


Abstract
In most bacteria, ribosomal RNA is transcribed as a single polycistronic precursor that is first processed by RNase III. This double-stranded specific RNase cleaves two large stems flanking the 23S and 16S rRNA mature sequences, liberating three 16S, 23S and 5S rRNA precursors, which are further processed by other ribonucleases. Here, we investigate the rRNA maturation pathway of the human gastric pathogen Helicobacter pylori. This bacterium has an unusual arrangement of its rRNA genes, the 16S rRNA gene being separated from a 23S-5S rRNA cluster. We show that RNase III also initiates processing in this organism, by cleaving two typical stem structures encompassing 16S and 23S rRNAs and an atypical stem–loop located upstream of the 5S rRNA. Deletion of RNase III leads to the accumulation of a large 23S-5S precursor that is found in polysomes, suggesting that it can function in translation. Finally, we characterize a cis-encoded antisense RNA overlapping the leader of the 23S-5S rRNA precursor. We present evidence that this antisense RNA interacts with this precursor, forming an intermolecular complex that is cleaved by RNase III. This pairing induces additional specific cleavages of the rRNA precursor coupled with a rapid degradation of the antisense RNA.
INTRODUCTION
The processing of ribosomal RNA (rRNA) is an important step in the biogenesis of ribosomes, leading to the production of mature rRNAs from large precursors. Our current understanding of bacterial rRNA processing is based primarily on studies using Escherichia coli (E. coli) and Bacillus subtilis (B. subtilis), the paradigms of Gram-negative and Gram-positive bacteria, respectively. In these two model organisms, like in many other bacterial species, the three rRNA genes are organized into operons and transcribed as a single precursor that contains the 16S, 23S and 5S rRNA species, sometimes interspersed with tRNA genes (1). The first processing reaction occurs co-transcriptionally on the nascent precursor and is performed by RNase III (1). This double-stranded specific endoribonuclease specifically cleaves the two large stem structures that are formed by the pairing of complementary sequences flanking the mature 16S and 23S rRNAs (2–5). This initial cleavage generates the precursor forms of 16S, 23S and 5S, which contain extra nucleotides (nt) at both their 5′ and 3′ ends (1,6). The three precursors are then further processed by a series of endo- and exonucleolytic cleavages to produce the mature species. The rRNA processing pathways and associated ribonucleases have been also investigated in other bacterial species, such as the α-proteobacteria (7,8). However, little is known for bacteria that have a different structural ribosomal gene organization. Indeed, unlinked operons in which either the 16S or the 5S gene is separated from a 23S-5S or a 16S-23S operon, respectively, have been reported in several bacteria (9). How processing occurs from the two ribosomal clusters, which RNases are involved, and whether there is a specific mechanism of regulation to ensure stoichiometric amounts of the three rRNAs, is not known.
The human bacterial pathogen Helicobacter pylori (H. pylori) has an unusual arrangement of its rRNA genes, with the 16S rRNA gene being separated from a 23S-5S rRNA operon (10,11). This Gram-negative, epsilon-proteobacterium colonizes the stomachs of about half of the human population. Infection by this bacterium generally causes asymptomatic gastritis but can progress, in rare cases, to more severe gastric diseases such as peptic ulcer or gastric adenocarcinoma (12). Although fragmentation of rRNA in aging cultures of H. pylori has been reported (13,14), nothing is known on rRNA maturation and ribosome biogenesis in this human pathogen. Among the RNases that play important roles in bacterial rRNA processing, H. pylori contains RNase III, RNase J and YbeY but lacks RNase E. Here, we have investigated whether RNase III is involved in H. pylori rRNA processing despite its unusual rRNA organization.
In addition to its role in bacterial rRNA processing, RNase III is also involved in the turnover of specific mRNAs by cleaving intramolecular stem–loop structures, or intermolecular structures formed between the mRNA and a small non-coding RNA (15). In H. pylori, RNase III has recently been shown to promote the degradation of the aapA1 toxin mRNA base-paired to IsoA1, a small RNA encoded on the opposite strand of the toxin gene (16). Hundreds of other cis-encoded RNAs, also called antisense RNA (asRNA), have been identified by transcriptome analysis of H. pylori (17). Whether they have a function and base-pair to their sense transcript is unknown. Interestingly, several of these asRNAs are encoded on the opposite strand of genes encoding stable RNA (17). Whereas some are complementary to transfer RNAs (tRNAs), others cover the 5′ precursor region of 16S and 23S rRNAs. Given their complementary with precursor sequences, these asRNAs could potentially interact with tRNA or rRNA precursors and affect their processing.
We have studied here in H. pylori the maturation of rRNA and characterized an asRNA that is transcribed at the 23S-5S rRNA locus. We show that, like in many other bacteria, RNase III also initiates rRNA processing in H. pylori, by cleaving two typical intramolecular stem regions encompassing the mature 16S and 23S rRNAs. Moreover, this enzyme also cleaves a stem–loop located upstream of the 5S rRNA, revealing a processing step that had not been reported in other bacteria. We also show, both in vivo and in vitro, that the asRNA interacts with the 5′ leader region of the 23S-5S precursor to form an intermolecular complex that is cleaved by RNase III. This pairing induces additional specific cleavages in the rRNA precursor coupled with a rapid degradation of the asRNA.
MATERIALS AND METHODS
Helicobacter pylori strains and culture conditions
The H. pylori strains used in this study were B128 (18–20) and X47-2AL (21). The corresponding strains deleted for the rnc gene were constructed by replacing the rnc Open Reading Frame (ORF) by the aphA-3 gene, as described in (16). Media and culture conditions were as described in (16). Liquid cultures were performed in brain–heart infusion (BHI) medium supplemented with 10% fetal bovine serum (SVF), at 37°C under microaerobic conditions (10% CO2, 6% O2, 84% N2) using an Anoxomat atmosphere generator. When required, antibiotics were used at the following concentrations: 20 μg/ml kanamycine, 8 μg/ml chloramphenicol and 30 μg/ml apramycin.
Plasmid constructions
Oligonucleotides used are given in Supplementary Table S1.
To construct the pET-rnc and pILL2157-rnc plasmids, the rnc ORF from B128 strain was polymerase chain reaction (PCR) amplified using FD378/FD380 primers and cloned into the NdeI-BamHI sites of pET15b (Novagen) and pILL2157 (22), respectively.
To construct pILL2159 plasmid, a derivative of pILL2150 (22) deleted for the lacI gene, pILL2150 was digested by HindIII and self-ligated.
The 23S asRNA promoter mutation TATAAT→TATAAC was constructed by site-directed mutagenesis using the FD689 and FD690 primers. The regions upstream and downstream of the promoter were amplified using B128 genomic DNA and the FD533/FD690 and FD689/FD708 pairs of primers, respectively. After assembly of the two PCR products, the final product was cloned into the BglII and NdeI sites of the pILL2159 vector, giving rise to the pILL2159-Pasmut plasmid.
The 23S rRNA promoter TATAAT→TATAAC mutation was constructed by site-directed mutagenesis using the FA250 and FA251 primers. The regions upstream and downstream of the promoter were amplified using B128 genomic DNA and the FD625/FA251 and FA250/FA155 pairs of primers. The two PCR fragments were annealed and re-amplified using the outer primers and the final product was cloned into pGEM®-T vector (Promega), resulting in the pGEMT-C2.7 vector.
The asRNA sequence was cloned under the control of the 23S rRNA promoter as follows: the 23S promoter and the upstream 250 nt was amplified from B128 strain using FD533/FD640 primers, whereas the asRNA sequence was amplified using FD660/FD662 primers. The two PCRs were annealed and re-amplified using external primers and cloned into BglII-KpnI sites of pILL2150 giving rise to the pILL2150-asRNA plasmid.
Construction of H. pylori strains
For complementation experiments, the B128 strain was transformed with either the pILL2157 or pILL2157-rnc plasmids, as described in (16).
All mutant strains were generated by homologous recombination and natural transformation of PCR-amplified cassettes carrying an antibiotic resistance gene flanked by ∼500 bp homology regions, as previously described (16). All strains were verified by PCR and the region of interest was sequenced. The kanamycin (kan) resistance aphA-3 gene, the chloramphenicol (cm) resistance catGC gene and the apramycine (apra) resistance aac(3)-IV gene were amplified from the pUC18K2 (23), pILL2150 (22) and the p1450 (24) plasmids, respectively. The oligonucleotides used for strain constructions are given in Supplementary Table S1.
Construction of the B128Δ23S-5S strain: One of the two 23S-5S clusters (rrn1 or rrn2, see Supplementary Figure S1A for the genomic location of the rrn genes) was replaced with a kanR cassette as follows. Regions upstream and downstream of the 23S-5S locus were PCR-amplified using FD625/FD626 and FD627/FD628 pairs of primers. The aphA-3 gene under the control of the RepG promoter was PCR amplified from strain CSS-0213 (25) using FD622/FD255 primers. The three overlapping fragments were assembled and used to transform B128 strain. KanR transformants deleted for either rrn1 or rrn2 were obtained. In this study, we used the BΔ23S-5S-1 strain that is deleted for the rrn1 operon.
Construction of the B128Δ23S-5S strains carrying either a wt or a mutated asRNA promoter (Supplementary Figure S8C): The 23S rRNA upstream region was amplified from B128 genomic DNA using FD625/FA840 primers; the cmR cassette was amplified from pILL2150 using FD232/FD841 primers; and the 23S leader region was amplified either from B128 genomic DNA (wt asRNA promoter) or from the pILL2159-asmut plasmid (mutated asRNA promoter) using FD533/FD842 primers. The three PCR fragments were annealed and amplified using FD625/FD842 primers. The resulting PCR product was transformed into the B128Δ23S-5S strain and kanR cmR transformants were selected. The resulting strains (BΔ23S-5S-29 for the wt asRNA promoter and BΔ23S-5S-30 for the mutated promoter) are deleted for the rrn1 locus (kanR cassette) and carry a cmR cassette ∼250 nt upstream of the 23S rRNA promoter at the rrn2 locus.
Construction of strains BC36 and BC41 (used in Figure 6): A kanR cassette was used to insert a mutation of the 23S rRNA promoter into one B128 rrn copy, and a cmR cassette was used to introduce a deletion of the asRNA into the other B128 rrn copy. The kanR cassette was introduced upstream of the 23S rRNA promoter at the rrn1 locus (see Supplementary Figure S1A) as follows: The region upstream of the 23S promoter was PCR amplified using primers FD625 and FA361 from B128 genomic DNA, the kanR cassette was amplified using primers FD622 and FD255 from strain BΔ23S-5S-1, and the promoter region was amplified with primers FA362 and FA155 from pGEMT-C2.7 plasmid that contains a single mutation in the 23S rRNA promoter. The three fragments were then annealed via their overlapping region and the fusion PCR product was transformed into B128 wt strain. KanR transformants were selected giving rise to the BC36 strain. The cmR cassette was introduced upstream of the 23S rRNA promoter at the rrn2 locus as follows: the cmR cassette from strain B128Δ23S-5S-29 was amplified using primers FD625/FA196. A PCR amplifying the 23S locus from nt 204 of the leader to nt 1800 of mature sequence was performed using FA195/FD842 primers. The annealing of the two PCR fragments created the deletion of the asRNA promoter and the first 160 nt of the asRNA transcript. The final PCR product was transformed into the BC36 strain and cmR transformants were selected. The resulting strain was called BC41.
Figure 6.

RNase III degradation of asRNA is strictly dependent on its interaction with its precursor complementary sequence. (A) Schematic view of the 5′ region of the two 23S-5S operons in B128 derivatives expressing either a wt or a truncated 23S-5S rRNA precursor. The ‘rrn1’ and ‘rrn2’ genes have been named arbitrarily (see Supplementary Figure S1A). Strain BC41 was constructed by inserting a point mutation that inactivates the rRNA promoter of one rrn copy and deleting the sequence of the asRNA (Δas) from the second rrn copy. In this strain, the asRNA cannot base-pair with the rRNA precursor. As a control, strain BC36 was constructed, in which the rrn2 copy is unchanged. Symbols are as in Figure 1A. (B) Northern blot analysis of asRNAs from BC36 (lanes ‘1’) and BC41 (lanes ‘2’) strains carrying or lacking the RNase III gene. Total RNA from BC36 and BC41 strains was extracted, separated on a denaturing acrylamide gel and transferred to a nylon membrane. A probe specific to the asRNA was used, as in Figure 5B. Reprobing with the 5S rRNA served as a loading control. (C) Proposed model to explain the different fates of asRNA expressed from strains BC36 and BC41. In strain BC36, as in a wt strain, the 175 and 130 nt asRNAs (‘as’) base-pair with the complementary sequence on the rRNA precursor (‘p23S’). The resulting intermolecular duplex is cleaved by RNase III, generating an upstream asRNA fragment of 70 nt, whereas the downstream fragment is degraded by endo- and/or exoribonucleases (depicted as scissors and pacman, respectively). In the absence of RNase III, the base-paired asRNAs are protected from degradation by other RNases, leading to their strong stabilization. In strain BC41, the asRNAs cannot base-pair with the truncated rRNA precursor (‘p23SΔas’). The free asRNAs are now exposed to other RNase activities (endo- and/or exoribonucleases) leading to their degradation, and no 70 nt long fragment is produced. The free 130 nt species is probably less sensitive to these RNases than the free 175 nt species, explaining its higher amount. This alternative degradation process occurs whether or not RNase III is present, explaining similar patterns in rnc+ or Δrnc backgrounds.
The BC36 and BC41 strains were then transformed with a rnc::apraR cassette and apramycin resistant clones were selected.
Deletion of the rnc gene with an apramycine resistance cassette. The aac(3)-IV apramycine resistance gene was used to create an in-frame deletion of the rnc gene. The regions upstream and downstream of the rnc gene were amplified from B128 strain using FD314/FA300 and FA301/FD319 primers, respectively. The aac(3)-IV gene was amplified from p1450 plasmid using FA302/FA304 primers.
RNA analysis
Total RNA was extracted from mid-exponential phase cultures (A600 nm 0.4–0.9) as described in (16). For northern blot analysis of large rRNA species, 8 μg of total RNA was separated on a 1% agarose gel in 0.5x TBE (Tris-Borate, ethylenediaminetetraacetic acid (EDTA)) buffer and vacuum-blotted onto a nylon membrane (HybondTM-N, GE Healthcare Life Science). For northern blot analysis of smaller RNAs, 5–10 μg of total RNA was separated on a denaturing 8% polyacrylamide gel and transferred as described in (16). Hybridization with 5′ end-labeled oligonucleotides was performed as in (16). For northern analysis of RNA extracted from polysome fractions, 1% agarose-formaldehyde gels in Tricine/Triethanolamine buffer were used (26).
Rifampicin experiments: B128 wt strains transformed with either pILL2150 or pILL2150-asRNA were grown in BHI medium supplemented with 10% SVF and chloramphenicol 8 μg/ml. At A600 nm 0.7–0.8, 80 μg/ml of rifampicin was added to the culture, cells were harvested at different time points of incubation and RNA was extracted.
Primer extension experiments: Total RNA from wt and Δrnc B128 strains was used for primer extension. After denaturation at 90°C, 4.5 μg RNA and 0.2 pmoles of 5′ end-labeled primer were annealed. Reverse transcription was performed with Superscript III (Invitrogen) at 52°C for 30 min. The extension products were separated on 6% polyacrylamide/7M urea gels and detected by autoradiography using a Pharos FX phosphorimager (Biorad).
Polysome profile analysis
Strains were grown in SVF containing BHI medium to an A600 nm of 0.8. Cells were rapidly cooled and collected by centrifugation. Cell pellets were resuspended in buffer A (10 mM Tris–HCl, pH 7.5, 60 mM KCl, 10 mM MgCl2) supplemented with 0.1 u/μl RNasin® Ribonuclease Inhibitor (Promega)) and lyzed with glass beads in a Precellys homogenizer (Bertin). Cleared lysates were loaded onto a 10–40% sucrose gradient in buffer B (10 mM Tris–HCl, pH 7.5, 50 mM NH4Cl, 10 mM MgCl2, 1 mM dithiothreitol (DTT)) and centrifuged in an SW41Ti rotor for 3h45 at 35 000 rpm. Gradients were analyzed with an ISCO UA-6 detector with continuous monitoring at 254 nm. Fractions (0.5 ml) were collected and RNA was precipitated by the addition of two volumes ethanol. Pellets were resuspended in 20 mM Na acetate, pH 5.2, 0.5% sodium dodecyl sulphate, 1 mM EDTA and RNA was extracted as in (16). One microgram of RNA from each fraction was then used for northern analysis. The peak observed between the top of the gradient and the 30S peak is probably due to the presence of genomic DNA since no DNase was used for the extract preparation (see Figure 4A).
Figure 4.

Distribution of the rRNA precursors across polysome profiles. (A) Polysome profiles of wt and Δrnc strains. Helicobacter pylori B128 extracts were fractionated on 10–40% sucrose gradients. The peaks of 30S, 50S, 70S and polysomes are indicated. The peak appearing between the top of the gradient and the 30S subunit probably contains genomic DNA (see ‘Material and Methods’ section). (B) RNA extracted from each gradient fraction was analyzed by northern blot. One microgram of RNA was separated on a denaturing 1% agarose gel and transferred to a nylon membrane. Methylene blue staining of the membrane revealed mature and precursor rRNAs. The ‘E’ lane corresponds to the extract before fractionation. Dotted vertical lines mean that two gels runned at the same time were brought together at this position. (C) The membranes shown in (B) were probed with the 5′ end-labeled oligonucleotide ‘a’ to reveal the 23S-5S p1 precursor and shorter processed products of ∼0.2–0.3 kb (highlighted by an asterisk). (D) The membranes shown in (B) were re-probed with the 5′ end-labeled oligonucleotide ‘g’ to reveal the 16S p4 and p5 precursors.
In vitro RNase III assays
The H. pylori His6-RNase III protein was overexpressed from pET-rnc plasmid in E. coli BL21(DE3) strain after 3 h isopropyl-1-thio-ß-D-galactopyranoside induction at 30°C. Cell pellets were resuspended in Binding Buffer (BB: 20 mM Tris–HCl, pH 7.9, 0.5 M NaCl) containing 5 mM imidazole, 5 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride (PMSF), 0.4 mg/ml of lysozyme and 10 μg/ml of DNase I. After sonication, the extract was centrifuged at 15 000 g for 10 min at 4°C. The supernatant was incubated on a Ni2+-NTA agarose column (Qiagen) equilibrated with BB buffer containing 5 mM imidazole. The column was washed with BB buffer containing 60 mM imidazole, then with 20 mM Tris–HCl, pH 7.9, 1 M NaCl, 60 mM imidazole. The His6-RNase III protein was then eluted with 20 mM Tris–HCl, pH 7.9, 1 M NaCl, 400 mM imidazole and dialyzed against 60 mM Tris–HCl, pH 7.9, 1 M NaCl, 1 mM EDTA, pH8.0, 1 mM DTT. The purified protein was kept in 30 mM Tris–HCl, 0.5 M NaCl, 0.5 mM EDTA, 0.5 mM DTT, 50% glycerol at −80°C.
For in vitro synthesis of the asRNA and 23S rRNA transcripts, DNA templates were amplified from B128 genomic DNA using the primer pairs FA197/FA234 (asRNA 178 nt) and FA88/FA156 (p280). The DNA template used for transcription of the p765 nt transcript was generated by fusing two PCR products amplified with FA88/FD373 and FA193/FA194. This construct is deleted for most of the mature 23S rRNA sequence, retaining only the 75 nt present at both the 5′ and 3′ ends. The transcript produced from this construction is expected to contain the RNase III processing stem. Each forward primer carries a T7 promoter sequence. In vitro transcription was performed using the MEGAscript® T7 Transcription kit (Invitrogen), according to the manufacturer’s protocol. Before use, each transcript was denatured and renatured and the p23S/asRNA complex formation was performed by mixing 0.7 μM of each transcript in digestion buffer (10 mM Tris–HCl pH 7.9; 50 mM NaCl; 10 mM MgCl2; 1 mM DTT) at room temperature for 10 min. For RNase III digestion, 1 μl of H. pylori His6-RNase III (∼3 pmoles) was added in a final volume of 10 μl for 5 min at 37°C. The reaction products were then analyzed by northern blot.
In vitro structure probing
For in vitro synthesis of the asRNA and the p280 nt rRNA transcripts, DNA templates were amplified from B128 genomic DNA using the primer pairs FA234/FA310 and FA311/FA156, respectively. Transcripts were 5′ end-labeled as in (16). The denaturation and renaturation steps of individual transcripts were performed as in (16). About 20 nM of labeled asRNA or 10 nM of labeled pre-23S were incubated with 290 nM pre-23S (280 nt) or 100 nM asRNA, respectively, in 1× Structure buffer (10 mM Tris–HCl, pH 7.0, 100 mM KCl, 10 mM MgCl2) for different times at 37°C (see Supplementary Figure S7A). The resulting complexes were then digested with 5 mM Pb Ac for 1 min at 37°C. The complexes that were formed after 30 min were digested either with 0.02–0.04 units of RNase T1 (Invitrogen), or with 0.25–0.5 units of RNase TA (Jena Biosciences), for 2 min at 37°C (in a final volume of 10 μl). For the ‘G’ lane, incubation with 0.2 units RNaseT1 was performed under denaturing conditions (20 mM Na Citrate, pH 5.0, 1 mM EDTA, 7 M urea) at 37°C for 5 min. Reactions were stopped and analyzed as in (16).
RESULTS
Role of RNase III in processing of H. pylori 23S-5S rRNA
In contrast to a large number of bacteria where the three rRNA genes form a single operon, H. pylori has an unusual rRNA gene organization, the 16S gene being separated from the 23S-5S operon (Supplementary Figure S1A and Figure 1A) (10,11,19). Since RNase III has been shown to be involved in rRNA maturation in several bacterial species, we examined whether it also participates in this process in H. pylori, despite this unusual rRNA organization. A precise in-frame deletion of the RNase III encoding gene, rnc, was generated in the B128 H. pylori strain (16). The parental and rnc-deleted strains will be referred to as wild-type (wt) and Δrnc, respectively. We observed that the doubling time of the Δrnc strain was only slightly increased (by ≤10%) compared to the wt strain (Supplementary Figure S1B). Total RNA was extracted from wt and Δrnc strains and analyzed on an agarose gel (Figure 1B). In addition to the mature 23S and 16S rRNAs, the Δrnc strain accumulated two high molecular weight species migrating above the 23S rRNA (Figure 1B, left panel). A northern blot using an oligonucleotide probe specific for the 5S rRNA (probe ‘c’) revealed that these two bands correspond to large rRNA precursors encompassing both the 23S and 5S rRNA mature sequences. Using the probe ‘a’ specific for the 5′ region of the precursor and other probes located at various positions on the operon, the upper band was attributed to a large 23S-5S precursor (p1, precursor 1) starting 374 nt upstream of the 23S rRNA mature 5′ end, whereas the lower band was attributed to a smaller 23S-5S precursor (p2) lacking most of the 5′ leader region (depicted as horizontal arrows in Figure 1A). The 5′ end of p1 coincides with the transcription start site (TSS) identified by RNA-seq in both B128 and 26695 H. pylori strains (Supplementary Figure S1C and (17)) and is consistent with the presence of a consensus TATAAT promoter sequence. The p1 precursor thus corresponds to the primary transcript of the cluster. The p2 precursor probably starts at or near the 5′ end of the mature 23S rRNA since it was not detected with a probe that covers the 18 nts located upstream of the mature 5′ terminus (Supplementary Figure S1D). Potential 5′ ends of this precursor mapped to 7–5 nt (nt 367–369) upstream of the mature 5′ end (see Figure 2A). In addition to the large p1 and p2 precursors, the Δrnc mutant also accumulated 5S rRNA precursor species (p5S) (see probe ‘c’). Of note, in both wt and Δrnc strains, the probe ‘a’ also detected shorter species of ∼0.2–0.3 kb (highlighted by an asterisk) that arise from processing cleavages in the leader region (see hereafter). Altogether, these results show that deletion of RNase III leads to strong processing defects of the primary 23S-5S rRNA precursor. The rRNAs that are not correctly processed are probably actively degraded, as suggested by the smear observed in the Δrnc strain (Figure 1B, probes a, b, c). This degradation may account for the slightly lower levels of the mature products. Nevertheless, mature 23S and 5S species are still produced, indicating an alternative RNase III-independent processing pathway.
Figure 1.

RNase III processes the 23S-5S rRNA precursor in vivo. (A) Schematic view of the Helicobacter pylori 23S-5S rRNA operon. The 23S and 5S rRNA mature sequences are represented as light gray boxes with nucleotide positions listed above (the +1 position corresponds to the transcriptional start site of the primary precursor). The dark gray box denotes the cis-antisense RNA. Promoters are represented by bent arrows and RNase III processing sites by vertical black arrows. The positions of the probes used in (B) are indicated as small black boxes on the upper horizontal line. Horizontal arrows below the operon indicate locations and sizes of the large rRNA precursors identified either in Δrnc or wt strains. (B) Northern blot analysis of rRNAs from wt and Δrnc strains. Total RNAs from B128 wt and Δrnc strains were separated on an agarose gel and stained with ethidium bromide (left). The gel was transferred on a membrane that was hybridized with probes c, a and b. The mature 23S, 16S and 5S rRNAs and the precursors (p1, p2, p3 and p5S) are indicated. The asterisks denote fragments of 0.2–0.3 kb processed from the leader region of the 23S-5S rRNA precursor. Their analysis on polyacrylamide gels showed that these fragments have a length of ∼280 and ∼210 nt in the wt and of ∼300 and ∼210 nt in the Δrnc strain (see Figure 8 and Supplementary Figure S8D).
Figure 2.

RNase III cleaves two stem structures of the 23S-5S rRNA precursor. (A) Total RNA extracted from B128 wt and Δrnc strains were subjected to primer extension analysis using 5′ end-labeled primers d, e and c (see location of the primers in (B)). The extension products were separated by electrophoresis on a denaturing polyacrylamide gel alongside a DNA sequence ladder generated with the same primer (GATC lanes). Sequence around the main RT stops generated from wt RNA is shown alongside the gel and arrows indicate positions of RNase III cleavage sites. ‘M’ corresponds to the 5′ end of the mature 23S rRNA and the asterisk denotes the mature 5′ end of 5S rRNA. The signal for both mature ends was saturated. In the left panel (primer d), two different RNA samples (1 and 2) from wt and Δrnc strains were used. (B) Secondary structure prediction of the 23S-5S pre-rRNA. The structure was obtained using the Mfold program (27). The mature 23S and 5S sequences are in gray, whereas the precursor sequences are represented as black lines. The coordinates refer to the rRNA precursor transcription start point. The locations of the primers used for reverse extension are indicated by gray arrows. Thick black arrows indicate positions of the RNase III cleavage sites, which were mapped in vivo (A). Thin arrows show RT stops obtained with RNA extracted from Δrnc strain. An enlargement of the processing stems with cleavage sites is shown on the right. The upper case nucleotides in the 5S stem correspond to the first nucleotides of the 5S rRNA mature sequence.
Whereas the p1 and p2 precursors were detected only in the Δrnc strain, another precursor (p3), revealed with the probe ‘b’, was observed only in the wt strain (Figure 1B, right panel). From the primer extensions experiments (see below) and the use of different probes for northern experiments, we showed that this precursor is the product of an RNase III cleavage occurring on both sides of 23S rRNA (depicted by vertical arrows in Figure 1A).
All of the 23S and 5S rRNA processing defects observed in the Δrnc strain were corrected by transforming the Δrnc strain with a vector expressing RNase III (Supplementary Figure S1E), indicating that they can be directly attributed to the absence of the RNase III encoding gene. Thus, our results show that RNase III is directly involved in the processing of both 23S and 5S rRNAs.
RNase III cleaves two stem structures of the 23S-5S rRNA precursor
Processing of pre-rRNA by RNase III in several bacteria has been shown to involve large stem structures that are formed by the pairing of complementary sequences flanking the 16S and 23S rRNAs (6). Prediction of the secondary structure of the H. pylori p1 precursor by Mfold (27) showed that the sequence flanking the mature 23S rRNA might base-pair within a long helical domain, generating a potential RNase III substrate (Figure 2B). To map RNase III cleavage sites, we performed primer extension experiments on total RNA isolated from wt and Δrnc strains. Extension with the 5′ end-labeled primer ‘d’ that is complementary to the 5′ region of the mature 23S rRNA revealed a reverse transcriptase (RT) stop at nt 283 (with respect to the transcription start point) in the wt strain (Figure 2A, left). This RT stop was not detected in the Δrnc strain, thus reflecting a potential cleavage site by RNase III after nt 282. Extension from this primer also revealed RT stops at other positions of the precursor (nt 356 and nt 369–367) both in the wt and Δrnc strains, probably reflecting processing cleavages by unknown RNase(s). Finally, it also mapped the 5′ end of the mature 23S rRNA (‘M’). Then, the use of the primer ‘e’ that anneals downstream the 3′ end of mature 23S rRNA identified a major RT stop at nt 3310 in the wt strain (Figure 2A, middle). The two cleavage sites observed at nt 282 and 3309 occur in the double-stranded region formed by sequences flanking the 23S rRNA and produce a two-nucleotide 3′ overhang, which is a hallmark of processing by RNase III (black arrows in Figure 2B). This cleavage generates a 3 kb product, which corresponds to the p3 precursor detected in the wt strain (Figure 1B). Finally, extension with the primer ‘c’ located in the 5S sequence revealed, along with a major RT stop at nt 3492 corresponding to the 5′ end of the 5S mature sequence, a RT stop at nt 3488 that is detected only in the wt strain (Figure 2A, right). This stop is located within a putative stem–loop structure and likely corresponds to a specific RNase III cut (Figure 2B). Indeed, a cleavage site on the opposite strand (at nt 3460) was inferred by comparing RNA-seq data from wt and Δrnc strains (Supplementary Figure S2). The cleavages at nt 3460 and 3487 generate a two-nucleotide 3′ overhang, characteristic of RNase III cleavage. Altogether, our results show that in vivo RNase III cleaves two stem structures of the large 23S-5S precursor, one flanking the mature 23S rRNA sequence and the other located just upstream of the mature 5S rRNA. Whereas the first stem is analogous to the large processing stalks found in other bacteria, the second one has not been reported and could be due to the specific rRNA gene organization of H. pylori.
The primer extension experiments also revealed several RT stops that were generated only in the Δrnc strain (thin black arrows in Figure 2B). Whereas some of them may be due to secondary structures blocking the RT enzyme, others may be due to alternative processing cleavages.
Role of RNase III in processing of H. pylori 16S rRNA
To determine whether RNase III is also involved in the maturation of 16S rRNA, the membrane used in Figure 1 was hybridized with a probe specific to the 5′ end region of the 16S rRNA precursor (probe ‘f’, Figure 3A). A transcript of around 2 kb strongly accumulated in the Δrnc strain (Figure 3B, middle), showing that RNase III also participates in the processing of the 16S rRNA precursor, despite its monocistronic arrangement. This p4 unprocessed precursor starts 454 nt upstream of the 16S rRNA mature 5′ end, which is consistent with the TSS identified by RNA-seq in both B128 and 26695 H. pylori strains (Supplementary Figure S3; 17). The ‘g’ probe located downstream of the ‘f’ probe, revealed another precursor, slightly shorter than p4, which is found in the wt strain (Figure 3B, right). This p5 species is likely the product of an RNase III cleavage occurring in the double-stranded structure flanking the 16S rRNA mature sequence (Figure 3C). Thus, our results show that RNase III is involved in the processing of the 16S rRNA precursor by cleaving a stem structure flanking the 16S rRNA mature sequence. As for the 23S-5S rRNA precursor, the absence of RNase III does not abolish 16S rRNA processing completely, indicating that alternative RNase III-independent processing pathways also exist to produce mature 16S rRNA.
Figure 3.

RNase III processes the 16S rRNA precursor in vivo. (A) Schematic view of the Helicobacter pylori 16S rRNA gene. The 16S rRNA mature sequence is shown as a light gray box with nucleotide positions listed above (the +1 position corresponds to the transcriptional start site of the primary precursor). The promoter is represented by a bent arrow and RNase III processing sites by vertical black arrows. The positions of the probes used in (B) are indicated as black boxes on the upper horizontal line. Horizontal arrows below the 16S rRNA locus indicate locations and sizes of the large rRNA precursors identified either in Δrnc or wt strains. (B) Northern blot analysis of rRNAs from B128 wt and Δrnc strains. The same membrane used in Figure 1B was hybridized with probes f and g. The 16S rRNA precursors (p4 and p5) are indicated. (C) Secondary structure prediction of the 16S pre-rRNA. The structure was obtained using the Mfold program (27). The mature 16S sequence is in gray, whereas the precursor sequences are represented as black lines. The proposed RNase III cleavage site is shown.
Unprocessed 23S-5S but not 16S rRNA precursors are found in polysomes
To assess whether the unprocessed rRNA precursors that accumulate in absence of RNase III are incorporated into ribosomes, we analyzed the rRNA content of the ribosomal particles from wt and Δrnc strains. After growth in rich medium at 37°C, cells were lyzed and ribosomal particles were separated by sucrose gradient sedimentation. Although both profiles looked similar, a smaller amount of 70S ribosomes and a slight decrease of the 50S/30S ratio was reproducibly observed in the Δrnc strain in comparison to the wt strain (Figure 4A). We then analyzed the rRNA species present in the different ribosomal particles by northern blotting. The methylene blue staining of the membrane revealed the major rRNA species (Figure 4B). Surprisingly, the two large p1 and p2 precursors of the Δrnc strain were found not only in the 50S ribosomal subunits, but also in the free 70S ribosomes and in polysomal fractions. Hybridization of the membrane with the probe ‘a’ specific for the p1 precursor confirmed the distribution of this large precursor in the 50S subunit fraction throughout the heaviest polysomal fractions (5 ribosomes per mRNA; Figure 4C). The 5S precursors (see ‘p5S’ Figure 1B) were also found in polysomes (Supplementary Figure S4). Thus, immature 23S-5S and 5S species are assembled into ribosomes that can associate with mRNAs. Whether these immature ribosomes are engaged in translation or are being processed within translating ribosomes will be discussed later.
The probe ‘a’ also revealed smaller transcripts of ∼0.2–0.3 kb (highlighted by an asterisk). In contrast to the large precursors, these transcripts were found mainly in the top of the gradient (Figure 4C) and are thus not strongly associated with ribosomal particles.
We then analyzed the distribution of the 16S rRNA precursors within the gradient. Using the probe ‘g’, the 16S precursors were found mainly in fractions 5–7 corresponding to the 30S subunit (Figure 4D). These precursors were even visible on the methylene-blue stained membrane whereas they were barely detected in the total RNA extract (compare lanes ‘E’ with fractions ‘5, 6, 7’ in Figure 4B). This result indicates that, in contrast to the 23S-5S precursors that follow the same distribution than the mature 23S rRNA, the 16S precursors have a very narrowed localization, being found almost exclusively in 30S fractions. Thus, the precursors of the small subunit are not engaged in translation and thus may not be functional in translation. Alternatively, it is also possible that the final processing steps for 16S rRNA are more efficient than those of 23S rRNA in the absence of RNase III.
A cis-antisense RNA complementary to the 23S-5S rRNA precursor is degraded by RNase III
Since the transcriptome analysis of H. pylori strain 26695 identified a cis-antisense RNA on the opposite strand of the 23S rRNA gene (17), we asked whether this transcript also exists in the B128 strain. To this end, total RNA from wt and Δrnc B128 strains were analyzed by northern blot using a probe specific for the antisense transcript (oligonucleotide complementary to the ‘a’ probe; see Figure 1A). This probe, named ‘asRNA probe’, revealed faint signals of about 175, 130 and 70 nt in the wt strain (Figure 5B). Remarkably, a dramatic increase of the two larger species was observed in the Δrnc strain, whereas the short 70 nt transcript was no longer detected (Figure 5B). Moreover, species ranging from ∼75 to ∼80 nt, which may correspond to degradation products of the larger transcripts, also appeared in absence of RNase III. All these transcripts (depicted as horizontal arrows, Figure 5A) were also revealed by a probe complementary to the first 20 nt of the antisense transcript, indicating that they share the same 5′ end (Supplementary Figure S5A). Hence, the longer asRNA overlaps the first 172 nt of the rRNA precursor. Using rifampicin experiments, we then showed that the higher abundance of the transcripts in the Δrnc strain is due to their strong stabilization rather than to an increased transcription (Figure 5B, right). These transcripts will be called hereafter 23S antisense RNAs or 23S asRNAs.
Figure 5.

RNase III degradation of a cis-encoded RNA antisense to the 23S-5S rRNA precursor. (A) Schematic view of the Helicobacter pylori 23S-5S rRNA operon and its cis-encoded antisense RNA (denoted ‘as’). Symbols are as in Figure 1A. (B) Northern blot analysis of asRNA from wt and Δrnc strains. (Left) Total RNAs from B128 wt and Δrnc strains were separated on a denaturing acrylamide gel. After transfer, the membrane was hybridized with an asRNA specific probe. The main transcripts of ∼175, 130 and 70 nt are indicated. The same membrane was reprobed with 5S rRNA as a loading control. (Right) Total RNA from wt and Δrnc strains were extracted at different times after rifampicin (rif) addition. The membrane was hybridized with asRNA and 5S rRNA probes.
Sequence alignments indicated that the promoter of the antisense RNA is strictly conserved among different H. pylori strains (Supplementary Figure S5B). Moreover, we also observed a strong accumulation of the 23S asRNA following RNase III deletion in another H. pylori strain, X47-2AL (Supplementary Figure S5C). These results indicate that both transcription and RNase III degradation of the asRNA are conserved in H. pylori, suggesting a functional relevance of this transcript.
In vivo evidence for the direct pairing of the cis-antisense RNA with the rRNA precursor
We have shown that RNase III is responsible for the rapid degradation of the asRNA (Figure 5). We then asked whether this enzyme cleaves an intramolecular structure of the asRNA or an intermolecular base-pairing formed between the asRNA and the complementary 172 nt of rRNA precursor. To answer this question, we examined the abundance of the asRNA in the absence of its complementary sequence, using a strain that expresses the asRNA and a truncated 23S-5S rRNA precursor lacking the asRNA sequence. We then analyzed whether the rnc deletion can still lead to the stabilization of the asRNA despite its inability to interact with the rRNA precursor. We took advantage of the presence of only two copies of each rRNA gene in H. pylori to create this strain. On one rrn copy, a single mutation in the promoter of the rRNA transcript was introduced, whereas the asRNA promoter was unaffected. We showed that the single point mutation strongly, although not fully, inhibited transcription (Supplementary Figure S6). On the second rrn copy, a deletion removing both the promoter and the first 160 nt of the asRNA was created, whereas the promoter of the rRNA transcript was unchanged (strain BC41, Figure 6A). As a control, a strain containing a single mutation in the rRNA promoter but no deletion in the asRNA region was constructed (strain BC36). RNA extracted from these two strains was analyzed by northern blot. The asRNA probe revealed the three 175, 130 and 70 nt transcripts in strain BC36, as observed in a wt strain (see Figure 5B), whereas only the 175 and 130 nt species could be detected in strain BC41 (Figure 6B). This result showed that the production of the 70 nt transcript depends on the presence of the rRNA complementary sequence. Introducing the rnc deletion in strain BC36 led to a strong accumulation of the 175 and 130 nt asRNAs, and appearance of the ∼80 nt fragments, as observed before (see Figure 5B). In contrast, it did not result in such accumulation in strain BC41 (Figure 6B). This result indicates that the degradation of asRNA by RNase III is due to its interaction with its complementary sequence on the rRNA precursor. This cleavage produces a 70 nt product that is seen in wt but not in Δrnc strains (Figure 6C). In the absence of RNase III, the asRNA is presumably protected from degradation by other RNases due to its interaction with the rRNA precursor. When this interaction cannot occur, as in strain BC41, the unbound asRNA is degraded by the activity of other RNases, whether the RNase III is present or not (Figure 6C). We noticed that, in the BC41 rnc+ strain, the 130 nt species was more abundant than the 175 nt species, suggesting a higher sensitivity of the unbound 175 nt species to these RNases. Moreover, we also observed a modest increase of both species in the BC41Δrnc strain, which may be due to their protection by the residual complementary transcripts arising from the mutated rrn1 promoter (Supplementary Figure S6). However, we cannot exclude that a certain fraction of unbound asRNAs adopt a secondary structure that would be cleaved by RNase III. This fraction would be stabilized in the absence of RNase III, explaining the observed increased amount of both transcripts. Nevertheless, the dramatic stabilization of both transcripts observed when the complementary sequence is present, clearly shows that the majority of the asRNA is base-paired with the rRNA precursor. In conclusion, our results showed that the asRNA interacts with the leader region of the rRNA precursor in vivo and this interaction creates a double-stranded structure that is cleaved by RNase III.
In vitro RNase III processing of the asRNA/rRNA precursor complex
The base-pairing between the asRNA and the rRNA precursor can potentially occur over 172 nt, creating a long duplex that could be cleaved by RNase III at numerous positions, leading to short RNA products. To analyze these cleavages, RNase III cleavage assays were performed in vitro. The H. pylori RNase III was purified and incubated with the in vitro synthesized transcripts. Two precursor transcripts were used, a 765 nt long one that mimics the primary transcript (i.e. presence of the processing stem) but which is deleted for the mature 23S sequence and a shorter one of 280 nt corresponding to the upstream RNase III cleavage product occurring at the processing stem and which could be a target for the asRNA. These transcripts were incubated with asRNA, RNase III, or both, and the reaction products were then analyzed by northern blot. Using the probe ‘h’, we observed that incubation of either the 280 nt or the 765nt transcript with asRNA resulted in a shift of migration, indicating that both precursor transcripts interact with asRNA to form a stable complex that resists the denaturing conditions of the gel (Figure 7A). While the free 280 nt transcript was not efficiently cleaved by RNase III, the pre-formed asRNA-p280 duplex was strongly cleaved, and two shorter fragments of about 175 and 125 nt were detected. In contrast to the 280 nt template, the 765 nt long transcript was efficiently cleaved by RNase III in the absence of asRNA, forming a ∼280 nt product, which arises from the cleavage in the processing stem. The pre-formed 765 nt–asRNA complex was also cleaved by RNase III, generating the 175 and 125 nt fragments, as observed for the 280 nt substrate. Thus, in vitro, RNase III cuts the pre rRNA–asRNA complex at two specific positions. Using the asRNA specific probe, we observed that the asRNA is efficiently cleaved in the presence of either precursor transcript, leading to the production of a ∼70 nt product that corresponds to the product observed in vivo (Figures 5B and 6B). This asRNA fragment results from the RNase III cleavage that also generates the 175 nt rRNA fragment (see below). We also observed that the free asRNA is cleaved by RNase III, giving rise to ∼125 and 70 nt fragments (Figure 6B, last lane). These cleavages can be explained by the self-structuring of the free asRNA (Supplementary Figure S7B). Nevertheless, the efficiency of the RNase III cleavages was clearly much stronger in the presence of the complementary pre-rRNA. Altogether, these results showed that in vitro, RNase III cleaves both the intramolecular stem flanking the 23S sequence and the asRNA/precursor duplex at limited and specific positions. Structural probing experiments were performed to further analyze the interaction between the asRNA and rRNA precursor. The transcripts, free or in a complex, were subjected to either enzymatic or lead(II) hydrolysis under limited digestion, and the cleavage products were analyzed (Supplementary Figure S7A). We observed that, after formation of the complex, the complementary regions of either the asRNA or the rRNA precursor became resistant to enzymatic cleavages, indicating that in vitro both transcripts could form a long duplex. However, a few residues were more exposed to lead cleavage, suggesting that the asRNA/pre-rRNA complex adopts a non-canonical structure, which might explain the restricted cleavages by RNase III.
Figure 7.
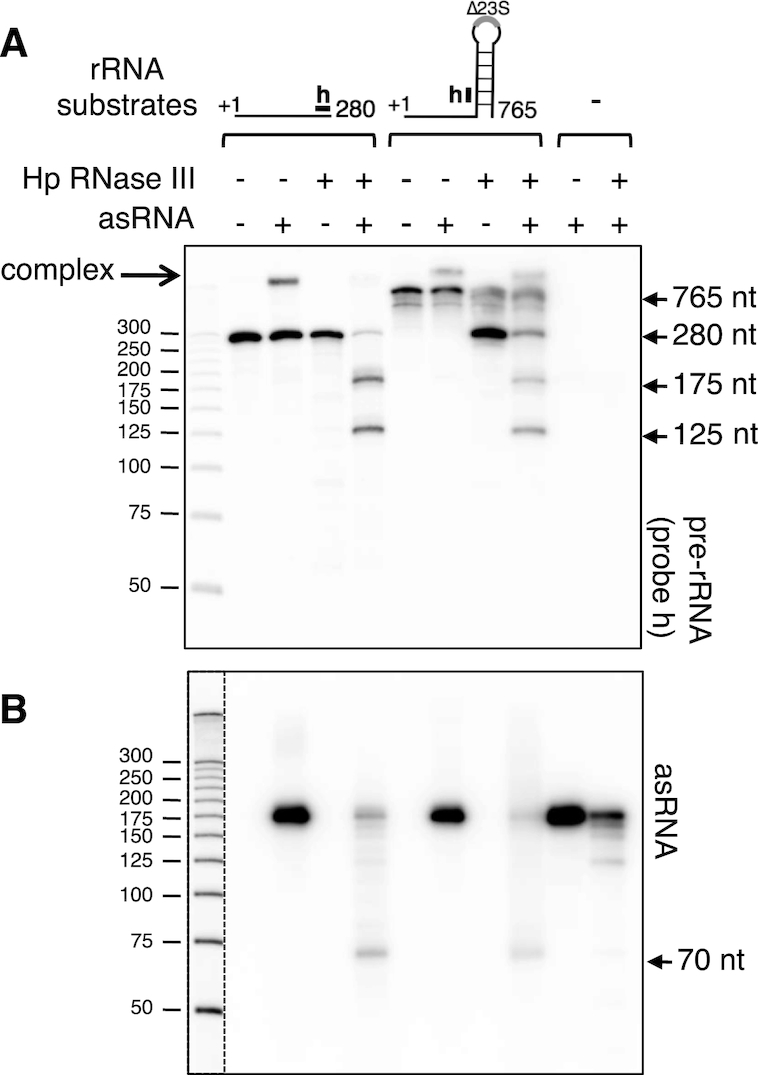
In vitro RNase III cleavage of the asRNA/rRNA precursor complex. (A) Two rRNA precursor transcripts were synthesized in vitro, the ‘280 nt’ one corresponding to the first 280 nt of the precursor, and the ‘765 nt’ one lacking the 23S rRNA mature sequence but retaining the stem structure flanking the 23S rRNA. These transcripts were incubated with the Helicobacter pylori RNase III, either in presence or absence of the in vitro transcribed asRNA. Controls in the absence of RNase III or with asRNA alone were also included. The reaction products were separated on a denaturing acrylamide gel and subjected to northern analysis. Probes were specific either to the rRNA precursor (the position of the probe ‘h’ is shown) or to the asRNA (B). The dotted vertical line represents two different expositions of the same gel.
Pairing with the asRNA affects the processing of the 23S-5S rRNA precursor in vivo
We have shown that the asRNA/rRNA precursor complex is a substrate for RNase III in vitro (Figure 7), and that the corresponding cleavages result in a rapid degradation of the asRNA in vivo (Figure 6). Here, we characterize, symmetrically, the effects of these RNase III cleavages on the processing pathway of the rRNA precursor in vivo. To facilitate the detection of such effects, we overexpressed the asRNA in a B128 wt strain. The 23S asRNA was cloned under the control of the 23S-5S rrn promoter, in a derivative of the pILL2150 H. pylori vector (22; Figure 8A). Using the asRNA specific probe, we showed that overexpression of the asRNA was effective as the strain transformed with this vector produces ∼25-fold more asRNA, compared to the same strain transformed with the empty plasmid (Figure 8B, upper panel). Interestingly, the 70 nt transcript, which results from an RNase III cleavage of the asRNA base-paired with the precursor, also increased, suggesting that the overexpressed asRNA interacts with the rRNA precursor similarly to the endogenous cis-encoded asRNA. The fact that the overproduced asRNA is almost completely processed into the 70 nt suggests that the asRNA is normally present in limiting quantities in the cell compared to the rRNA precursor. We next analyzed the consequences of this overexpression on the 23S-5S transcripts. Whereas no detectable effect on the amount of the mature 23S and 5S rRNAs was detected (Supplementary Figure S8A and panel ‘5S’ in Figure 8B), important changes in the precursor region were observed. The probe ‘a’ detected two main transcripts of about 280 and 210 nt (called p280 and p210) in the control strain (Figure 8B, middle panel). The larger one results from the RNase III cleavage in the processing stem at position 282 (see Figure 2), and it is therefore not detected in a Δrnc strain (see Supplementary Figure S6C). The smaller one is probably due to a cleavage by an unknown RNase at position ∼210, since it was also detected with a probe covering the 5′ end of the precursor. Overexpression of the asRNA resulted in a dramatic increase (∼50-fold) of a ∼75 nt pre-rRNA transcript (denoted p75). From primer extension experiments and RNA-seq data, the 5′ end of this p75 product was mapped at nt 106 of the rRNA precursor (Supplementary Figure S1C and S8B). This cleavage together with the intramolecular cleavage at nt 282 is expected to generate a fragment of ∼175 nt. However, this fragment is only observed in vitro (Figure 7A) indicating that it is further degraded into a ∼75 nt product in vivo. The symmetrical cleavage on the asRNA leading to the canonical 2 nt 3′overhang is expected to occur at nt 69 of the asRNA. The ‘70 nt’ asRNA detected in northern is thus likely the product of this cleavage (see Figure 8C), explaining the proportional increase of both p75 and the 70 nt asRNA. It is likely that the two p75 and 70 nt asRNA products remain base-paired after RNase III cleavage. This base-pairing would protect both transcripts from both 5′→3′ and 3′→5′ exonuclease activities, explaining their relative high stability compared to the other asRNA or precursor transcripts (Figure 8B). Using the downstream ‘h’ probe, we observed the asRNA-dependent induction of another transcript of 122 nt (Figure 8B, bottom panel). The 5′ end of this product was mapped at position 161 by primer extension analysis (Supplementary Figure S8B) and its size is consistent with a 3′ terminus at nt ∼280, which likely arises from the RNase III cleavage in the processing stem. This 122 nt fragment was also observed in vitro (‘125 nt’, Figure 7A). Altogether, these results indicate that the asRNA induces RNase III cleavages in the rRNA leader in at least two positions (nt 105 and 160) (Figure 8C), generating fragments of 75 and 122 nt. The absence of these latter species in a Δrnc strain or in a strain in which the asRNA promoter has been inactivated (Supplementary Figure S8C and S8D) confirms that they are generated by an asRNA-mediated RNase III cleavage. In a wt strain, these two asRNA-dependent processed transcripts are produced in low amounts compared to the other p280 and p210 asRNA-independent transcripts, due to the lower abundance of asRNA relative to that of the rRNA precursor. Therefore, only a small fraction of the precursor transcripts in the cell is paired to the asRNA.
Figure 8.

Overexpression of asRNA uncovers RNase III cleavages in the leader of 23S-5S rRNA precursor. (A) The B128 wt strain was transformed with the pILL2150 vector overexpressing the asRNA. As a control, the strain was also transformed with the empty vector. (B) Northern blot analysis of asRNA and precursor transcripts from strains overexpressing or not the asRNA. Total RNA was extracted from B128 strains transformed with either the asRNA overexpressing vector (pILL-asRNA) or the control vector (pILL), at different times after rifampicin addition. RNA was separated on a denaturing acrylamide gel and transferred to a nylon membrane, which was probed with oligonucleotides specific to either asRNA (upper panel) or precursor rRNA (middle and bottom panels, see panel (C) for the location of probes ‘a’ and ‘h’). A 5S rRNA probe served as a loading control. (C) The asRNA base-pairing with the leader of 23S-5S rRNA precursor induces specific RNase III cleavages. Cleavage at position 105 of the precursor leads to a product of 75 nt (p75), whereas the cleavage on the opposite strand occurs at nt 70 of the asRNA, and is thus responsible for the 70 nt product observed in northern blots. This intermolecular cleavage results in a 2-nt 3′ overhang characteristic of RNase III processing. The scissors indicate that the 3′ end of the p75 fragment can be generated either by an endoribonuclease or by a 3′ to 5′ exoribonuclease that would be stopped by the duplex. Cleavage at position 160 of the precursor produces a 122 nt precursor fragment (p122), whereas cleavage on the opposite strand is expected to produce an asRNA of 14 nt that is not detected in our conditions. Symbols are as in Figure 1A.
Interestingly, the RNase III cleavage at nt 105 does not occur concomitantly to the cleavage at nt 160, otherwise it would be impossible to detect the p75 product. Thus, RNase III cleaves at either position 105 or at position 160 of the precursor. The asRNA probably interacts with the p280 and p210 transcripts since their levels slightly decreased (∼2 fold) following asRNA overexpression. Although the overexpression of asRNA is high (∼25 fold), the decrease of precursor transcripts is not expected to be as high due to their large excess over the amount of available asRNA. It is also possible that the asRNA base-pairs to nascent rRNA transcripts, before the intramolecular processing at nt 282 occurs. Thus, the asRNA may interact at different steps during the maturation of the pre-rRNA.
DISCUSSION
Maturation of rRNA has been extensively studied in E. coli and B. subtilis, but there are fewer studies concerning this process in other bacteria. For instance, nothing was known regarding rRNA maturation in H. pylori, one of the most prevalent human bacterial pathogens. In this study, we provide a detailed characterization of the initial processing step by RNase III in this organism and found similarities and differences with E. coli and B. subtilis. We have also characterized a cis-encoded small RNA overlapping the 23S-5S rRNA precursor and found that its base-pairing with the precursor leads to additional RNase III processing coupled with a rapid degradation of the asRNA.
rRNA maturation in H. pylori: an example of atypical rrn gene organization
In most bacteria, the rRNA genes are organized into single operons containing 16S, 23S and 5S sequences, with the canonical order 5′ 16S-23S-5S 3′. The atypical arrangement of rRNA genes in H. pylori, where the 16S gene is separated from a 23S-5S rRNA gene cluster has been observed in several bacteria of different phyla (9,28), such as Thermus thermophilus (29), Borrelia burgdorferi (30), Pirellula marina (31), Mycoplasma gallisepticum (32) and Rickettsia species (33). A complete separation of the three rRNA genes has also been reported in Leptospira interrogans (34). Andersson et al. proposed that the disruption of the normal rRNA structure results from genomic rearrangements occurring during genome reduction and loss of some rRNA operons (9,33). Moreover, unusual rearrangements of rRNA genes have been also found more frequently in host-dependent than in free-living bacteria (28). Thus, it can be tentatively speculated that a free-living H. pylori ancestor having multiple rRNA operons underwent genomic rearrangements through intragenomic recombination, resulting in both a rRNA copy number decrease and disruption of the normal arrangement. Interestingly, the event of split rRNA operon in H. pylori did not occur in the phylogenetically closely related Campylobacter jejuni and Wolinella succinogenes (28).
Whether processing of rRNA occurs similarly in canonical and split operons is not known. For instance, one might wonder whether RNase III, which role is to separate the three precursors from the single polycistronic operon, is also involved in the processing of split rRNAs. Interestingly, the processing of the Borrelia burgdorferi rRNA, which has an rrn gene organization similar to H. pylori, has recently been studied. The authors proposed that RNase III is required to produce mature 23S rRNA, but not 16S and 5S rRNAs (35). Here, we show that in H. pylori, RNase III is involved in the initial processing of the three rRNAs. Thus, despite its atypical rRNA gene organization, this enzyme plays a similar role to bacteria having a canonical rRNA gene organization. Therefore, we hypothesize that the H. pylori ancestor contained a canonical polycistronic rrn operon and genomic rearrangements led to the separation of the 16S rRNA gene without loss of the processing stalks. Indeed, we show that RNase III specifically cleaves three stem structures of rRNA precursors: two long-range base-pairings flanking the 16S and 23S mature sequences and one short-range base-pairing located upstream the mature 5S sequence (Figures 2B and 3C). Whereas the long-range stems that bracket the mature rRNAs have been described in other species where they constitute specific targets for RNase III, the short-range stem–loop structure preceding mature 5S rRNA had never been reported yet. This additional RNase III cleavage may compensate the lack of efficient secondary enzymes such as RNase E and RNase M5 that are involved in 5S rRNA maturation in E. coli and in B. subtilis, respectively (36,37). Whereas RNase III cleavage generates a 5S rRNA precursor that is extended at its 5′ end by only 4 nts, it produces a large 23S precursor (p3) containing 92 and 55 extra nts at its 5′ and 3′ ends, respectively, and a large 16S rRNA precursor (p5) containing 374 and around 80 extra nts at its 5′ and 3′ ends, respectively. The enzymes responsible for the removal of these large precursor extensions are yet unknown.
rRNA processing in H. pylori: comparison with other bacteria
Functionality of immature precursors
Consistent with the role of RNase III in the initial processing step, the absence of RNase III results in the accumulation of 16S, 5S and 23S-5S rRNA precursors (Figures 1B and 3B). The large 23S-5S precursors, p1 and p2, are the most abundant, being visible on an ethidium bromide agarose gel, and represent a significant fraction of total rRNA (∼35% of total 23S molecules, see Figure 1B, left panel). Their high accumulation suggests that the secondary processing cleavages producing the mature 23S and 5S rRNAs are slowed down in the absence of RNase III. Nevertheless, accumulation of immature rRNAs does not significantly compromise ribosome activity since the growth rate of the Δrnc strain is only slightly affected in comparison to the wt strain. The presence of the p1 and p2 precursors in 70S ribosomes and in polysomes raises the question of their competency in translation. Indeed, they can be assembled into functional 50S ribosomal particles or their presence in polysomes reflects the fact that their maturation occurs after the 50S join in polysomes. These two interpretations have been previously proposed to explain the presence of E. coli and B. subtilis pre-rRNAs in polysomes (1). Indeed, the final processing steps of the E. coli 23S and 16S rRNA have been proposed to occur only after pre-ribosomes join in polysomes (38,39). On the other hand, it has also been shown that some rRNA precursors are functional in translation. For instance, in E. coli cells lacking RNase III, no mature 23S rRNA is formed and the immature 23S molecules are incorporated into functional ribosomes (40). Similarly, the B. subtilis 5S precursors that accumulate in the absence of RNase M5 are never converted into mature 5S and are assembled into translating ribosomes (37). In the case of H. pylori, the p1 and p2 precursors are found even in the heaviest polysomes, suggesting that they are incorporated not only in the initiating ribosomes but also in the translating ribosomes. Otherwise, the fraction of precursors should progressively decrease as the size of polysomes increases. Although this seems to be the case for the p1 precursor, the fraction of p2 appears to be constant in all polysome fractions. Thus, although the p1 and p2 precursors may be processed within polysomes, it is possible that the p2 species are at least partially functional. In contrast to the 23S-5S precursors, the 16S precursors were not found in significant amount in polysomes (Figure 4), suggesting that they are not functional or that the final processing reactions are completed before the 30S subunits are assembled into 70S. A different situation was observed for B. subtilis 16S precursors that accumulate in the absence of RNase J1. These precursors were found in polysomes and proposed to be at least partially functional (41).
Final maturation processing
Due to a different combination of RNases, the final processing steps in H. pylori are probably different compared with E. coli and B. subtilis. For example, in E. coli, the 5′ end maturation of 16S rRNA is catalyzed by the endoribonucleases RNase E and RNase G (6), which are absent in H. pylori. In B. subtilis, this reaction is performed by the 5′-3′ exoribonuclease activity of RNase J1 (41,42). Although H. pylori contains an RNase J homolog, it is not involved in rRNA maturation in this organism (43). The Mini-III and RNase M5, which are involved respectively in 23S and 5S rRNA maturation in B. subtilis (6), are also absent in H. pylori. In contrast, YbeY a ubiquitous endoribonuclease involved in the 16S rRNA 3′ end processing both in E. coli and B. subtilis (44,45) is also present in H. pylori. It would thus be interesting to determine whether it catalyzes the same reaction in H. pylori.
A cis-antisense RNA overlapping ribosomal RNA precursor
Recent genome-wide transcriptome analyses have revealed a plethora of chromosomally encoded cis-antisense RNAs in many bacterial species (46,47). Although a regulatory role has been shown for a few examples, the function and mechanism of action of most of them remain unknown. In the present work, we characterize one of the hundreds of small cis-antisense RNAs that were predicted by RNA-seq in the H. pylori strain 26695 (17). Our results confirm the existence of this asRNA in at least two other H. pylori strains (B128 and X47-2AL; Figure 5 and Supplementary Figure S5). We also present both in vitro and in vivo evidence, that this asRNA interacts with its complementary sequence on the sense strand, i.e. the leader of the 23S-5S rRNA precursor, forming an intermolecular complex that is cleaved by RNase III (Figures 6–8). In vivo, this cleavage impacts both the asRNA and the rRNA precursor: two main specific, mutually exclusive cleavages on the precursor generate the two p75 and p122 processed transcripts, whereas cleavage on the asRNA generates the 70 nt antisense transcript (see Figure 8C). We hypothesize that the complex adopts a non-canonical structure (see Supplementary Figure S7), limiting RNase III cleavages at specific positions. To date, asRNA targeting of stable RNA has been reported only in the chloroplast of Arabidopsis thaliana. In this plastid, the rRNA is transcribed as a polycistronic precursor containing all rRNAs and tRNAs. The AS5 antisense transcript, which is complementary to the 5S rRNA and tRNAArg region, was proposed to regulate 5S rRNA maturation. Indeed, over-accumulation of AS5 was correlated with decreased abundance of 5S rRNA in vivo and AS5 was shown to inhibit 5S rRNA maturation in vitro (48). What is the mechanism of regulation and whether the duplex between AS5 and pre-5S rRNA is targeted by an RNase III-like enzyme remain open questions. Later, Hotto et al. (49) identified another chloroplast antisense RNA overlapping the 4.5S-5S rRNA intergenic region. The authors proposed that the two transcripts could anneal and form a duplex that would be recognized by the plastid Mini-III enzyme, a member of the RNase III family lacking the double-stranded RNA binding domain (6).
Our results raise the question of whether transcription of the H. pylori 23S asRNA and its impact on pre-rRNA processing have a biological function. The conservation of the asRNA promoter and the expression of the asRNA in at least three H. pylori strains (Supplementary Figures S5B and S5C) are in favor of a functional relevance. Moreover, although in the conditions used (rich medium at 37°C), the asRNA does not seem to significantly affect growth or rRNA maturation (Supplementary Figure S9), other conditions may exist under which the synthesis of asRNA is sufficient to affect the rRNA processing pathway (cf. Figure 8). We propose that the asRNA may have fine-tuning roles in the rRNA maturation pathway, for example by facilitating either the degradation of processed fragments or the correct folding of rRNA. Indeed, the initial processing of the pre-rRNA produces several fragments, up to 300 nt, which need to be eliminated. Whereas some products can be degraded by exoribonuclease activities such as PNPase and RNase J, other fragments may be more difficult to degrade because of their structural conformation. In this hypothesis, the asRNA, by interacting with them, would destabilize their structure and facilitate their degradation by RNase III. Moreover, it has been shown that rRNA precursor sequences assist correct folding through transient interactions with the nascent rRNA in E. coli (see for example, 50,51). The asRNA could play a similar quality control role, its binding to nascent rRNA preventing the formation of unfavorable structures within the precursor.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Marc Dreyfus for critical reading of the manuscript. We also thank Cynthia M. Sharma and Konrad U. Förstner for generating the RNAseq data and authorizing the use of unpublished data as screen shots and Andrew Goldsborough for English correction. We thank anonymous referees for suggestions and constructive criticisms that helped to improve this manuscript. We thank H. de Reuse for the gift of the apramycin plasmid.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
INSERM [U1212]; CNRS [UMR5320]; Université de Bordeaux; Agence Nationale de la Recherche [ANR-12-BSV6-0007-asSUPYCO, ANR-12-BSV5-0025-Bactox1]. Funding for open charge: Institut National de la Santé et de la Recherche Médicale.
Conflict of interest statement. None declared.
REFERENCES
- 1. Srivastava A.K., Schlessinger D.. Mechanism and regulation of bacterial ribosomal RNA processing. Annu. Rev. Microbiol. 1990; 44:105–129. [DOI] [PubMed] [Google Scholar]
- 2. Young R.A., Steitz J.A.. Complementary sequences 1700 nucleotides apart form a ribonuclease III cleavage site in Escherichia coli ribosomal precursor RNA. Proc. Natl. Acad. Sci. U.S.A. 1978; 75:3593–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bram R.J., Young R.A., Steitz J.A.. The ribonuclease III site flanking 23S sequences in the 30S ribosomal precursor RNA of E. coli. Cell. 1980; 19:393–401. [DOI] [PubMed] [Google Scholar]
- 4. Loughney K., Lund E., Dahlberg J.E.. Ribosomal RNA precursors of Bacillus subtilis. Nucleic Acids Res. 1983; 11:6709–6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Herskovitz M.A., Bechhofer D.H.. Endoribonuclease RNase III is essential in Bacillus subtilis. Mol. Microbiol. 2000; 38:1027–1033. [DOI] [PubMed] [Google Scholar]
- 6. Deutscher M.P. Maturation and degradation of ribosomal RNA in bacteria. Prog. Mol. Biol. Transl. Sci. 2009; 85:369–391. [DOI] [PubMed] [Google Scholar]
- 7. Evguenieva-Hackenberg E., Klug G.. RNase III processing of intervening sequences found in helix 9 of 23S rRNA in the alpha subclass of Proteobacteria. J. Bacteriol. 2000; 182:4719–4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rische T., Klug G.. The ordered processing of intervening sequences in 23S rRNA of Rhodobacter sphaeroides requires RNase J. RNA Biol. 2012; 9:343–350. [DOI] [PubMed] [Google Scholar]
- 9. Andersson S.G., Kurland C.G.. Genomic evolution drives the evolution of the translation system. Biochem. Cell Biol. 1995; 73:775–787. [DOI] [PubMed] [Google Scholar]
- 10. Tomb J.-F., White O., Kerlavage A.R., Clayton R.A., Sutton G.G., Fleischmann R.D., Ketchum K.A., Klenk H.P., Gill S., Dougherty B.A. et al.. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997; 388:539–547. [DOI] [PubMed] [Google Scholar]
- 11. Alm R.A., Ling L.-S.L., Moir D.T., King B.L., Brown E.D., Doig P.C., Smith D.R., Noonan B., Guild B.C., deJonge B.L. et al.. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999; 397:176–180. [DOI] [PubMed] [Google Scholar]
- 12. Peek R.M., Blaser M.J.. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer. 2002; 2:28–37. [DOI] [PubMed] [Google Scholar]
- 13. Monstein H.J., Tiveljung A., Jonasson J.. Non-random fragmentation of ribosomal RNA in Helicobacter pylori during conversion to the coccoid form. FEMS Immunol. Med. Microbiol. 1998; 22:217–224. [DOI] [PubMed] [Google Scholar]
- 14. Monstein H.J., de la Cour C.D., Jonasson J.. Probing 23S ribosomal RNA cleavage sites in coccoid Helicobacter pylori. Helicobacter. 2001; 6:100–109. [DOI] [PubMed] [Google Scholar]
- 15. Nicholson A.W. Nicholson AW. Ribonuclease III and the role of double-stranded RNA processing in bacterial systems. Ribonucleases. Nucleic Acids and Molecular Biology. 2011; Berlin, Heidelberg: Springer; 269–297. [Google Scholar]
- 16. Arnion H., Korkut D.N., Gelo S.M., Chabas S., Reignier J., Iost I., Darfeuille F.. Mechanistic insights into type I toxin antitoxin systems in Helicobacter pylori: The importance of mRNA folding in controlling toxin expression. Nucleic Acids Res. 2017; 45:4782–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sharma C.M., Hoffmann S., Darfeuille F., Reignier J., Findeiß S., Sittka A., Chabas S., Reiche K., Hackermüller J., Reinhardt R. et al.. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010; 464:250–255. [DOI] [PubMed] [Google Scholar]
- 18. McClain M.S., Shaffer C.L., Israel D.A., Peek R.M., Cover T.L.. Genome sequence analysis of Helicobacter pylori strains associated with gastric ulceration and gastric cancer. BMC Genomics. 2009; 10:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Farnbacher M., Jahns T., Willrodt D., Daniel R., Haas R., Goesmann A., Kurtz S., Rieder G.. Sequencing, annotation, and comparative genome analysis of the gerbil-adapted Helicobacter pylori strain B8. BMC Genomics. 2010; 11:335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Noto J.M., Chopra A., Loh J.T., Romero-Gallo J., Piazuelo M.B., Watson M., Leary S., Beckett A.C., Wilson K.T., Cover T.L. et al.. Pan-genomic analyses identify key Helicobacter pylori pathogenic loci modified by carcinogenic host microenvironments. Gut. 2018; 67:1793–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Veyrier F.J., Ecobichon C., Boneca I.G.. Draft genome sequence of strain X47-2AL, a feline Helicobacter pylori isolate. Genome Announc. 2013; 1:e01095‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boneca I.G., Ecobichon C., Chaput C., Mathieu A., Guadagnini S., Prevost M.-C., Colland F., Labigne A., de Reuse H.. Development of inducible systems to engineer conditional mutants of essential genes of Helicobacter pylori. Appl. Environ. Microbiol. 2008; 74:2095–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Skouloubris S., Thiberge J.M., Labigne A., De Reuse H.. The Helicobacter pylori UreI protein is not involved in urease activity but is essential for bacterial survival in vivo. Infect. Immun. 1998; 66:4517–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Demarre G., Guérout A.-M., Matsumoto-Mashimo C., Rowe-Magnus D.A., Marlière P., Mazel D.. A new family of mobilizable suicide plasmids based on broad host range R388 plasmid (IncW) and RP4 plasmid (IncPα) conjugative machineries and their cognate Escherichia coli host strains. Res. Microbiol. 2005; 156:245–255. [DOI] [PubMed] [Google Scholar]
- 25. Pernitzsch S.R., Tirier S.M., Beier D., Sharma C.M.. A variable homopolymeric G-repeat defines small RNA-mediated posttranscriptional regulation of a chemotaxis receptor in Helicobacter pylori. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:E501–E510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mansour F.H., Pestov D.G.. Separation of long RNA by agarose-formaldehyde gel electrophoresis. Anal. Biochem. 2013; 441:18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003; 31:3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Merhej V., Royer-Carenzi M., Pontarotti P., Raoult D.. Massive comparative genomic analysis reveals convergent evolution of specialized bacteria. Biol. Direct. 2009; 4:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hartmann R.K., Ulbrich N., Erdmann V.A.. An unusual rRNA operon constellation: in Thermus thermophilus HB8 the 23S/5S rRNA operon is a separate entity from the 16S rRNA operon. Biochimie. 1987; 69:1097–1104. [DOI] [PubMed] [Google Scholar]
- 30. Schwartz J.J., Gazumyan A., Schwartz I.. rRNA gene organization in the Lyme disease spirochete, Borrelia burgdorferi. J. Bacteriol. 1992; 174:3757–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liesack W., Stackebrandt E.. Evidence for unlinked rrn operons in the Planctomycete Pirellula marina. J. Bacteriol. 1989; 171:5025–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen X., Finch L.R.. Novel arrangement of rRNA genes in Mycoplasma gallisepticum: separation of the 16S gene of one set from the 23S and 5S genes. J. Bacteriol. 1989; 171:2876–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Andersson S.G., Stothard D.R., Fuerst P., Kurland C.G.. Molecular phylogeny and rearrangement of rRNA genes in Rickettsia species. Mol. Biol. Evol. 1999; 16:987–995. [DOI] [PubMed] [Google Scholar]
- 34. Fukunaga M., Mifuchi I.. Unique organization of Leptospira interrogans rRNA genes. J. Bacteriol. 1989; 171:5763–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Anacker M.L., Drecktrah D., LeCoultre R.D., Lybecker M., Samuels D.S.. RNase III processing of rRNA in the Lyme disease spirochete Borrelia burgdorferi. J. Bacteriol. 2018; 200:e00035-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ghora B.K., Apirion D.. Structural analysis and in vitro processing to p5 rRNA of a 9S RNA molecule isolated from an rne mutant of E. coli. Cell. 1978; 15:1055–1066. [DOI] [PubMed] [Google Scholar]
- 37. Condon C., Brechemier-Baey D., Beltchev B., Grunberg-Manago M., Putzer H.. Identification of the gene encoding the 5S ribosomal RNA maturase in Bacillus subtilis: mature 5S rRNA is dispensable for ribosome function. RNA. 2001; 7:242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Srivastava A.K., Schlessinger D.. Coregulation of processing and translation: mature 5′ termini of Escherichia coli 23S ribosomal RNA form in polysomes. Proc. Natl. Acad. Sci. U.S.A. 1988; 85:7144–7148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mangiarotti G., Turco E., Ponzetto A., Altruda F.. Precursor 16S RNA in active 30S ribosomes. Nature. 1974; 247:147–148. [DOI] [PubMed] [Google Scholar]
- 40. King T.C., Sirdeshmukh R., Schlessinger D.. RNase III cleavage is obligate for maturation but not for function of Escherichia coli pre-23S rRNA. Proc. Natl. Acad. Sci. U.S.A. 1984; 81:185–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Britton R.A., Wen T., Schaefer L., Pellegrini O., Uicker W.C., Mathy N., Tobin C., Daou R., Szyk J., Condon C.. Maturation of the 5′ end of Bacillus subtilis 16S rRNA by the essential ribonuclease YkqC/RNase J1. Mol. Microbiol. 2007; 63:127–138. [DOI] [PubMed] [Google Scholar]
- 42. Mathy N., Bénard L., Pellegrini O., Daou R., Wen T., Condon C.. 5′-to-3′ exoribonuclease activity in bacteria: role of RNase J1 in rRNA maturation and 5′ stability of mRNA. Cell. 2007; 129:681–692. [DOI] [PubMed] [Google Scholar]
- 43. Redko Y., Aubert S., Stachowicz A., Lenormand P., Namane A., Darfeuille F., Thibonnier M., De Reuse H.. A minimal bacterial RNase J-based degradosome is associated with translating ribosomes. Nucleic Acids Res. 2013; 41:288–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jacob A.I., Köhrer C., Davies B.W., RajBhandary U.L., Walker G.C.. Conserved bacterial RNase YbeY plays key roles in 70S ribosome quality control and 16S rRNA maturation. Mol. Cell. 2013; 49:427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Baumgardt K., Gilet L., Figaro S., Condon C.. The essential nature of YqfG, a YbeY homologue required for 3′ maturation of Bacillus subtilis 16S ribosomal RNA is suppressed by deletion of RNase R. Nucleic Acids Res. 2018; 46:8605–8615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thomason M.K., Storz G.. Bacterial antisense RNAs: how many are there, and what are they doing. Annu. Rev. Genet. 2010; 44:167–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Georg J., Hess W.R.. Widespread antisense transcription in prokaryotes. Microbiol. Spectr. 2018; 6:doi:10.1128/microbiolspec.RWR-0029-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sharwood R.E., Hotto A.M., Bollenbach T.J., Stern D.B.. Overaccumulation of the chloroplast antisense RNA AS5 is correlated with decreased abundance of 5S rRNA in vivo and inefficient 5S rRNA maturation in vitro. RNA. 2011; 17:230–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hotto A.M., Castandet B., Gilet L., Higdon A., Condon C., Stern D.B.. Arabidopsis chloroplast mini-ribonuclease III participates in rRNA maturation and intron recycling. Plant Cell. 2015; 27:724–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Besançon W., Wagner R.. Characterization of transient RNA-RNA interactions important for the facilitated structure formation of bacterial ribosomal 16S RNA. Nucleic Acids Res. 1999; 27:4353–4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liiv A., Remme J.. Importance of transient structures during post-transcriptional refolding of the pre-23S rRNA and ribosomal large subunit assembly. J. Mol. Biol. 2004; 342:725–741. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.