

Abstract
Soluble model compounds, such as flavones, are frequently employed in initial and mechanistic studies under homogeneous conditions in the search for effective bleaching catalysts for raw cotton. The relevance of model substrates, such as morin and chrysin, and especially their reactivity with manganese catalysts [i.e. in combination with 1,4,7‐triazacyclononane (tacn) based ligands] applied in raw cotton bleaching with H2O2 in alkaline solutions is examined. We show that morin, used frequently as a model, is highly sensitive to oxidation with O2, by processes catalyzed by trace metal ions, that can be accelerated photochemically, although not involve generation of 1O2. The structurally related chrysin is not susceptible to such photo‐accelerated oxidation with O2. Furthermore, chrysin is oxidized by H2O2 only in the presence of a Mn‐tacn based catalyst, and does not undergo oxidation with O2 as terminal oxidant. Chrysin mimics the behavior of raw cotton's chromophores in their catalyzed oxidation with H2O2, and is likely a mechanistically relevant model compound for the study of transition metal catalysts for dye bleaching catalysts under homogeneous conditions.
Keywords: Manganese, Morin, Chrysin, Photochemistry, Bleaching, Oxidation
Introduction
The industrial bleaching of the cellulose based materials paper and raw cotton1 is of substantial economic and environmental importance.2 Since the early 1990s, chlorine free bleaching processes to avoid, e.g., the formation of dioxins,3 using atom efficient terminal oxidants such as O2 and H2O2, have been pursued through either decolorizing undesired pigments or increasing their aqueous solubility in water.4, 5 In contrast to hypochlorite (bleach), these oxidants require activation with catalysts and even with H2O2 temperatures in excess of 90–100 °C and pH > 11 are required.6 Transition metal (Mn,6, 7 Fe or Co complexes), are increasingly used to activate H2O2 or O2 and enhance stain removal at lower temperatures and at a reduced chemical cost.2
In the 1990s, several detergent companies patented a wide range of transition metal complexes for this purpose.2 For example, [Mn2 III,IV(µ‐O)2(µ‐CH3CO2)(Me4dtne)](PF6)2 (1) [where Me4dtne = 1,2‐bis(4,7‐dimethyl‐1,4,7‐triazacyclonon‐1‐yl)ethane], reported first by Wieghardt et al.,8, 9 was reported by Unilever together with analogous catalysts, e.g., [Mn2 IV,IV(µ‐O)3(tmtacn)2](PF6)2 (2) (where tmtacn = 1,4,7‐trimethyl‐1,4,7‐triazacyclononane) for laundry bleaching in the early 1990s (Figure 1).10, 11 More recently, these catalysts have been applied to raw cotton bleaching with H2O2,12 and the effectiveness of 1 shown to be highly dependent on conditions (e.g., pH, temperature, H2O2 and catalyst concentration),2 with optimum activity observed at pH > 10 and > 40 °C.10
Figure 1.
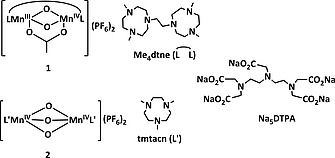
Structure of 1, 2, the ligands Me4dtne (L) and tmtacn (L′), and Na5DTPA.
Over the last decade, mechanistic studies on manganese catalyzed oxidations for the oxidation of a wide range of organic substrates in organic solvents has provided a considerable body of data on the mode of action of these catalysts and a broad understanding of the role of additives in such catalytic systems.10, 13, 14 However, extending these mechanistic insights to understanding the behavior of such catalysts in aqueous media15 under the highly heterogeneous conditions of cotton and wood pulp bleaching, is challenging; not least in efforts to understand the factors that influence catalysts activity, including other reaction components such as sequestrants and buffers. The complex mixture of substrates, hydrophobic waxes and the heterogeneity of raw cotton make the direct spectroscopic and kinetic study of bleaching reactions highly challenging. The availability of substrates that are stable at high pH and temperatures, and thus provide a homogeneous model system, is therefore essential in the elucidation of the mode of action of these catalysts and especially in understanding how other reaction components influence their activity.
The pigments responsible for the brown coloration of natural cotton include polyphenols, flavones and tannins, with flavonoid compounds being the primary source of coloration.16 Furthermore, the pigments in other substrates that require bleaching, e.g., paper pulp (i.e. lignin) and domestic laundry, and dishwashing applications (i.e. polyphenols in tea and wine stains), contain chemically similar structural motifs.2, 6 Therefore these substrate classes, and especially flavones, are employed primarily in mechanistic studies.17
Flavonoids share a C6–C3–C6 flavone skeleton, where the three‐carbon bridge is cyclized with oxygen and the aromatic rings bear various numbers of hydroxyl substituents (Figure 2). Morin, which has been used most widely as a model18 for the chromophores in raw cotton,7, 19 is a flavon‐3‐ol, i.e. it bears a hydroxyl group at the 3 position.20 It is the most reactive of the flavones, and reacts directly with O2 in the presence of catalysts,21, 22 including 2.23 Decoloration of cotton with O2, even with catalysts,2, 6 has not been observed and hence, its actual suitability to model the reactivity of the more stable naturally occurring dyes is questionable.
Figure 2.
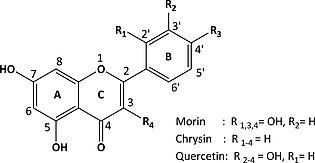
General structure and numbering scheme of flavonoids under investigation.
Furthermore, from a practical perspective, the pronounced pH dependence of the absorption spectrum of morin (pH 8–11) increases the complexity in studying the effect of pH on catalyst activity due to both changes in spectral shape and its susceptibility to oxidation.
Here we show that morin is in fact unsuitable as a model compound due to its photochemical instability even to the light used to monitor its conversion by UV/Vis absorption spectroscopy, which is the standard tool to monitor dye degradation. We show that it undergoes metal catalyzed oxidative photo‐accelerated degradation with O2 as terminal oxidant. This reactivity complicates kinetic analyses in bleaching studies with H2O2. The related flavone chrysin (Figure 2) does not undergo oxidation with O2 nor photochemically induced oxidations and is a more suitable model substrate. It is used in the study of the activity of catalyst 1 in the oxidation of chrysin at pH 10 and 11, and at 23, 40 and 60 °C, conditions relevant to industrial bleaching.
Chrysin is related structurally to morin, differing in the absence of hydroxyl groups at the 3‐position of the C ring and 2′ and 4′ positions of the B ring, and has been the subject of studies related to biological processes and physical chemistry.24, 25
Results and Discussion
The UV/Vis absorption spectra of morin and chrysin (Figure 3) are characterized by two resolved absorption bands. The transition in the (near) visible region (300 to 550 nm) is assigned to the B ring and the shorter wavelength transition (240 to 285 nm) assigned to the A ring (Figure 2).26 The Raman (λ exc 785 nm) and resonance Raman spectra (λ exc 266 nm) of chrysin in the solid state and in water, respectively, are shown in Figure 4. The spectra show expected differences (due to the resonance enhancement at 266 nm) in intensities of the bands, however, there are differences in the Raman shift of several bands also indicating that the compounds structure (conformation) in solution is different to that in the solid state.
Figure 3.
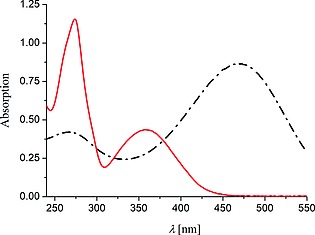
UV/Vis absorption spectra of morin (40 µm, black) and chrysin (40 µm, red) in water (with 10 mm NaHCO3) at pH 11.
Figure 4.
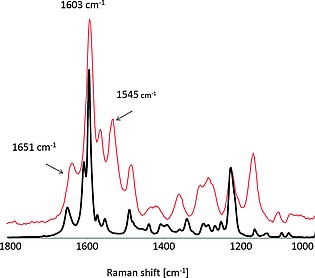
Raman spectrum of chrysin in the solid state (black, λ exc 785 nm), and resonance Raman spectrum in water (1 mm, pH 10, red, at λ exc 266 nm).
pH Dependence of the Absorption Spectra of Morin and Chrysin
The UV/Vis absorption spectra of phenols and flavonoids show pronounced pH dependence.27 Morin bears five hydroxyl groups, three of which are relatively acidic (pKa 5.2, 8.2 and 9.9).28 A bathochromic shift of the longest wavelength absorption band of morin is observed as the pH is increased (λ max = 394 nm at pH 8.2, 404 nm at pH 9.4, 412 nm at pH 10.2, and 417 nm at pH 11.4, Figure S1a). By contrast, chrysin, in which the pKa of hydroxyl groups at C5 and C7 of the A‐ring, are 8.0 and 11.9, respectively,28 does not exhibit significant changes in its UV/Vis absorption spectrum over the pH range of interest in the present study (i.e. pH 8.2 to 11.4, Figure S1b).
Photochemical Stability of Morin in the Presence of O2 and Metal Ions
An often overlooked complication in the use of flavones as model compounds in spectroscopic studies is their well‐known photochemistry,29 together with the ability of hydroxyaromatic compounds in general and flavonoids in particular to react rapidly with 1O2. Garcia et al. reported the photodecomposition of the flavonoids quercetin, morin, and rutin (Figure 2), under aerobic conditions.30 The oxidation of morin can be monitored conveniently by UV/Vis absorption spectroscopy, however, the propensity for morin to undergo photochemically driven degradation is such that the light used during monitoring can itself induce substantial oxidation. Indeed, comparison of the absorption spectra of morin in aqueous solution when monitored by UV/Vis absorption spectroscopy with continuous exposure to the monitoring light shows a 78 % decrease in visible absorption compared to a < 7 % decrease when held for the same period in the dark (Figure 5 and Figure S2). The extent of decrease in visible absorbance corresponds with [O2] (ca. 200–280 µm),31 i.e. O2 is the limiting oxidant under these conditions (Figure S3). Argon purged solutions of morin show no changes even under extended irradiation.
Figure 5.
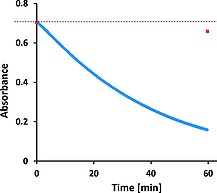
Absorbance at 412 nm of Morin (40 µm) in water (10 mm NaHCO3), pH 10.2, over time during repeated spectral acquisition (at 5 s intervals with 0.5 s exposure time, blue) and after 1 h for a sample held in dark (shown as a red square). The dashed line indicates the initial absorbance.
The photochemical degradation of morin in the absence of added catalysts is suppressed substantially by addition of the sequestrant Na5DTPA (5 µm, pentasodium diethylene‐triamine‐pentaacetate, Figure 1), indicating that trace metal ions present are involved in the observed photochemistry (Figure 6, Figure S4 and Figure S5). Furthermore it confirms that both metal ions and oxygen are necessary in order for oxidation of morin with O2 to proceed and that irradiation accelerates the reaction.
Figure 6.
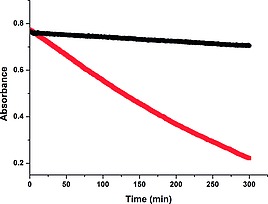
Absorbance at 412 nm of morin [40 µm] in water (10 mm NaHCO3) at pH 10.2, without (red), and with (black) Na5DTPA (5 µm). Spectra recorded at 30 s intervals (1 s exposure time) over 5 h. See also Figure S4.
In the presence of 1 (1 µm), a decrease in absorbance at 412 nm with a concomitant increase in absorbance at 321 nm is observed in air equilibrated solutions of morin at between 8.0 and 11.5, with the reaction rate increasing with pH (Figure 7 and Figure 8). Two isosbestic points, at 287 and 370 nm, were maintained over most of the reaction, suggesting the initial formation of a single oxidation product in which the chromophoric system is disrupted. The resonance Raman spectrum recorded at 355 nm, of the primary photoproduct (Figure 7), shows the appearance of a band at 1550 cm–1 and an intense set of bands at 1350 cm–1 which are consistent with the formation of arylcarboxylates.32 At later times, i.e. after morin is nearly completely consumed, the isosbestic points were lost and the band at 321 nm underwent a red shift to 331 nm together with a decrease in absorbance. These data indicate that the primary product reacts further, however, the subsequent reactions do not necessarily involve further oxidation but instead may be due to, e.g., hydrolysis.
Figure 7.
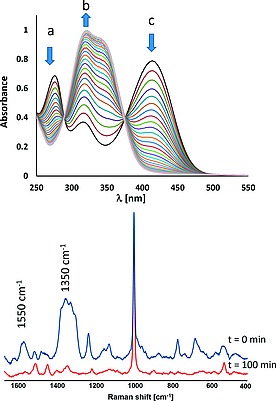
(Top) UV/Vis absorption spectra of morin (40 µm) with 1 (1 µm), over time (spectra recorded at 50 s intervals for 30 min, for clarity only the spectra at 100 s intervals are shown) at pH 11.4, (10 mm NaHCO3) at 23 °C. (Bottom) Raman spectra (λ exc 355 nm) of morin (40 µm) with 1 (1 µm) after 0 min (blue) and 100 min (red), at pH 10.
Figure 8.
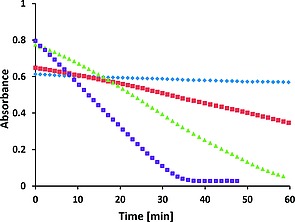
Absorbance at 405 nm (band c in Figure 7), at pH 8.2 (blue), 9.4 (red), 10.2 (green) and 11.4 (purple) over time of morin (40 µm) with 1 (1 µm), NaHCO3 (10 mm), 23 °C, and with O2 (200–350 µm) as terminal oxidant. For changes at ca. 275 nm, and 316 nm (band a and b, respectively, in Figure 7) see Figure S6.
The oxidation of morin with H2O2, catalyzed by 1 (Figure 9), is two to three times faster than with O2 as terminal oxidant alone (Figure 8). At pH 11 conversion was complete within ca. 10 min, while at pH 10 conversion was complete at ca. 30 min. Hence, under conditions relevant to bleaching, morin is shown, even in the absence of H2O2 and 1, to undergo a photochemically accelerated oxidation with O2 and metal ions that is of the same order of magnitude rate to that observed with H2O2.
Figure 9.
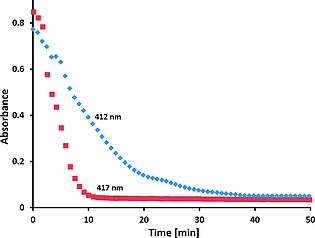
Absorbance at 412 nm (blue) and 417 nm (red) of morin (40 µm) with 1 (1 µm), NaHCO3(aq) (10 mm) at pH 10 and at pH 11, respectively, over time after addition of H2O2 (200 µm) at 23 °C.
Photochemical Stability of Chrysin
In contrast to morin, the UV/Vis absorption and resonance Raman spectra of chrysin did not change even under continuous visible and UV (λ exc 266 nm) irradiation in the presence of O2 (Figure S7). Furthermore, the UV/Vis absorption spectrum of chrysin is not significantly different at pH 11 compared to pH 10 (Figure 10). Continuous monitoring of a mixture of chrysin and morin (10:1), by UV/Vis absorption spectroscopy under air with 1 (Figure S8), shows changes (Figure S9) that are consistent with oxidation of morin only. These data indicate that the photochemistry of morin does not involve the production of species that react with chrysin.
Figure 10.
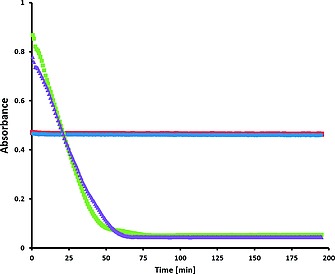
Absorbance at 359 nm of chrysin (40 µm, red at pH 10.2 and blue at pH 11.0) and at 412 and 417 nm of morin (40 µm, purple at pH 10.2 and green at pH 11.0, respectively) over time in air equilibrated solution with 1 (1 µm), in aqueous NaHCO3 (10 mm) at 23 °C.
Reaction of Chrysin and Morin with Singlet Oxygen
The stability of chrysin and morin in the presence of 1O2 (generated by irradiation of Rose Bengal at 532 nm) at pH 10 was examined in D2O. The absorbance of both substrates, as well as that of Rose Bengal, was bleached within several minutes (Figure S10). Addition of Na5DTPA had no effect on the changes observed, in contrast to that observed upon direct irradiation of the substrate in the absence of Rose Bengal. Furthermore, the final absorption spectra did not resemble the final spectra obtained with H2O2 or by irradiation in the absence of Rose Bengal. Hence, although both morin and chrysin are susceptible to degradation by 1O2, it is highly unlikely that 1O2 is responsible for the oxidation of morin (vide supra) in the absence of the sensitizer. Further, the bleaching of morin is only ca. twice as fast in D2O as in H2O (Figure S11), which is inconsistent with the ca. 22 fold increase33 in the lifetime of 1O2 in D2O compared with H2O.
Oxidation of Chrysin with H2O2 Catalyzed by 1
Complex 1 retains its catalytic activity in oxidations with H2O2 even at high pH (pH > 11).6, 10 At pH > 10, the µ‐acetato ligand of 1 dissociates partly and/or fully to form 1a and/or 1b, respectively (Scheme 1).15 Theses structural changes provide exchangeable sites for coordination of H2O2 and addition of carboxylato ligands, such as acetate, or (bi)carbonate, results in reversion to a carboxylate bridged complex (e.g., 1c) and a decrease in activity (Scheme 1).15
Scheme 1.
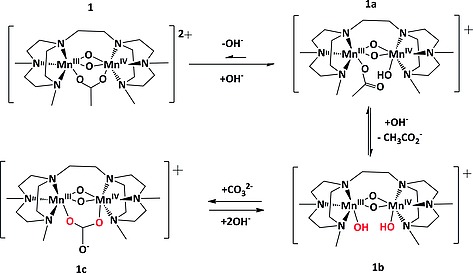
Equilibrium between 1 and the µ‐acetate dissociated forms (1a and 1b) formed at high pH.15 and the carbonate bound complex 1c.
Raw cotton bleaching with H2O2 is carried out at elevated temperatures (up to 90 °C) at pH 11–12, in the presence of surfactants, to remove hydrophobic components such as waxes and long chain fatty acids present on the cotton fibers. The temperature stability of chrysin with respect to oxidation with O2 in the presence of 1 even under UV irradiation, allows for the oxidation with H2O2 to be monitored spectroscopically at pH 10 and 11 at 23 and 60 °C (Figure S12 and Figure 11). The rate of the catalyzed oxidation of chrysin with H2O2 increased with temperature (Figure 12), and at all temperatures the oxidation was fastest at pH 11. In the absence of 1, chrysin was stable at room temperature and degraded only relatively slowly at higher temperatures (Figure 12). These data correspond well to observations made under industrial bleaching conditions with 1 and H2O2, i.e. in catalytic bleaching of raw cotton in the presence of H2O2.6
Figure 11.
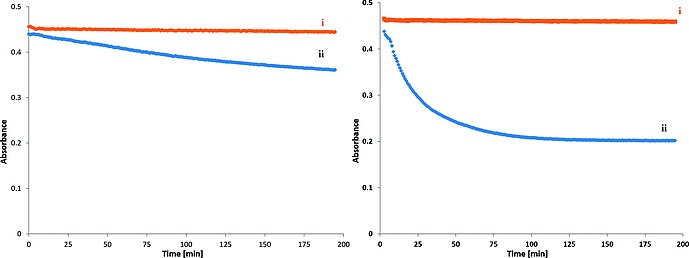
Absorbance at 359 nm vs. time showing the extent of oxidation of chrysin with O2 or H2O2 at pH = 10 (left), at pH = 11 (right), (I) 1 under air (red), (II) 1 with H2O2 (blue). Conditions: chrysin (40 µm), catalyst (1 µm), H2O2 (200 µm), in aqueous NaHCO3 (10 mm), at 23 °C.
Figure 12.
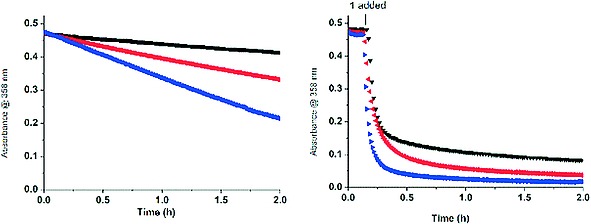
Change in absorption at 358 nm for chrysin [40 µm] with H2O2 [10 mm] in H2O at pH 10, (left) without catalyst and (right) with 1 [1 µm], at 23 °C (black), 40 °C (red), 60 °C (blue). For data at pH 11 see Figure S13. See also Figure S14.
Comparison of the bleaching of morin and chrysin at 40 °C and pH 10, in the absence of catalyst, shows that the rate of oxidation of morin with O2 as terminal oxidant is higher than of chrysin with H2O2. Indeed after 2 h, chrysin shows only a ≈ 25 % decrease in the absorbance with H2O2 as terminal oxidant whereas morin shows a ca. 40 % decrease in the absorbance with O2 as terminal oxidant.
Discussion
Model dyes, e.g., soluble analogues of naturally occurring insoluble dyes, such as those in raw cotton, are invaluable in understanding how changes in the conditions and variation in structural motifs affects the oxidation activity of catalysts. Naturally, UV/Vis absorption spectroscopy is an obvious method to study the bleaching of chromophores, however, the technique is not necessarily “innocent” as shown in the present study. The challenge is therefore to identify model compounds, which (i) mimic the reactivity observed with raw cotton, and (ii) are not affected by the measurement technique used to monitor reaction.
Morin, is a yellow colored flavone and has been used21, 34 as a soluble model compound for insoluble flavones and polyphenols as it undergoes oxidation relatively easily in comparison to related compounds such as chrysin. However, as shown here, the sensitivity of morin to light, metal ions, etc., is on a par with the rates that are to those observed with 1 and H2O2 alone. Hence, time dependent changes observed in model studies do not necessarily relate to the behavior of catalysts under bulk reaction conditions, e.g., in raw cotton bleaching. Indeed with less active catalysts the rates of reaction with oxygen may even be greater than with H2O2. From a fundamental perspective, however, the data reported here also offers deeper insight into the photochemistry of flavones and the role played by metal ions.
Oxidation of Flavones with 1O2
The photochemistry of flavones is well known, not least to the brewing industry, with the sensitization of 3O2 to generate 1O2 often inferred. The antioxidant properties of several flavonoids, including quercetin, morin and rutin, and their activity against reactive oxygen species (ROS) generated upon visible irradiation of riboflavin (a 1O2 generator) in methanol were described by Montana et al., whom noted morin as the most reactive with around 80 % of the 1O2–morin collisions resulting in oxidation.30
In the present study, sensitized generation of 1O2 in water is shown to degrade both morin and chrysin in water also.
Colombini et al. proposed two pathways for the photochemical degradation for morin upon direct irradiation (Scheme 2); involving (a) metal ions and (b) the generation of 1O2, although a definitive conclusion as to which pathway was followed under their conditions was not reached.35
Scheme 2.
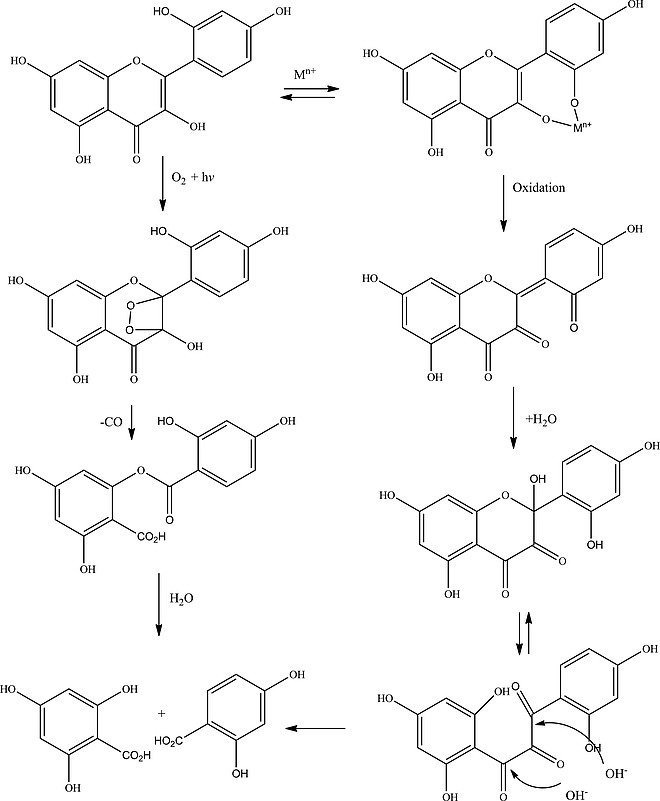
Degradation pathways for morin with O2 proposed by Colombini et al.35
The greater susceptibility of morin with regard to oxidation, presumed by 1O2, was ascribed to deprotonation of the phenolic moiety that is absent in the other flavonoids.36 Furthermore, the hydroxyl substituents increase its susceptibility towards electrophilic attack by 1O2. Hence differences in reactivity can be related to differences in the B‐ring, especially the presence of an 7‐OH group in morin,37 which, as noted by Agrawal, Musialik, and co‐workers, is the most acidic in the case of chrysin also.28, 38
Notably, Matsuura et al.39, 40 have shown that the photosensitized oxidation of 3‐hydroxyflavones is inhibited by methylation at the 3‐hydroxy group.
Mechanism of Photochemical Activation of O2 in Basic Aqueous Solutions
At high pH, as used here, 1O2 generated with Rose Bengal degrades both morin and chrysin rapidly, which is not inhibited by addition of a sequestrant (Na5DTPA). In the absence of Rose Bengal, however, the photodegradation of morin is unlikely to involve 1O2, but instead through oxidation of the photo‐excited morin by electron transfer to oxygen (vide infra).
The oxidation of the flavonoids, including morin, with H2O2 catalyzed by 2 was reported by Topalovic et al.23, 41 In those studies, morin was shown to undergo oxidation in basic solution with O2 as the terminal oxidant; a process which was catalyzed by 2 as well as MnII salts. A lag phase was observed with 2, which indicated that catalyst activation23 through direct electron transfer between the morin and 2 occurred, followed by reaction with O2 either with the reduced form of the catalyst or with the oxidized morin.23 The intermediacy of superoxide O2*– and H2O2 in the oxidation of morin by O2 catalyzed by 2 was confirmed through the inhibition observed with superoxide dismutase and with catalase.23 Hence, it was proposed that formation of superoxide by electron transfer from morin to O2 occurred, followed by dismutation of superoxide to H2O2.23 It was postulated that the 3‐hydroxyl group was key to this process as flavonoids that lacked this moiety (e.g., chrysin) were stable with regard to oxidation under the same conditions.34 It is notable, however, that all reactions reported were monitored by UV/Vis absorption (diode array) spectroscopy (vide infra).
The reported reactivity of morin with O2 is in stark contrast to raw cotton which does not undergo oxidation with O2 alone and requires high temperatures and H2O2 to undergo bleaching.6 Indeed, in the present study oxidation of morin in hydrogen carbonate buffer (in the dark) was found to be relatively slow, although non‐negligible, in comparison to chrysin, which is wholly unreactive even when under intense irradiation (10 mW cm–2) at 355 nm. Indeed the rapid oxidation of morin with O2, reported earlier,41 is found here to require both irradiation of the reaction mixture with light (either with the light from a spectrometer or monochromatic radiation at 355 or 405 nm) and the presence of metal ions (either catalysts 1 and 2, or simple salts).
The observation of slow oxidation of morin when held in the dark indicates that irradiation is accelerating an otherwise thermal process. Indeed the spectral changes observed upon oxidation with O2 and with H2O2 are remarkably similar and hence a largely common mechanism involved. The direct reaction of morin with 1 can be excluded by the lack of spectral changes when degassed (oxygen free) solutions of morin and 1 were irradiated. Furthermore, although the catalyst 1 is not essential to the process, metal ions must be available for the activity to be observed; addition of the sequestrant Na5DTPA prevents conversion.
Although 1O2 can degrade both morin and chrysin, under the present reaction conditions (vide supra) it is evident that 1O2 is not generated (Scheme 3). Nevertheless, the oxidation of morin with O2 requires the availability of metal ions, since sequestration of metal ions with Na5DTPA stops oxidation with both O2 and H2O2 as terminal oxidant. The inhibition observed by Topalovic et al.23, 41 upon addition of catalase and superoxide dismutase confirms that H2O2, and possibly the superoxide radical anion, are formed from O2. Hence, it is likely that excitation of morin leads to photoinduced electron transfer to 3O2 to generate the superoxide radical anion, which then undergoes disproportionation to H2O2 or reacts with the morin radical cation. In the presence of metal ions, the formation of metal peroxides is likely to occur, and hence the same chemical reactivity as observed upon addition of H2O2 would be expected.
Scheme 3.
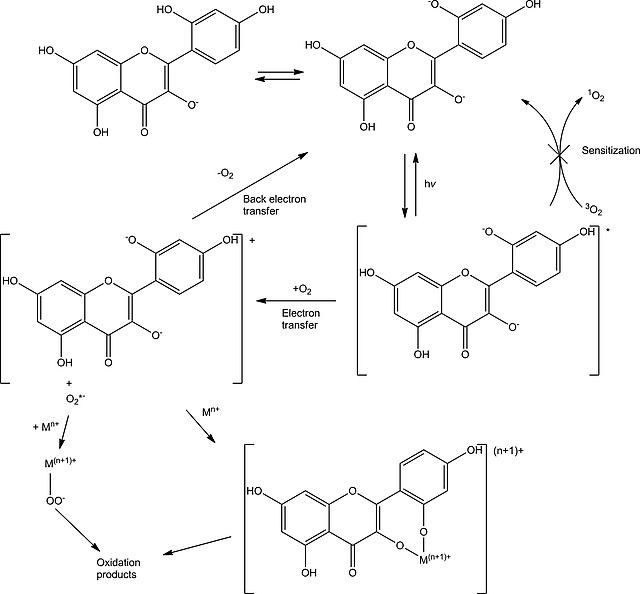
Degradation pathways for morin with O2 and H2O2.
Model Substrates for Raw Cotton Bleaching
The photo degradation of morin in aqueous hydrogen carbonate solution under irradiation together with its oxidation with O2 and the pH dependence of its UV/Vis absorption spectrum limit its applicability as a model compound for raw cotton bleaching in basic solutions. Furthermore, the two activating, i.e. electron donating, OH substituents of morin are deprotonated at high pH increasing susceptibility to electron transfer oxidation. In contrast, chrysin is not susceptible to oxidation with O2, neither thermally nor photochemically and shows relatively little, if any, pH dependence in the range of interest and as a consequence of it having fewer hydroxyl moieties, it is less nucleophilic and hence less easily oxidized.
Conclusions
In the present contribution the suitability of morin and chrysin as model compounds for homogeneous reactions used in mechanistic studies relating to raw cotton bleaching catalyzed by 1 is explored. The use of morin as a model compound is fundamentally flawed due to its photochemistry and sensitivity to O2, which do not reflect the chemistry of the colorants present in raw cotton. Even exposure of morin to the light of a UV/Vis absorption spectrometer is sufficient to induce rapid photochemical degradation in the presence of traces of metal ions. The oxidation seen with metal ions and O2 proceeds at a similar rate as with H2O2, which complicates the elucidation of the various mechanistic pathways involved in bleaching.
The data presented here indicate that chrysin is sufficiently stable and requires H2O2 and elevated temperatures for oxidation to occur at an appreciable rate. Taken together with the amenability of chrysin towards reaction monitoring by UV resonance Raman as well as UV/Vis absorption spectroscopy, it can be concluded that it provides a much more realistic homogeneous model on which to further the development of catalysts for raw cotton bleaching and understanding the mechanisms involved.
As a final remark, although chrysin is shown here to have many attributes that make it suitable as a homogeneous model for raw cotton bleaching studies, an important challenge, however, lies in the relatively weak visible absorption of chrysin in comparison to natural cotton colorants. Establishing an optimum model system for homogeneous studies would require elucidation of the precise molecular nature of the dyes present in raw cotton.
Experimental Section
Materials and Methods: MnSO4 was obtained from Fluka. Morin, chrysin, sodium hydroxide and sodium hydrogen carbonate were purchased from Sigma Aldrich. Hydrogen peroxide (50 % w/w in H2O) was obtained from Acros Organics. All chemicals were used as obtained without further purification. Doubly distilled water was used unless stated otherwise. Bicarbonate (10 mm) solutions of morin and chrysin were prepared freshly, and the pH was adjusted with NaOH or H2SO4 (1 m) before and, where necessary, after addition of H2O2. The required amount of a freshly prepared stock solution of catalyst was added to the cuvette containing the flavonoid solution and H2O2. All reactions were performed, at least, in triplicate at room temperature (23 ± 2 °C) and under air unless stated otherwise. The pH was measured at the end of the reactions, and in all cases there were no significant changes. Initial rates of reaction were obtained by fitting the linear portion of the plot of absorbance vs. time and the slope of this best fit line is reported as the maximum rate.
Instrumentation: UV/Vis absorption spectra were recorded with a HP8453 spectrophotometer or a Specord600 (AnalytikJena) in stoppered 1 cm pathlength quartz cuvettes unless stated otherwise. pH was determined using a Hanna Instruments pH 211 microprocessor pH meter previously calibrated with standard buffer solutions at 4.01, 7.01 and 10.00. Raman spectra were recorded at 785 nm using a Perkin–Elmer RamanStation and at 266 and 355 nm using a custom built system described earlier.43 Singlet oxygen was generated by irradiation at 532 nm (300 mW, Cobolt lasers), with the beam expanded to a diameter of 1 cm using a 5 cm focal length planoconvex lens.
[Mn2III,IV(µ‐O)2(µ‐CH3CO2)(Me4dtne)](PF6)2 (1): Elemental analysis (calcd. for Mn2C20H43N6O4P2F12): C 28.89 % (28.89 %), H 5.26 % (5.21 %), N 10.11 % (10.11 %).9, 42
General Method for Oxidation of Morin and Chrysin Catalyzed by 1 in Air Equilibrated Solutions: The pH of a freshly prepared solution of morin (40 µm) in NaHCO3(aq) was adjusted as necessary using NaOH (1 m) or H2SO4 (10 vol.‐%). 25 µL of a freshly prepared solution of the catalyst (100 µm) was added directly to a cuvette containing 2.5 mL of stock solution substrate.
General Method for Oxidation of Morin and Chrysin Catalyzed by 1 with H2O2: The pH of a freshly prepared solution of morin or chrysin (40 µm) in NaHCO3(aq) was adjusted as necessary. H2O2 (200 µm, 5 equiv. with respect to substrate) was added to a cuvette containing substrate, followed immediately by the addition of catalyst (final concentration; 1 µm).
General Method for Oxidation of Morin Catalyzed by 1 with O2 or H2O2 in the Presence of Na5DTPA: (Pentasodium diethylene triamine pentaacetate, Aldrich). The pH of a freshly prepared solution of morin (40 µm) in NaHCO3(aq) was adjusted as necessary. Na5DTPA (5 µm) was added to a cuvette containing morin. H2O2 (1 equiv. with respect to substrate) was added to a cuvette containing morin (40 µm) and Na5DTPA followed immediately by the addition of catalyst (1 µm).
Supporting information
Supporting Information
Acknowledgements
Financial support from the European Research Council (ERC‐2011‐StG‐279549, W. R. B.), the Netherlands Fund for Technology and Science STW (11059, S. A., J. W. d B., W. R. B.) the Ministry of Education, Culture and Science (Gravity program 024.001.035, W. R. B.) is acknowledged.
Contributor Information
Ronald Hage, Email: ronald.hage@catexel.com, http://www.catexel.com.
Wesley R. Browne, Email: w.r.browne@rug.nl, http://www.rug.nl/research/molecular-inorganic-chemistry/browne/.
References
- 1. Weissermal H. and Arpe K., in: Industrial Organic Chemistry, VCH‐Wiley, Weinheim, 1997. [Google Scholar]
- 2. Hage R. and Lienke A., Angew. Chem. Int. Ed., 2005, 45, 206–222; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2005, 117, 212–229. [Google Scholar]
- 3. Beychok M. R., Atmos. Environ., 1987, 21, 29–36. [Google Scholar]
- 4. Nakamata K. and Ohi H., J. Wood Sci., 2003, 49, 525–530. [Google Scholar]
- 5. Nakamata K., Motoe Y. and Ohi H., J. Wood Sci., 2004, 50, 242–247. [Google Scholar]
- 6. Hage R., de Boer J. W., Gaulard F. and Maaijen K., Adv. Inorg. Chem., 2013, 65, 85–116. [Google Scholar]
- 7. Rothbart S., Ember E. and van Eldik R., Dalton Trans., 2010, 39, 3264. [DOI] [PubMed] [Google Scholar]
- 8. Wieghardt K., Bossek U., Nuber B., Weiss J., Bonvoisin J., Corbella M., Vitols S. E. and Girerd J. J., J. Am. Chem. Soc., 1988, 110, 7398–7411. [Google Scholar]
- 9. Bossek U., Weyhermüller T. and Wieghardt K., J. Inorg. Biochem., 1991, 43, 371. [Google Scholar]
- 10. Saisaha P., de Boer J. W. and Browne W. R., Chem. Soc. Rev., 2013, 42, 2059–2074. [DOI] [PubMed] [Google Scholar]
- 11. Hage R., Iburg J. E., Kerschner J., Koek J. H., Lempers E. L. M., Martens R. J., Racherla U. S., Russell S. W., Swarthoff T., Vanvliet M. R. P., Warnaar J. B., Wanderwolf L. and Krijnen B., Nature, 1994, 369, 637–639. [Google Scholar]
- 12. Alves V., Capanema E., Chen C.‐L. and Gratzl J., J. Mol. Catal. A, 2003, 206, 37–51. [Google Scholar]
- 13. Angelone D., Abdolahzadeh S., De Boer J. W. and Browne W. R., Eur. J. Inorg. Chem., 2015, 2015, 3532–3542. [Google Scholar]
- 14. Sibbons K. F., Shastri K. and Watkinson M., Dalton Trans., 2006, 645–661. [DOI] [PubMed] [Google Scholar]
- 15. Abdolahzadeh S., Boyle N. M., Hoogendijk M. L., Hage R., de Boer J. W. and Browne W. R., Dalton Trans., 2014, 43, 6322–6332. [DOI] [PubMed] [Google Scholar]
- 16. Ardon O., Kerem Z. and Hadar Y., J. Biotechnol., 1996, 51, 201–207. [Google Scholar]
- 17. Blum H., Mayer B., Pegelow U., Catalytic Activator Complexes with N4 Ligands for Peroxygen Compounds, 1997, DE19620267 A1.
- 18. Sen P., Simsek D. K. and Yildiz S. Z., Appl. Organomet. Chem., 2015, 29, 509–516. [Google Scholar]
- 19. Dannacher J. J., J. Mol. Catal. A, 2006, 251, 159–176. [Google Scholar]
- 20. Wang F., Xu Y., Zhao J. and Hu S., Bioelectrochemistry, 2007, 70, 356–362. [DOI] [PubMed] [Google Scholar]
- 21. Medina‐Escriche J., Hernández F. H. and López Benet F. J., Analyst, 1985, 110, 1457–1461. [Google Scholar]
- 22. López Benet F. J., Hernández Hernández F., Medina Escriche J. and Marin Saez R., Analyst, 1986, 3, 1–6. [Google Scholar]
- 23. Topalovic T., in: Catalytic Bleaching of Cotton: Molecular and Macroscopic Aspects, [Google Scholar]
- 24. Lau K. S., Mantas A., Chass G. A., Ferretti F. H., Estrada M., Zamarbide G. and Csizmadia I. G., Can. J. Chem., 2002, 80, 845–855. [Google Scholar]
- 25. Arts M. J. T. J., Haenen G. R. M. M., Voss H.‐P. and Bast A., Food Chem. Toxicol., 2004, 42, 45–49. [DOI] [PubMed] [Google Scholar]
- 26. (Ed.) B. A. Bohm, in: Introduction to Flavanoids, Harwood Academic Publishers, Amsterdam, 1999, pp. 175–242. [Google Scholar]
- 27. Webb M. R. and Ebeler S. E., Biochem. J., 2004, 384, 527–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Musialik M., Kuzmicz R., Pawcowski T. S. and Litwinienko G., J. Org. Chem., 2009, 74, 2699–2709. [DOI] [PubMed] [Google Scholar]
- 29. Sisa M., Bonnet S. L., Ferreira D. and Van Der Westhuizen J. H., Molecules, 2010, 15, 5196–5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Montaña M. P., Massad W. A., Criado S., Biasutti A. and García N. A., Photochem. Photobiol., 2010, 86, 827–834. [DOI] [PubMed] [Google Scholar]
- 31. Montalti M. T. G. Marco, Credi Alberto and Prodi Luca, in: Handbook of Photochemistry, 2006. [Google Scholar]
- 32. Trout C. C., Tambach T. J. and Kubicki J. D., Spectrochim. Acta Part A, 2005, 61, 2622–2633. [DOI] [PubMed] [Google Scholar]
- 33. Ogilby P. R. and Foote C. S., J. Am. Chem. Soc., 1982, 104, 2069–2070. [Google Scholar]
- 34. De Boer J. W., Browne W. R., Brinksma J., Alsters P. L., Hage R. and Feringa B. L., Inorg. Chem., 2007, 46, 6353–6372. [DOI] [PubMed] [Google Scholar]
- 35. Colombini M. P., Andreotti A., Baraldi C., Degano I. and Łucejko J. J., Microchem. J., 2007, 85, 174–182. [Google Scholar]
- 36. Jovanovic S. V., Steenken S., Tosic M., Marjanovic B. and Simic M. G., J. Am. Chem. Soc., 1994, 116, 4846–4851. [Google Scholar]
- 37. Montaña P., Pappano N., Debattista N., Ávila V., Posadaz A., Bertolotti S. G. and García N. A., Can. J. Chem., 2003, 81, 909–914. [Google Scholar]
- 38. Agrawal P. K. and Schneider H.‐J., Tetrahedron Lett., 1983, 24, 177–180. [Google Scholar]
- 39. Matsuura T., Matsushima H. and Sakamoto H., J. Am. Chem. Soc., 1967, 89, 6370–6371. [DOI] [PubMed] [Google Scholar]
- 40. Matsuura T., Matsushima H. and Nakashima R., Tetrahedron, 1970, 26, 435–443. [Google Scholar]
- 41. Topalovic T., Nierstrasz V. A. and Warmoeskerken M. M. C. G., Fibers Polym., 2010, 11, 72–78. [Google Scholar]
- 42. Schäfer K. O., Bittl R., Zweygart W., Lendzian F., Haselhorst G., Weyhermüller T., Wieghardt K. and Lubitz W., J. Am. Chem. Soc., 1998, 120, 13104–13120. [Google Scholar]
- 43. Draksharapu A., Boersma A. J., Browne W. R. and Roelfes G., Dalton Trans., 2015, 44, 3656–3663. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information