

Supplemental Digital Content is available in the text
Keywords: family case, juvenile-onset myoclonus, SCN2A, single nucleotide variation
Abstract
Rationale:
The phenotypic spectrum caused by SCN2A mutations includes benign neonatal/infantile seizures, Ohtahara syndrome, infantile spasms, West syndrome, and other unclassified epileptic phenotypes. Mutations in SCN2A have been implicated in neonatal seizure cases. Here, we described a Chinese family with 2 members having juvenile-onset myoclonus and identified a novel SCN2A point mutation within this family.
Patient concerns:
The 21-year-old male proband suffered from frequent myoclonus at 11 years old with subsequent progressive ataxia. His elder maternal half-sister also experienced myoclonus. Genomic DNA of the patients was extracted from the peripheral blood cells of the proband, elder maternal half-sister, parents, and uncle of the proband. Targeted next-generation sequencing was used to screen gene mutations in the proband. The potential functional effects of mutations within SCN2A were predicted In silico analyses.
Diagnoses:
Genetic testing revealed a novel SCN2A variant, c.T4820C, which contains a highly conserved amino acid substitution within segment S5 (p.V1607A). This mutation was predicted to produce a dysfunctional Nav1.2 protein by Mutation Taster and Protein Variation Effect Analyzer (PROVEAN). Genotype–phenotype correlation showed an incomplete penetrance of p.V1607A.
Interventions:
The proband was treated by multiple antiepileptic drugs. These included carbamazepine, oxcarbazepine, valproate, and topiramate.
Outcomes:
The duration of follow up was 2 years, and the proband developed drug-resistant epilepsy.
Lessons:
The case gives us the lesson that SCN2A mutation can contribute to juvenile-onset myoclonus. Our findings extend the spectrums of SCN2A mutations and the clinical features of patients with SCN2A mutations.
1. Introduction
SCN2A encodes the alpha subunit of voltage-gated sodium channel type II (Nav1.2) protein and plays crucial roles in the initiation and conduction of action potentials in the neural system. The spectrum of phenotypic and related single nucleotide variant (SNV) studies has relatively expanded since the first description of the relationship between SCN2A mutations and benign neonatal/infantile seizures.[1,2] SCN2A-related disorders are common in patients with infantile epilepsies. Elderly patients with SCN2A-related epilepsies are rarely reported.[3,4] Here, we report a Chinese family with juvenile-onset myoclonus and identify a novel SCN2A point mutation within the genome of this family.
2. Methods
2.1. Ethical approval and patient consent
This study was conducted in compliance with the ethical standards of the First Affiliated Hospital to Guangxi Medical University. The patient and his family have provided informed consent for publication of the case.
2.2. Genetic analysis
Genomic DNA of the patients was extracted from the peripheral blood cells of the proband (III-1), elder maternal half-sister (III-2), parents (II-1 and II-2), and uncle (II-3) of the proband. Targeted next-generation sequencing (TNGS) was used to screen gene mutations in the proband. A custom-designed panel capturing the coding exons of 500 genes related to epilepsy was synthesized using NimbleGenSeqCap Target Enrichment (Supplementary file 1). Sanger sequencing was performed to analyze the parental origin of these variants. One-hundred Chinese ancestries (50% male) were recruited as healthy controls.
2.3. In silico analyses
The potential functional effects of mutations within SCN2A were predicted using Sorting Intolerant from Tolerant,[5] Mutation Taster,[6] Polymorphism Phenotyping,[7] and Protein Variation Effect Analyzer (PROVEAN).[8] To further investigate the effect of the novel missense mutation within SCN2A, we developed a 3D protein structures using I-TASSER[9] and UniProt.[10]
3. Case report
The 21-year-old male proband was the first-born child of unrelated parents. Teratogen exposure was not present during the pregnancy. His seizures began when he was 11 years old. The seizure pattern was myoclonus, which involved the neck and bilateral upper and lower limbs, and commonly appeared when he watched television or computer. His seizure attacks became frequent when he was treated with carbamazepine and oxcarbazepine. After receiving treatment with valproate and topiramate, the patient still had a seizure frequency of 20 times/day. He progressively developed uncoordinated movement after 2 years. Neurological examination shows that he has slow reaction time and mild memory disorders, which were accompanied by poor performance in finger-nose-finger test and rapid alternating movements. The boy presented photosensitivity response under 16-, 21-, and 27-Hz photic stimulation and ictal electroencephalogram (EEG) showed generalized spike and wave complexes (Fig. 1-A and B). The mini-mental state examination scale result was 22. No abnormality signal was found under conventional MRI scans.
Figure 1.
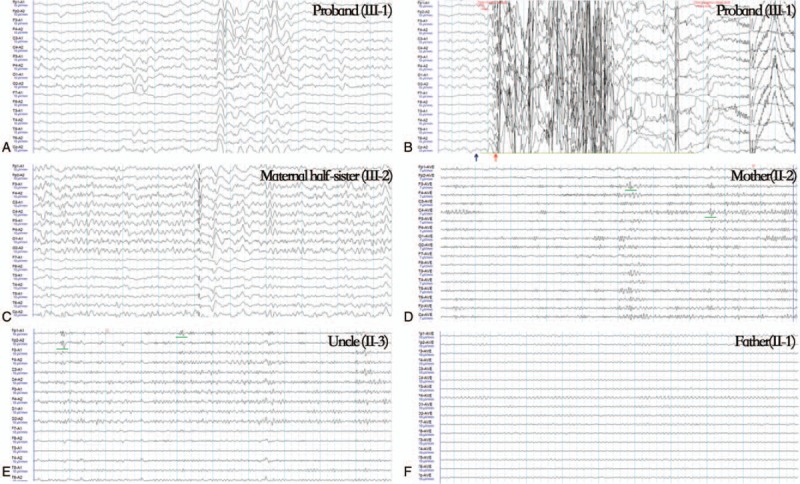
Interictal and ictal EEG features of the family with SCN2A mutation. Generalized seizure pattern was recorded when the proband received 16 Hz photic stimulation (B, blue arrow) with myoclonus (B, red arrow). The interictal EEG results of his mother and uncle showed an episodic frontal small spike sharp (D & E, green line). EEG = electroencephalogram.
The patient's maternal half-sister had experienced myoclonus since her youth and showed resistance to carbamazepine. The EEG results showed interictal generalized polyspike wave discharge at the time of this study (Fig. 1-C).
TNGS of the proband DNA was conducted with 99.79% coverage and 195.52 average read depth. Sanger sequencing was performed for pedigree analysis. The results revealed a heterozygous T>C missense mutation at nucleotide position 4820 (c.T4820C) in the SCN2A gene of the proband (Fig. 2). Similar heterozygous mutation was also detected in the maternal half-sister, mother, and uncle of the proband (Fig. 3). The c.T4820C mutation resulted in an amino acid change from valine (V) to alanine (A) at residue 1607 (p.V1607A).
Figure 2.
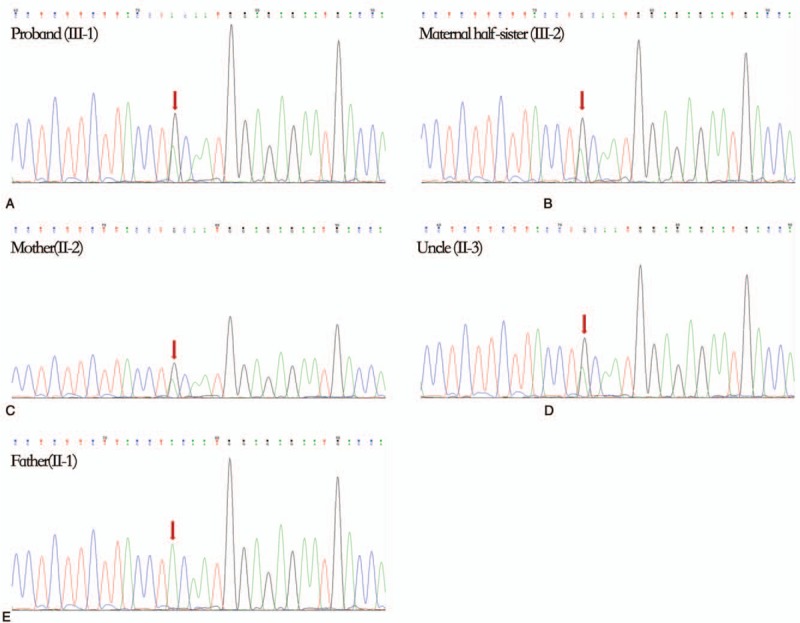
DNA sequencing chromatographs of the SCN2A gene. (A) Nucleotide sequence in the proband (III-1), (B) the maternal half-sister of the proband (III-2), (C) the mother of the proband (II-2), (D) the uncle of the proband (II-3), and (E) the father of the proband (II-1). The heterozygous missense mutation (red arrow) c.T4820C was identified in all the family members except the father.
Figure 3.
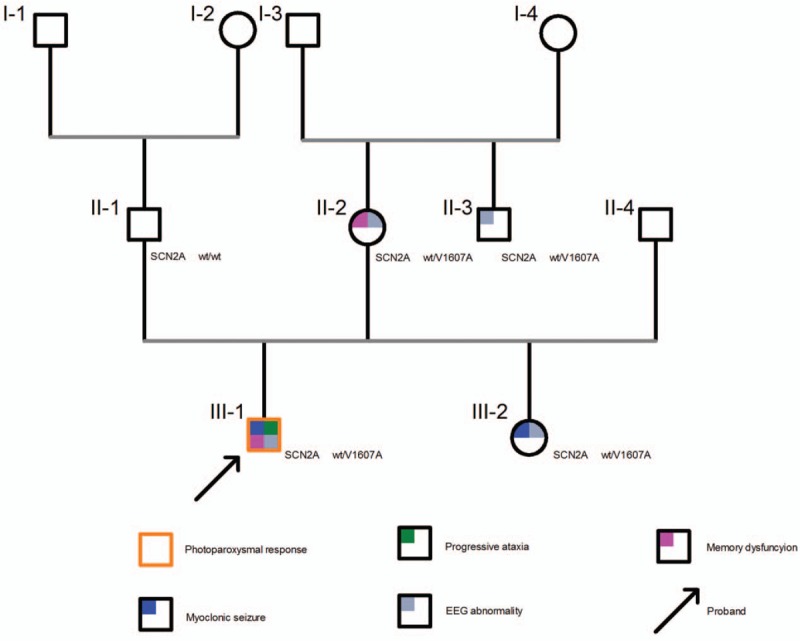
Pedigree diagram and clinical pictures of the family.
The mutation c.T4820C was absent in 100 Chinese health controls and was found at the extremely conserved positions in mammals (Fig. 4-B). The p.V1607A substitution was predicted to be “deleterious” by PROVEAN (score -3.235) and “disease-causing” by Mutation Taster. PolyPhen-2 and SIFT predicted that the p.V1607A mutation was functionally “possible damaging” (score 0.566; sensitivity 0.98, specificity 0.91) and “tolerated” (score 0.07), respectively.
Figure 4.
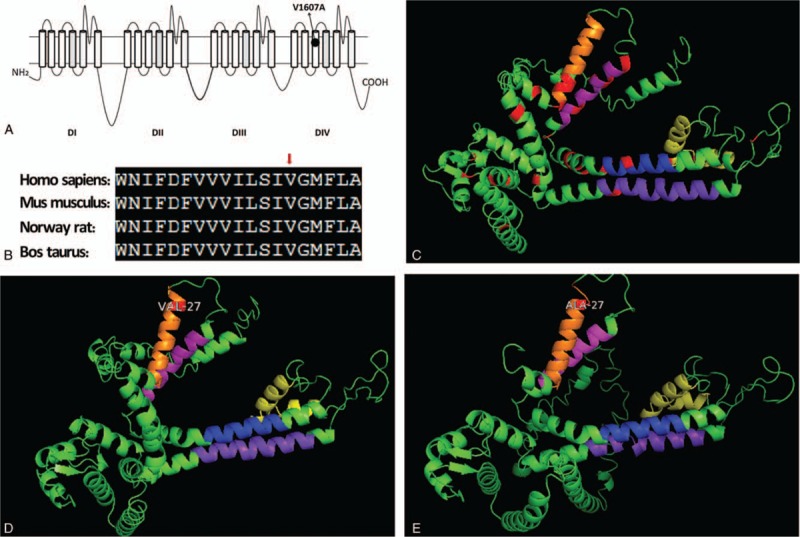
Topological analysis of Nav1.2. The alternation p.V1607A is located on S3 of dominant IV (A) and on extremely conserved positions in mammals (B). 3D structure of residues 1581 to 2005 of SCN2A as predicted by I-TASSER (C–E). Transmembrane segments, S3 (Orange), S4 (Peach), S5 (Blue), S6 (Purple), and pore forming area (Yellow) were marked. Previous missense mutations were highlighted in red (C). The replacement of V1607 (D) by alanine decreases the helix propensity, indicating that the helix is less stable in the mutant (E).
The topology of the structure was modeled using I-TASSER and UniProt. Both tools independently predict that V1607A is located in a transmembrane helix of SCN2A (Fig. 4-A). The replacement of V1607 by alanine reduced the helix propensity, indicating that the helix is less stable in the mutant (Fig. 4-E).
4. Discussions
SCN2A encodes the alpha subunit of Nav1.2, which plays a role in the initiation and conduction of action potentials. The phenotypic spectrum caused by SCN2A mutations includes benign neonatal/infantile seizures, Ohtahara syndrome, epilepsy of infancy with migrating focal seizures, infantile spasms, West syndrome, and other unclassified epileptic phenotypes.[11–13] SCN2A-related epileptic encephalopathy is the most common among SCN2A carriers and is characterized with refractory status epilepticus, cognitive dysfunction, and episodic ataxia.[2] The clinical outcome and the response to anti-epileptic drugs vary between early and late onset patients. Seizure reduction and good response to sodium channel blockers are often associated with early onset patients. These phenomena might result in mutations associated with increased sodium channel activity.[2,14]
To date, more than 150 SCN2A mutations have been reported in epilepsy. Among these mutations, 90% affect early development. Here, we reported a novel mutation, p.V1607A (c.T4820C), in a subject affected by juvenile-onset seizures. The SCN2A c.T4820C mutation that we identified was present in all affected members of a Chinese pedigree but was absent in the healthy controls. This finding indicated that the c.T4820C alteration is not a polymorphism. The c.T4820C mutation resulted in a missense mutation, a change from valine to alanine at codon 1607. Compared with nonsense and frameshift mutations that are usually pathogenic, missense mutations are commonly benign.[15] As a result, interpreting novel missense mutations is difficult. For further concerns, we employed in silico prediction tools such as PROVEAN and Mutation Taster to determine the pathogenicity of V1607A. The mutations in the pore area in SCN1A-related epilepsy are associated with most disabling epilepsies. By contrast, the disease-causing mutations of SCN2A are distributed widely. Such mutations were detected in the pore-forming area (S5-S6), transmembrane segments (S3, S4), and cytoplasmic residues of the Nav1.2 protein. In addition, the mutations in S5, Dominant IV of Nav1.2, such as Y1589C, G1593R, I1596S, F1597L, and D1598G were detected in benign neonatal/infantile seizures and epilepsy with migrating focal seizures[2] (Fig. 4-C). Moreover, the gain-of-function effects of the mutation F1597L were also confirmed via the whole cell patch clamping analysis. Combining the collected clinical and electrophysiological evidence, we suggested that the c.T4820C mutation possibly contributes to the dysfunction of Nav1.2 and SCN2A-related diseases. On the basis of topological structure, we speculate that this novel mutation combined with a specific pathological condition is caused by the disruption of the α-helical structure of the protein.
In this study, we identified 4 carriers (including 2 patients) from 2 generations of a family. The seizures were mainly myoclonic and occur in juveniles. Additional photosensitivity, ataxia, and cognitive dysfunction were observed in the proband. SCN2A mutations are most commonly detected in neonatal or infantile seizures. Some scholars insisted that SNVs in SCN2A might not be relevant in childhood- or juvenile-onset disorders because these variants play crucial roles in early cerebral development.[16] However, further histological and electrophysiological studies present different viewpoints. Nav1.2 is expressed in myelinated nerve fibers in early development and in unmyelinated inhibitory neurons.[17] Unmyelinated inhibitory neurons expressing Nav1.2 control the activity of excitatory neuron. As a result, dysfunctions in SCN2A possibly result in seizure by enhancing the excitability of excitatory neurons in early neonatal or infantile epilepsies and reducing the excitability of inhibitory neurons in adults.[2] Moreover, several studies indicated that movement disorder might be a key feature of SCN2A-related encephalopathy. In particular, Gorman and Johannesen reported that ataxia is followed by neonatal seizures in patients with SCN2A mutation.[18,19]
However, the novel mutation in this study was inherited from the mother who has not had seizures, suggesting that this mutation is causative with incomplete penetrance. In 2015, Bronwyn reported a family with SCN2A-associated neonatal seizures. The segregation analysis within this family revealed that only 50% (6/12) showed clinical manifestations.[20] In addition to the SCN2A mutation's contribution to epilepsy, phenotypic severity may also be affected by extrinsic factors.
There were some limitations in this study. First, as the prevalence of epilepsy may be altered with the follow-up period,[20] longer follow-up time is necessary to summarize the characteristics of seizures in this family. Second, further whole-cell patch clamping analysis is needed to be verified the function of the novel mutation in SCN2A.
5. Conclusions
In summary, we have identified a new heterozygous mutation site c.T4820C on SCN2A. This mutation can cause an amino acid change from valine to alanine at residue 1607, thus disrupting the α-helical structure of the protein. The causative mutation had an incomplete penetrance with clinical characteristics that varied from unaffected to progressive myoclonic epileptic conditions. This novel mutation adds up to a growing list of SCN2A mutations and may contribute to therapeutic development and prognostic purposes.
Author contributions
Data curation: Qi Huang, Yuan Wu.
Investigation: Qi Huang, Hengchang Qi.
Methodology: Lu Yu, Meigang Ma.
Software: Qi Huang.
Supervision: Yuan Wu.
Supplementary Material
Footnotes
Abbreviations: EEG = electroencephalogram, PROVEAN = Protein Variation Effect Analyzer, SNV = single nucleotide variant.
This work was supported by grants from National Natural Science Foundation, China (nos. 81760242 and 81660225).
The authors have no conflicts of interest to disclose.
References
- [1].Sugawara T, Tsurubuchi Y, Agarwala KL, et al. A missense mutation of the Na+ channel alpha II subunit gene Na (v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc Natl Acad Sci U S A 2001;98:6384–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wolff M, Johannesen KM, Hedrich UBS, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 2017;140:1316–36. [DOI] [PubMed] [Google Scholar]
- [3].Horvath GA, Demos M, Shyr C, et al. Secondary neurotransmitter deficiencies in epilepsy caused by voltage-gated sodium channelopathies: a potential treatment target. Mol Genet Metab 2016;117:42–8. [DOI] [PubMed] [Google Scholar]
- [4].Kobayashi K, Ohzono H, Shinohara M, et al. Acute encephalopathy with a novel point mutation in the SCN2A gene. Epilepsy Res 2012;102:109–12. [DOI] [PubMed] [Google Scholar]
- [5].Sim NL, Kumar P, Hu J, et al. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res 2012;40(Web Server issue):W452–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Schwarz JM, Rodelsperger C, Schuelke M, et al. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010;7:575–6. [DOI] [PubMed] [Google Scholar]
- [7].Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 2013;Chapter 7:Unit7.20. doi:10.1002/0471142905.hg0720s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015;31:2745–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 2010;5:725–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].UniProt: a hub for protein information. Nucleic Acids Res. 2015; 43(Database issue): D204-212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Allen NM, Conroy J, Shahwan A, et al. Unexplained early onset epileptic encephalopathy: exome screening and phenotype expansion. Epilepsia 2016;57:e12–17. [DOI] [PubMed] [Google Scholar]
- [12].Heron SE, Crossland KM, Andermann E, et al. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet 2002;360:851–2. [DOI] [PubMed] [Google Scholar]
- [13].Nakamura K, Kato M, Osaka H, et al. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology 2013;81:992–8. [DOI] [PubMed] [Google Scholar]
- [14].Scalmani P, Rusconi R, Armatura E, et al. Effects in neocortical neurons of mutations of the Na (v)1.2 Na+ channel causing benign familial neonatal-infantile seizures. J Neurosci 2006;26:10100–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Santos BPD, Marinho CRM, Marques T, et al. Genetic susceptibility in Juvenile Myoclonic Epilepsy: Systematic review of genetic association studies. PLoS One 2017;12: DOI: 10.1371/journal.pone.0179629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Liao Y, Deprez L, Maljevic S, et al. Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy. Brain 2010;133:1403–14. [DOI] [PubMed] [Google Scholar]
- [17].Micheva KD, Wolman D, Mensh BD, et al. A large fraction of neocortical myelin ensheathes axons of local inhibitory neurons. Elife 2016;5DOI: 10.7554/eLife.15784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gorman KM, King MD. SCN2A p.Ala263Val variant a phenotype of neonatal seizures followed by paroxysmal ataxia in toddlers. Pediatr Neurol 2017;67:111–2. [DOI] [PubMed] [Google Scholar]
- [19].Johannesen KM, Miranda MJ, Lerche H, et al. Letter to the editor: confirming neonatal seizure and late onset ataxia in SCN2A Ala263Val. J Neurol 2016;263:1459–60. [DOI] [PubMed] [Google Scholar]
- [20].Grinton BE, Heron SE, Pelekanos JT, et al. Familial neonatal seizures in 36 families: clinical and genetic features correlate with outcome. Epilepsia 2015;56:1071–80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.