

Abstract
Background and Objectives:
rpoS is a bacterial sigma factor of RNA polymerase which is involved in the expression of the genes which control regulons and play a critical role in survival against stresses. Few suitable vectors are available which could be maintained successfully in Flexibacter chinesis cells and could in particular be used as a suicide vector to make mutation in the rpoS gene. The aim of this study was to investigate if rpoS mutagenesis has impact on bacterial morphology in addition to cell division.
Materials and Methods:
A 0.603 kb BamHI-PstI fragment subclone of pICRPOS38Ω was cloned into linearized pLYLO3. The final construct, pLRPOS38 suicide vector, was introduced into Flexibacter chinesis. Then the cytoplasm of mutant strain and wild-type were investigated by transmission electron microscopy.
Results:
After successful subcloning of suicide vector into F. chinesis, based on TEM study, it was demonstrated that mutation in rpoS gene leads to decomposition of outer membrane of F. chinesis.
Conclusion:
A suitable vector to make suicide mutation in rpoS was constructed for F. chinesi.
Keywords: rpoS, Flexibacter chinesis, Transposon, Suicide vector, Transmission electron microscopy
INTRODUCTION
rpoS controls a large regulon (1, 2) and plays a critical role in cell survival in stress conditions such as near-UV exposure (3), thermal stress (4), prolonged starvation (5), oxidative stress (3) and low pH (6, 7). Also rpoS is important for genome instability (8), switching changes normally from error-free double-strand break repair into an error-prone process under stress (9), phase variation (8), mutagenic processes in growth-limiting environments of bacteria (10, 11) and spontaneous mutations (12). In some cases which several promoters involved in regulation of genes, only one gene is under control of δS (13, 14).
The rpoS gene is located in a structural gene with one operon common with nlpD encoding a lipoprotein. There is two promoters not regulated by growth phase and in experiential growth phase demonstrated a low level of rpoS expression (5). The rpoS transcription is controlled by the cAMP-CRP complex negatively. There is a transcriptional start site for rpoS gene, however there is no report of involvement of this site in rpoS transcriptional control (15, 16). Every attempt to manipulate a suitable construct, using the conventional cassettes and suicide vectors, which usually are used with E. coli and other Gram-negative bacteria failed.
In this study a new technique was reported which proved suitability for the genetic manipulation of gilding bacteria (17). In this study, the 5.97 kb pLYLO3 suicide vector was digested and a 0.603 kb Bam HI fragment subclone of pICRPOS38Ω was cloned into linearized pLYLO3. This construct was named pLRPOS38 and introduced into Flexibacter chinesis using conjugation method. Then the cytoplasm of mutant strain and wild-type were investigated by transmission electron microscopy.
MATERIALS AND METHODS
Bacterial growth media and conditions.
All bacterial strains were grown in Luria Agar (5 g/l NaCl, 15 g/l Agar, 10 g/l tryptone, 5 g/l yeast extract) or on Luria Broth (5 g/lNaCl, pH 7.2, 5 g/l yeast extract, 10 g/l Bacto tryptone).
Amplification of the rpoS gene.
Primers which were as follow: 5′GGGGGATCCCGTCAAGGGAT-CACGGGTAGGAGCCAC3′ (forward), and 5′GG-GGAATTCTTCAACCTGAATCTGGCGAACAC-GTTC3′ (Reverse) were used to amplify the rpoS gene in PCR assay. Amplification was carried out using High Fidelity PCR Master Kit (Roche) and a Perkin-Elmer (USA) DNA thermal cycler. Amplification conditions were 94°C for 45 sec (1 cycle), 94°C for 1 min, 65°C for 45 sec, 72°C for 30 sec (30 cycles). The PCR products were purified using High Pure Product Purification Kit (Roche).
Conjugation method.
The method described by Maniatis et al. 1989, was used with slight modification. E. coli S17-1 was used as the donor for the transfer of the recombinant plasmid, pLRPOS38 into F. chinensis. The plasmid RP4 is an IncP type plasmid which is integrated into the chromosome of E. coli S17-1 and the plasmid pLYLO3, contains an oriT (transfer origin) from PK2, an incP1 plasmid, which is recognized by IncPα plasmids, such as RP4, but not by Incpβ plasmids. The recombinant plasmid, pLRPOS38, was transferred into the E. coli S17-1 by transformation and the recombinant E. coli S17-1 was used as donor to introduce the recombinant plasmid, pLRPOS38 into F. chinensis as recipient, using bi-parental conjugation method. Then the trans-conjugated bacteria were cultivated onto LB agar containing 200 μg erythromycin to select for trans-conjugants. The plates were incubated for 2 to 3 days at 30°C (18, 19).
Transmission electron microscopy.
Samples were prepared and examined using the Jeol JEM-100S transmission electron microscope with an 80 kV accelerating voltage. Photographs were taken using Kodak Panasonic film, which was developed in Kodak D-19 developer at 20°C for 3 min and fixed in Kodak fixer.
RESULTS
Strategy for mutagenesis in the rpoS gene of F. chinesis.
The strategy for the mutation of the rpoS gene was the interruption of the gene by the insertion of an interposon (Ω) into the gene. A one kb EcoRI/BamHI fragment of the rpoS gene subclone of pBRPOS38 was cloned into pIC19H which provided some convenient poly cloning site for the construction. The construct was named pICRPOS38. The restriction map of the miniprep of the rpoS gene in this construct is shown in Fig. 1a. A unique Pml I site was approximately in the middle of the gene and unique for the cloning of pICRPOS38. The restriction digestion of Pml I site approximately located in the middle of the rpoS gene is shown in Fig. 1b. This site was used for insertion of a 0.8 kbp interposon into the rpoS gene.
Fig. 1.

a) Miniprep of pICRPOS38 was digested with a number of restriction enzymes. Lane 1 is λ ladder of 1 kb DNA as a molecular size. Lane 2 is pICRPOS38 digested with EcoRI and BamHI
b) Restriction digestion of pICRPOS38 at a unique PmlI site. Lane 1 is λ ladder of 1 kb DNA as a molecular size. Lane 2 is pICRPOS38 digested with PmlI. Lane 3 is pICRPOS38 digested with PmlI and EcoRI. Lane 4 is pICRPOS38 digested with PmlI and BamHI. Lane 5 is pICRPOS38 digested with PmlI, EcoRI and BamHI.
Interruption of the rpoS gene of F. chinesis
A 0.8 kb Ω fragment consisting of the CM (chloramphenicol) resistance gene of the pKRB10 plasmid was digested with Hinc II (which has recognition sites at both ends of the cassette) and purified. The constructed pICRPOS38 was linearized by digestion at the unique Pml I site. Both the linearized pI-CRPOS38 and Ω fragments were blunt ended through the digestion with Hinc II and Pml I. The Ω fragment was ligated into the Pml I site of pICRPOS38 yielding pKRPOS38Ω. This was transformed into E. coli TG1 and selection for CM resistance was made which confirmed that the Ω fragment had been inserted into the rpoS gene. pKRPOS38Ω was digested with BamHI with four sites. A nondigested sample was used as the control. In Fig. 2 the 0.8 kb bands in lanes 2 and 4 belong to the Ω fragment.
Fig. 2.

Restriction digestion of pICRPOS38Ω. Lane 1 is λ ladder of 1 kb DNA as a molecular size. Lane 2 is pICRPOS38Ω digested with PstI. Lane 3 is undigested pICRPOS38Ω as a control. Lane 4 is pICRPOS38Ω digested with HindII and Lane 5 is pICRPOS38Ω digested with BamHI.
Subcloning of the rpoS mutation into the suicide vector, pKNG101.
The mutated rpoS gene with the interposon digested from pICRPOS38Ω at the BglII and BamHI sites was introduced to a linearized pKNG101 digested at a BamHI and dephosphorylated to prevent self-ligation. The linearized vector and rpoS mutant gene were ligated and the construct was named pKRPOS38Ω. Restriction analysis was carried out to confirm the presence of a 1.8 kb fragment of the mutated rpoS gene in pKRPOS38Ω. As the BglII and BamHI sites had the same ends, they would ligate but neither BamHI nor BglII recognized the BamHI and BglII ligated site. pKRPOS38Ω was digested using four enzymes in two separate digestions. At first, digestion was carried out with SalI and BamHI. Due to the orientation of the insertion in the vector, two possibilities could occur with this digestion. Either the BglII downstream of the mutated rpoS gene could be ligated into the BamHI site located next to the SalI site in the vector, subsequent digestion of SalI and BamHI miniprep would result in a 1.8 kb insertion and 6.8 kb vector band; or the BglII end could be ligated to the BamHI site downstream of the rpoS mutated gene located next to Bst-BI of the vector, then digestion at the SalI and BamHI sites would not result in any bands. The digestion of SalI and BamHI resulted in 1.8 kb fragment which belonged to the mutated rpoS gene and 6.8 kb vector fragment which showed that the first possibility had occurred here (Fig. 3).
Fig. 3.

Restriction digestion of pKRPOS38Ω. Lane 1 is λ ladder of 1 kb DNA as a molecular size. Lane 2 is pKRPOS38Ω digested with SalI and BamHI.
Introduction of plasmid pKRPOS38Ω into F. chinesis cells.
Initially electroporation was attempted to transfer the DNA into F. chinesis. Subsequently, conjugation was employed for the introduction of plasmid by tri-parental mating. Every attempt to introduce pKRPOS38Ω by electroporation was unsuccessful, probably because of the restriction of transferred DNA by the F. chinesis restriction system. Conjugation was widely used to transfer pKRPOS38Ω into F. chinesis cells by a tri-parental mating method. One of the three parents was E. coli GJ342 which contains the helper plasmid, JC2926; the second parent was E. coli SM10λpir. The construct can only replicate in this strain because it supplies in Trans form of the π protein encoded by the λpir gene, such as in the E. coli strain lysogenized with a λpir transducing λ phage. All the attempts using conjugation to transfer pKRPOS38Ω into the F. chinesis were unsuccessful due to unknown factors.
Introduction of some plasmids, such as pBlue+, into F. chinesis was attempted alongside E. coli ML30 as a control. Electroporation, different methods of transformation and conjugation were used to transfer this plasmid into F. chinesis. Although all the methods were highly successful with the control, no F. chinesis colonies were found. The positive control proved that the transformation methods were successful, but due to unknown factors the plasmid was not maintained in F. chinesis.
Introduction of Tn4351 into F. chinesis on the broad host range IncPβ plasmid R751 (R751::Tn4351).
Attempts were made to introduce Tn4351 on the broad-host-ranged Incβ plasmid R751 (R751::Tn4351) into F. chinesis. A construct of R751::Tn4351 was selected for introduction into F. chinesis to discover if the introduction and insertion of the vector R751 and the transposition of T4351 into the F. chinesis chromosome by a tri-parental mating occurred. One parent was E. coli GJ342 which carried a helper plasmid, the second parent was E. coli HB101 which contained R751::Tn4351 and the third parent was the F. chinesis target strain. 189 colonies were isolated on LB agar plates which in passage in fresh media were able to grow in 200 μgml−1 erythromycin. The erythromycin resistance gene is carried by Tn4351. Erythromycin resistance colonies were transfer to LB agar containing 200 μgml−1 trimethoprim. None of the colonies could grow in this medium and no free vector (R751) was obtained in plasmid miniprep. This indicates that no replication of R751 occurred. Colony blot hybridization was done separately to discover if Tn4351 and/or R751 had inserted into the chromosome of F. chinesis. Duplicated blots were probed separately with radio-labeled pVOHI (for the detection of the Tn4351) and R751 (for the detection of the transposon delivery vector). pVOHI is a derivated of pBR328 that carries Tn4351 but there is no common sequence with R751. Approximately half of the colonies selected from the first screen were positive in the second screen for the detection of Tn4351 (Fig. 4.a). A few colonies were positive for the detection of R751 (Fig. 4.b). A few mutant defective in spreading were isolated (Fig. 4.c) and some auxotroph mutants were also isolated.
Fig. 4.

Colony blot hybridization for Tn4351 detection and a none-spreading mutant of F. chinesis.
a. DNA from erythromycin-resistance colonies was transferred to nylon membranes in duplicate and probed with radiolabeled pVOHI to detect Tn4351. The positive colons are arrowed.
b. DNA from erythromycin-resistance colonies was transferred to nylon membranes in duplicate and probed with radiolabeled R751 to detect the delivery vector. The positive controls are arrowed.
c. A none-spreading colony is arrowed.
Insertional mutagenesis in F. chinesis using the novel pUC19-based suicide vector (pLYO3).
After successfully cloning Tn4351 into F. chinesis and isolation of some auxotroph and none spreading mutants, a convenient suicide vector was chosen for disruption mutagenesis in F. chinesis. The identification of the ermF gene which was carried by Tn4351 and conferred erythromycin resistance in F. chinesis was an efficient method for showing that conjugation from E. coli to F. chinesis had been taken place. pLYLO3 and pUC19 based suicide vector for Bacteroides spp., contained ermF and also an origin of transfer which would allow conjugative transfer from E. coli. The ermF gene was not expressed in E. coli (20). pLYO3 was constructed originally from pFD160, which contains pUC19 and the cryptic bacteroides plasmid pBI143 and was the basis for the GUC vector. pBI143 also contained a mobilization region that was recognized by the IncP plasmid R751. The region encoding the GUS on a 1.9 kb fragment was removed from pBT101 and cloned into pFD160 which had been digested with BamHI and SstI and named pMJF-1. pMJF-2 was constructed by inserting a 2.9 kb ClaI fragment containing the ermF gene from the bacteroides transposon Tn4351 into pMJF1 (the 2.9 kb fragment was digested from a shuttle vector pVJR-1). The pBI143 segment from pMJF-2 was deleted and replaced with an oriT region from RK2. oriT was recognized by IncPα plasmid, such as RP4, but not by IncPβ plasmids, such as R751. This construct was named pCQW1 (21). Finally a 1.9 kb SmaI-SstI of the region encoding GUS was deleted from pCQW1 and the pCQW1 lacking the SmaI-sstI site was named pLYO3 (22, 23). Therefore, the 5.9 kb pLYO3 suicide vector was digested with BamHI and PstI sites and a 0.603 kb BamHI-PstI fragment subclone of pICRPOS38Ω was cloned into the linearized pLYLO3 digested with BamHI-PstI. Fig. 5 shows the results of the digestion of the 0.603 kb BamHI-PstI fragment from the construct, pLRPOS38.
Fig. 5.
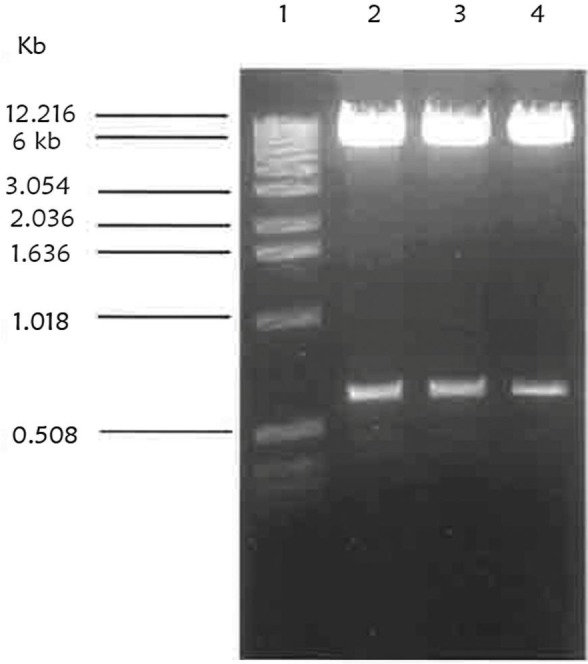
Restriction digestion of construct pLRPOS38. A miniprep of pICRPOS38 was digested. Lane 1 is λ ladder of 1 kb DNA as a molecular size. Lane 2, 3 and 4 show pLRPOS38 digested with BamHI and PstI sites resulting in a0.6 kb partial fragment of rpoS gene and 6 kb linearized pLYLO3 vector.
Introduction of the construct pLRpOS38 into F. chinesis cells by conjugation.
The construct, pLRPOS38 which contained oriT to allow conjugation was transferred into F. chinesis by a bi-parental conjugation method that one of the parents was E. coli S17-1 which contained a derivative of RP4 (RP4-2::Mu-Km::Tn7Tcr) integrated into the chromosome (24, 25). The other parent was F. chinesis as a recipient strain. The construct containing oriT carried the specific mob site which was recognized by IncPα plasmids, such as RP4, and facilitates transfer of the construct from E. coli S17-1. It also contained an ampicillin resistance gene which was expressed in E. coli and F. chinesis and an erythromycin resistance gene which was not expressed in E. coli but was expressed in F. chinesis. Therefore as the ermF gene did not confer erythromycin resistance to E. coli strain, the transformed construct in E. coli S17-1 was selected with ampicillin. After conjugation had taken place, as the ermF gene was not expressed in E. coli, the ex-conjugant cells were selected with erythromycin. 26 colonies were isolated on an erythromycin medium. This result demonstrated for the first time that pLYLO3 worked successfully as a suicide vector in gliding bacteria. The rpoS::pLYLO3 ex-conjugants were transferred into fresh erythromycin (200 μgml−1) medium to eliminate possible pseudo ex-conjugants.
Selection of recombinant F. chinesis.
24 erythromycin resistance colonies are single recombinants since the target gene was disrupted at one site. These cells would also been ampicillin resistant. The recombinant clone was F. chinesis carrying pLRPOS38 plasmid. Also the accuracy of cloning was confirmed by colony blot analysis (Fig. 4.a and 4.b).
Study of cell morphology using transmission electron microscopy (TEM).
The recombinant F. chinesis cells had a quite different shape to the starved wild-type cells and no shrinkage in the cytoplasm was observed (Data not shown). TEM study showed that the mutant strain had no outer membrane. This result suggested that the rpoS gene was important for composition of outer membrane in F. chinesis (Fig. 4.c).
DISCUSSION
The genetic studies of gliding bacteria had been limited with the exception of studies on one species, Myxococcus xanthus (26). Mutant of M. xanthus which were defective in motility had been isolated and a number of genes identified which were necessary for colony spreading (17). Following the development of technique for the genetic manipulation of Bacteroides spp., a number of techniques were developed to genetically manipulate gliding bacteria (17, 27).
Tn4351 was originally isolated from Bacteroides fragilis (28). The transposon was successfully introduced into Cytophaga succinicans, Flavobacterium meningosepticum, Flexibacter canadiansis, Flexibacter strain SFI and Sporocytophaga myxococcoides by conjugation (17). Tn4351 carried two antibiotic resistance genes. One of the genes coded for resistance to erythromycin and clindamycin was expressed in Bactroides but not in E. coli. The other gene coded resistance in tetracycline and was expressed in aerobically grown E. coli, but not in anaerobically grown E. coli or in Bacteroides. The transposon of Tn4351 was originally detected in E. coli which carried an unstable chimeric plasmid, pSS-2. The mobilization of pSS-2 from one strain of E. coli to another leaded to insertion of Tn4351 into R751 (R751::Tn4351) and less frequently to the insertion of Tn4351 into the chromosome. R751 also mobilized pE5-2 which contains a 3.8 kb EcoRI fragment of Tn4351 and can mobilize a cryptic bacteroides plasmid into Bacteroides spp. R751 was never detected in any of the Bacteroides trans-conjugants that carried pE5-2. This result demonstrates that R751 was not maintained in Bacteroides. In addition, there is some evidence that when R751 is inserted in to the bacteroides chromosome, it cannot mobilize out of the Bacteroides. Consequently it could be used as a suitable suicide vector in Bacteroides species.
Few suitable vectors are available which could be maintained successfully in F. chinesis cells and could, in particular, be used as a suicide vector to make mutation in the rpoS gene. In this study we used a novel method for mutagenesis of the rpoS gene in F. chinesis. Initially pKNG101 and pKRPOS38 plasmids were used to mutate the rpoS gene in the F. chinesis but the attempts were unsuccessful. Then In the bacteroides transposon, Tn4351 was cloned into R751 and transformed into F. chinesis successfully. In this method the 5.97 kb pLYLO3 suicide vector was digested and a 0.603 kb BamHI fragment subclone of pICRPOS38Ω was cloned into linearized pLYLO3. This construct was named pLRPOS38 and introduced into F. chinesis using conjugation method. The accuracy of cloning was approved by culturing of recombinant strain onto media containing erythromycin and ampicillin and using southern blot analysis. Electron microscopy of starved wild-type and starved recombinant F. chinesis showed that the cytoplasm of starved wild-type cells became clear and shrunk leaving cells which had an odd shape and appearance. In recombinant F. chinesis the cells had a quite different shape to the starved wild-type cells and no shrinkage in the cytoplasm was observed, previously in E. coli it was reported by Ozkanka (1993) (29) and in Hydrophila by Lim (1995) (30).
REFERENCES
- 1.Dong T, Kirchhof MG, Schellhorn HE. rpoS regulation of gene expression during exponential growth of Escherichia coli K12. Mol Genet Genomics 2008; 279: 267–277. [DOI] [PubMed] [Google Scholar]
- 2.Dong T, Schellhorn HE. Control of rpoS in global gene expression of Escherichia coli in minimal media. Mol Genet Genomics 2009; 281: 19–33. [DOI] [PubMed] [Google Scholar]
- 3.Sammartano LJ, Tuveson RW, Davenport R. Control of sensitivity to inactivation by H2O2 and broad-spectrum near-UV radiation by the Escherichia coli katF locus. J Bacteriol 1986; 168: 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hengge-Aronis R, Klein W, Lange R, Rimmele M, Boos W. Trehalose synthesis genes are controlled by the putative sigma factor encoded by rpoS and are involved in stationary-phase thermotolerance in Escherichia coli. J Bacteriol 1991; 173: 7918–7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lange R, Hengge-Aronis R. Identification of a central regulator of stationary-phase gene expression in Escherichia coli. Mol Microbiol 1991; 5: 49–59. [DOI] [PubMed] [Google Scholar]
- 6.Coldewey SM, Hartmann M, Schmidt DS, Engelking U, Ukena SN, Gunzer F. Impact of the rpoS genotype for acid resistance patterns of pathogenic and probiotic Escherichia coli. BMC Microbiol 2007; 7: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Small P, Blankenhorn D, Welty D, Zinser E, Slonczewski JL. Acid and base resistance in Escherichia coli and Shigella flexneri: role of rpoS and growth pH. J Bacteriol 1994; 176: 1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van den Broek D, Chin-A-Woeng TF, Bloemberg GV, Lugtenberg BJ. Role of rpoS and mutS in phase variation of Pseudomonas sp. PCL1171. Microbiology 2005; 151: 1403–1408. [DOI] [PubMed] [Google Scholar]
- 9.Ponder RG, Fonville NC, Rosenberg SM. A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation. Mol Cell 2005; 19: 791–804. [DOI] [PubMed] [Google Scholar]
- 10.Foster PL. Stress-induced mutagenesis in bacteria. Crit Rev Biochem Mol Biol 2007; 42: 373–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galhardo RS, Hastings PJ, Rosenberg SM. Mutation as a stress response and the regulation of evolvability. Crit Rev Biochem Mol Biol 2007; 42: 399–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Layton JC, Foster PL. Error-prone DNA polymerase IV is controlled by the stress-response sigma factor, rpoS, in Escherichia coli. Mol Microbiol 2003; 50: 549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gibson JL, Lombardo MJ, Aponyi I, Cruz DV, Ray MP, Rosenberg SM. Atypical role for PhoU in mutagenic break repair under stress in Escherichia coli. PLoS One 2015; 10(5): e0123315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamilton CM, Aldea M, Washburn BK, Babitzke P, Kushner SR. New method for generating deletions and gene replacements in Escherichia coli. J Bacteriol 1989; 171: 4617–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franchini AG, Ihssen J, Egli T. Effect of global regulators rpoS and cyclic-AMP/CRP on the catabolome and transcriptome of Escherichia coli K12 during carbon-and energy-limited growth. PLoS One 2015; 10(7):e0133793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lange R, Hengge-Aronis R. The cellular concentration of the sigma S subunit of RNA polymerase in Escherichia coli is controlled at the levels of transcription, translation, and protein stability. Genes Dev 1994; 8: 1600–1612. [DOI] [PubMed] [Google Scholar]
- 17.McBride MJ, Baker SA. Development of techniques to genetically manipulate members of the genera Cytophaga, Flavobacterium, Flexibacter, and Sporocytophaga. Appl Environ Microbiol 1996; 62: 3017–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sambrook J, Fritsch EF, Maniatis T. (1989). Molecular cloning: a laboratory manual. Cold Spring Harbor, NY, USA. [Google Scholar]
- 19.Clark MS. ed (2013). Plant molecular biology: a laboratory manual. Springer Science & Business Media; Berlin, Germany. [Google Scholar]
- 20.Reichenbach H. (2015). Myxococcales. In: Bergey's Manual of Systematics of Archaea and Bacteria. Eds, Whitman WB, Rainey F, Kämpfer P, Trujillo M, Chun J, DeVos P, Hedlund B, Dedysh S.). John Wiley & Sons, Inc. Hoboken, New Jersey, USA. [Google Scholar]
- 21.Rhodes RG, Samarasam MN, Van EG, McBride MJ. Mutations in Flavobacterium johnsoniae sprE result in defects in gliding motility and protein secretion. J Bacteriol 2011; 193: 5322–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veeranagouda Y, Husain F, Wexler HM. Transposon mutagenesis of the anaerobic commensal, Bacteroides fragilis, using the EZ:: TN5 transposome. FEMS Microbiol Lett 2012; 333: 94–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McBride MJ, Kempf MJ. Development of techniques for the genetic manipulation of the gliding bacterium Cytophaga johnsonae. J Bacteriol 1996; 178: 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feldhaus MJ, Hwa VI, Cheng QI, Salyers AA. Use of an Escherichia coli beta-glucuronidase gene as a reporter gene for investigation of Bacteroides promoters. J Bacteriol 1991; 173: 4540–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma L, Chen J, Liu R, Zhang XH, Jiang YA. Mutation of rpoS gene decreased resistance to environmental stresses, synthesis of extracellular products and virulence of Vibrio anguillarum. FEMS Microbiol Ecol 2009; 70: 130–136. [DOI] [PubMed] [Google Scholar]
- 26.Li X, Yang Q, Dierckens K, Milton DL, Defoirdt T. rpoS and indole signaling control the virulence of Vibrio anguillarum towards gnotobiotic sea bass (Dicentrarchus labrax) larvae. PloS One 2014; 9(10): e111801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrieres L, Hémery G, Nham T, Guérout AM, Mazel D, Beloin C, et al. Silent mischief: bacteriophage Mu insertions contaminate products of Escherichia coli random mutagenesis performed using suicidal transposon delivery plasmids mobilized by broad-host-range RP4 conjugative machinery. J Bacteriol 2010; 192: 6418–6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simon RU, Priefer U, Pühler A. A broad host range mobilization system for in vivo genetic engineering. Transposon mutagenesis in Gram negative bacteria. Biotechnol 1983; 1: 784–791. [Google Scholar]
- 29.Ozkanca R. (1993). Survival and physiological status of Escherichia coli in lake water under different nutrient conditions. Doctoral dissertation, University of Warwick. Coventry, UK. [Google Scholar]
- 30.Lim CH, Flint KP. The recovery of heat-stressed Escherichia coli in lake water microcosms. Lett Appl Microbiol 1995; 21: 364–367. [DOI] [PubMed] [Google Scholar]