing of precursor forms of carbamoyl-phosphate synthetase I. Biochemistry 24, 8107–8113 10.1021/bi00348a041 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.