

Abstract

Catecholaminergic polymorphic ventricular tachycardia (CPVT) is caused by mutations of cardiac calsequestrin (CASQ2) that impair its characteristic ability of Ca2+-induced polymerization–depolymerization. However, stabilizing the CASQ2 polymer by pharmacological agents to treat CPVT has not been reported so far. Here, we tested whether small molecules can stabilize CASQ2 polymers. We synthesized 24 glycinate/alaninate/acetate α-pyranone analogs and conducted the CASQ2 depolymerization assay. Most of the molecules of this class of compounds inhibited the depolymerization of the protein upon Ca2+ chelation by ethylene glycol tetraacetic acid. Structure–activity relationship studies revealed that the compounds with the 4-fluoro-phenyl group at the C-6 position of the pyranone ring and open-chain primary amine at C-4 are the most active of the class. This is the first report of an α-pyranone class of compounds with the ability to stabilize CASQ2 polymers and opens up the possibility to target Ca2+-release disorders via modulation of CASQ2 polymerization.
1. Introduction
Defects in Ca2+ release by the cardiac ryanodine receptor (RyR2) is associated with many heart diseases including catecholaminergic polymorphic ventricular tachycardia (CPVT), catecholaminergic idiopathic ventricular fibrillation, malignant hyperthermia, and central core disease.1−3 The first-line medications for most cardiac diseases include β-blockers or calcium channel blockers.4 However, these medications have serious limitations including proarrhythmic potential and drug toxicity. The newer targets for anti-arrhythmic treatment include acting upon intracellular Ca2+ handling in the cardiac myocyte, interference with the Ca2+/calmodulin-dependent protein kinase II, the Na+1/Ca2+ exchanger, RyR, and the late component of Na current (INa-Late).5 Ca2+ release from RyR2 is partly regulated by cardiac calsequestrin (CASQ2), the major Ca2+-buffering protein in the sarcoplasmic reticulum (SR).6 CASQ2 binds Ca2+ with high capacity and low affinity,7,8 which allows quick dissociation of Ca2+ for release via RyR2, thereby facilitating repetitive muscle contractions.9,10 CASQ2 interaction with RyR2 is complex, and different (both direct and indirect) mechanisms were proposed.6,11 Györke and Györke suggested that the action of the RyR is directed by CASQ, which can sense changes in luminal Ca2+ concentration in the range of 0.2–20 mM. Alternatively, CASQ2 might serve only as a fast source of Ca2+ ions near the RyR2 channel. In this case, CASQ2 would regulate the Ca2+ current flowing via the RyR2 channel. They further explained that it might be important to regulate Ca2+ release under normal and pathological conditions.12 Moreover, Terentyev et al. indicated that CASQ2 controls the magnitude and duration of Ca2+ release23 and thereby affects the luminal Ca2+-dependent RyR2. Approximately 15 mutations in CASQ2 were reported to cause a unique type of arrhythmia called CPVT. These mutations (L167H, D307H, P308L, and R33Q) affect Ca2+-induced CASQ2 polymerization that impairs RyR2-mediated Ca2+ release in the subsequent contraction–relaxation cycle leading to sudden cardiac death.13−21 Further, ablation of CASQ2 is known to cause CPVT in a mouse model.22 We therefore propose that targeting modulation of CASQ2 polymerization by a small molecule could be an attractive drug target for treating CPVT and possibly other forms of arrhythmia.23
Different classes of small molecules with varied biological significance were shown to bind to CASQ2 based on docking, X-ray crystallography, and biological studies.24−26 Some of the molecules bind to CASQ2 and cause destabilization of the polymeric state,24 while others induce polymerization.26 Anthracyclines (doxorubicin, doxorubicinol, daunorubicin, daunorubicinol) and phenothiazines (promethazine, thioridazine, chlorpromazine) are reported to have affinity for CASQ2, affect Ca2+-induced CASQ2 polymerization, and alter SR-mediated Ca2+ release.27,28 However, anthracyclines are known to cause cardiotoxicity.29 It was proven that addition of doxorubicin triggers Ca2+ release from cardiac SR vesicles30−32 Pessah et al. reported that the anthracyclines enhanced [3H]ryanodine binding to rat SR vesicles enriched in RyR2 channels. They further suggested that, with the interaction of anthraquinones, there is a receptor-mediated shift in the redox equilibrium of allosteric thiols in RyR.33 Anthracycline–RyR2 interaction is mediated by the presence of quinone and a side-chain carbonyl, which interacts with the cysteine residues in the cytosolic RyR2 face.34 Further, human carbonyl reductase induces one-electron reduction of quinone and two-electron reduction of side-chain carbonyl, and these are accompanied by iron and free radical reactions.35 The effect of the anthracycline metabolite is even more pronounced than the anthracycline itself. It decreases the extent of CASQ2 linked with RyR2, and it also interacts directly with CASQ2 to induce structural changes due to lower Ca2+ binding capacity.34 We aimed to design a molecule without a quinone moiety with the ability to interact with CASQ2 that does not induce oxidative stress forming metabolites, which could lead to toxicity. The ester/amide group requires harsh conditions for its reduction in comparison to aldehyde and ketone and hence is less likely to produce alcohol metabolites to interfere with the thiol residues of RyR2.
Herein, we report synthesis of derivatives based on a new scaffold, namely, 4,6-disubstituted-2H-pyranone-3-carbonyl-glycinate/alaninate/acetate, earlier unknown to interfere with CASQ2 polymerization. We hypothesize that molecules belonging to this novel class may stabilize the CASQ2 polymer in an arrhythmic condition.
2. Results and Discussion
2.1. Synthesis
The 6-aryl-4-methylthio-2H-pyranone-3-carboxylic acids (1a–e) were synthesized from various substituted acetophenones or 2-acetyl furan (Scheme 1) according to our reported procedure from ketene dithioacetals and diethyl malonate (DEM).36,37 Ethyl 6-aryl-4-(methylthio)-2-oxo-2H-pyran-3-carboxylate (1f) was formed as a minor product during the synthesis of 1b. Generally, in the synthesis, deprotonated DEM forms a new C–C bond with a ketene dithioacetal upon the elimination of the SMe– group and subsequent deprotonation followed by the release of the ethoxide anion leading to the formation of the ethyl ester derivative of pyranone carboxylic acid, which upon hydrolysis affords the corresponding 3-carboxylic acid derivatives (1a–e) with 36–54% yields (see Figure S1). Glycine/alanine ester was coupled with 1a–e to generate 6-aryl-4-methylthio-2H-pyranone-3-carbonyl glycinate/alaninate key intermediates (2a–e, details in the Supporting Information) in the presence of hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU) as the coupling agent and N,N-diisopropylethylamine (DIPEA) as the base with 85–91% yields. The reaction when performed using N,N′-dicyclohexylcarbodiimide (DCC) as a coupling reagent resulted in lower yields along with additional purification steps. The final 4,6-disubstituted-2H-pyranone-3-carbonyl-glycinates/alaninates/acetates (3a–x, Figure 1) were obtained by nucleophilic substitution of 2a–e/1f with appropriate amine in ethanol. All synthesized compounds were characterized (data in the Supporting Information) by melting point, IR, UV, MS, and 1H/13C NMR. In addition, one of the final compounds (3b) was analyzed by single-crystal X-ray diffraction. ORTEP diagram (Oak Ridge thermal ellipsoid plot) is presented in Figure 2. CCDC-150756 (The Cambridge Crystallographic Data Centre) contains the supplementary crystallographic data for this paper.
Scheme 1. Synthesis of 4,6-Disubstituted-2H-pyranone-3-carbonyl-glycinate/alaninate/acetate (3a–x).

Reagents and conditions: (i) glycine/alanine ester, HATU, DIPEA, DCM, rt, 3–4 h; (ii) aliphatic amine, ethanol, reflux, 2–6 h.
Figure 1.

Structures of synthesized pyranone derivatives.
Figure 2.
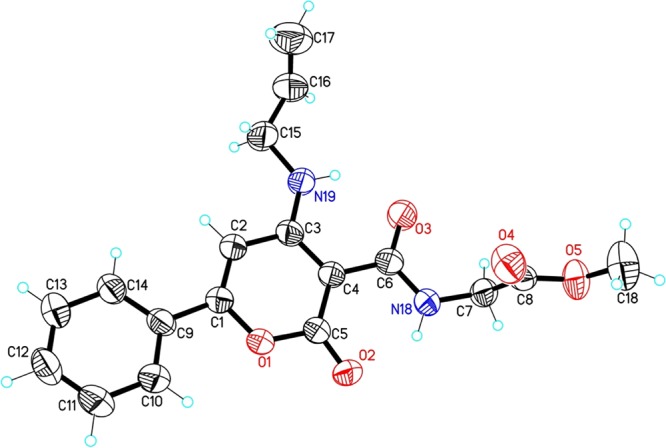
ORTEP diagram drawn with 50% thermal ellipsoidal probability with atom numbering scheme of compound 3b (CCDC No: 150756).
2.2. Biological Evaluation
A Ca2+-induced aggregation of CASQ2 followed by the ethylene glycol tetraacetic acid (EGTA)-mediated resolubilization/depolymerization assay20 was used for evaluating the ability of 3a–x to modulate the polymeric state of CASQ2 (Figure 3). EGTA has much higher affinity for Ca2+ than CASQ2. Therefore, Ca2+ chelation with EGTA compels CASQ2 polymers to release a portion of the bound Ca2+ with or without depolymerization. The concentrations of EGTA to resolubilize 50% of CASQ2 protein from aggregation (i.e., EC50 of EGTA-mediated resolubilization) in the absence and presence of any one of 3a–x at a time in a specified concentration were measured. Compounds at concentration 1 μM or less necessitating higher concentration of EGTA for resolubilization of CASQ2 are considered to inhibit its depolymerization (Figure 3). Percentage inhibition of CASQ2 depolymerization by a compound is the ratio of the percentage of CASQ2 left as polymer in its presence to the percentage of CASQ2 left as polymer in its absence at a particular EGTA concentration.
Figure 3.

Colored heatmap visualizing the results of EGTA-mediated resolubilization/depolymerization assay. Concentration values are scaled by row. Compound code is indicated in the x axis. Compounds 3e, 3u, and 3v have the highest potentials (>50% at 100 nM) to inhibit depolymerization of CASQ2.
2.3. Molecular Modeling
Blind docking of the most active molecules 3e, 3u, and 3v to the two dimers indicated the clear preference for the front-to-front dimer. Over 250 poses grouped into nearly 33 clusters were generated for each structure docked into both dimers. The top six clusters generated from docking into the front-to-front dimer suggested its binding into the interface of the two monomers (Figure 4). Meanwhile, we could not identify a specific preference for any binding site in the back-to-back dimer. We therefore examined the binding site at the interface of the front-to-front dimer in details. The CASQ2 monomer is composed of three thioredoxin folds connected by loops. The binding pocket is formed between the residues of the two monomers. The molecules were found to interact with the N-terminal, the first thioredoxin fold of the first monomer, and the third thioredoxin fold of the second monomer (Figure 4). The aliphatic amine forms a H-bonding interaction with His67 of one monomer, and Ar–F is involved in electrostatic interactions with Leu294 of the second monomer (Figure 4). It is further stabilized by the hydrophobic interactions with several residues including Ala297, Lys301, and Tyr298 of the first and Leu65, Val64, and Glu66 of the second monomer, respectively. The ester/amide group is projected outward from the protein without actually participating in the binding. The binding of the molecules provides additional arms to hold the monomers together and prevent the Ca2+ leak in an arrhythmic condition.
Figure 4.

Compound 3u is bound at the interface of CASQ2 dimer (1SJI). (A) Electrostatic surface image of the binding site. The ester group is projected out of the protein surface. (B) Binding mode of 3u in CASQ2 dimer. The amine group forms H-bonding interaction with the first monomer, and Ph–F is involved in electrostatic interaction with the second monomer. The molecule forms several hydrophobic interactions with Ala297, Lys301, and Tyr298 of the first and Leu65, Val64, and Glu66 of the second monomer holding the two monomers together. The model was created using Maestro. The ligand is represented using ball and stick, and the interacting residues are represented in line notation. The protein structure is represented in ribbon.
Fifteen molecules out of 3a–x (Figure 3) were able to inhibit depolymerization of CASQ2 at a concentration below 10 μM and considered active. Among them, molecules with an ability to inhibit more than 50% CASQ2 depolymerization at 100 nM were considered highly active. Compounds 3a–c with an unsubstituted phenyl ring at the C-6 position were found to be inactive. It was found that the 4-F-phenyl at the C-6 position enhances activity when the C-4 position contains a primary amine with acyclic alkyl group. 3e, 3u, and 3v (Figure 3) were able to inhibit more than 50% CASQ2 depolymerization at 100 nM, and 3m inhibited more than 50% CASQ2 depolymerization at 250 nM. When we replaced 4-F-phenyl with furan-2-yl, 4-Cl-phenyl, 4-Br-phenyl, or the primary amine at the C-4 position, we observed a considerable decrease in activity in the resulting compounds (Figure 3). Among the 24 molecules, only those molecules with open-chain amine at C-4 and 4-F-phenyl at the C-6 showed the high inhibitory effect (Figure 3). The structure–activity relationship (SAR) of the synthesized pyranone analogs is summarized in Figure 5.
Figure 5.
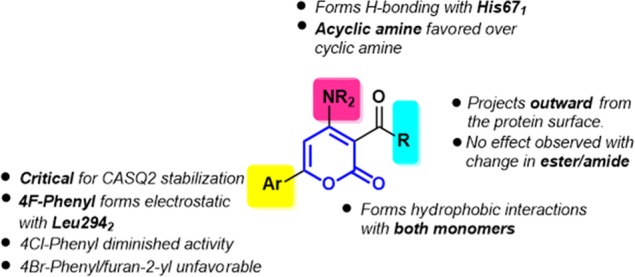
Structure–activity relationship of synthesized pyranone derivatives.
3. Conclusions
In conclusion, we have discovered the pyranone analogs as promising new chemotypes with the ability to stabilize the CASQ2 polymer. Twenty-four glycinate/alaninate/acetate pyranone analogs were synthesized and evaluated for CASQ2 polymer stabilization potential. SAR studies revealed that the 4-F-phenyl at the 6th position of the pyranone ring and open-chain primary amine at the 4th position were the most active of the class. Docking studies suggest that the molecules interact at the interface of the front-to-front dimer, which is the first key step in CASQ2 polymerization and is determinant of its rate. The present work opens an avenue for further research toward targeting CASQ2 polymerization to treat CPVT and other forms of arrhythmia by describing a new class of molecules and revealing the potential drug binding pocket in the CASQ2 that stabilizes its polymer.
4. Experimental Section
4.1. Chemistry
General: All the reactions were carried out in oven-dried glassware. The chemicals and solvents were purchased from Sigma-Aldrich, Spectrochem, or Acros and were used without any purification except THF (tetrahydrofuran), which was dried by refluxing over sodium followed by distillation. Reactions were monitored by TLC, which are precoated by silica gel (Kieselgel 60F 254, Merck), and the spots were detected under UV light (254 nm). Compounds were purified by column chromatography using silica gel (particle size 100–200 mesh). 1H and 13C NMR spectra were characterized in CDCl3 or DMSO-d6 solution by using (Wormhole-vnmrs/Bruker/Jeol-Delta/Varian) 400/300 MHz spectrophotometers. Chemical shifts are reported as ppm (δ) relative to TMS (δ 0.0) as the internal standard. 1H NMR data is recorded as follows: chemical shift [multiplicity, coupling constant(s) J (Hz), relative integral] where multiplicity is defined as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and bs (broad singlet). Mass spectra were recorded on a 6430 Triple quardrupole mass spectrometer for ESI and are given in m/z. FTIR spectra were recorded on a PerkinElmer 1600 series FTIR spectrophotometer. UV spectra were recorded on a PerkinElmer Lambda 25 UV–visible spectrophotometer (USA).
4.1.1. Synthesis of the Amides 2a–e
General Procedure: To a solution of appropriate 1a–e (2.02 mmol) in 10 mL of dichloromethane (DCM) at room temperature (rt, 15–35 °C), HATU (2.05 mmol) was added and stirred for 10 min. DIPEA (7.6 mmol) was added and stirred for another 15 min. Thereafter, glycine/alanine ester (2.05 mmol) was added and stirred for a further 2 h at rt. The reaction was monitored by TLC for completion and then was washed with water and brine subsequently. DCM was evaporated, and the crude product was purified by column chromatography by eluting with an ethyl acetate/hexane mixture to obtain 2a–e with >80% yields.
4.1.1.1. Methyl (4-(Methylthio)-2-oxo-6-phenyl-2H-pyran-3-carbonyl)glycinate (2a)
Yellow crystalline powder; yield 88%; mp: 175–176 °C; IR (KBr) (ν, cm–1) 3310 (N–H), 1744, 1686, and 1639 (C=O); UV (MeOH) λmax = 252.7, 336.8 nm; MS-ESI (m/z): [M+] = 333.9; 1H NMR (400 MHz, DMSO-d6) δ 2.60 (s, 3H, SMe), 3.66 (s, 3H, OCH3), 4.06–4.07 (d, J = 6.0 Hz, 2H, NCH2), 7.18 (s, 1H, pyranone-H), 7.59–7.62 (m, 3H, ArH), 8.02–8.05 (m, 2H, ArH), 9.23–9.26 (t, J = 6.0 Hz, 1H, NH amide); elemental analysis: C: 57.73, H: 4.62, N: 4.13, S: 9.43.
4.1.2. Synthesis of the Final Compounds 3a–x
To a solution of appropriate 2a–e/1f (1.5 mmol) in ethanol, corresponding amine (RNH2, 2.2 mmol) was added and refluxed for 2 h. The completion of the reaction was monitored by TLC. Ethanol was removed under reduced pressure (<100 mbar). The crude was dissolved in DCM and washed with water and brine, and the organic layer was concentrated under reduced pressure (<800 mbar). The crude was purified using column chromatography by eluting with a mixture of hexane and ethyl acetate to get 3a–x in 40–65% yields.
4.1.2.1. Methyl (6-(4-Bromophenyl)-4-(cyclopropylamino)-2-oxo-2H-pyran-3-carbonyl)glycinate (3p)
White powder; yield 65%; mp: 180–183 °C; IR (KBr) (ν, cm–1): 1759, 1682, 1639 (C=O stretching), 3333 (N–H stretching), 3078 (C–H); UV (MeOH) λmax = 227.9, 248.3, 311.7 nm; MS-ESI (m/z): [M+ + 1] 422.8; 1H NMR (400 MHz, CDCl3) δ 0.74–0.77 (m, 2H, CH2), 0.93–0.98 (m, 2H, CH2), 2.67–2.69 (m, 1H, CH), 3.77 (s, 3H, OCH3), 4.10–4.12 (d, J = 5.6 Hz, 2H, CH2), 6.91 (s, 1H, pyranone-H), 7.61–7.63 (m, 2H, ArH), 7.73–7.75 (m, 2H, ArH), 9.69–9.71 (t, J = 5.6 Hz, 1H, CONH), 11.06 (bs, 1H, NH); 13C NMR (75 MHz, CDCl3) 7.73, 24.54, 41.01, 52.29, 86.52, 94.11, 126.35, 127.66, 129.86, 132.28, 159.45, 162.74, 163.42, 169.22, 170.40.
4.2. Depolymerization Assay
The rat wild-type (WT) CASQ2 was cloned into a pET21a vector (Novagen) between Nde I and Xho I restriction sites. The protein was expressed in bacterial cells and purified using an earlier published method.18 To a 2.5 μM CASQ2 solution in 50 mM Tris-HCl buffer (pH 7.5) containing 20 mM NaCl at 25 ± 2 °C, 3a was added at specified concentration. To this solution, 10 mM CaCl2 was added to polymerize the protein. The solution was left to stand for 1 h, thereafter it was mixed by pipetting and aliquot of 500 μL were pipetted into new test tubes. One of these test tubes was centrifuged at 20000g for 1 h, and absorbance was recorded at 350 nm. The soluble fraction was estimated for protein concentration by the Bradford reagent (Bio-Rad) following the manufacturer’s protocol. This was performed to confirm that more than 75% of proteins were polymerized. To the polymerized solution aliquots in several test tubes, 2–10 μL of EGTA (0.1–0.5 M) was added and allowed to equilibrate for 30 min. Then absorbance was recorded at 350 nm, and the percentage of protein still polymerized in different tubes was calculated as above. The concentration of EGTA to resolubilize 50% of CASQ2 protein from precipitation (i.e., EC50 of EGTA-mediated resolubilization) was calculated. EC50 of 3a at specified concentration was compared to EC50 in sample buffer. This procedure was repeated for the rest of the compounds (3b–x). The percentage of inhibition was calculated by taking the ratio between the percentage left as polymer with and without the chemical compound at a particular EGTA concentration.
4.3. Molecular Modeling
In silico molecular docking was performed to understand the possible binding site of our designed molecules. CASQ2 polymers are formed via front-to-front (N-terminal) and back-to-back (C-terminal) contacts.38 The structure for the front-to-front dimer is established; however, not much information is available for the back-to-back dimer (C-terminal contact points) due to the absence of a crystal structure. In order to facilitate polymerization, it is plausible that these molecules interact at the polymeric interface. Since there was no prior knowledge about the binding site of the molecules, blind docking was performed using web server Swiss Model39 with the accurate mode. The crystal structure of the front-to-front dimer 1SJI was downloaded from the RCSB protein data bank (http://www.rcsb.org/). The structure is devoid of intrinsically disordered C-terminal residues; however, it does not make a significant contribution in the protein folding.40 The protein structure was prepared using the default parameters of Protein Preparation Wizard of the Schrödinger Suite.41 As there was no information about any conserved water molecules, the water molecules were deleted. The structures of the most active molecules 3e, 3u, and 3v were provided after minimization with the OPLS3e force field, run for a maximum of 25000 iterations using the MacroModel module of the Schrödinger Suite. To examine if the molecules bind to the back-to-back interface of the CASQ2 structure, we built the homology model of the back-to-back dimeric CASQ2 structure. We used the sequence of 1SJI for building the model. The 3D-homology model of the CASQ2 back-to-back dimer was built using the Prime module of the Schrödinger Suite. Chains B and C of the crystal structure of human CASQ1 (PDB code 3UOM) was chosen as the template. 3UOM is the polymeric structure of CASQ, which is very likely to resemble the CASQ2 polymeric structure. The sequence alignment shows 68% identity and 84% positives. 3UOM is a hexamer composed of six chains A–F. The dimeric units AB, CD, and EF are involved in the front-to-front contact, and the dimeric units BC and DE have the back-to-back contacts. The homology model obtained was minimized, and loops were refined using the default parameters of Prime minimization and loop refinement. The structure hence obtained was used for blind docking using accurate mode of Swiss Dock. The results were analyzed using UCSF Chimera (https://www.cgl.ucsf.edu/chimera/). Binding mode was further analyzed using the Discovery Studio Visualizer.42
Acknowledgments
This work was supported in part by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number K01DK102772 and Ramalingaswami Fellowship from DBT, India to N.C.B. Authors are thankful to BIT for NMR facility (DST FIST: SR/FST/CSI-242/2012) and Central Instrument Facility for X-ray and analytical data.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b01113.
Analytical data and experimental procedures for all the compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Tang Y.; Tian X.; Wang R.; Fill M.; Chen S. R. W. Abnormal termination of Ca2+ release is a common defect of RyR2 mutations associated with cardiomyopathies. Circ. Res. 2012, 110, 968–977. 10.1161/CIRCRESAHA.111.256560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano M.; Ikeda Y.; Matsuzaki M. Altered intracellular Ca2+ handling in heart failure. J. Clin. Invest. 2005, 115, 556–564. 10.1172/JCI24159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrens X. H. T.; Lehnart S. E.; Marks A. R. Intracellular calcium release and cardiac disease. Annu. Rev. Physiol. 2005, 67, 69–98. 10.1146/annurev.physiol.67.040403.114521. [DOI] [PubMed] [Google Scholar]
- Pascual I.; Moris C.; Avanzas P. Beta-blockers and calcium channel blockers: first line agents. Cardiovasc. Drugs Ther. 2016, 30, 357–365. 10.1007/s10557-016-6682-1. [DOI] [PubMed] [Google Scholar]
- Driessen H. E.; Bourgonje V. J. A.; van Veen T. A. B.; Vos M. A. New antiarrhythmic targets to control intracellular calcium handling. Neth. Heart J. 2014, 22, 198–213. 10.1007/s12471-014-0549-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaburjakova M.; Bal N. C.; Gaburjakova J.; Periasamy M. Functional interaction between calsequestrin and ryanodine receptor in the heart. Cell. Mol. Life Sci. 2013, 70, 2935–2945. 10.1007/s00018-012-1199-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa A.; Podini P.; Clegg D. O.; Pozzan T.; Meldolesi J. Intracellular Ca2+ stores in chicken Purkinje neurons: differential distribution of the low affinity-high capacity Ca2+ binding protein, calsequestrin, of Ca2+ ATPase and of the ER lumenal protein, Bip. J. Cell Biol. 1991, 113, 779–791. 10.1083/jcb.113.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemoto N.; Ronjat M.; Meszaros L. G.; Koshita M. Postulated role of calsequestrin in the regulation of calcium release from sarcoplasmic reticulum. Biochemistry 2002, 28, 6764–6771. 10.1021/bi00442a033. [DOI] [PubMed] [Google Scholar]
- Beard N. A.; Laver D. R.; Dulhunty A. F. Calsequestrin and the calcium release channel of skeletal and cardiac muscle. Prog. Biophys. Mol. Biol. 2004, 85, 33–69. 10.1016/j.pbiomolbio.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Park H.; Park I. Y.; Kim E.; Youn B.; Fields K.; Dunker A. K.; Kang C. Comparing skeletal and cardiac calsequestrin structures and their calcium binding. J. Biol. Chem. 2004, 279, 18026–18033. 10.1074/jbc.M311553200. [DOI] [PubMed] [Google Scholar]
- Beard N. A.; Wei L.; Dulhunty A. F. Ca 2+ signaling in striated muscle: the elusive roles of triadin, junctin, and calsequestrin. Eur. Biophys. J. 2009, 39, 27. 10.1007/s00249-009-0449-6. [DOI] [PubMed] [Google Scholar]
- Györke I.; Györke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys. J. 1998, 75, 2801–2810. 10.1016/S0006-3495(98)77723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahat H.; Pras E.; Olender T.; Avidan N.; Ben-Asher E.; Man O.; Levy-Nissenbaum E.; Khoury A.; Lorber A.; Goldman B.; Lancet D.; Eldar M. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am. J. Hum. Genet. 2001, 69, 1378–1384. 10.1086/324565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle G.; Galla D.; Nori A.; Priori S. G.; Gyorke S.; De Filippis V.; Volpe P. Catecholaminergic polymorphic ventricular tachycardia-related mutations R33Q and L167H alter calcium sensitivity of human cardiac calsequestrin. Biochem. J. 2008, 413, 291–303. 10.1042/BJ20080163. [DOI] [PubMed] [Google Scholar]
- Rizzi N.; Liu N.; Napolitano C.; Nori A.; Turcato F.; Colombi B.; Bicciato S.; Arcelli D.; Spedito A.; Scelsi M.; Villani L.; Esposito G.; Boncompagni S.; Protasi F.; Volpe P.; Priori S. G. Unexpected structural and functional consequences of the R33Q homozygous mutation in cardiac calsequestrin. Circ. Res. 2008, 103, 298–306. 10.1161/CIRCRESAHA.108.171660. [DOI] [PubMed] [Google Scholar]
- di Barletta M. R.; Viatchenko-Karpinski S.; Nori A.; Memmi M.; Terentyev D.; Turcato F.; Valle G.; Rizzi N.; Napolitano C.; Gyorke S.; Volpe S.; Priori S. G. Clinical phenotype and functional characterization of CASQ2 mutations associated with catecholaminergic polymorphic ventricular tachycardia. Circulation 2006, 114, 1012–1019. 10.1161/CIRCULATIONAHA.106.623793. [DOI] [PubMed] [Google Scholar]
- Kim E.; Youn B.; Kemper L.; Campbell C.; Milting H.; Varsanyi M.; Kang C. Characterization of human cardiac calsequestrin and its deleterious mutants. J. Mol. Biol. 2007, 373, 1047–1057. 10.1016/j.jmb.2007.08.055. [DOI] [PubMed] [Google Scholar]
- Kalyanasundaram A.; Bal N. C.; Franzini-Armstrong C.; Knollmann B. C.; Periasamy M. The calsequestrin mutation CASQ2 D307H does not affect protein stability and targeting to the junctional sarcoplasmic reticulum but compromises its dynamic regulation of calcium buffering. J. Biol. Chem. 2010, 285, 3076–3083. 10.1074/jbc.M109.053892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirksen W.; Lacombe V.; Chi M.; Kalyanasundaram A.; Viatchenko-Karpinski S.; Terentyev D.; Zhou Z.; Vedamoorthyrao S.; Li N.; Chiamvimonvat N.; Carnes C. A.; Franzini-Armstrong C.; Györke S.; Periasamy M. A mutation in calsequestrin, CASQ2D307H, impairs sarcoplasmic reticulum Ca2+ handling and causes complex ventricular arrhythmias in mice. Cardiovasc. Res. 2007, 75, 69–78. 10.1016/j.cardiores.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal N. C.; Sharon A.; Gupta S. C.; Jena N.; Shaikh S.; Gyorke S.; Periasamy M. The catecholaminergic polymorphic ventricular tachycardia mutation R33Q disrupts the N-terminal structural motif that regulates reversible calsequestrin polymerization. J. Biol. Chem. 2010, 285, 17188–17196. 10.1074/jbc.M109.096354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal N. C.; Jena N.; Sopariwala D.; Balaraju T.; Shaikh S.; Bal C.; Sharon A.; Gyorke S.; Periasamy M. Probing cationic selectivity of cardiac calsequestrin and its CPVT mutants. Biochem. J. 2011, 435, 391–399. 10.1042/BJ20101771. [DOI] [PubMed] [Google Scholar]
- Knollmann B. C.; Chopra N.; Hlaing T.; Akin B.; Yang T.; Ettensohn K.; Knollmann B. E.; Horton K. D.; Weissman N. J.; Holinstat I. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca 2+ release, and catecholaminergic polymorphic ventricular tachycardia. J. Clin. Invest. 2006, 116, 2510–2520. 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terentyev D.; Viatchenko-Karpinski S.; Györke I.; Volpe P.; Williams S. C.; Györke S. Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: mechanism for hereditary arrhythmia. Proceedings of the National Academy of Sciences 2003, 100 (20), 11759–11764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subra A. K.; Nissen M. S.; Lewis K. M.; Muralidharan A. K.; Sanchez E. J.; Milting H.; Kang C. Molecular mechanisms of pharmaceutical drug binding into calsequestrin. Int. J. Mol. Sci. 2012, 13, 14326–14343. 10.3390/ijms131114326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlier H. A.; Olson R. D.; Thornock C. M.; Mercer W. K.; Olson D. R.; Broyles T. S.; Muhlestein D. J.; Larson C. L.; Cusack B. J.; Shadle S. E. Investigations of calsequestrin as a target for anthracyclines: comparison of functional effects of daunorubicin, daunorubicinol, and trifluoperazine. Mol. Pharmacol. 2005, 67, 1505–1512. 10.1124/mol.104.005728. [DOI] [PubMed] [Google Scholar]
- Sanchez E. J.; Hayes R. P.; Barr J. T.; Lewis K. M.; Webb B. N.; Subramanian A. K.; Nissen M. S.; Jones J. P.; Shelden E. A.; Sorg B. A.; Fill M.; Schenk J. O.; Kang C. Potential role of cardiac calsequestrin in the lethal arrhythmic effects of cocaine. Drug Alcohol Depend. 2013, 133, 344–351. 10.1016/j.drugalcdep.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson R. D.; Li X.; Palade P.; Shadle S. E.; Mushlin P. S.; Gambliel H. A.; Fill M.; Boucek R. J. Jr; Cusack B. J. Sarcoplasmic reticulum calcium release is stimulated and inhibited by daunorubicin and daunorubicinol. Toxicol. Appl. Pharmacol. 2000, 169, 168–176. 10.1006/taap.2000.9065. [DOI] [PubMed] [Google Scholar]
- Kang C.; Nissen M. S.; Sanchez E. J.; Lam K.-S.; Milting H. Potential adverse interaction of human cardiac calsequestrin. Eur. J. Pharmacol. 2010, 646, 12–21. 10.1016/j.ejphar.2010.08.001. [DOI] [PubMed] [Google Scholar]
- Shan K.; Lincoff A. M.; Young J. B. Anthracycline-induced cardiotoxicity. Ann. Intern. Med. 1996, 125, 47–58. 10.7326/0003-4819-125-1-199607010-00008. [DOI] [PubMed] [Google Scholar]
- Kim D. H.; Landry A. B. III; Lee Y. S.; Katz A. M. Doxorubicin-induced calcium release from cardiac sarcoplasmic reticulum vesicles. J. Mol. Cell. Cardiol. 1989, 21, 433–436. 10.1016/0022-2828(89)90782-7. [DOI] [PubMed] [Google Scholar]
- Ondrias K.; Borgatta L.; Kim D. H.; Ehrlich B. E. Biphasic effects of doxorubicin on the calcium release channel from sarcoplasmic reticulum of cardiac muscle. Circ. Res. 1990, 67, 1167–1174. 10.1161/01.RES.67.5.1167. [DOI] [PubMed] [Google Scholar]
- Saeki K.; Obi I.; Ogiku N.; Shigekawa M.; Imagawa T.; Matsumoto T. Doxorubicin directly binds to the cardiac-type ryanodine receptor. Life Sci. 2002, 70, 2377–2389. 10.1016/S0024-3205(02)01524-2. [DOI] [PubMed] [Google Scholar]
- Pessah I. N.; Durie E. L.; Schiedt M. J.; Zimanyi I. Anthraquinone-sensitized Ca2+ release channel from rat cardiac sarcoplasmic reticulum: possible receptor-mediated mechanism of doxorubicin cardiomyopathy. Mol. Pharmacol. 1990, 37, 503–514. [PubMed] [Google Scholar]
- Hanna A. D.; Lam A.; Tham S.; Dulhunty A. F.; Beard N. A. Adverse effects of doxorubicin and its metabolic product on cardiac RyR2 and SERCA2A. Mol. Pharmacol. 2014, 86, 438–449. 10.1124/mol.114.093849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menna P.; Salvatorelli E.; Gianni L.; Minotti G.. Anthracycline cardiotoxicity. In Anthracycline Chemistry and Biology II; Springer: 2007; pp 21–44, 10.1007/128_2007_11. [DOI] [PubMed] [Google Scholar]
- Karampuri S.; Bag P.; Yasmin S.; Chouhan D. K.; Bal C.; Mitra D.; Chattopadhyay D.; Sharon A. Structure based molecular design, synthesis and biological evaluation of α-pyrone analogs as anti-HSV agent. Bioorg. Med. Chem. Lett. 2012, 22, 6261–6266. 10.1016/j.bmcl.2012.07.098. [DOI] [PubMed] [Google Scholar]
- Konreddy A. K.; Toyama M.; Ito W.; Bal C.; Baba M.; Sharon A. Synthesis and Anti-HCV Activity of 4-Hydroxyamino α-Pyranone Carboxamide Analogues. ACS Med. Chem. Lett. 2013, 5, 259–263. 10.1021/ml400432f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Trumble W. R.; Liao H.; Wesson C. R.; Dunker A. K.; Kang C. Crystal structure of calsequestrin from rabbit skeletal muscle sarcoplasmic reticulum. Nat. Struct. Mol. Biol. 1998, 5, 476–483. 10.1038/nsb0698-476. [DOI] [PubMed] [Google Scholar]
- Arnold K.; Bordoli L.; Kopp J.; Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Bal N. C.; Jena N.; Chakravarty H.; Kumar A.; Chi M.; Balaraju T.; Rawale S. V.; Rawale J. S.; Sharon A.; Periasamy M. The C-terminal calcium-sensitive disordered motifs regulate isoform-specific polymerization characteristics of calsequestrin. Biopolymers 2015, 103, 15–22. 10.1002/bip.22534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestro; Schrödinger, LLC: New York, NY, 2017.
- Visualizer D. S.Release 3.5.; Accelrys Inc:San Diego, CA, USA,2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.