

Abstract
Background
Rhabdomyosarcoma (RMS) is the most common soft-tissue sarcoma in children. Pathogenesis of RMS is associated with aggressive growth pattern and increased risk of morbidity and mortality. There are two main subtypes or RMS: embryonal and alveolar. The embryonal type is characterized by distinct molecular aberrations, including alterations in the activity of certain protein kinases. Focal adhesion kinase (FAK) is a non-receptor tyrosine kinase that plays a vital role in focal adhesion (FA) assembly to promote cytoskeleton dynamics and regulation of cell motility. It is regulated by multiple phosphorylation sites: tyrosine 397, Tyr 576/577, and Tyr 925. Tyrosine 397 is the autophosphorylation site that regulates FAK localization at the cell periphery to facilitate the assembly and formation of the FA complex. The kinase activity of FAK is mediated by the phosphorylation of Tyr 576/577 within the kinase domain activation loop. Aberrations of FAK phosphorylation have been linked to the pathogenesis of different types of cancers. In this regard, pY397 upregulation is linked to increase ERMS cell motility, invasion, and tumorigenesis.
Methods
In this study, we have used an established human embryonal muscle rhabdomyosarcoma cell line RD as a model to examine FAK phosphorylation profiles to characterize its role in the pathogenies of RMS.
Results
Our findings revealed a significant increase of FAK phosphorylation at pY397 in RD cells compared to control cells (hTERT). On the other hand, Tyr 576/577 phosphorylation levels in RD cells displayed a pronounced reduction. Our data showed that Y925 residue exhibited no detectable change. The in vitro analysis showed that the FAK inhibitor, PF-562271 led to G1 cell-cycle arrest induced cell death (IC50, ~ 12 µM) compared to controls. Importantly, immunostaining analyses displayed a noticeable reduction of Y397 phosphorylation following PF-562271 treatment. Our data also showed that PF-562271 suppressed RD cell migration in a dose-dependent manner associated with a reduction in Y397 phosphorylation.
Conclusions
The data presented herein indicate that targeting FAK phosphorylation at distinct sites is a promising strategy in future treatment approaches for defined subgroups of rhabdomyosarcoma.
Keywords: Focal adhesion kinase (FAK), Rhabdomyosarcoma, Phosphorylation, Cell migration
Background
Rhabdomyosarcoma (RMS) is the most common form of soft-tissue sarcoma in pediatric oncology (Malempati and Hawkins 2012). Despite the recent advances in the development of therapeutic and clinical care of RMS patients, subgroups of RMS remain with high risk of morbidity and mortality (Ruymann and Grovas 2000). There are two biologically distinctive subtypes of RMS: embryonal (ERMS) and alveolar (ARMS) (Shern et al. 2015). ERMS is characterized by various molecular aberrations, such as loss of heterozygosity (LOH), genetic mutations, and changes of gene expression profiles (Kohsaka et al. 2014; Szuhai et al. 2014; Nishimura et al. 2013; De Pitta et al. 2006). The previous reports showed that the oncogenic process of RMS involves dysregulation of tyrosine kinase phosphorylation and activity (Cen et al. 2007; Crose and Linardic 2011). Focal adhesion kinase (FAK) is a non-receptor tyrosine kinase that plays essential function(s) in integrin-mediated signaling to promote cytoskeleton dynamics, cell adhesion, and regulation of membrane protrusions and cell motility (Zhao and Guan 2011; Mitra et al. 2005). It has been linked to multiple types of human carcinomas (Fujii et al. 2004; Lark et al. 2005; Giaginis et al. 2009; Sood et al. 2004; Canel et al. 2006; Beierle et al. 2008; Theocharis et al. 2003; Ocak et al. 2012). Central to this study, prolonged activation and/or dysregulation of FAK has also been linked to RMS tumor progression and development (Yan et al. 2009; Liu et al. 2008). The activation of this kinase requires autophosphorylation of Tyrosine 397, which serves as a docking site for Src homology 2 (SH2) binding (Schlaepfer et al. 2004; Seong et al. 2011). This binding leads to the activation of Src kinase activity, inducing the phosphorylation of Tyr 576/577 within the kinase domain activation loop (Zhou et al. 2015). Autophosphorylation (pY397) is a pre-requisite for the assembly of focal adhesion complex at the periphery of cell membrane (Israeli et al. 2010; Wozniak et al. 2004; Oh et al. 2009). Thus, FAK phosphorylation is integral to the recruitment of other key regulatory proteins to this complex, following integrin-mediated cell adhesion (Gilmore and Burridge 1996). In addition, FAK cellular functions are not limited to focal adhesion complex formation, but FAK has been shown to mediate multiple other nuclear functions, such as scaffolding of distinct regulatory proteins and regulation of gene expression (Lim et al. 2008; Mei and Xiong 2010; Luo et al. 2009). These data suggest a potentially important role(s) of FAK in ERMS cell survival and migration. In this study, we set out to examine the phosphorylation status of FAK in an ERMS cell model using the RD cell line. This cell line was originally established from the embryonal rhabdomyosarcoma tumor specimen from 7-year-old Caucasian female patient and has been shown to express myoglobin, myosin ATPase genes (McAllister et al. 1969). It has been used as a valid experimental model to study pathways of RMS tumor growth and proliferation as well as to investigate novel therapeutic targets for future treatments (Zhou et al. 2018; Kinn et al. 2016; Li et al. 2018). Using this cell model, we provide proof-of-concept data to describe that activation landscape of FAK in RMS cell survival and migration and describe the activity of the small molecule inhibitor PF-566227 to validate the targetability of FAK in future therapeutic strategies.
Materials and methods
Cell lines and cell culture
RD (ATCC® CCL-136™) human embryonal rhabdomyosarcoma cell line was maintained in Opti-MEM media (Gibco, Invitrogen Corporation, Burlington, ON) supplemented with 5% fetal bovine serum and 100 units/ml penicillin and 100 units/ml streptomycin (Gibco). Immortalized primary fibroblast cells (hTERT) (ATCC® CRL-2846™) were used as a control in this study. All cell cultures were maintained at 37 °C in a humidified incubator with 5% CO2. PF-562271 compound was purchased from Selleck (Cedarlane, Burlington, Ontario, Canada). Stock solutions of PF-562271 were prepared as 10 mM in DMSO and stored in aliquots at − 20 °C.
Cytotoxicity assay
1 × 104 of RD and hTERT cells were cultured in 100 µl of Opti-MEM per well in 96 well plates. Cells were treated with a PF-562271 to a final concentration ranging from 1 × 10−3 to 100 µM. Cultured cells were incubated in the presence or absence of the drug for 96 h. Then, total cell viability (% cytotoxicity) was assessed by Alamar blue assay. Cells were incubated with 5% Alamar blue for 4 h, and then, the absorbance at 570–620 nm was measured (Opsys MR Plate Reader, Dynex Technologies, Chantilly, Virginia). Cell survival (%) was calculated by normalizing the absorbance ratio of the treated (drug) well to the vehicle control (DMSO).
Western-blot analyses
Whole cell extracts were prepared using RIPA buffer [50 mM Tris–HCl (pH 8), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulphate (SDS)] supplemented with 1% phosphatase inhibitor (Sigma-Aldrich), 1% protease inhibitor (Sigma-Aldrich), and 1% sodium orthovanadate (Alfa Aesar, Ward Hill, Massachusetts). Total protein content was quantified using the Bicinchoninic Acid (BCA) Protein Assay Kit (Pierce, Rockford, Illinois). Cellular extracts were resolved by SDS-PAGE and transferred to 0.2 μm nitrocellulose membranes in a Tris/glycine transfer buffer containing 10% (v/v) methanol. Non-specific binding sites were blocked with 5% (w/v) nonfat dry milk in Tris-buffered saline with Tween [TBST, 25 mM Tris–HCl, 137 mM NaCl, 3 mM KCl, and 0.05% (v/v) Tween-20]. Membranes were washed and incubated overnight with primary antibody at 1:1000 dilution with 1% (w/v) nonfat dry milk in TBST. Membranes were then incubated for 50 min with horseradish peroxidase (HRP)-conjugated secondary antibody (dilution 1:10,000) in TBST and developed with ECL reagent. Antibodies to pTyr397-FAK, pTyr576/577-FAK, and pTyr925-FAK were purchased form Abcam (Cambridge, United Kingdom); antibodies to FAK and β-actin were from Cell Signaling (Danvers, MA);Goat anti-mouse IgG (H + L) cross-adsorbed secondary antibody, Alexa Fluor 568 and goat anti-rabbit IgG (H + L) cross-adsorbed secondary antibody, and AlexaFluor488-phalloidin were purchased from ThermoFisher Scientific (Waltham, Massachusetts, United States).
Immunocytochemistry
RD cells were plated at 5 × 104 in Opti-MEM media with 5% (v/v) FBS at 37 °C with 5% CO2. Cells were fixed for 15 min in 4% (v/v) paraformaldehyde in PBS and then permeabilized with 0.5% Tween-20 for 15 min. Cells were then incubated at 4 °C overnight with primary antibody diluted 1:200 in blocking serum (0.3% BSA, 5% goat serum, 0.3% Triton X-100 in PBS, pH 7.4). Then, Cells were washed three time with PBS before adding Alexa Fluor 568-conjugated secondary antibody (1:300) for 1 h at room temperature. For F-actin visualization, Alexa Fluor 488-phalloidin was diluted 1:50 in 1% (w/v) BSA in PBS and then incubated with the cells for 1 h at room temperature. Cells were rinsed with PBS, counterstained with DAPI for 5 min to detect nuclei, and then visualized with an InCell 6000 Imaging System (GE Healthcare). For visualization, the InCell 6000 Imaging System was programmed to complete whole-well scanning for fluorescent (Immunocytochemistry data) using a high-resolution scientific-grade system.
Flow cytometry
5 × 105 RD cells were treated with 10 μM PF-562271 or DMSO (vehicle control) for 24 h. Cells were harvested and fixed in ice-cold 70% (v/v) ethanol and then stored at 4 °C. Prior to analysis, cells were washed with PBS, resuspended in 500 µl PBS with 50 µg/ml propidium iodide (Sigma) supplemented with and 25 µg/ml RNAse A (Sigma), and incubated at 37 °C for 1 h. Analysis of cell cycle was completed using propidium iodide and analyzed by FACScan (Becton–Dickinson). Cell number in each phase was expressed as the percentage of the total cell number.
Migration assay
RD cells were cultured in 12-well culture plates (TransWell; Corning Inc., Lowell, MA) with 8-μm micropore inserts. PF-562271 or vehicle-treated cells (7 × 103) were placed into the upper well and allowed to migrate overnight to the bottom side. The inserts were then fixed with 1% crystal violet in 95% ethanol for 1 min. Cell migration (%) was quantified by counting the stained cells that were adherent to the lower side of the membranes in five random fields of view (at 10 × magnification). Percent migration was normalized to vehicle control treatment.
Statistical analysis
Data are presented as the mean ± SEM, with n indicating the number of independent experiments. Data were analyzed by Student’s t test, and p < 0.05 was considered to indicate statistical significance. All statistical analyses were performed using the GraphPad Prism 6.0 program.
Results
Focal adhesion kinase (FAK) phosphorylation status in RD cells
Dysregulation of FAK phosphorylation is highly associated with survival, progression, and invasion of various types of solid tumors (Sulzmaier et al. 2014; Lee et al. 2012; Wang et al. 2016). We set out to examine the phosphorylation status of the key phosphorylation sites of FAK protein in RD cells. In our examination, we have profiled three phosphorylation sites: Tyr 397, Tyr 576/677, and Tyr 925. Western-blot analysis showed that FAK autophosphorylation (pY397) site was significantly increased in RD, five times higher than phosphorylation levels detected in hTERT cells as observed in western blots (Fig. 1). FAK-pY397 autophosphorylation promotes the assembly and recruitments of other focal adhesion key proteins into the focal adhesion sites at the cell periphery. In contrast, phosphorylation of p[Y576/577]-FAK located in the activation loop of the kinase domain showed lower levels in RD compared to hTERT (Fig. 1). Furthermore, no detectable changes in the phosphorylation of FAK at Y925 site was noted (Fig. 1).
Fig. 1.

Focal adhesion kinase (FAK) phosphorylation status in RD and hTERT cells. RD or hTERT cells were collected and analyzed by Western blot. Protein phosphorylation was detected using anti-phospho-specific antibodies (pY397, pY576/577, and pY925). Bands were quantified by scanning densitometry and normalized to total FAK protein and to the loading control. Values represent means ± SEM for n = 4 independent experiments. *Significantly different from the vehicle control (Student’s t test, p < 0.05)
PF-562271 suppresses FAK-pY397 in RD cells
FAK autophosphorylation at tyrosine 397 is strongly associated with formation of focal adhesion complex which plays a key role in the regulation of cytoskeleton dynamics (Wu 2007; Xu et al. 2012; Hamadi et al. 2005). This phosphorylation promotes complex assembly with various SH2-domain containing proteins, such as SRC-family kinases (Luo et al. 2009). Tyr-397 phosphorylation has been implicated in promoting cell motility and invasion events through the activation of downstream signaling pathways (Hsia et al. 2003). To explore whether phosphorylation of FAK at Tyr 379 associated with ERMS, we examined pY397-FAK phosphorylation status in RD cells using immunocytochemistry. RD cells were treated with PF-562271 (10 µM) or DMSO and then visualized to assess phosphorylation levels. Our investigation demonstrated that a significant amount of pY397-FAK phosphorylation was eliminated after 24 h of PF-562271 treatment (Fig. 2a). Immunocytochemical analysis of the subcellular localization of FAK phosphorylation at tyrosine 397 in the control RD cells revealed a predominant co-localization of pY397 with F-actin stress fibers (red arrows). However, immunofluorescence signal for pY397 showed a minor signal localized in the nucleus (Fig. 2b, left panel). Our data showed that a substantial amount of FAK pY397 was found to be significantly eliminated from the cell edges/periphery after PF-562271 treatment (10 µM) (Fig. 2b, right panel). In addition, morphological changes were observed in RD cells following FAK inhibition. These changes may be due to the elimination of a major pool of pY397 from the cell edge after PF-562271 treatment.
Fig. 2.

Immunocytochemistry of RD cells for pY397 FAK following treatment with PF-562271. RD cells fixed and stained with anti-pY397-FAK (red channel) to examine phosphorylation dynamics following vehicle control (DMSO) and PF-562271 (10 μM) (a). Cytoskeletal localization of pY397-FAK was detected by staining F-actin cytoskeleton with AlexaFluor488-phalloidin (green channel). b Higher resolution images were taken for pY397 following vehicle control (left panel) and PF-562271 (10 μM) (right panel). Arrows indicated signal localization in the nucleus and F-actin cytoskeleton. For each independent plate of cells, 10 random visual fields were acquired from each whole-well scan, and cells in 8 images were quantified from each field. Scale bars 30 μm
PF-562271-induced cell cytotoxicity in RD cells
We have examined the effects of FAK inhibition on RD cell viability. PF-562271 is a potent, and selective ATP-competitive, reversible inhibitor of FAK (Luzzio et al. 2007). In vitro cell cytotoxicity assay of RD and hTERT (control) cells was achieved by applying a gradient concentration [1 × 10−9 to 1 × 10−4, μM] of PF-562271 compound. These data demonstrated that inhibition of FAK by PF-562271 leads to RD cell growth inhibition with IC50 values in the micromolar range (Fig. 3a). In addition, the maximum concentration used in this analysis (100 µM) displayed an acceptable level of cytotoxicity (~ 30%) on hTERT cells. These data demonstrated a pronounced anti-proliferative activity of PF-562271 compound against RD cells with a therapeutic window (Fig. 3a). Western-blot analysis was achieved by treating RD cells with a gradient concentration [1 nM–100 µM] of PF-562271. These data showed a gradual decrease of FAK-pY397 over increase of PF-562271 concentration (Fig. 3b). FAK phosphorylation signal was normalized to total FAK and β-actin protein levels.
Fig. 3.

PF-562271-induced cell cytotoxicity in RD cells. a Human rhabdomyosarcoma (RD) cell lines or hTERT cells were treated with increasing concentration of PF-562271 [1 × 10−4 to 100, μM] for 96 h. Cellular viability was calculated as (%) by comparing the absorbance ratio (percentage) of the treated cells normalized to the control (DMSO) treated cells. b Western-blot analysis of FAK-pY397 in treated RD cells with a concentration gradient of (1 nM–100 µM) of PF-562271. FAK phosphorylation signal was normalized to total β-actin loading control protein
PF-562271 induces cell-cycle arresting in RD cells
Recent reports have suggested a role for FAK and its associated signaling pathways in the regulation of cell-cycle progression through integrins signaling (Zhao et al. 1998). In this study, we examined whether if FAK inhibition can modulate cell-cycle components in RD cells. Cell-cycle analysis using flow cytometry in treated RD cells (10 µM of PF-562271) or control RD cells (vehicle, DMSO) showed that inhibition of FAK resulted in G1 arresting in RD cells following FAK inhibition (Fig. 4a). PF-562271 induced G1 arresting by 14% in the treated cells compared to vehicle control (Fig. 4b).
Fig. 4.

PF-562271 effects on RD cell cycle. DNA histogram of the RD cell cycle was analyzed using flow cytometry with PI staining following PF562271 [10 µM] or vehicle control (DMSO) treatments (a). Phases represent G1%, S%, and G2% phases were analyzed for both conditions (b)
PF-562271 diminishes RD cell migration
Integrin signaling through FAK has been proposed to play a role in the regulation of integrin-mediated cell migration (Huttenlocher and Horwitz 2011; Xie et al. 2001; Gupta and Vlahakis 2009). This key regulatory mechanism has been broadly implicated in metastasis and invasion activities in cancer signaling (Jiang et al. 2015). We aimed to investigate the impact of FAK inhibition using PF-562271 on RD cell migration. Our in vitro examination has employed a trans-well assay to detect and quantify migration events. An increasing concentration [0–25, µM] of PF-562271 resulted in a dose-dependent suppression of RD cell migration (Fig. 5, upper panel). Furthermore, we have profiled the phosphorylation levels of the tyrosine 397 of FAK using the similar concentrations of PF-562271. Western-blot analysis showed a pronounced decrease of FAK phosphorylation at tyrosine 397 with increasing PF-562271 concentration (Fig. 5, lower panel). These data suggest that the reduction of cell migration with exposure to PF-562271 was accompanied with a decrease of pY397-FAK phosphorylation levels. These findings suggest that phosphorylation of FAK at Y397 FAK is a key element of RD cell-migration mechanism, and its inhibition leads to the reduction of RD cell migration.
Fig. 5.
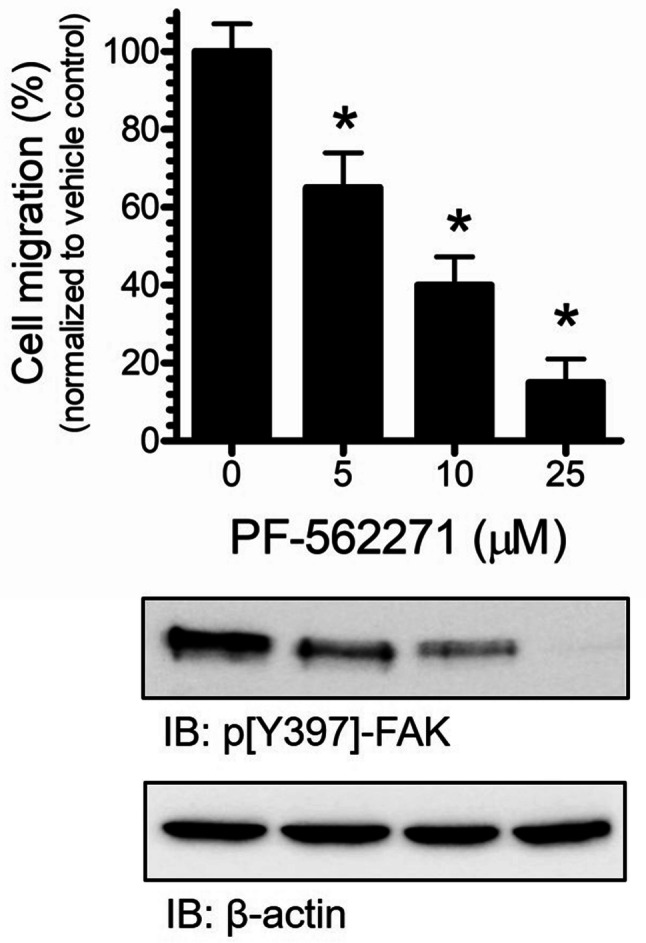
RD cell migration and FAK phosphorylation (pY397) were suppressed by FAK inhibition. Cell-migration assay was completed by incubating RD cells (7 × 103) with increasing concentration of PF-562271 [0–25 µM] for 24 h. Cell migration was calculated and expressed as the percentage of number of migrated cells into the total number of cells. % Migration was normalized to vehicle control treatment (upper panel). FAK phosphorylation was analyzed by SDS-PAGE with anti-pY397-FAK and normalized to total β-actin loading control protein (lower panel). Values represent means ± SEM for n = 4 independent experiments. *Significantly different from the vehicle control (Student’s t test, p < 0.05)
Discussion
In this study, we have explored FAK protein phosphorylation status with respect to regulation of RD cells’ viability and motility. We have employed an embryonic muscle rhabdomyosarcoma cancer cells (ERMS) to examine the role of FAK in this cell line. Regulation of FAK phosphorylation and localization are highly dynamic mechanisms that promote various numbers of cellular activities under physiological conditions (Cohen and Guan 2005). FAK is a cytoplasmic non-receptor protein tyrosine kinase that transmits upstream extracellular signals (e.g., growth factors) to regulate cellular activities, such as cell proliferation and migration (Schaller 2001). FAK plays an important role during different developmental stages and is highly linked to the pathogenesis of various tumors (Gabarra-Niecko et al. 2003). FAK autophosphorylation and activation is highly dynamic mechanism that regulates protein localization at the focal adhesion sites located at the edges of the plasma membrane. This dynamic recruitment of FAK to focal adhesions is a key regulatory step towards the formation of focal adhesion (FA) complex and activation of the downstream signaling phosphorylation cascade (Hu et al. 2014). Phosphorylation of Y397 serves as a docking site for Src binding, which leads to a conformational change and activation of Src and subsequent phosphorylation of FAK at multiple sites (Schlaepfer and Hunter 1996). Y576/Y577 phosphorylation residues are located within the activation loop at the kinase domain, which results in a full activation of FAK kinase activity (Westhoff et al. 2004). The phosphorylation of FAK at Y925 is possibly important to regulate the FA turnover via regulation of protein interaction with other FA key regulatory proteins (Deramaudt et al. 2011). In this study, RD cells displayed a notable elevation of FAK protein phosphorylation at tyrosine 397 compared to hTERT (control) cells. Remarkably, we have also observed a major pool of pY397 phosphorylation that has mainly co-localized with F-actin stress fibers at the cell membrane/periphery. These data are consistent with previously defined roles of FAK activities in the regulation of cell migration and invasion events detected in different types of cancer cell models. In a recent study, Waters et al. showed that the inhibition of FAK by small molecule inhibitors and/or small interference RNA (siRNA) resulted in suppression of rhabdomyosarcoma cancer cell migration, invasion, and survival (Waters et al. 2016). Our data showed a reduced phosphorylation levels of Y576/Y577 located in the kinase domain of FAK. The phosphorylation of Tyr 576 and Tyr 577 is subsequently achieved following Src binding and activation (Schlaepfer and Hunter 1996). These data indicate a possible mechanism which leads to suppression of full activation of FAK in RD cells that might be attributed to aberrations or dysfunction of Src signaling. Currently, very little data exist with respect to Y576/Y577 phosphorylation of FAK in cancer, whether if there is cross-talk with other proteins or protein complexes to promote FAK protein autoinhibition leading to the attenuation of FAK phosphorylation in the kinase domain. Further investigations are required to examine the details of Y576/Y577 phosphorylation dynamics in rhabdomyosarcoma. Our examination showed no detectable changes of Y925 phosphorylation site in RD cells compared to hTERT (Fig. 1).
In this study, we employed a selective and potent FAK small molecule inhibitor, PF-562271. This compound has been previously characterized by Wiemer et al. during their examination of FAK activity in T-cell activation and proliferation (Wiemer et al. 2013). Our in vitro characterization showed an inhibitory effect of PF-562271 on RD cell viability at micromolar concentrations. Our findings are similar with the in vitro cytotoxicity data obtained by Waters et al. using another FAK small molecule inhibitor (PF-573228) or small RNA interference molecule (siRNA) (Waters et al. 2016).
Cell-cycle analysis revealed that FAK inhibition induced G1 arresting in RD cells. This effect is likely to be associated with cytoskeleton-dependent mechanisms and strongly associated with FAK activity to stimulate the p42/p44 MAPKs pathway and/or transcriptional regulation of cyclin D1 (Zhao et al. 2001; Margadant et al. 2007). Our immunostaining examinations displayed a nuclear localization of FAK which is possibly regulated by the nuclear localization signal (NLS) located at the N-terminus that is responsible to shuttle the protein towards the nucleus (Serrels et al. 2015; Ossovskaya et al. 2008). This nuclear localization of FAK may be important to promote regulation of cell cycle in RD cells.
FAK-pY397 suppression was achieved in a dose-dependent fashion using a gradient concentration of PF-562271 and was associated with diminishing cell motility in RD cells. Fundamentally, FAK phosphorylation at FAK-pY397 and localization at the focal adhesion and/or association with F-actin stress fibers is integral to promote cell motility, adhesion, and regulation of cytoskeleton dynamics (Zhao and Guan 2011; Westhoff et al. 2004; Katz et al. 2003). Phosphorylation of FAK at Tyr397 has been repeatedly linked to cell migration and metastatic events in cancers (Kolli-Bouhafs et al. 2014; Higuchi et al. 2013; Zhao and Guan 2009). Morphological changes observed in RD cells following treatment with PF-562271 can be associated with the disruption of the F-actin assembly and/or modulation of cytoskeletal architecture. These results strongly suggest that FAK tyrosine phosphorylation at Tyr 397 is highly integral to promote cell migration of RD cells.
Conclusions
Data presented herein demonstrate the efficacy of FAK inhibition using PF-562271 to suppress RD cell-migration mechanism by attenuating FAK phosphorylation at Tyr397 site. This finding points to the presence of aberrant phosphorylation of FAK in rhabdomyosarcoma. Overall, the data presented in this in vitro study suggest a key role for FAK in cell-proliferation signals, cell-cycle regulation, and cell-migration activities of RMS. Based on these findings, additional more details can be formulated to provide the essential preclinical data for effective early phase clinical trials for RMS in the future.
Abbreviations
- DMSO
Dimethyl sulfoxide
- FAK
Focal adhesion kinase
- hTERT
ATCC humantelomerase reverse transcriptase immortalized cell lines
- IC50
The half maximal inhibitory concentration
- RD
Rhabdomyosarcoma cells
- siRNA
Small interference RNA
Author contributions
AAG wrote the manuscript, collected the data, and analyzed the majority of the data. DOQ contributed to the writing of this manuscript, Fig. 2 analysis, and figures preparation. MA contributed in collecting microscopy data presented in Fig. 2, and figures preparation. AN conceived and coordinated the study. All authors reviewed the results and approved the final version of the manuscript.
Funding
This work was supported in part by a research grant from the POETIC Foundation.
Availability of data and materials
The data sets used and/or analyzed during the current study are available from the corresponding author on request.
Compliance with ethical standards
Conflict of interest
The authors declare no potential conflict of interest.
Ethics of approval
N/A.
Consent for publication
Not applicable.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Beierle EA et al (2008) Focal adhesion kinase expression in human neuroblastoma: immunohistochemical and real-time PCR analyses. Clin Cancer Res 14(11):3299–3305 [DOI] [PubMed] [Google Scholar]
- Canel M et al (2006) Overexpression of focal adhesion kinase in head and neck squamous cell carcinoma is independent of fak gene copy number. Clin Cancer Res 12(11 Pt 1):3272–3279 [DOI] [PubMed] [Google Scholar]
- Cen L et al (2007) Phosphorylation profiles of protein kinases in alveolar and embryonal rhabdomyosarcoma. Mod Pathol 20(9):936–946 [DOI] [PubMed] [Google Scholar]
- Cohen LA, Guan JL (2005) Mechanisms of focal adhesion kinase regulation. Curr Cancer Drug Targets 5(8):629–643 [DOI] [PubMed] [Google Scholar]
- Crose LE, Linardic CM (2011) Receptor tyrosine kinases as therapeutic targets in rhabdomyosarcoma. Sarcoma 2011:756982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pitta C et al (2006) Gene expression profiling identifies potential relevant genes in alveolar rhabdomyosarcoma pathogenesis and discriminates PAX3-FKHR positive and negative tumors. Int J Cancer 118(11):2772–2781 [DOI] [PubMed] [Google Scholar]
- Deramaudt TB et al (2011) FAK phosphorylation at Tyr-925 regulates cross-talk between focal adhesion turnover and cell protrusion. Mol Biol Cell 22(7):964–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii T et al (2004) Focal adhesion kinase is overexpressed in hepatocellular carcinoma and can be served as an independent prognostic factor. J Hepatol 41(1):104–111 [DOI] [PubMed] [Google Scholar]
- Gabarra-Niecko V, Schaller MD, Dunty JM (2003) FAK regulates biological processes important for the pathogenesis of cancer. Cancer Metastasis Rev 22(4):359–374 [DOI] [PubMed] [Google Scholar]
- Giaginis CT et al (2009) Expression and clinical significance of focal adhesion kinase in the two distinct histological types, intestinal and diffuse, of human gastric adenocarcinoma. Pathol Oncol Res 15(2):173–181 [DOI] [PubMed] [Google Scholar]
- Gilmore AP, Burridge K (1996) Molecular mechanisms for focal adhesion assembly through regulation of protein–protein interactions. Structure 4(6):647–651 [DOI] [PubMed] [Google Scholar]
- Gupta SK, Vlahakis NE (2009) Integrin α9β1 mediates enhanced cell migration through nitric oxide synthase activity regulated by Src tyrosine kinase. J Cell Sci 122(Pt 12):2043–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamadi A et al (2005) Regulation of focal adhesion dynamics and disassembly by phosphorylation of FAK at tyrosine 397. J Cell Sci 118(Pt 19):4415–4425 [DOI] [PubMed] [Google Scholar]
- Higuchi M et al (2013) Akt1 promotes focal adhesion disassembly and cell motility through phosphorylation of FAK in growth factor-stimulated cells. J Cell Sci 126(Pt 3):745–755 [DOI] [PubMed] [Google Scholar]
- Hsia DA et al (2003) Differential regulation of cell motility and invasion by FAK. J Cell Biol 160(5):753–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu YL et al (2014) FAK and paxillin dynamics at focal adhesions in the protrusions of migrating cells. Sci Rep 4:6024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher A, Horwitz AR (2011) Integrins in cell migration. Cold Spring Harb Perspect Biol 3(9):a005074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israeli S et al (2010) Abnormalities in focal adhesion complex formation, regulation, and function in human autosomal recessive polycystic kidney disease epithelial cells. Am J Physiol Cell Physiol 298(4):C831–C846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang WG et al (2015) Tissue invasion and metastasis: molecular, biological and clinical perspectives. Semin Cancer Biol 35(Suppl):S244–S275 [DOI] [PubMed] [Google Scholar]
- Katz BZ et al (2003) Targeting membrane-localized focal adhesion kinase to focal adhesions: roles of tyrosine phosphorylation and SRC family kinases. J Biol Chem 278(31):29115–29120 [DOI] [PubMed] [Google Scholar]
- Kinn VG, Hilgenberg VA, MacNeill AL (2016) Myxoma virus therapy for human embryonal rhabdomyosarcoma in a nude mouse model. Oncolytic Virother 5:59–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohsaka S et al (2014) A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat Genet 46(6):595–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolli-Bouhafs K et al (2014) FAK competes for Src to promote migration against invasion in melanoma cells. Cell Death Dis 5:e1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lark AL et al (2005) High focal adhesion kinase expression in invasive breast carcinomas is associated with an aggressive phenotype. Mod Pathol 18(10):1289–1294 [DOI] [PubMed] [Google Scholar]
- Lee S et al (2012) FAK is a critical regulator of neuroblastoma liver metastasis. Oncotarget 3(12):1576–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y et al (2018) HNRNPH1 is required for rhabdomyosarcoma cell growth and survival. Oncogenesis 7(1):9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ST et al (2008) Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell 29(1):9–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L et al (2008) Rapamycin inhibits F-actin reorganization and phosphorylation of focal adhesion proteins. Oncogene 27(37):4998–5010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo SW et al (2009) Regulation of heterochromatin remodelling and myogenin expression during muscle differentiation by FAK interaction with MBD2. EMBO J 28(17):2568–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzzio M et al (2007) Design, synthesis, activity and properties of selective focal adhesion kinase inhibitors which are suitable for advanced preclinical evaluation: the discovery of PF-562271. Can Res 67(9 Supplement):5432 [Google Scholar]
- Malempati S, Hawkins DS (2012) Rhabdomyosarcoma: review of the Children’s Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr Blood Cancer 59(1):5–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margadant C, van Opstal A, Boonstra J (2007) Focal adhesion signaling and actin stress fibers are dispensable for progression through the ongoing cell cycle. J Cell Sci 120(Pt 1):66–76 [DOI] [PubMed] [Google Scholar]
- McAllister RM et al (1969) Cultivation in vitro of cells derived from a human rhabdomyosarcoma. Cancer 24(3):520–526 [DOI] [PubMed] [Google Scholar]
- Mei L, Xiong WC (2010) FAK interaction with MBD2: a link from cell adhesion to nuclear chromatin remodeling? Cell Adhes Migr 4(1):77–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD (2005) Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol 6(1):56–68 [DOI] [PubMed] [Google Scholar]
- Nishimura R et al (2013) Characterization of genetic lesions in rhabdomyosarcoma using a high-density single nucleotide polymorphism array. Cancer Sci 104(7):856–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocak S et al (2012) Expression of focal adhesion kinase in small-cell lung carcinoma. Cancer 118(5):1293–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MA et al (2009) Specific tyrosine phosphorylation of focal adhesion kinase mediated by Fer tyrosine kinase in suspended hepatocytes. Biochim Biophys Acta 1793(5):781–791 [DOI] [PubMed] [Google Scholar]
- Ossovskaya V et al (2008) FAK nuclear export signal sequences. FEBS Lett 582(16):2402–2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruymann FB, Grovas AC (2000) Progress in the diagnosis and treatment of rhabdomyosarcoma and related soft tissue sarcomas. Cancer Investig 18(3):223–241 [DOI] [PubMed] [Google Scholar]
- Schaller MD (2001) Biochemical signals and biological responses elicited by the focal adhesion kinase. Biochim Biophys Acta 1540(1):1–21 [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Hunter T (1996) Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol Cell Biol 16(10):5623–5633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Mitra SK, Ilic D (2004) Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochim Biophys Acta 1692(2–3):77–102 [DOI] [PubMed] [Google Scholar]
- Seong J et al (2011) Detection of focal adhesion kinase activation at membrane microdomains by fluorescence resonance energy transfer. Nat Commun 2:406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrels A et al (2015) Nuclear FAK controls chemokine transcription, Tregs, and evasion of anti-tumor immunity. Cell 163(1):160–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shern JF, Yohe ME, Khan J (2015) Pediatric rhabdomyosarcoma. Crit Rev Oncog 20(3–4):227–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood AK et al (2004) Biological significance of focal adhesion kinase in ovarian cancer: role in migration and invasion. Am J Pathol 165(4):1087–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzmaier FJ, Jean C, Schlaepfer DD (2014) FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer 14(9):598–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szuhai K et al (2014) Transactivating mutation of the MYOD1 gene is a frequent event in adult spindle cell rhabdomyosarcoma. J Pathol 232(3):300–307 [DOI] [PubMed] [Google Scholar]
- Theocharis SE et al (2003) Focal adhesion kinase expression is not a prognostic predictor in colon adenocarcinoma patients. Eur J Surg Oncol 29(7):571–574 [DOI] [PubMed] [Google Scholar]
- Wang B et al (2016) Expression of pY397 FAK promotes the development of non-small cell lung cancer. Oncol Lett 11(2):979–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters AM et al (2016) Targeting focal adhesion kinase suppresses the malignant phenotype in rhabdomyosarcoma cells. Transl Oncol 9(4):263–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhoff MA et al (2004) SRC-mediated phosphorylation of focal adhesion kinase couples actin and adhesion dynamics to survival signaling. Mol Cell Biol 24(18):8113–8133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemer AJ et al (2013) The focal adhesion kinase inhibitor PF-562271 impairs primary CD4+ T cell activation. Biochem Pharmacol 86(6):770–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak MA et al (2004) Focal adhesion regulation of cell behavior. Biochim Biophys Acta 1692(2–3):103–119 [DOI] [PubMed] [Google Scholar]
- Wu C (2007) Focal adhesion: a focal point in current cell biology and molecular medicine. Cell Adhes Migr 1(1):13–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie B et al (2001) Focal adhesion kinase activates Stat1 in integrin-mediated cell migration and adhesion. J Biol Chem 276(22):19512–19523 [DOI] [PubMed] [Google Scholar]
- Xu B et al (2012) RhoA/ROCK, cytoskeletal dynamics, and focal adhesion kinase are required for mechanical stretch-induced tenogenic differentiation of human mesenchymal stem cells. J Cell Physiol 227(6):2722–2729 [DOI] [PubMed] [Google Scholar]
- Yan D et al (2009) MicroRNA-1/206 targets c-Met and inhibits rhabdomyosarcoma development. J Biol Chem 284(43):29596–29604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Guan JL (2009) Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev 28(1–2):35–49 [DOI] [PubMed] [Google Scholar]
- Zhao X, Guan JL (2011) Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Deliv Rev 63(8):610–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JH, Reiske H, Guan JL (1998) Regulation of the cell cycle by focal adhesion kinase. J Cell Biol 143(7):1997–2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Pestell R, Guan JL (2001) Transcriptional activation of cyclin D1 promoter by FAK contributes to cell cycle progression. Mol Biol Cell 12(12):4066–4077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J et al (2015) Mechanism of focal adhesion kinase mechanosensing. PLoS Comput Biol 11(11):e1004593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z et al (2018) Prohibitin 2 localizes in nucleolus to regulate ribosomal RNA transcription and facilitate cell proliferation in RD cells. Sci Rep 8(1):1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data sets used and/or analyzed during the current study are available from the corresponding author on request.