

Abstract
This review focuses on recent developments in the physicochemical profiling of morphine and other opioids. The acid‐base properties and lipophilicity of these compounds is discussed at the microscopic, species‐specific level. Examples are provided where this type of information can reveal the mechanism of pharmacokinetic processes at the submolecular level. The role of lipophilicity in quantitative structure–activity relationship (QSAR) studies of opioids is reviewed. The physicochemical properties and pharmacology of the main metabolites of morphine are also discussed. Recent studies indicate that the active metabolite morphine‐6‐glucuronide (M6G) can contribute to the analgesic activity of systemically administered morphine. The unexpectedly high lipophilicity of M6G partly accounts for its analgesic activity. When administered parenterally, another suspected minor metabolite, morphine‐6‐sulfate (M6S) has superior antinociceptive effects to those of morphine. However, because sulfate esters of morphine derivatives cannot cross the blood‐brain barrier these esters may be good candidates to develop peripheral analgesic drugs.
Keywords: basicity, glucuronide conjugates, lipophilicity, physicochemical profiling, opioids
Who is in charge? In solutions, naloxone exists in four differently charged forms. Its overall lipophilicity profile indicated by the broad black line is the sum of the contributions of its four forms. Although the non‐charged form of naloxone is less lipophilic than naltrexone, its relative concentration at physiological pH is higher. This explains the observation that naloxone has a more rapid onset for antagonist activity and a shorter duration of action.

1. Acid‐Base Properties
The development of therapeutic agents that are able to reach and interact with target molecules in the central nervous system (CNS) is a challenging task, due to the highly sophisticated system evolved to protect the CNS from external xenobiotics. An important component of this system is the blood‐brain barrier (BBB) which has to be crossed by the agents acting in the CNS, including many opioid compounds.1, 2
Various physicochemical properties, including acid‐base properties and lipophilicity of therapeutically utilized opioid compounds account for many differences in their pharmacokinetic and pharmacodynamic behaviour.3, 4, 5
Acid‐base properties are among the most fundamental ones for drug action, as they quantify the charge of a species at a given pH, influencing thus other physicochemical properties, such as lipophilicity, solubility and permeability. Moreover, the site‐specific acid‐base properties for molecules with several protonation sites, characterize even the localization of charges within the molecule, which is especially important for passive membrane transport.5 Consideration of acid‐base properties together with other molecular properties is essential in drug discovery, as pharmacodynamically active drug candidate molecules with unfavourable pharmacokinetic properties cannot even reach the intended target molecule.6 Highly basic compounds are unsuitable for acting in the CNS as they are liable to be substrates of the active efflux transporter P‐glycoprotein (P‐gp, or MDR1).7 Acid‐base properties also play a decisive role in the formulation of drug molecules for both intravenous and oral dosage forms.8
In this review we regard protonation processes from the viewpoint of proton association and use protonation constants (K) or their logarithm (log K) to characterize them. log K data can help to predict pharmacokinetic properties, including absorption and distribution of potential drugs in the entire human gastrointestinal tract, where pH varies in the interval of 1.7 to 8.0.9
Morphine, and many of its derivatives, such as the opioid antagonist naloxone are amphoteric molecules, containing an acidic phenol and a basic amino site. In highly alkaline solutions naloxone has two basic sites, the non‐protonated phenolate site with a negative charge and the neutral tertiary amino group. In solutions naloxone exists in four microscopic protonation forms (microspecies), namely the anionic, zwitterionic, non‐charged and cationic ones. The protonation scheme of naloxone is depicted in Figure 1, showing the two different protonation isomers of the monoprotonated, neutral form as well. Figure 1 also shows the microscopic protonation constants (microconstants) that quantify the basicity of individual protonation sites, when the protonation states of all other sites are definite in the molecule.6 Indices N and O denote the nitrogen atom in the amino group and the oxygen atom in the phenolate group, respectively. Superscripts of microconstants indicate the group protonating in the given process, whereas a group already holding a proton is shown in the subscript. The concentration ratio of protonation isomers (kz, the tautomeric ratio) is independent of both the total concentration and the pH.
Figure 1.

The protonation scheme and equilibria of naloxone in terms of macroscopic (K 1, K 2) and microscopic (k N, k°, , ) protonation constants.
In the knowledge of the pH of the environment and the macro‐ and microconstants the pH‐dependent distribution of macro‐and microspecies can be calculated. Microspeciation has shown that it is not necessarily the major species that is reactive in biochemical processes.6, 10
Protonation constants can also be used to calculate the pH‐dependent charge of a species, which influences all other physicochemical properties. The localization of charges within a covalent skeleton dramatically influences the affinity of the drug molecule for target molecules, including various receptors and enzymes.
A decade ago we published a review about the physicochemical properties of morphine and some of its derivatives, where we tabulated all the literature data on their acid‐base properties, lipophilicity and solubility.4 However, at that time, the physicochemical properties of only morphine were characterized at the microscopic level by several research groups. In recent years we determined the protonation macro‐ and microconstants of several therapeutically important opioids,11, 12, 13 and these constants are collected in Table 1. The structure of the investigated compounds can be seen in Figure 2. These compounds include agonists codeine, oxymorphone, oxycodone, hydromorphone, hydrocodone, dihydromorphine, dihydrocodeine; and compounds used as antagonists, like naloxone and naltrexone. Nalbuphine and nalorphine are mixed agonists‐antagonists, they both have agonist activity at the kappa‐receptor and antagonist activity at the mu‐receptor.14, 15
Table 1.
Protonation macro‐and microconstants of morphine derivatives.
|
compound |
log K 1 |
log K 2 |
log k N |
log k° |
log k O N |
log k N° |
log k z |
|---|---|---|---|---|---|---|---|
|
morphine |
9.49 |
8.16 |
8.90 |
9.36 |
8.29 |
8.75 |
−0.46 |
|
codeine |
8.22 |
|
8.22 |
|
|
|
|
|
N‐methylmorphine |
8.75 |
|
|
8.75 |
|
|
|
|
nalorphine |
9.12 |
7.70 |
8.36 |
9.04 |
7.78 |
8.46 |
−0.68 |
|
N‐methylnalorphine |
8.46 |
|
|
8.46 |
|
|
|
|
dihydromorphine |
9.77 |
8.46 |
9.47 |
9.47 |
8.76 |
8.76 |
0.00 |
|
dihydrocodeine |
8.76 |
|
8.76 |
|
|
|
|
|
N‐methyldihydromorphine |
8.72 |
|
|
8.72 |
|
|
|
|
hydromorphone |
9.38 |
8.04 |
9.02 |
9.13 |
8.29 |
8.40 |
−0.11 |
|
hydrocodone |
8.18 |
|
8.18 |
|
|
|
|
|
N‐methylhydromorphone |
8.40 |
|
|
8.40 |
|
|
|
|
oxymorphone |
9.51 |
8.19 |
9.38 |
8.92 |
8.78 |
8.32 |
0.46 |
|
oxycodone |
8.78 |
|
8.78 |
|
|
|
|
|
N‐methyloxymorphone |
8.23 |
|
|
8.23 |
|
|
|
|
naloxone |
9.10 |
8.00 |
8.73 |
8.86 |
8.24 |
8.37 |
−0.13 |
|
O‐methylnaloxone |
8.29 |
|
8.29 |
|
|
|
|
|
N‐methylnaloxone |
8.37 |
|
|
8.37 |
|
|
|
|
naltrexone |
9.82 |
8.33 |
9.69 |
9.22 |
8.93 |
8.46 |
0.47 |
|
O‐methylnaltrexone |
8.93 |
|
8.93 |
|
|
|
|
|
nalbuphine |
10.28 |
8.64 |
10.20 |
9.52 |
9.40 |
8.72 |
0.68 |
|
O‐methylnalbuphine |
9.40 |
|
9.40 |
Figure 2.

The structure of some important semisynthetic opioid compounds.
The general structure‐activity relationships (SARs) of morphine and 4,5‐epoxymorphinans have recently been reviewed.16, 17 The pharmacophore consists of a weakly basic tertiary amino function protonated at physiologic pH, separated by a short carbon linker from an aromatic ring. The potency is enhanced when the C3 phenol group is intact, as this imitates the N‐terminal tyrosine of endogenous opioid peptides (enkephalins, endorphins). Alkylation of the C3 phenol group excludes the formation of hydrogen bonds with the complementary receptor moiety and generally results in lower‐affinity mu‐receptor ligands. Substituents on the amino group influence efficacy and potency, e. g. allyl or cyclopropylmethyl substituents produce antagonists.
Buprenorphine and etorphine are semi‐synthetic opioids derived from oripavine, the major metabolite of thebaine. Buprenorphine is about 30 times more potent than morphine, has partial agonist activity at mu‐opioid receptors, but it acts as a weak antagonist on kappa‐opioid receptors. Etorphine acts on both mu‐, delta‐ and kappa‐receptors, and is about 1000 times more potent than morphine.14, 15
The basicity of the amino and phenolate site is within an order of magnitude in each molecule. The non‐charged form is the dominant one for morphine, nalorphine, hydromorphone and naloxone, whereas the zwitterionic form has the higher concentration in solutions of oxymorphone, naltrexone and nalbuphine. The two protonation sites of dihydromorphine have practically the same basicity.
The differences in the basicities can be explained by field and inductive effects from moieties in the vicinity of the protonating group. The replacement of the N‐methyl group with an N‐allyl one lowers the log k N in case of the oxymorphone‐naloxone and morphine‐nalorphine pairs, but the antagonist substituent cyclopropylmethyl group shows the opposite effect for the oxymorphone‐naltrexone pair. The comparison of vs. k N values for the morphine vs. codeine, hydromorphone vs. hydrocodone and naloxone vs. O‐methylnaloxone pairs reveals that the alkylation of the C‐3 phenolic hydroxyl group has only a small effect (smaller than 0.11 log units) on the basicity of the tertiary nitrogen. The replacement of the C‐6 hydroxyl group by the powerful electron withdrawing keto group lowers the log k for the phenolate site at C‐3 (see the dihydromorphine‐hydromorphone pair). A bit surprisingly, introduction of a hydroxyl group at C‐14 increase the basicity of the nitrogen in the hydromorphone‐oxymorphone and nalorphine‐naloxone pairs. This increment in k N is probably caused by intramolecular hydrogen bonding between the hydroxyl and tertiary amino groups. In the morphine‐dihydromorphine pair saturation of the C7–C8 double bond increases the electron density and thus k N of the tertiary amino group.
The determination of microconstants makes the construction of microspecies distribution diagrams feasible, shown here for naloxone (Figure 3).
Figure 3.
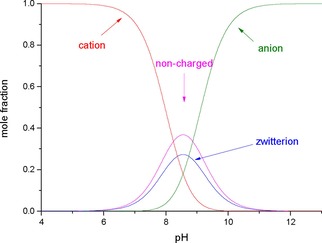
Distribution of naloxone microspecies, as a function of pH.
Figure 3 shows that in the pH range 8.24–8.86 naloxone mainly exists with mole fraction >0.336 in the non‐charged form (i. e. >33.6 %). In more acidic pH values, the cationic form predominates, its mole percent in the blood plasma (pH 7.40) is 79.6 %. At the isoelectric point (pH 8.55) the concentration of the zwitterionic form reaches its maximum, amounting to 27.3 %.
2. Lipophiliciy
Passive absorption through biological membranes, including the blood‐brain barrier is influenced by the lipophilicity and concentration of the various microscopic forms of the species.5, 18, 19 Lipophilicity is typically quantified by log P, the logarithm of the partition coefficient, the concentration ratio of a species in a given electrical state, in equilibrium between two immiscible solvents, namely water and an organic one, which is octanol in most cases. The analogous partition parameter for microspecies is designated with lower case p.5
Several papers were published to correlate analgesic activity of opioids with their lipophilic character. Kutter et al.20, 21 attempted to correlate the analgesic potency of 11 structurally diverse opioids with their lipophilic properties. Partition coefficients were determined between heptane and phosphate buffer pH 7.4, and the logarithm of partition coefficient (log P) was used. Analgesic activity was determined in rabbits by means of the tooth‐pulp test, opioids were administered intravenously and intraventricularly. For correlation analysis the logarithm of the reciprocal of ED50 was used. After intravenous administration the rank order of the reciprocal of ED50 was etorphine>fentanyl>levorphanol>methadone>dihydromorphinone>pethidine>dihydromorphine>morphine>normorphine, where the compounds are arranged in order of increasing effectiveness by the iv. route. Using intraventricular administration the rank order is somewhat different, etorphine>fentanyl>dihydromorphinone>morphine>dihydromorphine>normorphine>levorphanol>methadone>pethidine. For correlation studies the authors applied the activity quotient which is the ratio of intraventricular and intravenous activity (iventr./iv.). The correlation was significant between log P and the activity quotient. From the regression equations the linear function has a correlation coefficient of 0.832; however the parabolic function results in better correlation, with a 0.970 correlation coefficient. These results highlighted that lipophilic compounds can penetrate the blood‐brain barrier with more ease. Initial experiments sometimes used other lipophilic solvents, like refined olive oil.22 Jacobson et al.23, 24 found a quantitative relationship between receptor affinity and lipophilicity of ten structurally diverse opioid analgesics. Receptor binding affinity was expressed as IC50 values and lipophilicity was determined in heptane‐phosphate buffer pH 7.4. In the regression equation logarithm of the reciprocal value of the binding affinity (log 1/IC50) and log P were applied. The correlation coefficient was 0.91. McQuay et al.25 studied the relationship between analgesic potency and lipid solubility. The authors selected only four compounds (morphine, normorphine, pethidine and methadone) and they used the partition coefficients of Kutter et al. Analgesic activity was determined in an electrophysical model in rats, utilizing intrathecal administration of the studied compounds. A significant correlation was found between logED50 and the heptane‐buffer pH 7.4 partition coefficients, the correlation coefficient was 0.988. The relationship was inverse, the least lipid soluble compounds (morphine and normorphine) were the most potent and the highly lipid soluble methadone was considerably the least potent. These results can be criticized because the number of selected compounds was small to derive exact relationships.
In the last decades octanol became the organic solvent of choice in lipophilicity and quantitative structure‐activity relationship (QSAR) studies.2, 9 Dose‐response relationships of intrathecal opioids in rats could be established with the help of the octanol/water partition coefficient, where 14‐hydroxydihydromorphine, the one with the lowest octanol/pH 7.4 buffer distribution coefficient, had the lowest rate of uptake into the spinal cord.26 The lipophilicity of opioids was also determined by a radiolabelled ligand displacement micromethod from opioid receptors in brain membranes.27 Cyclic voltammetry at the interface of 1,2‐dichloroethane and water has been used to determine the ionic partition coefficients of the monocationic forms of several opioid compounds and correlated to metabolic pathways.28 A critical comparison of the most widespread lipophilicity determination methods, i. e. shake‐flask, potentiometric and chromatographic methods has been published last year.29
Recently the Conformationally Sampled Pharmacophore (CSP) method was applied to develop a consensus SAR of the efficacy of mu‐opioid receptor ligands.30
When differently charged forms of a species co‐exist, only the pH‐dependent distribution coefficient (D) can be measured directly, which is the sum of the pH‐independent species‐specific partition coefficients (p), weighted by their pH‐dependent mole fractions in the aqueous phase. The species‐specific partition coefficient of the non‐charged microspecies shows good correlation with the blood‐brain distribution coefficient.31 The lipophilicity profile (the variation of log D as a function of the aqueous pH) of a drug is essential in understanding its pharmacokinetic and pharmacodynamic properties.32 Optimal log D values for BBB uptake should be in the range of 1−4.33 log D and molecular weight are the most important factors in determining the permeability of drug candidates.34
The lipophilicity of ionizable molecules has been underrepresented in the literature until recently, due mainly to the lack of reliable methods to determine the partition coefficients of the ionic forms. This is especially true for the zwitterionic and non‐charged protonation isomers of amphoteric compounds. The lipophilicity of species in low concentrations can be determined with the deductive method, where they are mimicked by model compounds of the closest possible similarity. In a subsequent step, the typically minor effect of chemical modification is taken into account.5 In recent years we introduced a deductive method to determine the species‐specific partition coefficients of the protonation isomers of therapeutically important opioids. The results are collected in Table 2 alongside with partition coefficients of the cationic and anionic forms.11, 12, 13
Table 2.
The logarithm of microscopic partition coefficients of opioid compounds studied in the octanol/water system at 0.15 M ionic strength.
|
compound |
log p N°n |
log p Zwi |
log p Cat |
log p Ani |
|---|---|---|---|---|
|
morphine |
0.93 |
−2.10 |
−2.11 |
−2.01 |
|
codeine |
1.20 |
|
−1.90 |
|
|
N‐methylmorphine |
|
−2.40 |
−2.41 |
|
|
nalorphine |
1.87 |
−2.31 |
−1.73 |
−1.27 |
|
N‐methylnalorphine |
|
−2.76 |
−2.18 |
|
|
dihydromorphine |
1.13 |
−1.90 |
−2.29 |
−1.93 |
|
dihydrocodeine |
1.25 |
|
−2.11 |
|
|
hydromorphone |
1.16 |
−1.87 |
−1.82 |
−2.06 |
|
hydrocodone |
1.34 |
|
−1.69 |
|
|
oxymorphone |
1.22 |
−1.81 |
−2.24 |
−1.90 |
|
oxycodone |
2.03 |
|
−1.96 |
|
|
naloxone |
2.18 |
−0.85 |
−1.95 |
−1.09 |
|
O‐methylnaloxone |
2.38 |
|
−1.65 |
|
|
naltrexone |
2.24 |
−0.79 |
−1.76 |
−0.60 |
|
O‐methylnaltrexone |
2.63 |
|
−1.44 |
|
|
nalbuphine |
3.13 |
−1.05 |
−1.61 |
0.34 |
|
O‐methylnalbuphine |
3.44 |
|
−1.33 |
The comparison of log p values in Table 2 allows us to draw some interesting structure‐lipophilicity relationships. The substitution of the tertiary amino function with a longer side chain markedly increases the lipophilicity of these molecules in all possible protonation forms. This effect is proportional to the number of carbon atoms in the side chain, with nalbuphine showing the highest partition coefficient. The O‐methylation of the non‐charged or cationic forms increases their partition coefficient, typically with around 0.30 log units. The N‐methylation and concomitant permanent positive charge on morphine and nalorphine makes them less lipophilic than their parent compounds.
The partition coefficient for hydromorphone is almost twice that of morphine, which explains why hydromorphone is approximately 6–8 times more potent than morphine whereas the binding affinity is only 3 times greater.35
The onset and duration of narcotic agonist and antagonist activity have also been shown to be related to lipid solubility. Although the non‐charged form of naloxone is slightly less lipophilic than naltrexone, its relative concentration at physiological pH is 11.5 % as opposed to the 2.6 % of naltrexone.12 This fact explains the pharmacologic observation that naloxone has a more rapid onset for antagonist activity and a shorter duration of action.36, 37
All amphoteric opioids display an approximately parabolic lipophilicity‐pH profile, with a maximum at the isoelectric point, shown here for naloxone (Figure 4). The broad black line is the overall lipophilicity profile of the molecule, the sum of the contributions of its four microspecies.
Figure 4.

The lipophilicity profile of naloxone (broad black line), and the contribution of its four microspecies (thin lines) to the overall lipophilicity.
The contribution of the non‐charged form of naloxone is overwhelming in the pH range 4.12–12.12, but at lower or higher pH values the monoionic forms dominate the value of the distribution coefficient. The maximum of log D at the isoelectric point reaches 1.75. In the vicinity of the isoelectric point the contribution of the zwitterionic form to D exceeds those of the monoionic forms. Parallel lines on the graph show the pH‐independent contribution ratio of the non‐charged to the zwitterionic form, whose value is 1450 for naloxone.
The solvation parameter model developed by Abraham applied to the octanol/water partitioning system include Σα2 H and Σβ2 H, the effective or summation hydrogen bond acidity and hydrogen bond basicity of the solute.38 Based on that study the octanol/water partition coefficient is dominated by the hydrogen bond basicity and the size of the solute. In a subsequent study hydrogen bond descriptors for the neutral form of morphine were calculated and resulted in Σα2 H=0.42 and Σβ2 H=1.86. The low value of 0.42 for Σα2 H strongly indicates an intramolecular hydrogen bond between the phenolic OH group and the oxygen atom in the epoxide ring.31 The current procedures for the estimation of Σα2 H and Σβ2 H values have recently been reviewed.39 These two parameters, alongside log P oct were successfully used to predict the passive blood‐brain distribution coefficient.31
3. Metabolites of Morphine
Morphine, a substrate for organic cation transporter type 1 (OCT1),40 is almost completely absorbed from the gastrointestinal tract after oral administration. However, extensive hepatic first‐pass metabolism results in a mean bioavailability of only 20–30 %. Morphine is mainly metabolised by conjugation with glucuronic acid into morphine‐3‐glucuronide (M3G) and morphine‐6‐glucuronide (M6G). M3G is the main (45–55 %) morphine metabolite, while 10–15 % of morphine is converted into the analgesically active M6G.41, 42, 43, 44, 45
The differently protonated forms of M6G can be seen in Figure 5. This compound, just as its M3G isomer, exists predominantly in its zwitterionic form in physiological pH values. Figure 6 shows the distribution diagram of the macrospecies of M6G, as a function of pH.
Figure 5.

The protonation scheme of M6G.
Figure 6.

The distribution diagram of M6G macrospecies, as a function of pH.
Despite the fact that morphine glucuronides are highly polar metabolites, they appear to be able to cross the BBB, although they are expected to be markedly less permeable than morphine.46, 47, 48 The blood brain permeability of morphine and its glucuronides was compared under identical conditions using intravenous injection in rats following HPLC determination of the metabolites. The active metabolite M6G was found 32 times, whereas M3G 25 times less permeable than morphine.49 Reversed‐phase HPLC studies showed M6G and to a lesser extent M3G were far more lipophilic than expected.50 Force‐field and quantum mechanical calculations indicate that the two glucuronides exist in conformational equilibrium between folded and extended forms, and as a molecular chameleon can adapt their polarity to that of the medium.50, 51 The extended, polar conformer is dominant in water, whereas the folded conformer predominates in lipophilic environments. Avdeef et al. measured the partition coefficients of the variously charged species of M6G and M3G in the octanol/water system,3 the observed order of lipophilicities (log D, pH 7.4) was morphine (−0.07), M6G (−0.79) and M3G (−1.12).
Figure 7 shows the lipophilicity profile of M6G, alongside the contributions of its macrospecies.
Figure 7.

Contribution of the four macrospecies of M6G to the lipophilicity profile (broad black line).
Another research group proposed that two molecules of morphine‐6‐glucuronide could form a double ion pair, in which the carboxylate group of one molecule pairs with the protonated tertiary nitrogen of the other.
This would produce electronically neutral zwitterionic dimers, which could also passively diffuse across the BBB.52 It was also established that two 6‐acetylmorphine molecules can form a stable complex by two water molecules.53
Active transport can also play a part in the uptake of M6G from blood to brain, as this molecule is a substrate of organic anion transport protein 2 (OATP2)54 and glucose transporter GLUT‐1.55
The oral bioavailability of morphine is higher than that of M6G. However, whereas the low bioavailability of morphine is due to the extensive first‐pass metabolism after an almost complete absorption in the gastrointestinal tract, the low bioavailability of M6G is due to poor absorption from the gastrointestinal tract.44, 49 Compared to morphine, M6G has higher delta‐receptor binding affinity, but lower mu‐receptor affinity.56 M6G has a slower onset, but longer duration of action (>12 h) than morphine.57 M6G proved to be slightly more potent analgesic than morphine in mice after systemic administration, but M6G was even more effective when administered directly into the central nervous system. After intraventricular or intrathecal administration the potency of M6G was 60–650 times greater than that of morphine depending on the analgesic test.58, 59 Later on, it was recognized that M6G can contribute to the analgesic effects of morphine in humans.60, 61 The extent to which M6G contributes to the observed analgesic effects of oral morphine is controversial.44 A recent study indicates that irrespective of the route of administration of morphine to patients, the analgesic effect is mainly caused by M6G instead of morphine itself.62 Thus M6G is an attractive alternative to morphine in the treatment of severe postoperative pain.
A minor metabolite of morphine in humans is morphine‐6‐sulfate (M6S). When administered parenterally, the minor metabolite morphine‐6‐sulfate (M6S) has superior antinociceptive effects to those of morphine.63 However, because M6S is produced at very low concentration levels,64 it is unclear whether this metabolite contributes substantially to the observed pharmacologic effects of orally administered morphine.
Sulfate esters of opioids carry a negative charge at their sulfate group at any pH and compartment in the human body, even in the stomach. Since morphine derivatives possess a basic tertiary amino group, these compounds are zwitterionic at physiological pH values. Thus, these sulfate esters cannot cross the blood‐brain barrier, and consequently are good candidates to develop peripheral analgesic drugs.63, 65
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Károly Mazák received his degree in chemistry in 1997 from the Eötvös Loránd University, after which he joined the Department of Pharmaceutical Chemistry, Semmelweis University. Under the supervision of Prof. Béla Noszál he received his Ph. D. degree in 2005. He spent a year as Research Associate with Prof. C. K. Larive at the Chemistry Department of University of California, Riverside, USA. His research interests include microspeciation of biomolecules, physicochemical profiling of drug molecules and the chemistry of membrane penetration and receptor‐binding.

Biographical Information
Béla Noszál graduated as a pharmacist at the Semmelweis University in Budapest. He received his Ph. D. degree from the Eötvös Loránd University in 1975. He worked at the Department of Inorganic and Analytical Chemistry there until 1994, when he moved to the Semmelweis University to become Head of Department of Pharmaceutical Chemistry. He spent several years as Research Associate with Prof. R. B. Martin at the Chemistry Department of University of Virginia, USA and with Prof. D. L. Rabenstein at the Chemistry Department of University of California, Riverside, USA. He obtained his Doctor of Science degree from the Hungarian Academy of Sciences in 1993. He was Dean of the Faculty of Pharmacy from 2002 to 2013. His research interests include microspeciation of biomolecules, metal‐complexation of bioligands, biological chemistry of drug molecules, chirality and other isomerisms, NMR spectroscopy.

Biographical Information
Sándor Hosztafi graduated as a chemist at the Kossuth L. University in Debrecen. He received his Ph. D. degree from the Organic Chemistry Department of Kossuth L. University. He worked for the Alkaloida Chemical Company in Tiszavasvári from 1982 to 2004, then he moved to the Department of Pharmaceutical Chemistry of Semmelweis University, Budapest where he is presently a research fellow. His research interests include the structure‐activity relationship studies of morphine derivatives, N‐demethylation reactions in the field of morphine alkaloids, and the study of the Diels‐Alder reactions of morphinan dienes.

Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the National Research Fund of Hungary, OTKA T 73804.
K. Mazák, B. Noszál, S. Hosztafi, ChemistryOpen 2019, 8, 879.
Contributor Information
Prof. Károly Mazák, Email: mazak.karoly@pharma.semmelweis-univ.hu, FAX: +36‐1‐2170891.
Prof. Béla Noszál, Email: noszal.bela@pharma.semmelweis-univ.hu.
Sándor Hosztafi, Email: hosztafi.sandor@pharma.semmelweis-univ.hu.
References
- 1. Liu X., Smith B. J., Chen C., Callegari E., Becker S. L., Chen X., Cianfrogna J., Doran A. C., Doran S. D., Gibbs J. P., Hosea N., Liu J., Nelson F., Szewc M. A., Deusen J. V., J. Pharmacol. Exp. Ther. 2005, 313, 1254–1262. [DOI] [PubMed] [Google Scholar]
- 2. Rankovic Z., J. Med. Chem. 2015, 58, 2584–2608. [DOI] [PubMed] [Google Scholar]
- 3. Avdeef A., Barrett D. A., Shaw P. N., . Knaggs R. D., Davis S. S., J. Med. Chem. 1996, 39, 4377–4381. [DOI] [PubMed] [Google Scholar]
- 4. Mazák K., Hosztafi S., Rácz Á., Noszál B., Mini-Rev. Med. Chem. 2009, 9, 984–995. [DOI] [PubMed] [Google Scholar]
- 5. Mazák K., Noszál B., Eur. J. Pharm. Sci. 2014, 62, 96–104. [DOI] [PubMed] [Google Scholar]
- 6. Mazák K., Noszál B., J. Pharm. Biomed. Anal. 2016, 130, 390–403. [DOI] [PubMed] [Google Scholar]
- 7. Wager T. T., Chandrasekaran R. Y., Hou X., Troutman M. D., Verhoest P. R., Villalobos A., Will Y., ACS Chem. Neurosci. 2010, 1, 420–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Manallack D. T., Prankerd R. J., Yuriev E., Oprea T. I., Chalmers D. K., Chem. Soc. Rev. 2013, 42, 485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.A. Avdeef in Absorption and Drug Development: Solubility, Permeability, and Charge State, 2nd ed.; Wiley & Sons: Hoboken 2012.
- 10. Noszál B., Rabenstein D. L., J. Phys. Chem. 1991, 95, 4761–4765. [Google Scholar]
- 11. Mazák K., Noszál B., Eur. J. Pharm. Sci. 2012, 45, 205–210. [DOI] [PubMed] [Google Scholar]
- 12. Mazák K., Hosztafi S., Noszál B., Eur. J. Pharm. Sci. 2015, 78, 1–7. [DOI] [PubMed] [Google Scholar]
- 13. Mazák K., Hosztafi S., Kraszni M., Noszál B., J. Pharm. Biomed. Anal. 2017, 135, 97–105. [DOI] [PubMed] [Google Scholar]
- 14. Yaksh T. L., Wallace M. S., Goodman & Gilman's The Pharmaceutical Basis of Therapeutics (Eds.: L. L. Brunton, B. A. Chabner, B. C. Knollmann), McGraw-Hill Medical 2010, pp. 481–527. [Google Scholar]
- 15. Friel C. J., Lu M. C., Wilson and Gisvold's Textbook of Organic Medicinal and Pharmaceutical Chemistry (Eds.: J. M. Beale, J. H. Block), Lippincott Williams & Wilkins, Baltimore 2011, pp. 776–818. [Google Scholar]
- 16. Fürst S., Hosztafi S., Acta Physiol. Hung. 2008, 95, 3–44. [DOI] [PubMed] [Google Scholar]
- 17. Devereaux A. L., Mercer S. L., Cunningham C. W., ACS Chem. Neurosci. 2018, 9, 2395–2407. [DOI] [PubMed] [Google Scholar]
- 18. Mazák K., Noszál B., Hosztafi S., Curr. Med. Chem. 2017, 24, 3633–3648. [DOI] [PubMed] [Google Scholar]
- 19. Avdeef A., Testa B., Cell. Mol. Life Sci. 2002, 59, 1681–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kutter E., Herz A., Teschemacher H. J., Hess R., J. Med. Chem. 1970, 13, 801–805.5458362 [Google Scholar]
- 21. Herz A., Teschemacher H. J., Advances in Drug Research, vol. 6. (Eds.: N. J. Harper, A. B. Simmonds), Academic Press, London and New York 1971, pp. 79–119. [Google Scholar]
- 22. Oldendorf W. H., Proc. Soc. Exp. Biol. Med. 1974, 147, 813–5. [DOI] [PubMed] [Google Scholar]
- 23. Jacobson A. E., Klee W. A., Dunn W. J., Eur. J. Med. Chem. 1977, 12, 49–52. [Google Scholar]
- 24. Jacobson A. E., NIDA Res. Monogr. 1978, 22, 129–45. [PubMed] [Google Scholar]
- 25. McQuay H. J., Sullivan A. F., Smallman K., Dickenson A. H., Pain 1989, 36, 111–115. [DOI] [PubMed] [Google Scholar]
- 26. Plummer J. L., Cmielewski P. L., Reynolds G. D., Gourlay G. K., Cherry D. A., Pain 1990, 40, 339–47. [DOI] [PubMed] [Google Scholar]
- 27. Medzihradsky F., Emmerson P. J., Mousigian C. A., J. Pharmacol. Toxicol. Methods 1992, 27, 67–69. [DOI] [PubMed] [Google Scholar]
- 28. Gulaboski R., Cordeiro M. N., Milhazes N., Garrido J., Borges F., Jorge M., Pereira C. M., Bogeski I., Morales A. H., Naumoski B., Silva A. F., Anal. Biochem. 2007, 361, 236–243. [DOI] [PubMed] [Google Scholar]
- 29. Port A., Bordas M., Enrech R., Pascual R., Rosés M., Ràfols C., Subirats X., Bosch E., Eur. J. Pharm. Sci. 2018. , 122, 331–340. [DOI] [PubMed] [Google Scholar]
- 30. Shim J., Coop A., MacKerell A. D., J. Phys. Chem. B. 2011, 115, 7487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abraham M. H., Takács-Novák K., Mitchell R. C., J. Pharm. Sci. 1997, 86, 310–315. [DOI] [PubMed] [Google Scholar]
- 32. Pagliara A., Carrupt P. A., Caron G., Gaillard P., Testa B., Chem. Rev. 1997, 97, 3385–3400. [DOI] [PubMed] [Google Scholar]
- 33. Van de Waterbeemd H., Camenisch G., Folkers G., Chretien J. R., Raevsky O. A., J. Drug Targeting 1998, 6, 151–165. [DOI] [PubMed] [Google Scholar]
- 34. Waring M. J., Bioorg. Med. Chem. Lett. 2009, 19, 2844–51. [DOI] [PubMed] [Google Scholar]
- 35. Inturrisi C. E., Clin. J. Pain. 2002, 18, S3-13. [DOI] [PubMed] [Google Scholar]
- 36. Fishman J., Hahn E. F., Norton B. I., Life Sci. 1975, 17, 1119–1126. [DOI] [PubMed] [Google Scholar]
- 37. Kaufman J. J., Semo N. M., Koski W. S., J. Med. Chem. 1975, 18, 647–655. [DOI] [PubMed] [Google Scholar]
- 38. Abraham M. H., Chadha H. S., Whiting G. S., Mitchell R. C., J. Pharm. Sci. 1994, 83, 1085–1100. [DOI] [PubMed] [Google Scholar]
- 39. Pallicer J. M., Pascual R., Port A., Rosés M., Rafols C., Bosch E ., Eur. J. Pharm. Sci. 2013, 48, 484–493. [DOI] [PubMed] [Google Scholar]
- 40. Tzvetkov M. V., dos Santos Pereira J. N., Meineke I., Saadatmand A. R., Stingl J. C., Brockmoller J., Biochem. Pharmacol. 2013, 86, 666–678. [DOI] [PubMed] [Google Scholar]
- 41. Aasmundstad T. A., Mørland J., Paulsen R. E., J. Pharmacol. Exp. Ther. 1995, 275, 435–41. [PubMed] [Google Scholar]
- 42. Van Dorp E. L. A., Romberg R., Sarton E., Bovill J. G., Dahan A., Anesth. Analg. 2006, 102, 178–197. [DOI] [PubMed] [Google Scholar]
- 43. Dahan A., van Dorp E., Smith T., Yassen A., Eur. J. Pain. 2008, 12, 403–411. [DOI] [PubMed] [Google Scholar]
- 44. De Gregori S., De Gregori M., Ranzani G. N., Allegri M., Minella C., Regazzi M., Metab. Brain Dis. 2012, 27, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sverrisdóttir E., Lund T. M., Olesen A. E., Drewes A. M., Christrup L. L., Kreilgaard M., Eur. J. Pharm. Sci. 2015, 74, 45–62. [DOI] [PubMed] [Google Scholar]
- 46. Murphey L. J., Olsen G. D., J. Pharmacol. Exp. Ther. 1994, 271, 118–124. [PubMed] [Google Scholar]
- 47. Milne R. W., Nation R. L., Somogyi A. A., Drug Metab. Rev. 1996, 28, 345–472. [DOI] [PubMed] [Google Scholar]
- 48. Wu D., Kang Y. S., Bickel U., Pardridge W. M., Drug Metab. Dispos. 1997, 25, 768–771. [PubMed] [Google Scholar]
- 49. Bickel U., Schumacher O. P., Kang Y. S., Voigt K., J. Pharmacol. Exp. Ther. 1996, 278, 107–13. [PubMed] [Google Scholar]
- 50. Carrupt P. A., Testa B., Bechalany A., El Tayar N., Descas P., Perrissoud D., J. Med. Chem. 1991, 34, 1272–1275. [DOI] [PubMed] [Google Scholar]
- 51. Gaillard P., Carrupt P. A., Testa B., Bioorg. Med. Chem. Lett. 1994, 4, 737–742. [Google Scholar]
- 52. Preechagoon D., Smith M. T., Prankerd R., Int. J. Pharm. 1998, 163, 191–201. [Google Scholar]
- 53. Schwarzinger S., Hartmann M., Kremminger P., Muller N., Bioorg. Med. Chem. Lett. 2001, 11, 1455–1459. [DOI] [PubMed] [Google Scholar]
- 54. Bourasset F., Cisternino S., Temsamani J., Scherrmann J. M., J. Neurochem. 2003, 86, 1564–1567. [DOI] [PubMed] [Google Scholar]
- 55. Dagenais C., Rousselle C., Pollack G. M., Scherrmann J. M., J. Cereb. Blood Flow Metab. 2000, 20, 381–386. [DOI] [PubMed] [Google Scholar]
- 56. Osborne P. B., Chieng B., Christie M. J., Br. J. Pharmacol. 2000, 131, 1422–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yoshimura H., Ida S., Oguri K., Tsukamoto H., Biochem. Pharmacol. 1973, 22, 1423–1430. [DOI] [PubMed] [Google Scholar]
- 58. Paul D., Standifer K. M., Inturrisi C. E., Pasternak G. W., J. Pharmacol. Exp. Ther. 1989, 251, 477–83. [PubMed] [Google Scholar]
- 59. Frances B., Gout R., Monsarrat B., Cros J., Zajac J. M., J. Pharmacol. Exp. Ther. 1992, 262, 25–31. [PubMed] [Google Scholar]
- 60. Lötsch J., Geisslinger G., Clin. Pharmacokinet. 2001, 40, 485–499. [DOI] [PubMed] [Google Scholar]
- 61. Lötsch J., Curr. Opinion in Anaesthesiology 2004, 17, 449–453. [DOI] [PubMed] [Google Scholar]
- 62. Klimas R., Mikus G., Br. J. Anaesth. 2014, 113, 935–944. [DOI] [PubMed] [Google Scholar]
- 63. Yadlapalli J. S. K., Ford B. M., Ketkar A., Wan A., Penthala N. R., Eoff R. L., Prather P. L., Dobretsov M., Crooks P. A., Pharm. Res. 2016, 113, 335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Andersson M., Bjorkhem-Bergman L., Ekstrom L., Bergqvist L., Lagercrantz H., Rane A., Beck O., Pharmacol. Res. Perspect. 2014, 2, e00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lackó E., Riba P., Giricz Z., Váradi A., Cornic L., Balogh M., Király K., Csekő K., Mousa S. A., Hosztafi S., Schäfer M., Zádori Z. S., Helyes Zs., Ferdinandy P., Fürst S., Al-Khrasani M., J. Pharmacol. Exp. Ther. 2016, 359, 171–181. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary