


Abstract
We present Nucleosome Dynamics, a suite of programs integrated into a virtual research environment and created to define nucleosome architecture and dynamics from noisy experimental data. The package allows both the definition of nucleosome architectures and the detection of changes in nucleosomal organization due to changes in cellular conditions. Results are displayed in the context of genomic information thanks to different visualizers and browsers, allowing the user a holistic, multidimensional view of the genome/transcriptome. The package shows good performance for both locating equilibrium nucleosome architecture and nucleosome dynamics and provides abundant useful information in several test cases, where experimental data on nucleosome position (and for some cases expression level) have been collected for cells under different external conditions (cell cycle phase, yeast metabolic cycle progression, changes in nutrients or difference in MNase digestion level). Nucleosome Dynamics is a free software and is provided under several distribution models.
INTRODUCTION
Eukaryotic chromatin is organized in a hierarchical manner, where the basic structural units are repetitive elements named nucleosomes. Each of them is defined by around 147 base pairs of DNA wrapped around a protein octamer, the histones. The position of the nucleosomes in the cell is not random and recurrent patterns have been detected in cell populations (1–3), indicating a maintenance of the nucleosome architecture which seems to be crucial for a correct regulation of genome activity (4). The protein octamer serves as an anchoring point for proteins recognizing histone epigenetic signals, while unwrapped DNA is targeted by transcription factors and enhancers (5,6). Thus, nucleosomes shifting due to alterations in the sequence (7), DNA methylation (8) or the action of chromatin remodelers (5,9–13) can result in dramatic changes in gene expression. Characterizing such changes is crucial for the understanding of the connection between chromatin structure and genome functionality (6).
Experimental determination of nucleosome positioning is typically performed by treating a group of cells (in the range 106-109) with enzymes acting on nucleosome-free DNA. ATAC-seq (14) uses a hyperactive transposase for tagging nucleosome-free DNA segments for sequencing (the linkers). MNase-seq, the most widely used technique for nucleosome localization, uses Micrococcal nuclease to degrade linker DNA preserving the DNA segments wrapped in the nucleosomes, which are then sequenced. Both MNase-seq and ATAC-seq, after filtering nucleosomal reads by size (14), provide at the end the same type of information: DNA reads that need to be grouped into individual nucleosomes using a variety of computational approaches (15–18), which in all cases suffer from the intrinsic dispersion in read coverage. The resulting nucleosome maps show well defined depleted regions (the nucleosome free regions, NFR), well-positioned (W) nucleosomes, and a large number of ‘fuzzy’ (F) nucleosomes giving partial protection signals longer than 147 bps (19). Fuzzy positioning signals are the result of nucleosomes not being in exactly the same genomic position in the cell population, and are intrinsically difficult to annotate by any nucleosome calling algorithms (15,17). While it is known (20,21) that this technique can be affected by MNase concentration and sequence-preference biases that affect the detection of the so called ‘fragile’ nucleosomes, it is still the most widely used to detect nucleosome positioning for its versatility and accuracy. In 2012, Brogaard et al. developed a chemical cleavage method that provides a very accurate positioning of nucleosomes (22). However, this technique requires to do genetic engineering replacing the endogenous histone H4 (or H3 (23)) by a mutated version, therefore restricting its use (24–27). Moreover, it has been shown that the MNase sequence bias can be corrected using digested naked DNA as baseline (20,21), obtaining more pronounced nucleosome coverage peaks.
The noisy nature of experimental data such as MNase-seq, makes very difficult to compare nucleosome architecture in two samples, as the signal is masked by the intrinsic fuzziness of the maps. Methods available such as DANPOS and Dimnp (15,28) can detect only a limited number of changes affecting large percentage of the cells, as they work at the level of the fragment coverage, missing the opportunity to work with the raw data: the fragments themselves.
We present here Nucleosome Dynamics, a complete virtual framework to characterize the structure and dynamics of nucleosome architectures. The package consists of two main blocks: an improved version of our nucleR algorithm for nucleosome location (17), and NucDyn, an algorithm specifically created to detect changes (shifts, evictions and insertions) in nucleosome architectures based on the direct processing of raw data (the sequencing reads) obtained from pairs of experiments. The Nucleosome Dynamics package (available under the Apache 2.0 License) can be installed from the source code, obtained from BioConductor (29), or run as a web tool hosted by the MuGVRE workspace (30) as well as in a Galaxy server (31), where additional analysis algorithms, browsers and visualization tools are included.
MATERIALS AND METHODS
Package overview
The input data for Nucleosome Dynamics is one or several files containing sequence reads aligned to the reference genome and stored in BAM format. The user can select (see Figure 1) between: (i) processing a single file to define the consensus nucleosome architecture using an extended version of nucleR (17) or (ii) detecting changes in nucleosome distribution between two experiments, by comparing pairs of mapped sequence files using the newly developed NucDyn module. For a complete description of the parameters of the available functionalities, see Supplementary Table S1. In the MuGVRE implementation (see Table 1), the user has access to a wide range of analysis and visualization tools to characterize the nucleosome patterns, their changes across different conditions, and to put all the data in the context of other information mapped to the genome (genome structure, expression, epigenetic signals, etc). We have evaluated the performance of nucleR and NucDyn generating synthetic nucleosome maps and have tested their descriptive power using publicly available nucleosome positioning experimental data.
Figure 1.

Analysis pipeline for Nucleosome Dynamics. A single MNase-seq experiment can be analysed, obtaining: nucleosome calls with nucleR, their fuzzy/well-positioned classification and stiffness estimation, Nucleosome Free Regions location, classification of TSS according to −1 and +1 nucleosomes, and nucleosome phasing along the gene body. Comparing two MNase-seq experiments, NucDyn identifies hotspots of changes (SHIFT +, SHIFT -, INCLUSION and EVICTION), and reports a significance score of the difference in the coverage profiles at base-pair level. Summary statistics per gene as well as genome-wide are also reported for each calculation.
Table 1.
Implementation models for Nucleosome Dynamics
| Code distribution | ||
| Standalone installation | Nucleosome Dynamics CLI | https://github.com/nucleosome-dynamics/nucleosome_dynamics |
| nucleR R package | https://github.com/nucleosome-dynamics/nucleR Bioconductor: http://bioconductor.org/packages/nucleR/ | |
| NucDyn R package | https://github.com/nucleosome-dynamics/NucDyn Bioconductor: (in review) | |
| Containerized installation | Docker | https://github.com/nucleosome-dynamics/docker Docker-hub: mmbirb/nucleosome-dynamics |
| Singularity | https://github.com/nucleosome-dynamics/nucleosome_dynamics_singularity Singularity-hub: https://singularity-hub.org/collections/2579 | |
| Platforms in use | ||
| MuG Virtual Research Environment | https://vre.multiscalegenomics.eu/workspace/?from=nucldynwf | |
| Galaxy Platform | https://dev.usegalaxy.es (in development) Galaxy ToolShed: https://toolshed.g2.bx.psu.edu/repository?repository_id=822e9c879cf92fd0 | |
Single experiment analysis
Nucleosome positioning and coverage
BAM files from a single MNase-seq experiment are processed to define nucleosome coverage, which can be directly visualized using a genome browser (Figure 2A) or processed to obtain nucleosome positions. Accordingly, following signal theory the read coverage is described as a combination of periodic waves, which are then subjected to Fast Fourier Transformation (FFT) to remove the high frequency components responsible for the noise (see Supplementary Figure S1). The parameters for FFT filtering can be adjusted taking into account the nucleosome repeat length and noise level of each organism and cell type (see Supplementary Table S1). Clean profiles are processed to annotate the nucleosome dyads (located at the local maxima of the distributions). Putative nucleosomes are then scored based on the shape of the associated peaks (see Supplementary Figure S1). Those leading to sharp signals are labelled as well-positioned (W) nucleosomes (high localization score), while flat peaks are labelled as ‘fuzzy’ (F) nucleosomes (low localization score). Once all nucleosomes are localized, the software analyses the nucleosome architecture (see Supplementary Figure S2A) around the transcription start sites (TSS) and classifies the nucleosome architecture for each gene based on (19): the extension of the nucleosome free region (NFR) around the core promoter (open (o), closed (c) or missing −1 or +1 nucleosome) and the degree of localization of the +1 (downstream the TSS) and −1 (upstream the TSS) nucleosomes (see Supplementary Figure S2A). Data are presented at the individual gene level as well as summarized at global level (Figure 2). Nucleosome Dynamics performs a global detection of all NFRs, as these regions usually are the main recognition sites for effector proteins, and well-defined and extended NFRs typically signal active regions in the genome.
Figure 2.

Visualization of Nucleosome Dynamics results in MuGVRE. (A) Nucleosome positioning along ACE2 (YLR131C) gene from S. cerevisiae between G2 and M cell cycle phases (a) YLR131C open reading frame; (b) ACE2 full length transcripts; (c), (c') coverage of MNase-seq reads aligned to reference genome (in G2 and M phase, respectively); (d), (d') nucleosome calls obtained with nucleR (G2 and M phase, respectively); (e) NFR coordinates in G2 phase; (f) prediction of the nucleosome coverage along each gene, using signals from +1 and -last nucleosomes; (g) genes shown as coloured boxes according to the phasing between the +1 and -last nucleosomes; (h) TSS classification based on nucleosomes −1 and +1 (W, F, missing) and the distance between them (open or close) represented as coloured boxes, with an arrow indicating the direction of the gene; (i) nucleosomes are coloured by their apparent stiffness value: darker blue nucleosomes are more stiff and lighter are less stiff; (j) significance of the differences in nucleosome coverage between G2 and M phases (-log10 of the p-value) (k) movement hotspots represented as colour coded boxes: purple for shift +, blue for shit -, green for inclusion and red for eviction; (l,m, n, o) Tracks from publicly available data representing (l) TATA elements (Rhee et al., 2012), (m) TFIIB binding sites (Mayer et al., 2010), (n) H3K4me3 histone mark enrichment (Liu et al., 2005) and (o) gene expression changes during cell cycle (Deniz et al., 2016). (B) Detailed view around a hotspot identified by NucDyn in chrXII. (C) Genome wide statistics of NFR width around TSS, in G2 phase. (D) Genome wide frequency of changes detected by NucDyn between G2 and M.
Periodicity at coding regions
The software evaluates the periodicity in the nucleosome pattern inside the genes, following signal propagation theory from two ‘emitting sites’ located at well-positioned nucleosomes. The first signal comes from the 5′ end of the gene (the +1 nucleosome located just downstream the TSS) and the second from the 3′ end of the gene (the –last nucleosome located just upstream the transcription termination site; TTS). We assume that both signals proceed in opposite directions (from +1 to –last nucleosome) following an exponential decay periodic function (32). We found out that when the +1 and –last originated waves are in phase the signals sum up and nucleosomes are well located inside the gene body, while when they are in antiphase the signals cancel out and the gene typically shows fuzzy nucleosomes. The periodicity (T) of the signal is obtained by maximizing the autocorrelation function (Equation 1: see an example in Supplementary Figure S2):
 |
(1) |
where X1 and X2 are the intervals of the window, I(x) stands for the coverage. This value will be dependent on the nucleosome repeat length of each species and cell type (see Supplementary Table S2 for suggested T in different cell types).
A ‘phased’ gene is defined when the distance between +1 and –last nucleosome is close to a multiple of T (Supplementary Figure S2B). An ‘antiphased’ gene is defined when the modulus of the ratio between the distance of the +1 and –last nucleosomes and T is close to T/2 (Supplementary Figure S2C). The package provides a theoretical nucleosome map based on signal propagation theory with +1 and –last nucleosomes as emitting sites. Comparison of the predicted and the real nucleosome maps helps to detect anomalies in the gene nucleosome distribution emerging from interacting proteins or from the effect of the remodelling machinery.
Nucleosome stiffness
Nucleosome Dynamics also analyses the sliding propensities of nucleosomes by computing its apparent resistance to be displaced along the sequence. For this purpose, we map the original reads around located nucleosomes and estimate the normalized Gaussian that better fits the distribution of reads (see Supplementary Figure S2) from which stiffness is derived by the elastic approximation as shown in Equation (2):
 |
(2) |
where 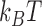 is the thermal energy at room temperature and
is the thermal energy at room temperature and  is the standard deviation of the Gaussian fitted to reads associated to the nucleosome.
is the standard deviation of the Gaussian fitted to reads associated to the nucleosome.
Defining changes in nucleosome distribution
Pairs of BAM files are processed to determine changes in the nucleosome architecture between two experiments. For this purpose (see Supplementary Figure S3) the program pairs the reads obtained from one experiment to the other to discard those that are unchanged. It also removes reads that share the same starting or ending point or those that can be fitted in longer read in the paired experiment, as they are likely to be generated by spurious differences in nuclease degradation activity. The remaining reads are then paired between the two experiments using a dynamic programming algorithm designed to maximize: (i) the number of matches, (ii) the proximity in the middle points of the paired reads, (iii) the assignment of the paired reads to the same nucleosome. To achieve these objectives the dynamic programming highly penalizes gaps and scores read pairs inversely proportional to their distance, with a –infinite score when the distance between the middle point of the reads is longer than half the length of the nucleosome. The final output of the procedure is a set of read pairs shifted in one experiment with respect to the other. These shifts are accumulated to define hotspots that are further analysed to determine their statistical significance as markers of shifts in the nucleosome architecture.
A second type of changes detected by the program is related to differences in occupancy (insertions and evictions) between the two experiments, that are determined directly from the coverage. To reduce the impact of experimental noise we analyse the coverage data by computing a Z-score for every position x across the genome, normalizing it in 10 000 bp windows, which allows us to find locally normalized differences in coverage (Equation 3).
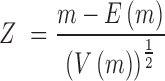 |
(3) |
where  is the number of reads covering position x in experiment 1,
is the number of reads covering position x in experiment 1, 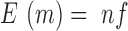 (with
(with 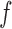 being the fraction of total reads in the window (N) that corresponds to experiment 1 (M) and n is the number of reads covering position x in both experiments) and
being the fraction of total reads in the window (N) that corresponds to experiment 1 (M) and n is the number of reads covering position x in both experiments) and 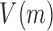 is the expected variance of a hypergeometric distribution, i.e.
is the expected variance of a hypergeometric distribution, i.e.  . Positive Z-score peaks mean that at that point the read coverage found at experiment 1 is higher than the coverage at experiment 2 and an eviction hotspot is annotated. Similarly, negative Z- peaks signal inclusions.
. Positive Z-score peaks mean that at that point the read coverage found at experiment 1 is higher than the coverage at experiment 2 and an eviction hotspot is annotated. Similarly, negative Z- peaks signal inclusions.
The statistical significance of the detected hotspots (shifts, inclusions and evictions) is scored using the P-values derived from Fisher's test from a contingency table between the reads in each experiment (columns) and the reads at a given position compared to reads within the window (rows):
| Exp 1. | Exp. 2 | Total | |
|---|---|---|---|
| Covering x | M– m | N– M– n+ m | N– n |
| Not covering x | m | n– m | n |
| Total | M | N– M | N |
Software availability and implementation
The Nucleosome Dynamics package is available in different deployment models to fulfil the needs of different users. Moreover, it is also offered as a service in two different research platforms. All available distributions are explained at Nucleosome Dynamics landing page: http://mmb.irbbarcelona.org/NucleosomeDynamics/, and summarized in Table 1.
Code distributions
Nucleosome Dynamics is written in R and composed of two packages (nucleR and NucDyn), and a series of R wrappers providing a unified interface to such core functionalities and other additional analyses (TSS classification, NFR, Phasing, Stiffness, etc., see above). Source code and documentation are available for standalone installation (see Table 1). Both nucleR and NucDyn packages are also distributed via BioConductor. Although the native R interface is recommended for experienced R users, other deployments built on top of the R software are also provided for further accessibility and portability.
Nucleosome Dynamics package depends on a series of other R packages and helper applications. To minimize the possibilities of collision with existing installations, and to avoid installation issues to the non-experts, the packages are offered as software containers in both, the well-known Docker implementation and the Singularity format, the latter intended for multi-user systems where running Docker containers natively is not trivial – i.e. HPC systems. A single container allows the user to obtain all functions of the package directly from the command-line, and additionally, the launcher is able to accept a list of Nucleosome Dynamics analysis commands in bash to orchestrate a custom workflow. Furthermore, the use of the containers allows seamless software update. The images are registered at the corresponding hubs (see Table 1).
Use in research platforms
MuG Virtual Research Environment (MuGVRE) is an integrated workspace designed to put together a series of applications related to the study of 3D/4D genomics (30). The MuGVRE workspace allows to combine data, either uploaded to the workspace or obtained from public repositories such as ArrayExpress (33). MuGVRE includes applications covering a wide range of levels in the study of chromatin, from atomistic simulation or protein-nucleic acids docking to coarse-grained simulation of large nucleic acids molecules or chromatin fibers, as well as the analysis of Hi-C data. All those applications share a common data space where interoperability is assured through a common data model, and a specific protocol to incorporate new tools. MuGVRE is a cloud-based application that simplifies the deployment and provides user access to visualization tools, additional data in external repositories, and to a variety of other programs for the analysis of chromatin at different levels of resolution.
The server provides a graphical interface based on an embedded sequence browser, Jbrowse (34), that allows visualization of nucleosome architectures in the context of other omics data (see Figure 2A). For this purpose, the outputs of all calculations are generated in GFF3 or bigWig format. Nucleosomes are represented as boxes coloured with different tones of blue according to their positioning score, with regions where nucleosome are too fuzzy displayed in a lighter colour. Similarly, nucleosome-free regions are highlighted by yellow boxes in a different row (see Figure 2A). In both cases, numerical information (scores, characteristics of the nucleosome or NFR) can be obtained by clicking on the corresponding boxes. The nucleosome architecture around TSS is classified based on the length of the NFR and the location score of the +1 and −1 nucleosome (see above). The results are shown as boxes between the −1 and +1 nucleosome dyads, color-coded by the corresponding architecture class. The analysis of nucleosome phasing generates a bigWig file with the theoretical prediction of the nucleosome positions inside the gene body, based on periodicity considerations and the +1 and –last nucleosomes (see above), and a GFF3 file which is displayed as a coloured box indicating whether the gene shows ‘phased’, ‘antiphased’ or intermediate nucleosome phasing (see Figure 2A). Similarly, the stiffness associated to the nucleosomes is represented (through a GFF3 file) as a box mapping to the nucleosome, coloured according to the estimated stiffness (see Figure 2A). Nucleosome Dynamics data can be put into genomic context by a series of additional tracks (Supplementary Table S3) providing gene annotations and relevant literature data. Further analyses can be obtained from the web server, such as detailed plots of nucleosome coverage and changes in nucleosome distribution between the two experiments (Figure 2B), genome-wide statistics of nucleosome architecture around TSS (Figure 2C), and overall frequency of inclusions, evictions and shifts between the two experiments (Figure 2D). Finally, Nucleosome Dynamics also generates a table listing the number of nucleosomes, their status (fuzzy/well-positioned), the identified nucleosome changes (inclusions, evictions, shifts), the classification of the promoter and the width of the NFR at the TSS for every single gene in the genome (Supplementary Table S4). These analyses are useful to test the effect of a treatment/growth conditions on nucleosome positioning both globally, or at gene level.
Running Nucleosome Dynamics on the galaxy platform: Galaxy is a web-based scientific analysis platform widely used by scientists to analyse biomedical datasets such as genomics, proteomics, metabolomics or imaging (31). Nucleosome Dynamics docker has been wrapped in a series of Galaxy tools, one for each analysis. Users can launch them individually, or as part of a Galaxy workflow, building a custom pipeline that may integrate other Galaxy applications. The tools are published in the Galaxy ToolShed (see Table 1) and adopted by the ELIXIR_ES Galaxy server (currently in development phase), together with a complete ready-to-use Nucleosome Dynamics workflow. The output calculations, mainly GFF3 and bigWig files, are treated in the platform as any other sequence annotation file. Plain files like GFFs are locally displayed using a column-based visualization, while the genomic-context analysis is based on the UCSC genome browser (35). Galaxy transparently loads the data to the central UCSC browser service, and there, the sequences are loaded as custom tracks and visualized together with the other UCSC annotations.
Benchmarking data sets
The ability of the package to determine the location and nature of nucleosomes and nucleosome architectures was evaluated using synthetic maps from single cell nucleosome architectures that are combined to create in silico MNase digestion maps approaching closely to those found in real yeast MNase-seq experiments. For this purpose, we created multiple single cell nucleosome architectures by first placing NFRs at specific positions separated by ∼2000 bp (the typical range of NFR-NFR distances in yeast) in a 10 kb DNA fragment. As the NFR are highly conserved, their positions are located with small noise in the different cells. Once the NFRs for a single cell have been placed, we defined windows for nucleosome positioning using the known average nucleosome periodicity (165 bp for yeast in the experiments simulated here). Each window was associated to either a W or F nucleosome following probability functions reproducing their expected populations at different distances from NFR. Windows that (in a given cell) appeared to be associated to W nucleosomes have a high probability to be occupied by a nucleosome, which is placed within a narrow range from the centre of the window (see Supplementary Figure S4). On the contrary, windows associated to F nucleosomes have a higher probability to be empty in a given cell, and once the nucleosome is assigned, its position is variable within the window. Once obtained, the population of in silico cells was processed by in silico MNase digestion repeating this process many times, introducing ‘digestion noise’ to reproduce the distributions of read lengths observed in typical yeast experiments. The integration of the reads for the entire population provides an in silico MNase-seq map where we know exactly the real population and fuzziness of all nucleosomes at all positions in the pool of cells. This constitutes an unambiguous benchmark to validate the performance of nucleosome annotation software. The probability functions used to generate the different cell nucleosome architectures were adjusted to qualitatively reproduce reads obtained in real MNase-seq experiments (Supplementary Figure S4). Synthetic data simulating ATAC-seq experiments can be derived in a very similar manner.
The synthetic data obtained as explained above were the starting point to generate pairs of in silico experiments simulating changes in nucleosome architectures. To this end, a percentage of the reads was either shifted or removed for a given nucleosome. Shifts from 1 to 5 turns of DNA (1 DNA turn = 10 bp) were introduced generating 100 replicates in each case to evaluate the sensitivity of the method to detect shifts of different lengths and affecting different percentage of the total population.
RESULTS
Performance using in silico datasets
NucleR
We explored the ability of the software to position nucleosomes using as reference our highly controlled synthetic data (see Methods). As a control, we repeated the exercise using another widely used program for nucleosome annotation, DANPOS (15). Both packages show a good ability to represent the nucleosome architecture using MNase-seq data. In terms of occupancy DANPOS performs slightly better than the nucleR module implemented in our Nucleosome Dynamics package (R2 = 0.97 for DANPOS versus R2 = 0.93 for nucleR), while nucleR can detect better the nucleosome fuzziness (R2 = 0.94 for nucleR versus R2 = 0.87 for DANPOS; see Supplementary Methods. for description of the metrics). The location of W nucleosomes is nearly identical in both methods, but for F nucleosomes the results are quite different, as DANPOS annotates a wide region of sequence reads as a single nucleosome positioned with a large uncertainty, while nucleR can assign several nucleosomes to the wide signal, even when in some cases the two nucleosomes can partially overlap (see Figure 3A). As a result, DANPOS provides, probably, the best ‘average’ distribution of nucleosomes, but nucleR provides a more realistic picture of the cellular variability, capturing the presence of alternative nucleosome architectures in the cellular population. As it can be seen in Figure 3A, where selected examples of DANPOS and nucleR nucleosome distributions are compared with the real nucleosome architecture existing in our synthetic data; in Figure 3B, where we compare the ability of DANPOS and nucleR to detect the presence of a percentage of cells showing a different nucleosome architecture and in Figure 3C, where we report the average distance between the real position of the dyads of the synthetic nucleosomes and those located by DANPOS or nucleR.
Figure 3.

Performance of nucleR and NucDyn. (A) Coverage of three synthetic nucleosome maps (shown in grey), containing well-positioned (dark blue) and fuzzy nucleosomes (light blue). Two possible nucleosome families generate different nucleosome positioning in fuzzy regions (Fam1 and Fam2). Predicted nucleosome positions using nucleR and Danpos are shown in green and purple, respectively. (B) Comparison of nucleR and Danpos for detection of a second family of nucleosomes (light blue nucleosome in the bottom-right panel). Y-axis shows the number of cells required in the second family in order to be detected by the algorithm. (C) Distance between the dyads identified by nucleR (green) and DANPOS (purple) to the dyad position in the true synthetic nucleosome map for fuzzy and well positioned nucleosomes. (D) Sensitivity of the EVICTION prediction for NucDyn, DANPOS and Dimnp. Evictions were simulated removing reads from a given percentage of families (10%, 20%, …, 90%) and were identified from DANPOS output as a nucleosome with point_log2FC < −1 and point_diff_FDR < 0.01 (point with highest difference in the two samples, as reported by the software), and with default parameters for Dimnp. (E) Sensitivity of the SHIFT prediction computed on synthetic nucleosome maps. Shifts were introduced displacing reads from 1 to 5 DNA turns and modifying different percentages of the families (10%, 20%, …, 90%).
NucDyn
We tested the ability of our method to detect rearrangements in the nucleosome architecture using again our controlled in-silico MNase-seq data, simulating displacement (shift), insertion or eviction of one nucleosome, occurring in a different percentage of the cells. Sizeable changes such as nucleosome insertion or eviction are detected with good sensitivity by our method, even when they affect a relatively small percentage of cells (Figure 3D), while DANPOS or Dimnp only detect such changes when affecting a very large proportion of cells. Small nucleosome shifts (implying less than one turn of DNA) are not detectable by our algorithm unless they occur in a large percentage of the cells; while shifts implying a displacement of at least two turns of DNA (20 base pairs) are detectable with good sensitivity, even when affecting less than half of the cellular population (see Figure 3E). In this case, the comparison with other programs is difficult, as only DANPOS (15) allows an indirect way to detect nucleosome shifts by looking at distances between nucleosome peaks in both experiments. Unfortunately, with our synthetic data, DANPOS achieved poor sensitivity (less than 0.20 for 5 turns of DNA shift in 70% of cells and <0.1 for 3 turns shifts affecting also 70% of cells; see Supplementary Figure S5).
In summary, analysis of well-controlled in silico data shows that the Nucleosome Dynamics package (including NucDyn and nucleR) is not only a very powerful tool to define nucleosome families from MNase-seq experiments performed with a population of cells, but also a powerful approach to detect subtle changes in nucleosome architecture affecting a percentage of the cells in the studied sample.
Test cases
In order to illustrate the information derived from Nucleosome Dynamics, we applied our method in different real cases where experimental MNase-seq data were available. It is important to mention that the biological relevance of this type of comparison depends on the quality of the data and especially on the similar level of MNase digestion of the samples being compared. Indeed, several groups, including ours, have demonstrated the impact of the level of MNase digestion on the final nucleosome maps in several organisms, essentially at the level of the so-called ‘fragile’ nucleosomes (19,36–38). To illustrate this observation, we took advantage of the extensive study made by (36) and used Nucleosome Dynamics to compare nucleosome positioning in the input of two H2B and two H4 MNase-ChIP-seq samples, one under-digested and one over-digested (50U and 400 U of MNase respectively for H2B; 25 U and 300 U MNase respectively for H4). First, we focused on the H2B-input samples and confirmed that the number of nucleosome detected by nucleR decreases as the amount of MNase increases (from 80 160 down to 72 775, Supplementary Table S5) which is corroborated by the detection by NucDyn of 3559 evictions genome wide (Supplementary Table S6). At the promoter level, the proportion of W-open-W TSS increases from 123 to 2026 while the W-close-W TSS decreases from 2656 down to 346 (Supplementary Figure S6A). Regarding the phasing analysis, the percentage of phased genes does not change significantly due to the level of MNase digestion (Supplementary Figure S6B). Similar numbers were obtained for the H4-input samples. Hence, it is important to control MNase digestion level before using Nucleosome Dynamics package. Another technique that is not influenced by the level of MNase digestion is chemical cleavage mapping. NucleR can be applied to map nucleosome positions using the coverage obtained from these experiments (Supplementary Figure S7),
Cell cycle
The first example comes from the analysis of the changes in nucleosome organization occurring along the cell cycle in yeast, using our own previously published data (39). As shown in Figure 4A, the nucleR module of the Nucleosome Dynamics package suggests that nucleosomes tend to be fuzzier (F) in S and M phases compared to G1 and G2 phases. The increase in fuzziness in S and M phases impacts on the promoter classification as the number of W-open-W and W-closed-W promoters decreases compared to G1 and G2 (Figure 4B), but overall the ratio of closed/open NFRs (nucleosome free regions) is not dramatically altered along cell cycle (Supplementary Figure S8). Very interestingly, the changes in nucleosome architecture detected by NucDyn are not randomly distributed along the genome, but appear to be localized in specific families of genes, which are related to cell cycle progression, as shown by Gene Ontology (GO) Enrichment Analysis (40) in Figure 4C. Examples of the detailed information provided by Nucleosome Dynamics for some specific genes are shown in Figure 4D, where we report nucleosome maps of PRY2 (a gene related to lipid transport), whose expression peaks in G1 phase, YHP1 (involved in negative regulation of transcription of certain cell cycle genes), and GIC1 (a GTPase-interaction component involved in mitosis regulation) expressed in S phase (39). In the three cases, expression changes correlate with significant variation in nucleosome architecture at the promoter region between two stages of cell cycle. Typically, eviction or shifts reducing the presence of nucleosome in the core promoter region are related to active states of the genes (4,19,28,41,42).
Figure 4.

Nucleosome Dynamics along the cell cycle. (A) Percentage of fuzzy and well-positioned nucleosomes and (B) promoter classification (number of genes in each class) for every cell cycle stage. (C) GO terms enriched in genes with nucleosome changes between G1 and S detected by NucDyn. (D) Example of three cell-cycle dependent genes that present differential nucleosome architectures between G1 and S. In gray, the normalized coverage from the BAM files of the two cell cycle stages, 500 bp upstream and 1000 bp downstream the TSS. Below each BAM file, the nucleosome calls obtained with nucleR are represented (dark blue for well-positioned nucleosomes, light blue for fuzzy nucleosomes). The fifth track contains shifts (yellow for positive, blue for negative), inclusions (green) and evictions (red) identified by NucDyn.
Yeast metabolic cycle
A second example of use of our tool was the comparison of nucleosome architecture amongst cells at different stages of the yeast metabolic cycle (YMC). We took advantage here of high resolution experiments (43) in which the authors analysed simultaneously gene expression and MNase-seq maps at regularly spaced periods of time after adding fresh culture media. At two of these time points (T9 and T12 in Nocetti and Whitehouse 2016) dramatic changes of expression in genes related to reductive charging (poorly transcribed at T9 and highly transcribed at T12) and oxidative phase (highly transcribed at T9 and poorly transcribed at T12) have been detected. Analysis of global nucleosome architecture shows moderate changes between T9 and T12 (Figure 5A), but differences are more noticeable when the analysis is focused on Ox-genes (involved in amino acid synthesis, sulphur metabolism, ribosome and RNA metabolism (44), which are expressed in T9 and repressed in T12) and R/C genes (involved in non-respiratory metabolism, protein degradation, autophagy and vacuole (44), which are expressed in T12 and repressed in T9). Nucleosome Dynamics allows the detection and quantification of the alterations in nucleosome architecture coupled to such changes in expression. Thus, for Ox-genes (Figure 5B), the fuzziness at the −1 nucleosome decreases when moving from T9 to T12, in agreement with the general rule that reduced NFR upstream the well positioned +1 nucleosome correlates with inactive genes. On the contrary, for R/C genes (Figure 5C) the NFR upstream well positioned +1 nucleosome enlarges, since the −1 nucleosomes become fuzzier, again in perfect agreement with the changes of expression. To discard any biases resulting from the MNase digestion conditions, we confirmed that the length of the sequenced fragments was comparable in both samples (Supplementary Figure S9). Examples of the detailed information provided by Nucleosome Dynamics for three Ox-genes (NSR1, RRP4 and SNU13, all of them related to ribosomal biogenesis and RNA metabolism) are shown in Figure 5D, where upon T9→T12 transition, shifts and even insertions are shown leading to a reduction in the width of the NFR upstream the TSS: a fingerprint of gene deactivation. Similarly, Figure 5E provides the same type of information for three R/C genes (CTA1, SUE1 and SAF1), which are associated respectively with peroxisome, cytochrome C degradation and proteasome (see above). In the three cases, T9→T12 transition is coupled with massive nucleosome eviction upstream the TSS, leading to open configurations of NFR, typical of highly expressed genes.
Figure 5.

Nucleosome Dynamics in two points of the YMC. Promoter classification in the two time points (T9 and T12) for (A) all genes, (B) genes from the Ox cluster and (C) genes from the R/C cluster. (D and E) Example of three genes from Ox and 3 from R/C clusters that present differential nucleosome architectures between T9 and T12. In grey, the normalized coverage from the BAM files of the two time points, 500 bp upstream and 1000 bp downstream the TSS. Below each BAM file, the nucleosome calls obtained with nucleR are represented (dark blue for well-positioned nucleosomes, light blue for fuzzy nucleosomes). The fifth track contains shifts (yellow for positive, blue for negative), inclusions (green) and evictions (red) identified by NucDyn.
Changes in nutrients
As a last test, we applied Nucleosome Dynamics to explore the modifications in nucleosome architecture in yeast, linked to the change in the media from glucose-rich to either galactose-rich or ethanol-rich (45). Changes in the TSS nucleosome architecture classification occur among the three conditions (Figure 6A). There are not complete expression data in Kaplan et al. 2009, but we expected that replacement of glucose by galactose in the media would imply changes in expression in genes related to carbohydrate metabolism and transport which encouragingly, are those where sizeable changes in nucleosome architecture are detected by Nucleosome Dynamics (Figure 6B). Similarly, replacement of glucose by ethanol was expected to have an impact on the cell through: (i) expression of stress response genes, (ii) changing completely hexose metabolism in the absence of hexoses and (iii) eliminating ethanol through oxidation generating changes in the redox state of the cell which need to be corrected (46). Very encouragingly again, genes involved in stress response, hexose metabolism and redox activities are those for which the largest changes in nucleosome architecture have been detected (Figure 6C).
Figure 6.

Nucleosome Dynamics under different nutrient conditions. (A) Promoter classification in glucose, galactose and ethanol rich media. (B and C) GO terms enriched in genes with nucleosome changes detected by NucDyn, changing the medium from glucose to galactose or ethanol, respectively. (D and E) Example of three genes involved in galactose and ethanol metabolism, respectively, that present differential nucleosome architectures depending on the carbon source. In gray, the normalized coverage from the BAM files of the two cell cycle stages, 500 bp upstream and 1000 bp downstream the TSS. Below each BAM file, the nucleosome calls obtained with nucleR are represented (dark blue for well-positioned nucleosomes, light blue for fuzzy nucleosomes). The fifth track contains shifts (yellow for positive, blue for negative), inclusions (green) and evictions (red) identified by NucDyn.
We analysed in detail some genes which are expected to change dramatically their expression upon glucose→galactose substitution, as they are crucial to integrate galactose in normal hexose metabolism: GAL1, coding for a Galactokinase, GAL10, coding for the UDP-glucose-4-epimerase, and GAL7, coding for the galactose-1-phosphate uridyl transferase (Figure 6D). In the three cases evictions and shifts (in some cases noticeably) generate wider NFRs upstream the gene, changes that in some cases extend to the coding regions and that signal a pronounced increase in expression of these galactose-related genes.
A similar detailed analysis was made for three genes which are expected to be overexpressed when ethanol substitutes glucose as energy source: two stress response genes HSP26 and HSP12, and HXK1, a hexokinase activated when cells are shifted to a non-fermentable carbon source such as ethanol (47). Results in Figure 6E illustrate the magnitude of the changes (mainly evictions) detected by Nucleosome Dynamics, which affect the NFR, and even in some cases the coding regions.
DISCUSSION
Different studies demonstrated that nucleosome architecture is coupled to gene function (4,43,48) and that transcriptional activity and nucleosome architecture are tightly coupled. Unfortunately, detecting changes in nucleosome architecture is complex as nucleosomes are dynamic and even a population of ‘identical’ well synchronized and grown under identical conditions cells might have nucleosomes placed at different positions. This, combined with the intrinsic uncertainties of MNase- or ATAC-seq experiments, generate noisy data which are difficult to process for precisely locating nucleosomes and even more difficult to detect significant changes in nucleosome arrangements due to internal or external stimuli. The suite of programs incorporated in Nucleosome Dynamics allows not only a robust location of nucleosomes, even in cases of heterogeneous pools of cells, but also the detection of changes in nucleosome arrangements, even those affecting a moderate population of cells. To increase its utility, Nucleosome Dynamics is integrated into a powerful virtual research environment, where it is combined with different tools for analysis of data and visualization in the context of genomic metadata, which help the user not only to analyse nucleosome architecture and dynamics, but also to put them in the context of known genomic information (Figure 2).
We validated the methodology using synthetic data that mimic typical MNase-seq maps, in which the positions of the nucleosome in the different cells are unambiguously known. The two main modules of Nucleosome Dynamics (nucleR and NucDyn) perform very well capturing cellular diversity and detecting shifts, evictions and inclusions that affect a moderate percentage of the cellular population. Furthermore, we tested the power of the methodology by exploring nucleosome rearrangements occurring along cell cycle, yeast metabolic cycle, and those linked to the change in the source of energy from glucose to galactose or ethanol. In the tested cases, Nucleosome Dynamics provides accurate global and local descriptions of nucleosome structure and dynamics and deciphers the nature of the connection between nucleosome organization and gene expression.
DATA AVAILABILITY
Raw MNase-seq datasets reported as test cases in this study were obtained from the ENA-SRA website (http://www.ebi.ac.uk/ena) and the GEO repository under accession numbers: PRJEB6970 for the cell cycle data, GSE77631 for the yeast metabolic cycle, GSE13622 for the nucleosome maps from yeast cultivated in glucose, galactose and ethanol media, and GSE83123 for the different levels of MNase digestion. Processed chemical cleavage data was obtained from GSE97290.
The processed test data and benchmarking synthetic data supporting the conclusions of this article are available in Zenodo repository (10.5281/zenodo.2632999), and can be incorporated to both MuGVRE and Galaxy Nucleosome Dynamics installations for additional testing.
Supplementary Material
ACKNOWLEDGEMENTS
We are indebted all members of the MuG consortia for help and for acting as a beta-tester of the software.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
M.O. is an ICREA (Institució Catalana de Recerca i Estudis Avancats) academia researcher; Spanish Ministry of Science [RTI2018-096704-B-100]; Catalan Government [2017-SGR-134]; Instituto de Salud Carlos III–Instituto Nacional de Bioinformática, the European Union's Horizon 2020 research and innovation program, and the Biomolecular and Bioinformatics Resources Platform [ISCIII PT 17/0009/0007 co-funded by the Fondo Europeo de Desarrollo Regional FEDER; Grants Elixir-Excelerate: 676559 and BioExcel2: 823830; ERC:812850; MuG-676566]; MINECO Severo Ochoa Award of Excellence from the Government of Spain (awarded to IRB Barcelona). Funding for open access charge: Spanish Ministry of Science [RTI2018-096704-B-100].
Conflict of interest statement. None declared.
REFERENCES
- 1. Barski A., Cuddapah S., Cui K., Roh T.-Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K.. High-Resolution profiling of histone methylations in the human genome. Cell. 2007; 129:823–837. [DOI] [PubMed] [Google Scholar]
- 2. Mavrich T.N., Jiang C., Ioshikhes I.P., Li X., Venters B.J., Zanton S.J., Tomsho L.P., Qi J., Glaser R.L., Schuster S.C. et al.. Nucleosome organization in the Drosophila genome. Nature. 2008; 453:358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yuan G.-C. Genome-Scale Identification of Nucleosome Positions in S. cerevisiae. Science. 2005; 309:626–630. [DOI] [PubMed] [Google Scholar]
- 4. Lai W.K.M., Pugh B.F.. Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat. Rev. Mol. Cell Biol. 2017; 18:548–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rando O.J., Ahmad K.. Rules and regulation in the primary structure of chromatin. Curr. Opin. Cell Biol. 2007; 19:250–256. [DOI] [PubMed] [Google Scholar]
- 6. Jiang C., Pugh B.F.. Nucleosome positioning and gene regulation: advances through genomics. Nat. Rev. Genet. 2009; 10:161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Raveh-Sadka T., Levo M., Shabi U., Shany B., Keren L., Lotan-Pompan M., Zeevi D., Sharon E., Weinberger A., Segal E.. Manipulating nucleosome disfavoring sequences allows fine-tune regulation of gene expression in yeast. Nat. Genet. 2012; 44:743–750. [DOI] [PubMed] [Google Scholar]
- 8. Collings C.K., Anderson J.N.. Links between DNA methylation and nucleosome occupancy in the human genome. Epigenet. Chromatin. 2017; 10:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kubik S., O’Duibhir E., de Jonge W.J., Mattarocci S., Albert B., Falcone J.-L., Bruzzone M.J., Holstege F.C.P., Shore D.. Sequence-directed action of RSC remodeler and general regulatory factors modulates +1 nucleosome position to facilitate transcription. Mol. Cell. 2018; 71:89–102. [DOI] [PubMed] [Google Scholar]
- 10. Mellor J., Morillon A.. ISWI complexes in Saccharomyces cerevisiae. Biochim. Biophys. Acta (BBA) - Gene Struct. Expression. 2004; 1677:100–112. [DOI] [PubMed] [Google Scholar]
- 11. Knight B., Kubik S., Ghosh B., Bruzzone M.J., Geertz M., Martin V., Dénervaud N., Jacquet P., Ozkan B., Rougemont J. et al.. Two distinct promoter architectures centered on dynamic nucleosomes control ribosomal protein gene transcription. Genes Dev. 2014; 28:1695–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Whitehouse I., Flaus A., Cairns B.R., White M.F., Workman J.L., Owen-Hughes T.. Nucleosome mobilization catalysed by the yeast SWI/SNF complex. Nature. 1999; 400:784–787. [DOI] [PubMed] [Google Scholar]
- 13. Whitehouse I., Stockdale C., Flaus A., Szczelkun M.D., Owen-Hughes T.. Evidence for DNA translocation by the ISWI chromatin-remodeling enzyme. Mol. Cell Biol. 2003; 23:1935–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J.. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013; 10:1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen K., Xi Y., Pan X., Li Z., Kaestner K., Tyler J., Dent S., He X., Li W.. DANPOS: Dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res. 2013; 23:341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen W., Liu Y., Zhu S., Green C.D., Wei G., Han J.-D.J.. Improved nucleosome-positioning algorithm iNPS for accurate nucleosome positioning from sequencing data. Nat. Commun. 2014; 5:4909. [DOI] [PubMed] [Google Scholar]
- 17. Flores O., Orozco M.. nucleR: a package for non-parametric nucleosome positioning. Bioinformatics. 2011; 27:2149–2150. [DOI] [PubMed] [Google Scholar]
- 18. Teif V.B. Nucleosome positioning: resources and tools online. Brief. Bioinform. 2016; 17:745–757. [DOI] [PubMed] [Google Scholar]
- 19. Flores O., Deniz O., Soler-Lopez M., Orozco M.. Fuzziness and noise in nucleosomal architecture. Nucleic Acids Res. 2014; 42:4934–4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deniz Ö., Flores O., Battistini F., Pérez A., Soler-López M., Orozco M.. Physical properties of naked DNA influence nucleosome positioning and correlate with transcription start and termination sites in yeast. BMC Genomics. 2011; 12:489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gutiérrez G., Millán-Zambrano G., Medina D.A., Jordán-Pla A., Pérez-Ortín J.E., Peñate X., Chávez S.. Subtracting the sequence bias from partially digested MNase-seq data reveals a general contribution of TFIIS to nucleosome positioning. Epigenet. Chromatin. 2017; 10:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brogaard K., Xi L., Wang J.-P., Widom J.. A map of nucleosome positions in yeast at base-pair resolution. Nature. 2012; 486:496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chereji R.V., Ramachandran S., Bryson T.D., Henikoff S.. Precise genome-wide mapping of single nucleosomes and linkers in vivo. Genome Biol. 2018; 19:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Voong L.N., Xi L., Sebeson A.C., Xiong B., Wang J.-P., Wang X.. Insights into nucleosome organization in mouse embryonic stem cells through chemical mapping. Cell. 2016; 167:1555–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thakur J., Talbert P.B., Henikoff S.. Inner kinetochore protein interactions with regional centromeres of fission yeast. Genetics. 2015; 201:543–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moyle-Heyrman G., Zaichuk T., Xi L., Zhang Q., Uhlenbeck O.C., Holmgren R., Widom J., Wang J.-P.. Chemical map of Schizosaccharomyces pombe reveals species-specific features in nucleosome positioning. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:20158–20163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Henikoff S., Ramachandran S., Krassovsky K., Bryson T.D., Codomo C.A., Brogaard K., Widom J., Wang J.-P., Henikoff J.G.. The budding yeast Centromere DNA Element II wraps a stable Cse4 hemisome in either orientation in vivo. eLife. 2014; 3:e01861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu L., Xie J., Sun X., Luo K., Qin Z.S., Liu H.. An approach of identifying differential nucleosome regions in multiple samples. BMC Genomics. 2017; 18:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. R Core Team. R-A Language and Environment for Statistical Computing. 2016; Vienna: R Foundation for Statistical Computing. [Google Scholar]
- 30. Codó L., Bayarri G., Cid-Fuentes J.A., Conejero J., Hospital Adam, Royo R., Repchevsky D., Pasi M., Meletiou A., McDowall M.D. et al.. MuGVRE. A virtual research environment for 3D/4D genomics Bioinformatics. 2019;
- 31. Afgan E., Baker D., Batut B., van den Beek M., Bouvier D., Čech M., Chilton J., Clements D., Coraor N., Grüning B.A. et al.. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018; 46:W537–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Giancoli D.C. Physics for Scientists & Engineers with Modern Physics. 2000; 3rd ednUpper Saddle River: Prentice Hall. [Google Scholar]
- 33. Kolesnikov N., Hastings E., Keays M., Melnichuk O., Tang Y.A., Williams E., Dylag M., Kurbatova N., Brandizi M., Burdett T. et al.. ArrayExpress update—simplifying data submissions. Nucleic Acids Res. 2015; 43:D1113–D1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Buels R., Yao E., Diesh C.M., Hayes R.D., Munoz-Torres M., Helt G., Goodstein D.M., Elsik C.G., Lewis S.E., Stein L. et al.. JBrowse: a dynamic web platform for genome visualization and analysis. Genome Biol. 2016; 17:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler A. D.. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chereji R.V., Ocampo J., Clark D.J.. MNase-sensitive complexes in yeast: nucleosomes and non-histone barriers. Mol. Cell. 2017; 65:565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chereji R.V., Kan T.-W., Grudniewska M.K., Romashchenko A.V., Berezikov E., Zhimulev I.F., Guryev V., Morozov A.V., Moshkin Y.M.. Genome-wide profiling of nucleosome sensitivity and chromatin accessibility in Drosophila melanogaster. Nucleic Acids Res. 2016; 44:1036–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jeffers T.E., Lieb J.D.. Nucleosome fragility is associated with future transcriptional response to developmental cues and stress in C.elegans. Genome Res. 2017; 27:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Deniz Ö., Flores O., Aldea M., Soler-López M., Orozco M.. Nucleosome architecture throughout the cell cycle. Sci. Rep. 2016; 6:19729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Falcon S., Gentleman R.. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007; 23:257–258. [DOI] [PubMed] [Google Scholar]
- 41. Teif V.B., Vainshtein Y., Caudron-Herger M., Mallm J.-P., Marth C., Höfer T., Rippe K.. Genome-wide nucleosome positioning during embryonic stem cell development. Nat. Struct. Mol. Biol. 2012; 19:1185–1192. [DOI] [PubMed] [Google Scholar]
- 42. Chen J., Li E., Zhang X., Dong X., Lei L., Song W., Zhao H., Lai J.. Genome-wide nucleosome occupancy and organization modulates the plasticity of gene transcriptional status in maize. Mol. Plant. 2017; 10:962–974. [DOI] [PubMed] [Google Scholar]
- 43. Nocetti N., Whitehouse I.. Nucleosome repositioning underlies dynamic gene expression. Genes Dev. 2016; 30:660–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tu B.P., Kudlicki A., Rowicka M., McKnight S.L.. Logic of the yeast metabolic Cycle: Temporal compartmentalization of cellular processes. Science. 2005; 310:1152–1158. [DOI] [PubMed] [Google Scholar]
- 45. Kaplan N., Moore I.K., Fondufe-Mittendorf Y., Gossett A.J., Tillo D., Field Y., LeProust E.M., Hughes T.R., Lieb J.D., Widom J. et al.. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009; 458:362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stanley D., Bandara A., Fraser S., Chambers P.J., Stanley G.A.. The ethanol stress response and ethanol tolerance of Saccharomycescerevisiae. J. Appl. Microbiol. 2010; 109:13–24. [DOI] [PubMed] [Google Scholar]
- 47. Rodríguez A., Cera T. de la, Herrero P., Moreno F.. The hexokinase 2 protein regulates the expression of the GLK1, HXK1 and HXK2 genes of Saccharomyces cerevisiae. Biochem. J. 2001; 355:625–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bai L., Morozov A.V.. Gene regulation by nucleosome positioning. Trends Genet. 2010; 26:476–483. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw MNase-seq datasets reported as test cases in this study were obtained from the ENA-SRA website (http://www.ebi.ac.uk/ena) and the GEO repository under accession numbers: PRJEB6970 for the cell cycle data, GSE77631 for the yeast metabolic cycle, GSE13622 for the nucleosome maps from yeast cultivated in glucose, galactose and ethanol media, and GSE83123 for the different levels of MNase digestion. Processed chemical cleavage data was obtained from GSE97290.
The processed test data and benchmarking synthetic data supporting the conclusions of this article are available in Zenodo repository (10.5281/zenodo.2632999), and can be incorporated to both MuGVRE and Galaxy Nucleosome Dynamics installations for additional testing.