


Abstract
Modification dependent restriction endonucleases (MDREs) often have separate catalytic and modification dependent domains. We systematically looked for previously uncharacterized fusion proteins featuring a PUA or DUF3427 domain and HNH or PD-(D/E)XK catalytic domain. The enzymes were clustered by similarity of their putative modification sensing domains into several groups. The TspA15I (VcaM4I, CmeDI), ScoA3IV (MsiJI, VcaCI) and YenY4I groups, all featuring a PUA superfamily domain, preferentially cleaved DNA containing 5-methylcytosine or 5-hydroxymethylcytosine. ScoA3V, also featuring a PUA superfamily domain, but of a different clade, exhibited 6-methyladenine stimulated nicking activity. With few exceptions, ORFs for PUA-superfamily domain containing endonucleases were not close to DNA methyltransferase ORFs, strongly supporting modification dependent activity of the endonucleases. DUF3427 domain containing fusion proteins had very little or no endonuclease activity, despite the presence of a putative PD-(D/E)XK catalytic domain. However, their expression potently restricted phage T4gt in Escherichia coli cells. In contrast to the ORFs for PUA domain containing endonucleases, the ORFs for DUF3427 fusion proteins were frequently found in defense islands, often also featuring DNA methyltransferases.
INTRODUCTION
Modification dependent restriction endonucleases (MDREs) are defined as enzymes that restrict (cleave) DNA much better or only if the substrate is modified. Traditionally, such enzymes are divided into Type IIM and Type IV REases (1). Type IIM restriction endonucleases, such as DpnI or BisI, cleave modified DNA in the immediate vicinity of one or several modified bases (2,3). In contrast, Type IV endonucleases, such as EcoKMcrBC, SauUSI or GmrSD, cleave at a (variable) distance from the site of modification (4–6). Typically, such enzymes are dependent on ATP or GTP, which is thought to power translocation from modification to cleavage sites.
MDREs that have both Type IIM and Type IV properties have recently attracted much attention. These enzymes cleave DNA at a distance from one or several modification sites, and they do so without requiring ATP or GTP hydrolysis. Architecturally, they are fusion proteins that combine a modification dependent DNA binding domain and a catalytic domain. With few exceptions, such as EcoK-McrA (7), most studies have been focused on fusion proteins featuring a domain classified as SRA, and a catalytic domain of the PD-(D/E)XK or HNH type. Examples include the SRA-PD-(D/E)XK endonucleases MspJI, AspBHI or LpnPI, the PD-(D/E)XK-SRA endonucleases PvuRts1I or AbaSI (8,9), and the SRA-HNH endonucleases ScoA3IV (Sco5333) and TagI (10,11). All these enzymes cleave DNA containing modified cytosine bases, in some cases dependent on sequence context (12). The precise requirements for cytosine modifications differ. Some enzymes preferentially cleave DNA containing 5-methylcytosine (5mC) or 5-hydroxymethylcytosine (5hmC) (11), but not glucosyl-5-hydroxymethylcytosine (g5hmC), whereas others cleave DNA containing 5hmC and g5hmC, but not 5mC (8,9). Crystal structures show that SRA domains extrude a modified base from the DNA and scrutinize it in a dedicated pocket (13–16).
The SRA domains in typical MDREs belong to a larger family of nucleic acid binding domains, collectively termed PUA superfamily (Figure 1). The name stems from two enzymes acting on RNA, PseudoUridine synthase and Archaeosine transglycosylase (17). According to the INTERPRO database the superfamily comprises the PUA domains in the strict sense, EVE domains (IPR002740), SRA domains (IPR040674) and SRA-YDG (IPR003105) domains (18). All of them are built around a five-stranded mixed β-sheet, which is sometimes termed a ‘pseudobarrel’ (19) (Supplementary Figure S1). Despite the overall similar fold, the subgroups within the PUA family are structurally clearly distinguishable (Supplementary Figures S2 and S3). Unlike SRA domains, PUA domains (in the strict sense) and EVE domains are considered as RNA binding domains (17,19,20). However, a possible exception emerged when the EVE-domain protein Thy28 was identified in a SILAC screen for proteins that bind 5hmC in DNA (21).
Figure 1.
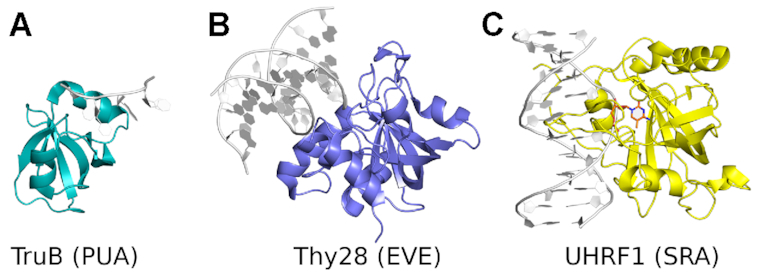
Structure of representative PUA superfamily domains. Proteins are shown in ribbon representation in color, nucleic acids are shown in grey. (A) PUA domain of TruB (53). (B) EVE domain of Thy28 (PDB ID: 5J3E, unpublished). (C) SRA domain of UHRF1 (13). Specifically recognized modified base (5mC) is highlighted in color.
SauUSI is an ATP-dependent Type IV MDRE that restricts 5mC and 5hmC modified DNA (S5mCNGS or S5hmCNGS) with variable cleavage locations. SauUSI contains three functional domains: N-terminal PLD family endonuclease domain, ATPase/DNA helicase domain in the middle, and C-terminal specificity domain (TRD, target recognition domain, DUF3427) (5). SauUSI-like restriction enzymes are widespread in bacteria. In some cases, the PLD endonuclease catalytic domain is fused to an ATPase/helicase domain, and the specificity domain is located in proximity as a separate protein.
Here, we report the results of a search for proteins featuring a putative modification sensing domain of either the PUA superfamily or the DUF3427 family, and an HNH or PD-(D/E)XK catalytic domain in sequenced microbial genomes and metagenomes. Several new groups of potentially modification dependent restriction endonucleases could be identified in multiple species. Representative cases were chosen for experimental characterization, and modification dependent activity, in vitro and in phage restriction assays, could be demonstrated for several protein families. The genome of Streptomyces coelicolor A3(2) is rich in predicted MDREs. Candidates were also chosen from this species, and the genomic DNA was analyzed for DNA methylation using PacBio real-time sequencing.
MATERIALS AND METHODS
Enzymes, strains and expression vectors
DNA restriction endonucleases (REases), methyltransferases (MTases), Phusion, Q5 and Taq DNA polymerases were provided by New England Biolabs, Inc (NEB). Escherichia coli K strains NEB 5α (Dcm+ RecA−), 10β (Dcm+ RecA−), Turbo (Dcm+ RecA+); and E. coli B strain C2566 (T7 Express), Dam-deficient T7 expression strain ER2948 were from Dr Elisabeth Raleigh's collection (NEB). Streptomyces coelicolor A3(2) strain was purchased from ATCC and its genomic DNA was prepared by a Qiagen genomic DNA kit. The IMPACT™ protein expression and purification system (pTXB1 vector and chitin beads) was provided by NEB (22). The target genes (PCR product or synthetic gene blocks from IDT) were inserted into pTXB1 (NdeI and XhoI digested) in fusion with an intein and a chitin-binding domain (CBD) using NEBuilder HiFi DNA assembly kit (NEB). Target proteins were purified through a chitin affinity column and DTT cleavage in the elution buffer (16–32 h). A few restriction genes were also cloned into T7 expression vector pET21b (NdeI and XhoI) with a C-terminal 6xHis tag for the purpose of testing phage restriction in vivo.
Escherichia coli ER2948 (Dam−) cells were made competent by CaCl2 treatment (23). The plasmid carrying cells were grown in antibiotic selective broth (phage broth with 0.1 mg/ml Amp) to late log phase and concentrated 10-fold by centrifugation. Cells were plated with soft agar. Diluted phages were spotted onto the cell lawns and incubated overnight at 37°C. Reduced plaque formation indicates restriction of phage propagation inside the cells.
Restriction buffers, modified and unmodified DNA substrates
Restriction buffers (NEBuffers) 1.1 (low salt), 2.1 (medium salt), 3.1 (high salt), and CutSmart™ buffer (buffer 4 + BSA) (www.neb.com) were used in all digestions unless specified otherwise. NEBuffer 1.1 (restriction buffer 1.1) contains 10 mM bis–Tris–propane–HCl, 10 mM MgCl2, 0.1 mg/ml BSA, pH 7.0 at 25°C. NEBuffer 2.1 (restriction buffer 2.1) contains 50 mM NaCl, 10 mM Tris–HCl, 10 mM MgCl2, 0.1 mg/ml BSA, pH 7.9 at 25°C. NEBuffer 3.1 (restriction buffer 3.1) contains 100 mM NaCl, 50 mM Tris–HCl, 10 mM MgCl2, 0.1 mg/ml BSA, pH 7.9 at 25°C. CutSmart buffer contains 50 mM KAc, 20 mM Tris-Ac, 10 mM MgAc2, 0.1 mg/ml BSA, pH 7.9 at 25°C (www.neb.com). To test divalent cation requirement of a particular REase, the medium salt buffer containing 10 mM Tris–HCl (pH 7.5), 50 mM NaCl and 1 mM DTT was used and supplemented with various divalent cations such as MnCl2 or CoCl2 (1 mM). The 5mC-modified DNA substrates included: Dcm+ pBR322 (C5mCWGG), pBRFM+ (pBR322 carrying the fnu4HIM gene, G5mCNGC), CpGM pBR322 (M.SssI, 5mCpG), 5mC-PCR fragment amplified by Q5 DNA polymerase (supplemented with GC enhancer) in dNTP mix with 5m-dCTP (NEB) replacing dCTP. The 5hmC-modified DNA substrates included: phage T4gt DNA prepared by a standard CsCl2 and phenol extraction procedure (23), PCR DNA fragment amplified by Phusion DNA polymerase in dNTP mix with 5hmdCTP (Zymo Research) replacing dCTP. WT phage T4 DNA contains g5hmC. T4GT7 with unmodified cytosines was a gift from Dr. Geoffrey Wilson (NEB). Phage λ DNA (partially modified by Dcm methyltransferase) and λvir were provided by NEB. Bacillus SP8 phage containing 5hmdU was a gift from Dr Peter Weigele (NEB). 5hmdU-PCR DNA (1.2 kb) was amplified from pBR322 using Taq DNA polymerase in dNTP mix with 5hmdUTP (TriLink Biotechnologies) replacing dTTP.
Adenine methylated plasmid substrates
M.EcoGII (NEB) was used to methylate adenine bases in DNA irrespective of the sequence context. The efficiency of the methylation reaction was monitored by digestion with a panel of restriction endonucleases. HpaII (CCGG) should cut irrespective of the adenine methylation status. Activity of MluCI (AATT) and MnlI (CCTC, bottom strand GAGG) should be impaired by the presence of 6mA, and DpnI (G6mATC) should be dependent on adenine methylation for DNA cleavage (24). The digestion results for M.EcoGII modified pBR322 plasmid were consistent with the expected widespread, context independent methylation. As an alternative to in vitro methylation, we used in vivo self-methylation of a vector carrying the M.EcoGII methyltransferase gene. As already shown earlier, the pRRS-ecoGIIM (high copy number plasmid) with ecoGIIM gene under Plac promoter converted ∼86–88% of adenines to 6mA in overnight cell culture with Amp selection (25).
Mapping of cut/nick sites by sequencing and consensus compilation with WebLogo
DNA run-off sequencing was carried out to map the cut/nick sites in digested plasmid or PCR DNA, using a BigDye™ terminator V3.1 cycle DNA sequencing kit from ABI (Thermo-Fisher) (26). Taq DNA polymerase adds an extra adenine base at the position where the template is cleaved (e.g. by restriction or nicking endonuclease), thus creating G/A, C/A or T/A doublets. The A/A doublet is underrepresented in partial digestions, unless there is sharp drop-off in sequencing peaks after the A peak. In some cases, the A/A doublet was inferred from the extra high peak of A compared to the peak height of uncut template DNA. Cut/nick site consensus sequences were compiled with WebLogo server (https://weblogo.berkeley.edu) (27).
SMRT bell library construction and SMRT sequencing
Genomic DNA was purified from cultured S. coelicolor A3(2) cells using the genomic DNA (gDNA) purification kit (Qiagen). The genome was sequenced using the Pacific Biosciences (PacBio) RSII sequencing platform as previously described (28,29). Briefly, SMRT bell libraries were constructed from a gDNA sample sheared to ∼10–20 kb using the G-tubes protocol (Covaris). The sheared ends were repaired and ligated to PacBio hairpin adapters according to the manufacturer's protocols. Incompletely formed SMRT bell templates and linear DNA were digested by Exonucleases III and VII (NEB). DNA quantification and the quality of the library was analyzed using the Qubit fluorimeter (Invitrogen, Eugene, OR) and 2100 Bioanalyzer (Agilent Technology). Two 18-kb SMRT bell libraries were prepared according to PacBio sample preparation protocols for 20 kb libraries and sequenced using C2-P4 chemistry (5 SMRT cells, 120 min collection times). Software provided by Pacific Biosciences or developed in house was used to detect modified bases (Interpulse duration, IPD ratios) in DNA sequencing. An IPD ratio >1 means that the sequencing polymerase slowed down (relative to the control) at this base position.
Genetic neighborhood mapping
The INTERPRO and UniProt databases were used to identify all listed combinations of SRA, EVE and PUA domains with HNH domains (18). The INTERPRO identifiers were then mapped to EBI identifiers, which in turn were used to query the NCBI Nuccore database using the efetch utility in batch mode (https://www.ncbi.nlm.nih.gov/books/NBK179288). Genomic regions were defined as ranging from 3 kb upstream of the start codon to 3 kb downstream of the stop codon of the open reading frame of interest. Genetic neighborhoods were obtained as annotated GeneBank (gb) entries and analyzed semi-manually relying on gb annotations.
Structure modeling and conservation analysis
Sequence comparisons were done using NCBI, UniProt or BLAST tools. Domain architecture searches were done using the INTERPRO or CDD databases (18,30). The weak structural similarity of the N-terminal domain of ScoA3V and PUA-superfamily domains was identified using the Phyre2 server (31). Structural classification of the PUA superfamily was done with the help of the DALI server using all against all comparisons (32). Sequence based classification was done using the CLANS program (33). Residue conservation scores within a domain group were calculated and mapped to the surface of the structural models using the ConSurf server in automatic mode (34). Multiple amino acid sequence alignment was carried out using PROMALS3D method at the server: http://prodata.swmed.edu/promals3d/promals3d.php (35).
DALI heat maps for PUA superfamily domain structure comparisons
All structures of proteins containing annotated PUA superfamily domains were downloaded from the PDB. The selection was performed with the help of PFAM database and included 377 entries. The domains were isolated based on the PFAM chain IDs and domain boundaries. The domain sequences were clustered using CD-HIT into 69 clans. The low resolution and NMR structures were eliminated from the domain families with largest numbers of representatives to bring the number of structures down to 64 (maximum allowed in the next step). The 64 domains were then structurally compared using DALI server that generated both the tree as well as the histogram. The domain subfamilies were based on the PFAM assignment.
RESULTS
PUA-superfamily fusions with HNH and PD-(D/E)XK domains
Previous bioinformatics analysis suggested that EVE-HNH fusions may correspond to MDREs (20). We decided to expand the search for combinations of PUA-like and endonuclease domains. Inspection of the INTERPRO database indicated that fusions of PUA-superfamily domains with HNH endonuclease domains are common (Supplementary Table S1). Using UNIPROT annotations and BLASTP searches, we were able to further extend the list of candidate PUA-HNH enzymes. Moreover, we found at least one instance of a PUA-superfamily domain fused to a PD-(D/E)XK catalytic domain, YenY4I. In between the N-terminal PUA-like domain and the C-terminal PD-(D/E)XK domain, the protein has a DUF3427 domain. While the EVE-DUF3427-PD-(D/E)XK domain combination is rare, the DUF3427-PD-(D/E)XK combination is not. DUF3427 domains have previously been associated with modification dependent DNA recognition in the context of SauUSI endonuclease. DUF3427 domains have similar secondary structure elements to PUA-like domains, but it is currently unclear whether they belong to the PUA superfamily. Although this was not a requirement of our searches, the putative modification dependent domains were found to be N-terminal, and the putative catalytic domains C-terminal.
We subjected sequences of proteins of known structure with established family assignment, and the N-terminal, putatively modification sensing domains of candidate endonucleases to CLANS analysis (33) to determine family relationships (DUF3427 domains were included despite their uncertain relationship to PUA superfamily). The new fusion proteins (or rather, their putative modification sensing domains) clustered in several families, frequently close to established PUA families (Figure 2).
Figure 2.
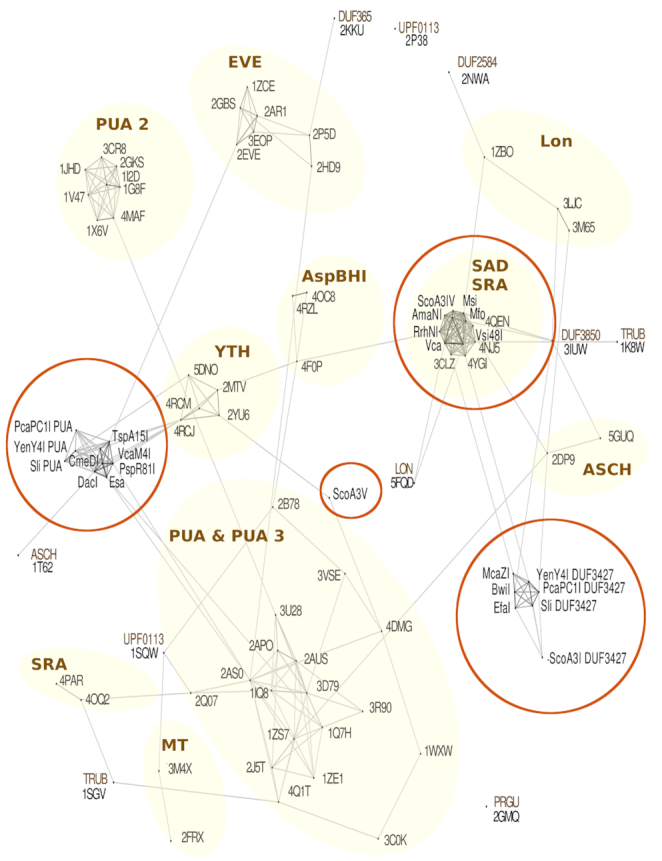
Clans analysis of putative modification dependent restriction endonucleases (MDREs). Sequences of PUA superfamily domains that have been confidently assigned to families within the PUA superfamily were obtained from the Protein Data Bank (PDB), and they are designated by four letter PDB IDs. Amino acid sequences of putative modification sensing domains of enzymes in this study were added. The combined sequences were subjected to CLANS analysis (33) to visualize the extent of similarity between domains. The clusters of sequences that share the same domain family annotation were marked with shaded background.
TspA15I group of HNH endonucleases
Enzymes in the TspA15I group have N-terminal domains with sequence similarity to EVE, YTH and PUA domains according to the CLANS analysis, and C-terminal HNH domains.
A proteobacterial representative (GenBank code: WP_020146688) from Thioalkalivibrio sp. ALJ15 (NZ_KB912059), named TspA15I based on the findings below, was chosen for further characterization. A hexa-histidine tagged TspA15I was expressed in the E. coli T7 Express strain (C2566) under the control of an inducible T7 promoter. Even when the protein was highly overexpressed, we did not observe significant toxicity, suggesting that the enzyme was at most mildly active on non-cytosine modified DNA in vivo. TspA15I was purified from overexpression lysates by nickel and heparin column chromatography.
TspA15I endonuclease activity was assayed using genomic DNA of T4GT7, T4gt and wild-type (WT) T4 phage. Phage T4 itself contains g5hmCs instead of cytosines in its genome. The T4GT7 variant was originally isolated by Wilson and coworkers for generalized transduction and carries mutations that prevent the replacement of cytosine bases, so that its genomic DNA contains non-modified cytosines (36). The T4gt phage differs from the WT T4 by glucosyltransferase deficiencies (α–,β–GT−), and therefore its gDNA contains 5hmCs (37).
TspA15I displayed endonuclease activity in medium salt buffer supplemented by Mg2+ or Mn2+ cations (Figure 3). In the presence of Mg2+ ions, even at high enzyme concentration, the activity on non-modified and g5hmC modified DNA was very weak and only 5hmC containing DNA was cleaved efficiently. TspA15I activity was much higher when Mn2+ was used as a co-factor. In Mn2+ containing buffer, concentrated TspA15I degraded all three gDNA types. Activity differences were apparent from the dilution series: less enzyme was required for ‘full’ degradation of 5hmC modified DNA than for g5hmC containing and non-modified substrates (Figure 3A). Phage restriction activity also showed the same (see below). TspA15I was most active in 1–10 mM Mn2+. When both Mn2+ and Mg2+ were present in the reaction buffer, the activity of the enzyme was slightly lower than with Mn2+ only, but still significantly higher on 5hmC modified gDNA (Supplementary Figure S4). The specific activity of TspA15I (0.5 to 1 × 103 U/mg protein) is relatively low compared to typical Type II REases (in the range of 104 to 106 U/mg protein) (38). Here, one unit of TspA15I is defined as the amount of protein to degrade 0.5 μg of phage T4gt DNA into fragments of less than 0.5 kb in 1 h at 37°C.
Figure 3.
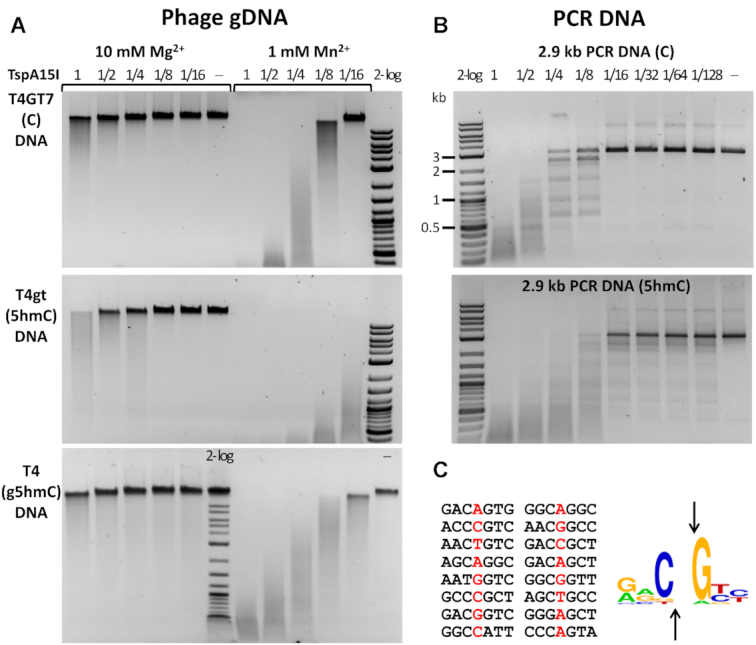
TspA15 restriction activity in vitro. (A) TspA15I digestion of phage T4GT7 (unmodified C), T4gt (5hmC) and WT T4 (g5hmC) gDNAs. Digestions were carried out in 10 mM Mg2+ or 1 mM Mn2+ buffer. (B) TspA15I digestion of C- and 5hmC-containing PCR DNA. 0.5 μg of PCR DNA (∼5.3 nM) was incubated with TspA15I enzyme in 2-fold serial dilutions, starting at 1 μg (280 nM) for 1 h in Mn2+ buffer. At 1/16 to 1/128 dilutions (∼17.5 nM to 2.2 nM range) the enzyme cleaves modified DNA more efficiently. The 2 log DNA ladder (2-log, 0.1 to 10 kb) was used for size marker in all presented digestions. (C) Mapping of cut/nick sites. The cut/nick sites were sequenced from 5hmC-modified PCR DNA digested by TspA15I in Mn2+ buffer (the base before the cut site is marked in red) and the 5hmCNG logo was generated with WebLogo (27).
We next evaluated TspA15I activity on DNA made by PCR from a 2′-deoxynucleoside triphosphate mix containing dCTP, 5mdCTP, or 5hmdCTP. The phage SP8 gDNA with 5hmdU was also included (5hmdU replacing all T in gDNA). TspA15I displayed higher activity on 5hmC containing DNA than on non-modified and 5mC-modified DNA (Figure 3B and Supplementary Figure S5). It cleaved 5hmdU containing DNA with efficiency similar to that observed for 5hmC (Supplementary Figure S5C). We conclude from these experiments that TspA15I behaves as a (g)5hmC and 5hmdU enhanced restriction endonuclease in vitro.
Run-off sequencing of 5hmC-modified PCR DNA digested by TspA15I in Mn2+ buffer indicated frequent double-stranded (ds) cuts on at 5hmCN↓G (Figure 3C, Supplementary Figure S6). The digestion of a pBR322 (Dcm+) plasmid in the Mg2+ buffer suggested that the nuclease domain makes dsDNA cleavage at the GACN↓GTC symmetric sites and single-stranded (ss) nicks at the related but asymmetric sites (Supplementary Figure S7). Even though the enzyme displays dsDNA cleavage and nicking activity on non-modified sites, it is not lethal to E. coli host in the absence of methyltransferase protection. Escherichia coli cells C2566 carrying the expression plasmid pTXB1-tspA15IR form colonies of typical size, likely as a result of low activity of TspA15I in vivo.
To test phage restriction activity in vivo, we spotted phages λvir, T4gt (5hmC), and T4 (g5hmC) on E. coli cells C2566 [pTXB1-tspA15IR] (uninduced, no IPTG added in the plate). The enzyme restricted T4gt infection strongly, WT T4 partially and left λvir unaffected (Figure 4). The control TagI (SRA-HNH endonuclease) restricted phage T4gt only as expected (39). The in vivo phage restriction data support the conclusion drawn from in vitro experiments that TspA15I has (g)5hmC enhanced endonuclease activity. The restriction activity against WT T4 infection appears weaker in vivo than cleavage activity in vitro on T4 DNA, probably due to the availability of Mg2+ versus Mn2+ ions.
Figure 4.
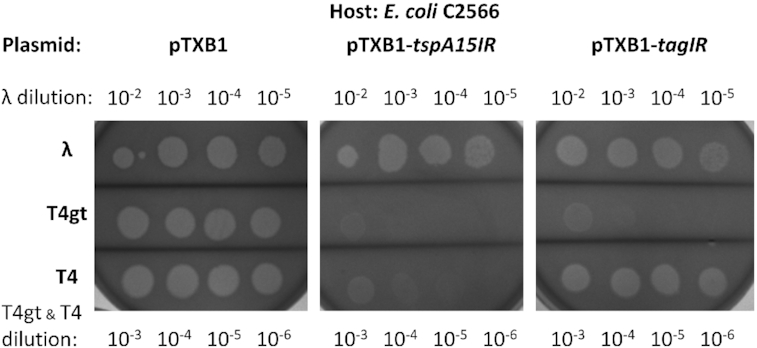
TspA15I phage restriction assay (phage spot test). E. coli C2566 [pTXB1-tspA15IR] cell lawns were spotted with 8 μl of the diluted phages λvir (10−2 to 10−5 dilutions), T4gt and WT T4 (10−3 to 10−6 dilutions). Negative control: C2566 [pTXB1] vector. Positive control: C2566 [pTXB1-tagIR], expressing TagI known to restrict phage T4gt.
We next expressed and purified two other endonucleases from this group with ∼46%–50.0% sequence identity to TspA15I: VcaM4I from Vibrio campbellii (GenBank ID WP_010645282) and CmeDI from compost metagenome (UniProt ID A0A3R2GY07, possibly an Aeromonas sp.) (Supplementary Figure S8). Both enzymes are active in Mg2+ and Mn2+ buffers, and partially active in Ni2+ and Co2+ buffers. Both enzymes prefer to cleave 5hmC-modified over unmodified and g5hmC containing substrates in vitro. They show promiscuous activity on 5hmdU modified phage SP8 DNA. It is not clear whether the activity on 5hmdU-DNA is due to relaxed star activity or the intrinsic properties of the enzymes.
In order to perform phage restriction assay, we subcloned the vcaM4IR and cmeDIR restriction genes into pET21b with C-terminal 6xHis tag (to eliminate the possibility that the endonuclease fused to intein and CBD might have diminished activity). Similar to TspA15I, both enzymes strongly restricted 5hmC containing phage T4gt. VcaM4I only moderately and CmeDI strongly restricted g5hmC modified WT T4. Moreover, CmeDI showed stronger restriction activity in vivo than in vitro on phages T4gt and WT T4, suggesting that a low level of expression (uninduced) is sufficient to cause substantial restriction. Due to the lack of a coliphage containing 5hmdU, we have not examined restriction of 5hmdU-modified phages in vivo. Inverse PCR to incorporate 5hmdU into PCR product in a small plasmid was hampered by the amplified product size (so far, we were only able to amplify a 1.2 kb fragment with 5hmdUTP replacing dTTP in dNTP mix and Taq DNA polymerase). Therefore, restriction of 5hmdU-containing plasmid DNA in plasmid transformation remains to be tested.
The results for four putative endonucleases in this group, Saccharomonospora paurometabolica protein (WP_007024064), Deinococcus yavapaiensis protein (WP_110885659), Delftia acidovorans protein (WP_097205143) and Mycolicibacter algericus protein (WP_083037766) were inconclusive (all four ORFs contain predicted EVE and HNH domains). The partially purified proteins showed low activity on cytosine modified DNA in Mn2+ buffer (data not shown). Their modification dependent activity, if any, remains to be investigated.
ScoA3V (Sco5330) group of HNH endonucleases
The Phyre2 server (31) suggested that ScoA3V (Sco5330) from S. coelicolor A3(2) may contain a PUA-like domain. BLAST searches detected ScoA3V close homologs only in Actinomycetales (in Steptomyces, Nocardiopsis and Micromonospora species), but did not suggest close similarity of enzymes in this group to endonucleases in the TspA15I, VcaM4I and CmeDI group of proteins. CLANS analysis confirmed the separate status of the N-terminal domain of ScoA3V, which did not cluster with the N-terminal domains of the TspA15I, VcaM4I, and CmeDI group of proteins.
ScoA3V (Sco5330) was chosen as a representative for further study. The enzyme was expressed in E. coli T7 Express strain from the pTXB1 vector as a fusion with intein and chitin binding domain (CBD). Transformation of the plasmid with insert was rather inefficient, perhaps due to nuclease activity in cells. ScoA3V enzyme was partially purified by affinity chromatography on a chitin resin, and after intein/DTT cleavage by chromatography on a heparin column.
In preliminary experiments, we detected only nicking activity of ScoA3V against DNA containing modified cytosines (Dam+ Dcm+ pBR322). As some PUA-superfamily members bind purines or purine-like bases, we suspected that the protein may preferentially interact with DNA containing N6-methyladenine (6mA). Partially purified ScoA3V fractions (after chitin or heparin chromatography) were tested for activity against Dam+ Dcm+ pBR322 and against this plasmid further methylated either by a sequence context independent adenine methyltransferase (M.EcoGII) or by M.SssI methylating cytosines in the CpG context. The first substrate remained mostly intact, but substantial degradation was observed for the further methylated DNA (Figure 5A and Supplementary Figure S9A). Similar result was obtained for unmodified (Dam−) and M.EcoGII-modified λDNA (Supplementary Figure S9B). The 6mA-stimulated activity was enhanced in low salt buffers and diminished in the presence of 0.1 M NaCl (Supplementary Figure S9C). Run-off sequencing indicated the nicks in both substrates, with a CCA↓GT preference in unmodified DNA (Supplementary Figure S9D) and more relaxed CW↓G or SW↓S selectivity in the more efficiently cleaved highly methylated DNA (Figure 5B). We conclude from the data that ScoA3V exhibits 6mA stimulated endonuclease activity. It is currently unclear whether single methylation sites induce DNA nicks that appear as double-strand breaks when modifications are dense enough, or whether a single modification can guide the nuclease domain to make a double strand break in DNA.
Figure 5.
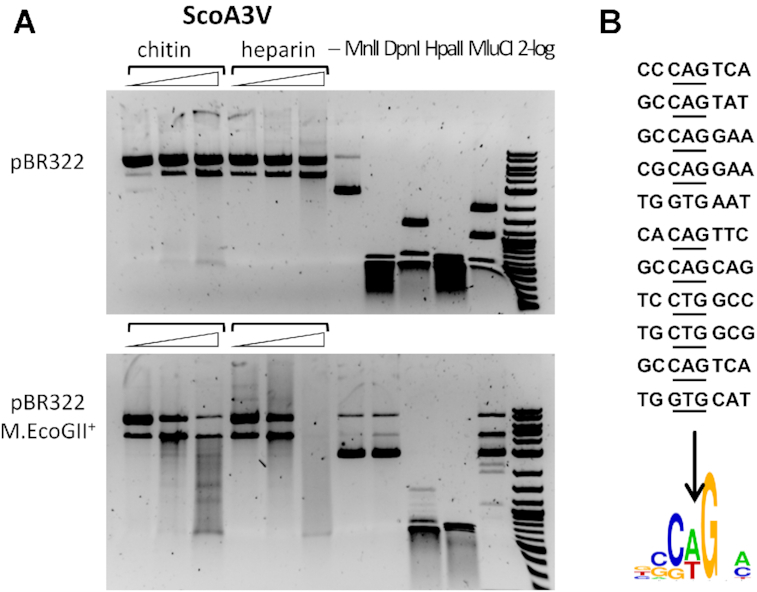
ScoA3V digestion of pBR322 and 6mA-modified pBR322. (A) pBR322 (Dam+ G6mATC; Dcm+ C5mCWGG) and M.EcoGII (6mA) modified pBR322 were digested by ScoA3V in CutSmart buffer. The plasmids were also digested by MnlI (GAGG), DpnI (G6mATC), HpaII (CCGG), and MluCI (AATT), respectively as controls. The modified plasmid was partially resistant to MnlI and MluCI digestion. (B) Nicking sites and sequence logo derived from ScoA3V digestion of M.EcoGII-modified pBR322.
In order to test in vivo restriction activity of ScoA3V against highly adenine methylated plasmid, we attempted to compare the plasmid transformation efficiency into E. coli either expressing ScoA3V or not. Unfortunately, the highly adenine methylated plasmid transformed poorly into E. coli cells even in the absence of the potential ScoA3V restriction barrier. Real-time sequencing shows that at least some DNA polymerases pause at sites of adenine methylation (29), which may hamper replication of densely methylated plasmids.
ScoA3IV (Sco5333) group of HNH endonucleases
ScoA3IV (Sco5333) is the prototype of SRA-HNH endonucleases (Type IV) (10). Previous results indicate that ScoA3IV binds to 5mC modified oligoduplexes in vitro, restricts 5mC-modified plasmid transformed into Dcm+E. coli host, and is severely toxic to RecA-deficient E. coli strains (10,11). Here, we show that pTXB1-scoA3IVR even kills the Dcm+ RecA−E. coli 10β host, which expresses ScoA3IV only at low levels because it lacks T7 RNA polymerase. ScoA3IV inflicted damage is better dealt with in RecA+ strains, since both NEB Turbo (Dcm+ RecA+) and T7 Express (Dcm− RecA+) strains can tolerate pTXB1-scoA3IVR (Supplementary Figure S10A).
The N-terminal SRA domain of ScoA3IV (aa residues 1–150) is predicted to be responsible for the 5mC recognition. The structure of a similar domain from the TagI enzyme was solved recently and confirmed the prediction (39). We found that the expression of the ScoA3IV SRA domain alone (SRAN168, SRA domain + linker) can partially attenuate T4gt phage infection (data not shown). A fusion of the domain with the DNA nicking domain (gp74) derived from HK97 phage (40,41) was constructed and purified by affinity chromatography on a chitin column (SRAN168-gp74). The activity of the fusion enzyme was tested with 5mC-modified plasmids DNA in Mn2+ buffer (Figure 6). At high concentration, the enzyme digested plasmid DNA into small fragments (due to relaxed specificity). But under limited digestion conditions, the supercoiled plasmid was mostly converted to nicked circular form. The nicking sites were mapped by run-off sequencing. Supplementary Figure S11 shows two examples of the bipartite recognition of the G5mCN4–7AG↑CGG sequence by the fusion NEase (G5mC by the SRA domain and AG↑CGG by the nicking domain) and sequence consensus of the nicking sites (the up arrow ↑ indicates the bottom strand is nicked).
Figure 6.
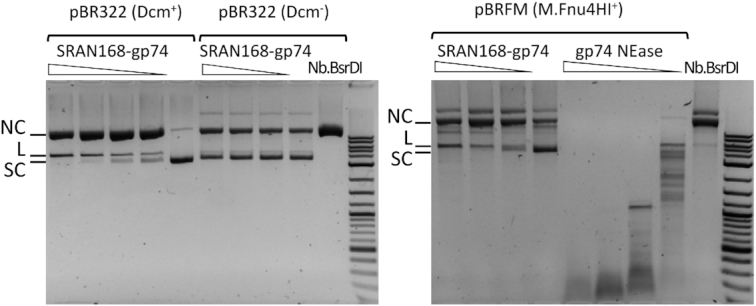
Nicking activity of SRAN168-gp74 fusion endonuclease. Digestion of pBR322 (Dcm modified in C5mCWGG context), pBR322 (Dcm−), and pBRFM+ (M.Fnu4HI modified in G5mCNGC context) with the SRAN168-gp74 fusion protein, and control NEases: gp74 (phage HK97) in Mn2+ and Nb.BsrDI in Mg2+ buffer. SC, L, NC: supercoiled, linear, nicked circular DNA, respectively.
Using the ScoA3IV sequence as a query, we searched GenBank and protein database and found over 160 homologous SRA-HNH proteins (e.g. SAV_3746 from S. avermitilis, 515 aa), mostly in Gram+ and GC rich Actino- and Proteobacteria. We expressed and purified seven of them: MfoEI, MmnI, MsiJI, RrhNI, Vsi48I, Vvu009I and VcaCI. All were capable of cleaving 5hmC modified DNA (Supplementary Figure S12). The enzymes were fully active in Mn2+ buffer and showed weak nicking activity in Mg2+ buffer, except for VcaCI, which was active also in Mg2+ buffer (albeit less so than in the presence of Mn2+), similarly as the previously characterized TagI endonuclease (39).
MsiJI preferentially digested 5mC or 5hmC containing DNA over unmodified DNA in PCR fragment mixtures in 1 mM Mn2+ buffer (Figure 7A). The enzyme showed strong activity in a medium salt buffer supplemented with Mn2+ or Co2+, moderate activity in Ni2+, and low activity in Mg2+ or Zn2+ buffer (Supplementary Figure S12B). MsiJI expression in vivo is toxic to Dcm+E. coli host and only Dcm-deficient host is receptive to pTXB1-msiJIR plasmid transformation (Supplementary Figure S10B), consistently with the previous results for TagI (39).
Figure 7.
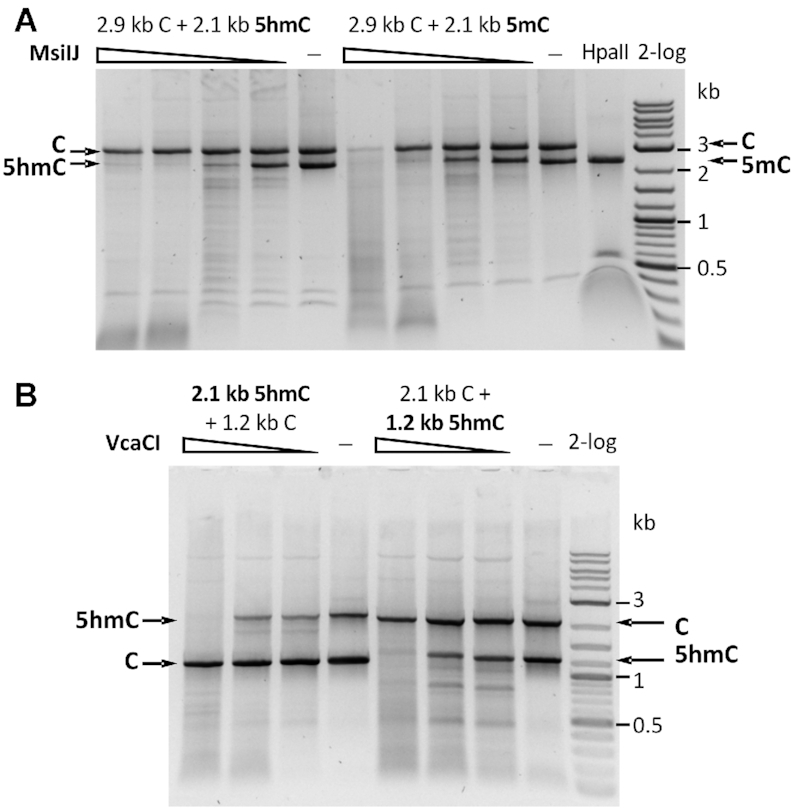
Digestion of mixed PCR fragments. (A) MsiJI digestion of mixed PCR DNA substrates (unmodified DNA mixed with either 5hmC or 5mC modified DNA) in 1 mM Mn2+. Arrows indicate the starting DNA fragments. HpaII (CCGG) endonuclease cleaves unmodified DNA only. (B) VcaCI digestion of mixed DNA substrates (unmodified DNA mixed with 5hmC containing DNA) in 10 mM Mg2+.
VcaCI showed a weak activity on 5hmC-modified DNA in mixed substrates in Mg2+ buffer (Figure 7B). VcaCI activity was much higher in Mn2+ buffer (data not shown). While TagI and MsiJI are toxic to Dcm+ cells, pTXB1-vcaCIR plasmid could be transformed into NEB Turbo (λDE3, Dcm+) and NEB 10β (Dcm+ RecA−) competent cells (Supplementary Figure S10C). It is possible that VcaCI is less toxic to the Dcm+ hosts due to its low activity in vivo in the presence of Mg2+ or Ca2+ ions.
AmaNI from Actinomadura macra (WP_084264606, 540 aa) is an interesting SRA-HNH protein, with additional N-terminal caspase domain (Pfam00656). In mammalian cells, caspases (cysteine protease family) play multiple roles in programmed cell death, inflammatory response and antiviral immunity (42). The caspase domain of AmaNI is presumably involved in regulating its restriction activity by the post-translational peptidase cleavage or abortive infection. The partially purified AmaNI enzyme displayed low DNA nicking activity in Mn2+ buffer (data not shown). Plasmid carrying the AmaNI restriction gene could be transferred into both Dcm+ or Dcm− competent cells, indicating low activity (toxicity) in vivo (Supplementary Figure S10D).
YenY4I group of PD-(D/E)XK endonucleases
YenY4I endonuclease and six close homologs (595 to 601 aa in length) contain an N-terminal domain which clusters with the N-terminal domains of the TspA15I, VcaM4I, and CmeDI family of endonucleases, a DUF3427 (pfam11907) middle domain, and C-terminal PD-(D/E)XK catalytic domain (Figure 8A). A DUF3427 domain has previously been identified as the target recognition domain (TRD) in the PLD-type endonuclease SauUSI, which cleaves preferentially at modified sites (S5mCNGS or S5hmCNGS, S = C or G). Hence, YenY4I and its homologues may consist of two modification sensing domains, and a C-terminal catalytic domain. YenY4I and its homologues, unlike SauUSI, do not contain a helicase domain and are therefore not expected to be ATP-dependent.
Figure 8.

YenY4I activity assays. (A) Schematic diagram of the YenY4I domain organization (N-terminal PUA superfamily domain (PUA-sf), SauUSI-like specificity domain (DUF3427), and C-terminal PD-(D/E)XK endonuclease catalytic domain (DUF3883)). (B) YenY4I digestion of mixed PCR fragments (C+5mC, top panel; C+5hmC, bottom panel) in Mg2+ (buffer 2.1) or Mn2+ buffer (1 mM MnCl2). Controls: HpaII digestion of regular C PCR fragment (2.9 kb); MspJI (5(h)mCNNR) digestion of 5mC or 5hmC PCR fragment (2.1 kb). YenY4I showed modified cytosine dependent activity in Mn2+ buffer at high enzyme concentration (2, 1, 0.5 and 0.25 μg input protein corresponding to 294, 147, 74, and 37 nM, respectively vs. 0.45 μg PCR DNA at ∼6 nM). (C) Phage restriction activity by YenY4I. Diluted λvir, T4gt, WT T4 phages were spotted onto the cell lawns of C2566 [pTXB1] and C2566 [pTXB1-yenY4IR]. Arrows indicate the partial restriction of phage T4gt by YenY4I. Controls: phage T4gt was strongly restricted by the expression of TspA15I or TagI. WT T4 was partially restricted by TspA15I, not restricted by TagI.
A synthetic YenY4I gene was cloned into pTXB1 and over-expressed in E. coli C2566. YenY4I was partially purified by affinity purification through a chitin column. The purified enzyme was used in digestion of mixed PCR fragments (C+5mC or C+5hmC PCR DNAs). YenY4I displayed very low activity in Mg2+ buffer on both modified and unmodified DNA (Figure 8B). However, the modification dependent activity was detected in Mn2+ buffer at high enzyme concentration (Figure 8B) in the absence of ATP (5mC and C, top panel; 5hmC and C, bottom panel). In control digestions, HpaII (CCGG) cleaved the unmodified DNA, and MspJI (5mCNNR or 5hmCNNR) digested the modified PCR fragment only.
The pTXB1 plasmid carrying the YenY4I gene could be transformed not only into the Dcm− strain E. coli C2566, but also into the Dcm+ strain NEB Turbo (λDE3). However, for the latter, colonies were smaller than for the former, indicating mild toxicity. In a phage restriction assay, YenY4I showed a weak restriction activity on phage T4gt and did not restrict WT T4. Phage λvir was also restricted to some degree. The positive control enzymes TspA15I and TagI strongly restricted T4gt (Figure 8C).
McaZI group of PD-(D/E)XK restriction endonucleases
The combination of two potentially modification sensing domains with a catalytic PD-(D/E)XK domain, as in YenY4I, is relatively rare. In contrast, combinations of only DUF3427 (pfam11907) and PD-(D/E)XK restriction endonucleases, without additional modification sensing domains, or helicase domains, are quite frequent (439 putative enzymes in UNIPROT). These ORFs are annotated as DUF3427 domain proteins and modified cytosine restriction system McrB variants (e.g. McrB2-4) in REBASE (24) (by aa sequence similarity, they are not true McrB/GTPase homologs). The exact function or activity of this family of enzymes has not been verified by experimentation.
We expressed three such fusion proteins with a chitin binding domain tag and purified the enzymes from chitin columns: McaZI (WP_064610770, 338 aa, found in Moraxella catarrhalis), BwiMMI (WP_000732259, 336 aa, found in Bacillus wiedmannii), and EfaL9I (WP_010818586, 323 aa, found in Enterococcus faecalis). Possible modification dependent activity was assayed by comparing the efficiencies of transformation into Dcm− and Dcm+ cells. Plasmids pTXB1 carrying McaZI or EfaL9I restriction genes could be transferred into Dcm-deficient T7 expression hosts such as C2566 and C3013 (lacIq, LysY) with 105 to 106 cfu/μg, but were toxic to Dcm+ NEB Turbo (λDE3) cells (no AmpR colonies after transformation of 50 ng plasmid DNA, corresponding to less than 1 cfu/50 ng or 20 cfu/μg). The pTXB1 plasmid carrying the BwiMMI restriction gene could be transferred into the Dcm+ strain NEB Turbo (λDE3) efficiently, but the transformants formed small to medium size colonies compared to the empty vector transformants (data not shown), indicating that BwiMMI was also somewhat toxic to the host under non-inducing conditions.
We next examined McaZI, BwiMMI and EfaL9I activity in vitro. On 5hmC-modified PCR DNA, McaZI showed a weak cleavage activity in Mn2+ buffer and also formed a brownish precipitate in Co2+ buffer (Supplementary Figure S13A). BwiMMI showed no apparent cleavage activity in a medium salt buffer supplemented with five different divalent cations (10 mM Mg2+, 1 mM Mn2+, 1 mM Ni 2+, 1 mM Co2+ or 5 mM Ca2+) although some binding activity was observed in Co2+ buffer (forming a brownish precipitate). EfaL9I behaved in a similar manner as BwiMMI and showed no apparent cleavage activity in the test tube (data not shown). When a mixture of a 5mC-modified PCR fragment and of unmodified DNA was used as the substrate, McaZI exhibited weak endonuclease activity on 5mC-PCR DNA at high enzyme concentration in Mn2+ buffer. No cleavage activity was observed in NEB buffer 2.1 (10 mM MgCl2) (Supplementary Figure S13B). Proteinase K treatment was necessary to detect the cleavage products, otherwise the modified DNA would be shifted upwards. McaZI displayed strong DNA binding activity to the 5mC-modified fragment when the digestion was not treated with Proteinase K; The modified DNA fragment appeared to be shifted (retarded) in the agarose gel while the unmodified fragment was not affected (Supplementary Figure S13C). BwiMMI and EfaL9I enzymes also bound to 5mC or 5hmC modified DNA fragments tightly in Mg2+ buffer and caused DNA mobility shift in agarose gels (data not shown).
We next used phage spot test to confirm phage attenuation (restriction) activity. The McaZI, BwiMMI, and EfaL9I expression plasmids were transferred into T7 expression cells C2566. CmeDI expression plasmid (pET21b-cmeDIR) was used as a positive control and the empty vectors pTXB1 and pET21b were negative controls. Diluted phages λvir, T4gt, and WT T4 were spotted onto the cell lawns expressing the restriction genes (uninduced condition with Amp selection). McaZI, BwiMMI and EfaL9I were able to attenuate phage T4gt infection. They had minimal effects on λvir and WT T4 infections (Supplementary Figure S13D). It was concluded that McaZI, BwiMMI, and EfaL9I could be considered as bone fide modification-dependent antiphage systems (phage attenuation systems), despite their poor cleavage activity on 5mC or 5hmC modified DNA in vitro. We have not determined the exact binding sequence or their phage attenuation activity in conjunction with ATP/GTP binding proteins (McaZI is co-localized with an ATP/GTP binding protein (NTPase) in the host genome).
Methylome study of S. coelicolor A3(2) genome by SMRT sequencing
Single molecule SMRT sequencing can identify 6mA and 4mC bases in bacterial genomes (29). It has been reported that the S. coelicolor A3(2) strain encodes both 5mC and 6mA-dependent REases and phage attenuation system (ScoMcrA, ScoA3IV and Pgl system) (43,44). To identify the modified nucleotides we re-sequenced the S. coelicolor A3(2) genome. A total of 277 520 sequence reads were obtained (with mean subread length of 2232 bp). The reads were used for modified sequence motif analysis against the reference genome with default quality and read length parameters. The new sequencing data shows 99.998% consensus concordance with the reference. The results are summarized in Table 1 (data deposited in REBASE). Two 4mC-containing motifs were detected (AAGC4mCCG and TGGC4mCGGC), but only 61.7% and 45.2% of sites were modified. PacBio modification and sequence motif analysis showed the average IPD ratios across all sites for each base in AAGCCCG and TGGCCGGC motifs (Supplementary Figure S14).
Table 1.
Summary of 4mC-modified motifs identified in the S. coelicolor A3(2) genome by SMRT sequencing and PacBio software
| Sequence motif | Fraction (%) | # of sites detected | # of sites in the genome | Mean IPD ratio |
|---|---|---|---|---|
| AAGCCCG | 61.7 | 823 | 1334 | 4.0430 |
| TGGCCGGC | 45.3 | 947 | 2093 | 3.7487 |
The modified cytosines are shown in bold and underlined. Putative N4mC MTases are Sco3104, Sco3510 and Sco6885. The underlined cytosines are modified N4mC detected by SMRT sequencing. An IPD ratio greater than 1 means that the sequencing polymerase slowed down at this base position, relative to the unmodified bases (29).
We have not identified the MTases that are responsible for methylation of the two sites, but the candidate genes can be narrowed down to three ORFs with amino-MTase conserved motifs (Sco3104, Sco3510 and Sco6885). Sco3104 is adjacent to Sco3105, a predicted PD-(D/E)XK endonuclease. Sco3510 is associated with Sco3509, a predicted protein with a catalytic site similar to RecB nuclease. Sco6885 appears to be an orphan MTase. Despite the presence of at least two predicted adenine DNA methyltransferases, PglX and Sco5331, 6mA was not detected in the genome. Thus, they are either tightly suppressed or have acquired inactivating mutations. The existence of a large number of 4mC modified sequence motifs in the genome suggests that EcoKMrr like activity (i.e. restriction of 6mA and 4mC modified DNA) is likely to be absent in this strain. The ScoA3Mrr activity would be suicidal and cause self-restriction.
ScoA3I, ScoA3II, and ScoA3III from S. coelicolor A3(2) do not show modification-dependent activity in vitro
ScoA3I
The predicted MDREs (ScoA3I to III) from the S. coelicolor A3(2) strain are based on genetic evidence in restriction of modified plasmid in DNA transformation (44). ScoA3I shows strong protein homology to SauUSI that cleaves 5mC and 5hmC modified DNA. To evaluate ScoA3I activity, a C-terminal 6xHig-tagged ScoA3I (Sco2863, 945 aa, from a TTG start codon) was purified through nickel agarose and heparin column chromatography. The purified enzyme was assayed for activity on modified DNA substrates such as: pBR322 (Dcm+), pBRFM+ (G5mCNGC), M.GpC-modified pBR322, pET21b-BceSVM (multi-specificity C5 MTase), 5hmC-PCR, phage T4gt (5hmC). ScoA3I did not show any endonuclease activity in NEB buffer 2.1 on the substrates described above in the presence or absence of ATP (data not shown). Neither did C2566 [pET21b-scoA3IR] cells show any phage restriction activity when phages λvir, T4gt (5hmC), and T4 (g5hmC) were spotted on the cell lawn under induced or non-induced condition (data not shown). Expression of a slightly longer version of ScoA3I with extra 46-aa residues from an upstream GTG start codon also failed to produce an active enzyme (data not shown). Closer inspection of the PLD endonuclease active site showed that one conserved active site residue Asp (D) in the catalytic motif HXKXD has been altered in this ORF. SMRT sequencing of the S. coelicolor A3(2) genome confirmed the same mutation. The S. coelicolor A3 genome already encodes two active m5C-dependent REases (ScoA3IV and ScoMcrA) so that a third 5mC-dependent endonuclease might be redundant. It was reported previously that Sco2863 (ScoA3I) expressed in S. lividans was able to restrict Dcm+-modified shuttle vector by about a thousand-fold in transformation efficiency (44). The discrepancy of ScoA3I expression in the two heterologous hosts remains to be studied.
ScoA3II
The annotated ORF (Sco3261) encoding ScoA3II is 431-aa long with aa sequence homology to AAA ATPases. A longer version (ScoA3II-long) with an upstream start codon (GTG) encodes a larger protein with 187 extra aa residues (431+187 = 618 aa). The additional 187-aa domain shows a weak homology to Abi protein (Abi, abortive infection protein). The synthetic gene blocks encoding ScoA3II-long was cloned into E. coli expression host. ScoA3II-long was somewhat toxic to E. coli host since mutants were selected (five out of seven alleles sequenced carry mutations). E. coli cells carrying ScoA3II-long plasmid formed small to medium size colonies. Partially purified ScoA3II-long appears as a 60 kDa protein, 8 kDa smaller than expected and it showed no endonuclease activity in vitro in digestion of modified or unmodified DNA substrates (data not shown).
ScoA3III
Sco3262 is located adjacent to ScoA3II in the genome. It is predicted to be an HNH endonuclease but it lacks any PUA superfamily domain at the N-terminus. Sco3262 is not co-localized with a predicted modified nt sensing protein. There are over hundreds of HNH endonuclease homologs in GenBank with 30% to 100% aa sequence identity to ScoA3III (many more homologs with less aa similarity). Partially purified ScoA3III is an endonuclease and it digested pBR322, 5mC- and 5hmC-PCR DNA into small fragments (100–200 bp) (Supplementary Figure S15A). Under limited digestion, however, the endonuclease displays certain sequence preference at the nicking sites SC↓R or SA↓R (S = C/G, R = A/G) (Supplementary Figure S15BC). Addition of ScoA3II-long to ScoA3III digestion did not enhance ScoA3III activity (data not shown). A His to Ala substitution in the predicted catalytic site (first His in H-N-H) in ScoA3III greatly improved the transformation efficiency of the encoding plasmid and alleviated its toxicity (XYH, unpublished result). ScoA3II and ScoA3III together are reminiscent of the Septu antiphage system consisting of two genes (ATPase + HNH nuclease) described by Doron et al. (45). The Septu bacterial defense system is widespread and found among 4.1% of sequenced microbial genomes. The antiphage activity of the Septu systems is probably rendered through host cell killing to limit the release of mature phage particles to the neighboring cells. The tight regulation of the ScoA3III to avoid self-killing in the absence of phage infection remains to be investigated. We concluded that ScoA3II and ScoA3III are not MDREs in vitro although they are somewhat toxic to the expression host. ScoA3III is a DNA nicking enzyme with 2–3 bp specificity.
Conservation of a putative base binding pocket in SRA and EVE, but not PUA domains
There are no enzyme:DNA complex structures for any modification dependent PUA-superfamily HNH endonucleases yet. However, for SRA-HNH endonucleases, confident models are possible. For the prototypical SRA-HNH endonuclease TagI, we have previously reported a crystal structure in the absence of DNA (39). Based on the homology with domains that have been crystallized with modified DNA (13–15), it was possible to build a model of the TagI–DNA complex, and to identify a pocket for the (putatively) flipped modified base (39). Interestingly, a candidate pocket for a flipped base is also present in EVE, but not genuine PUA domains. In order to verify the conservation of the putative pockets, we used the ConSurf server (34). This server automatically generates alignments starting from a seed sequence, and then maps conservation scores to the protein surface. Inspection shows that the putative pocket is more highly conserved than the overall protein surface, for both SRA and EVE domains, whereas higher conservation was not seen in this region for the PUA domain (Figure 9).
Figure 9.
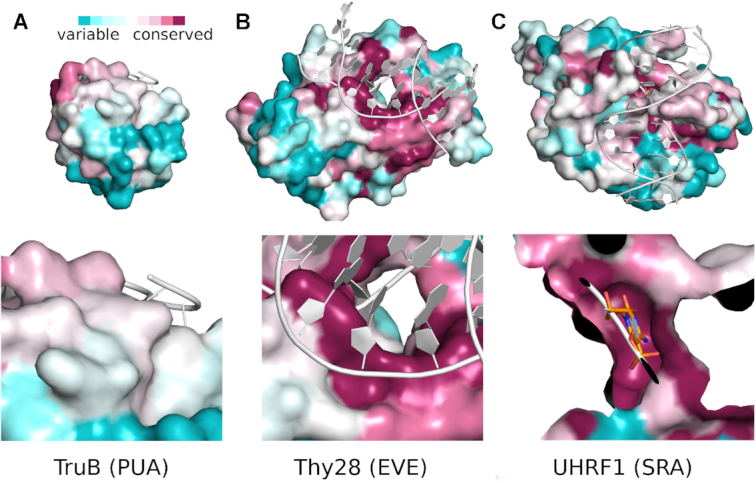
Conservation scores for PUA superfamily domains mapped to the protein surface. The ConSurf server (34) in automatic mode was used to calculate conservation scores for: (A) PUA; (B) EVE and (C) SRA domains. The top panels show a complete view of the active site, the bottom panels a close-up on the putative binding site for the domain (EVE and SRA domains), or the structurally equivalent region (PUA domain).
We conclude that SRA-HNH and EVE-HNH, but not PUA-HNH endonucleases, are likely to interrogate modified bases in DNA in a similar manner. Our conclusion that EVE and PUA domains bind their nucleic acid partners differently is in line with a similar conclusion reached previously in the context of a discussion of the RNA binding properties of the domains (19).
DISCUSSION
DNA modifications that are normally protective can become an Achilles heel
DNA base methylation renders DNA resistant against Type II restriction. Protection of DNA by modifications inhibiting Type I, II and III endonucleases provides an incentive for phages and other mobile genetic elements to acquire such modifications, either by passage through a modification competent host, or by acquisition of host enzymes. Indeed, hypermodified phage genomes M6, ViI, phi W-14 and 9g are resistant to 48%, 71%, 69%, and 71% of Type II restrictions, respectively (46,47). Phage T4gt and WT T4 genomes show even higher resistance against Type II restriction (Flodman and Xu, unpublished). This in turn creates opportunities for hosts, which do not modify their own DNA, to treat modification marks as hallmarks of non-self, and specifically degrade such foreign DNA. In this work, we have used a bioinformatics approach to look for such enzymes as first predicted by the Aravind group (20). The newly discovered modification-dependent enzymes are listed in Table 2. These restriction genes were expressed in heterologous E. coli host. Therefore, the phage restriction activity of the native hosts against phage infection has not been examined.
Table 2.
Modification-dependent or modification-stimulated REases described in this work
| Representative enzymes | GeneBank Accession # or UniProt ID | Other domains | Putative modification sensing domain | Putative catalytic domain | DNA modification stimulated in vitro activity | Modification dependent restriction (in vivo assays) | Double strand cleavage or nicking activity |
|---|---|---|---|---|---|---|---|
| TspA15I | WP_020146688 | PUA-like | HNH | (g)5hmC, 5hmdU > 5mC | restriction of 5hmC (T4gt) > g5hmC (T4) | ds cleavage (5hmCNG) | |
| VcaM4I | WP_010645282 | 5hmC > g5hmC, 5hmdU | restriction of 5hmC (T4gt) > g5hmC (T4) | ||||
| CmeDI | A0A3R2GY07 (UniProt) | 5hmC, 5hmdU > g5hmC, 5mC | restriction of 5hmC (T4gt) and g5hmC (T4) | ||||
| ScoAIV (Sco5333) | NP_629473.1 | SRA (SAD-SRA) | HNH | 5mC | toxicity to Dcm+ Rec− strains | Strong binding to 5mC | |
| MsiJI | WP_065019168 | 5mC or 5hmC | toxicity to Dcm+ strains | ||||
| VcaCI | WP_045378060 | 5hmC | low toxicity to Dcm+ strains | low activity | |||
| AmaNI | WP_084264606 | Caspase | low toxicity to Dcm+ strains | low nicking activity | |||
| MfoEI, MmaNI, RrhNI, Vsi48I, Vvu009I | WP_064867438 | 5hmC (5mC not tested) | (?/Unknown) | ||||
| WP_084259214 | |||||||
| WP_063758401 | |||||||
| WP_052237964 | |||||||
| WP_060534244 | |||||||
| YenY4I | WP_077293998 | DUF3427 and PUA-like | PD-(D/E)XK | 5mC or 5hmC | restriction of 5hmC (T4gt), λ some toxicity to Dcm+ strains | Low activity in Mn2+ | |
| McaZI | WP_064610770 | DUF3427 | PD-(D/E)XK | binding to 5mC or 5hmC, low activity | restriction of 5hmC (T4gt) toxicity to Dcm+ strains | ||
| BwiMMI | WP_000732259 | binding to 5mC or 5hmC | restriction of 5hmC (T4gt) some toxicity to Dcm+ strains | ||||
| EfaL9I | WP_010818586 | binding to 5mC or 5hmC | restriction of 5hmC (T4gt) toxicity to Dcm+ strains | ||||
| ScoA3V (Sco5330) | NP_629471.1 | distantly related to PUA | HNH | 6mA > A, 5mC | (?/Unknown) | nicking activity (S6mAS) | |
| SRA-nicking domain fusion | SRA domain of ScoA3IV | HNH (gp74) | 5mC > C | Nicking near 5mC |
More stringent modification dependence in vivo than in vitro
Many enzymes described in this work share the property that they can distinguish stringently between modified and non-modified DNA in vivo, and restrict phages with modified DNA efficiently, while exhibiting only modest modification dependence, and moderate activity, particularly in the presence of Mg2+ ions, in vitro. The physiological relevance of higher activity in the presence of Mn2+ is unusual and unclear, at least for HNH endonucleases. Moderate activity in vitro may be due to features of DNA in cells that are not present in our assays (such as supercoiling), may indicate that the proteins exert their anti-phage activity in vivo by tight binding to DNA, rather than by DNA cleavage, or could be due to a missing interaction partner in the in vitro assays. Limited discrimination between modified and non-modified DNA in vitro, in turn, could be due to the high enzyme concentrations that are used in the assays. We note that the discrepancy between in vitro and in vivo efficacies is not unique to the enzymes in this study. Similar observations have also been reported for SRA-HNH endonucleases (10,39), and for EcoKMcrA (RglA, HNH endonuclease) that shows strong binding to modified sites 5(h)mCGR in DNA and weak restriction activity in vitro (48,49).
Conservation of a putative base binding pocket in SRA and EVE, but not PUA domains
The binding of modified DNA bases is structurally well described only for the SRA domains in the PUA domain superfamily. SRA domains accommodate a modified cytosine base in a dedicated pocket in the domain (13–15). A candidate pocket for a flipped base is also present in EVE, but not PUA domains. Closer similarities of the N-terminal domains of MDREs to SRA and EVE domains on the one hand, or PUA domains on the other, may therefore explain the preferences for modified cytosines or adenines in different endonuclease families in this study.
ORFs of PUA domain containing endonucleases are rarely nearby methyltransferase ORFs
MDREs need not be accompanied by DNA methyltransferases to protect the host genome, and may even get in conflict with such methyltransferases. For example, the TspA15I group of endonucleases, mild conflicts could occur with C5-methyltransferases, and could be aggravated by coupling with Tet/JBP-like enzymes that can convert 5mC to 5hmC (20).
We searched the neighborhoods of 350 ORFs of fusion proteins made from an N-terminal PUA superfamily domain and a C-terminal catalytic domain. Neighborhoods were defined as the coding sequences for the endonuclease itself, and a region of 3 kb each upstream and downstream of the endonuclease open reading frame. Methyltransferase ORFs were found with considerable frequency. However, almost all of these were clearly not relevant to restriction biology. After excluding predicted RNA methyltransferases, protein methyltransferases, small molecule methyltransferases, and O-methyltransferases, <6% of all candidate endonuclease ORFs had a methyltransferase ORF within a 3 kb region on either side. When endonuclease-methyltransferase gene pairs on opposite DNA strands were additionally excluded, the list of exceptions could be narrowed down even further (Supplementary Table S2). In the remaining exceptional cases, the DNA methyltransferase was often not directly adjacent, or clearly part of another R-M system. We conclude that analysis of genomic neighborhoods strongly supports the modification dependence of endonucleases featuring a PUA superfamily domain.
McaZI group endonucleases tend to occur in defense islands
In contrast to the endonucleases featuring a PUA domain, those featuring a DUF3427 domain are frequently associated with DNA methyltransferases. In the genome of Yersinia enterocolitica Y4fallowdeer1A strain, the YenY4I restriction gene is probably located on a prophage where some phage related genes (integrase, phage adenine methylase, intracellular stress sensing protein, helix-turn-helix protein/transcription repressor) are clustered. In a search for YenY4I homologs by BlastP, only six close homologs of similar size (595–601 aa) are annotated in GenBank. McaZI-like DUF3427-DUF3883 fusions lacking the N-terminal domain are more widespread in bacterial genomes and annotated as putative McrB derivatives in REBASE.
There are three major gene organizations for the DUF3427-DUF3883 fusion endonucleases: 1) stand-alone restriction gene not closely associated with other bacterial host defense systems; e.g. PcaPC1 McrBP and EsaRB McrB2P (see REBASE annotation); 2) adjacent to a putative C5 DNA MTase with GATC specificity and 1–2 ORFs encoding predicted Sau3AI or MutH-like endonuclease (GATC); e.g. BwiMMI and EfaL9I. It is reasoned that modified DNA containing GAT5mC by the putative C5 MTase would not likely cause self-restriction by the associated MDRE as long as the modified cytosines are in different sequence context (e.g. S5mCNGS vs GAT5mC); 3) adjacent to a predicted NTPase (DUF2075), e.g. McaZI. In some bacterial genomes, DUF2075 (Pfam09848) domain in turn is fused to a DNA helicase, GIY-YIG or PD-(D/E)XK catalytic domain or HsdR-N(terminal) domain, DUF2075 proteins being similar to AAA DNA helicase, Type III restriction enzyme ATPase, RecD and RuvB helicase. This suggests that a subgroup of DUF3427-DUF3883 fusion enzymes might be coupled to NTPase/helicases. In human and mouse genomes, the DUF2075 proteins belong to the Schlafen family DNA helicase/P-loop NTPase (SLFN members 5, 8, 9, 11, 13). They serve as inhibitors of DNA replication that promote cell death in response to extensive DNA damage and act as a guardian of the genome by killing cells with defective replication (50).
The close association of DUF3427-DUF3883 fusions to bacterial defense systems suggests that they might be involved in restriction/attenuation of phages with modified gDNA. They may be involved in restriction of mobile genetic elements carrying Tet/JBP like enzymes that convert 5mC to 5hmC as first predicted by Aravind group (20). In future studies, the activity of DUF3427-DUF3883 fusion enzymes may be assayed with DUF2075 domain proteins (or ATP-dependent RecQ-like DNA helicases) and ATP to determine whether they form an active complex in restriction of modified DNA. DUF2075 domain protein fusion to DNA helicase and nuclease catalytic domains also points its potential role in phage attenuation and abortive infections.
Other domain combinations implying modification dependent activity
Prior work has shown that SRA and HNH domain combinations are frequent, in either order (51,52). In this work, we have specifically focused on fusion proteins featuring HNH or PD-(D/E)XK nuclease domain and a modified nt sensing domain of the PUA superfamily, expected to endow more fusion proteins with modification dependent (enhanced) specificity. In the cases that we have identified so far (not involving an SRA domain), the fusions always involve an N-terminal modification sensing domain, and a C-terminal HNH domain or PD-(D/E)XK catalytic domain. Interestingly, the linker length between domains is also not very variable, suggesting that there may be interdomain communication, and that the modification sensing domains do more than just drag the nuclease domains into the proximity of DNA modification sites.
PUA-superfamily domains can be found not only in association with HNH endonucleases, but also with other putative catalytic domains. According to domain classification tools, PUA superfamily domains can also be fused to Pin nuclease (RNase), Mrr-like catalytic domain (PD-QXK)-McrB (GTPase), or very short patch repair (Vsr) nuclease domains. There are also fusion proteins involving an PUA superfamily domains that are not expected to be endonucleolytic, including oxo-glutarate dependent dioxygenases or an acetyltransferase domain. It remains to be investigated whether these fusion proteins play a role in phage restriction biology.
CONCLUSION
We used computational tools (BlastP, Phyre2, PROMALS3D, CLANS) to identify and classify proteins featuring a PUA superfamily (SRA, EVE, genuine PUA) or DUF3427 domain, together with a predicted catalytic domain of HNH or PD-(D/E)XK type. For enzymes featuring a PUA-superfamily domain, we show that TspA15I and its close homologs prefer to cleave 5hmC modified DNA and strongly restrict phage T4gt. We further demonstrate that seven SRA-HNH endonucleases cleave 5mC or 5hmC modified DNA and some of the encoding plasmids are toxic to Dcm+ cells. ScoA3V, also featuring a PUA-superfamily domain, but of a different clade, is a natural DNA NEase with enhanced activity on 6mA modified substrates. YenY4I, featuring an N-terminal PUA superfamily domain, a middle DUF3427 domain (related to the TRD of Type IV restriction enzyme SauUSI), and a PD-(D/E)XK catalytic domain also prefers to cleave 5mC and 5hmC modified DNA. Our work provides the first evidence that not only the SRA family domains, but also other PUA superfamily domains, serve as nt sensing domains for modified DNA. The pseudobarrel structure may have been adapted to recognize modified bases in many occasions in restriction biology. It is anticipated that the PUA-related domains fused to other DNA binding domains or nuclease catalytic domains may play important roles in RNA/DNA modifications and in DNA restriction and repair.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Andrew Gardner, Elizabeth Raleigh, William Jack and Richard Roberts for critical comments and discussions; Donald Comb, James Ellards, Richard Roberts and Thomas Evans for support; Richard Roberts for advice. We are grateful to Iain Murray, Richard Morgan, Elizabeth Raleigh, Peter Weigele and Geoffrey Wilson for providing research materials.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The Small Business Innovation Research Program (NIGMS) of the National Institutes of Health [R44GM105125 to R.J.R. (NEB)]; Polish National Agency for Academic Exchange [PPI/APM/2018/1/00034 to IIMCB]; Polish National Science Centre [2011/02/A/NZ1/00052, 2014/13/B/NZ1/03991, 2014/14/M/NZ5/00558, 2017/27/L/NZ2/03234 to M.B.]; Foundation for Polish Science/EU Regional Development Fund [POIR.04.04.00-00-5D81/17-00 to M.B.]; Thomas Lutz, Kiersten Flodman and Alyssa Copelas were supported by NEB internship program. Funding for open access charge: New England Biolabs Inc.
Conflict of interest statement. S.Y.X., A.F. and M.M. are employees of New England Biolabs, Inc., a company that commercializes enzyme reagents for molecular biology applications.
REFERENCES
- 1. Roberts R.J., Belfort M., Bestor T., Bhagwat A.S., Bickle T.A., Bitinaite J., Blumenthal R.M., Degtyarev S., Dryden D.T., Dybvig K. et al.. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003; 31:1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Geier G.E., Modrich P.. Recognition sequence of the dam methylase of Escherichia coli K12 and mode of cleavage of Dpn I endonuclease. J. Biol. Chem. 1979; 254:1408–1413. [PubMed] [Google Scholar]
- 3. Xu S.Y., Klein P., Degtyarev S., Roberts R.J.. Expression and purification of the modification-dependent restriction enzyme BisI and its homologous enzymes. Sci. Rep. 2016; 6:28579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sutherland E., Coe L., Raleigh E.A.. McrBC: a multisubunit GTP-dependent restriction endonuclease. J Mol. Biol. 1992; 225:327–348. [DOI] [PubMed] [Google Scholar]
- 5. Xu S.Y., Corvaglia A.R., Chan S.H., Zheng Y., Linder P.. A type IV modification-dependent restriction enzyme SauUSI from Staphylococcusaureus subsp. aureus USA300. Nucleic Acids Res. 2011; 39:5597–5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bair C.L., Black L.W.. A type IV modification dependent restriction nuclease that targets glucosylated hydroxymethyl cytosine modified DNAs. J Mol. Biol. 2007; 366:768–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Czapinska H., Kowalska M., Zagorskaitė E., Manakova E., Slyvka A., Xu S.-y., Siksnys V., Sasnauskas G., Bochtler M.. Activity and structure of EcoKMcrA. Nucleic Acids Res. 2018; 46:9829–9841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang H., Guan S., Quimby A., Cohen-Karni D., Pradhan S., Wilson G., Roberts R.J., Zhu Z., Zheng Y.. Comparative characterization of the PvuRts1I family of restriction enzymes and their application in mapping genomic 5-hydroxymethylcytosine. Nucleic Acids Res. 2011; 39:9294–9305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Szwagierczak A., Brachmann A., Schmidt C.S., Bultmann S., Leonhardt H., Spada F.. Characterization of PvuRts1I endonuclease as a tool to investigate genomic 5-hydroxymethylcytosine. Nucleic Acids Res. 2011; 39:5149–5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Han T., Yamada-Mabuchi M., Zhao G., Li L., Liu G., Ou H.Y., Deng Z., Zheng Y., He X.. Recognition and cleavage of 5-methylcytosine DNA by bacterial SRA-HNH proteins. Nucleic Acids Res. 2015; 43:1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kisiala M., Copelas A., Czapinska H., Xu S.Y., Bochtler M.. Crystal structure of the modification-dependent SRA-HNH endonuclease TagI. Nucleic Acids Res. 2018; 46:10489–10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sasnauskas G., Zagorskaite E., Kauneckaite K., Tamulaitiene G., Siksnys V.. Structure-guided sequence specificity engineering of the modification-dependent restriction endonuclease LpnPI. Nucleic Acids Res. 2015; 43:6144–6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arita K., Ariyoshi M., Tochio H., Nakamura Y., Shirakawa M.. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. 2008; 455:818–821. [DOI] [PubMed] [Google Scholar]
- 14. Avvakumov G.V., Walker J.R., Xue S., Li Y., Duan S., Bronner C., Arrowsmith C.H., Dhe-Paganon S.. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature. 2008; 455:822–825. [DOI] [PubMed] [Google Scholar]
- 15. Hashimoto H., Horton J.R., Zhang X., Bostick M., Jacobsen S.E., Cheng X.. The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature. 2008; 455:826–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu G., Fu W., Zhang Z., He Y., Yu H., Wang Y., Wang X., Zhao Y.L., Deng Z., Wu G. et al.. Structural basis for the recognition of sulfur in phosphorothioated DNA. Nat. Commun. 2018; 9:4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aravind L., Koonin E.V.. Novel predicted RNA-binding domains associated with the translation machinery. J. Mol. Evol. 1999; 48:291–302. [DOI] [PubMed] [Google Scholar]
- 18. Mitchell A.L., Attwood T.K., Babbitt P.C., Blum M., Bork P., Bridge A., Brown S.D., Chang H.Y., El-Gebali S., Fraser M.I. et al.. InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019; 47:D351–D360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bertonati C., Punta M., Fischer M., Yachdav G., Forouhar F., Zhou W., Kuzin A.P., Seetharaman J., Abashidze M., Ramelot T.A. et al.. Structural genomics reveals EVE as a new ASCH/PUA-related domain. Proteins. 2009; 75:760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Iyer L.M., Zhang D., Burroughs A.M., Aravind L.. Computational identification of novel biochemical systems involved in oxidation, glycosylation and other complex modifications of bases in DNA. Nucleic Acids Res. 2013; 41:7635–7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Spruijt C.G., Gnerlich F., Smits A.H., Pfaffeneder T., Jansen P.W., Bauer C., Munzel M., Wagner M., Muller M., Khan F. et al.. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013; 152:1146–1159. [DOI] [PubMed] [Google Scholar]
- 22. Xu M.Q., Paulus H., Chong S.. Fusions to self-splicing inteins for protein purification. Methods Enzymol. 2000; 326:376–418. [DOI] [PubMed] [Google Scholar]
- 23. Sambrook J., Fritsch E.F., Maniatis T.. Molecular Cloning, A Laboratory Manual. 1989; 2nd ednCold Spring Harbor Laboratory Press. [Google Scholar]
- 24. Roberts R.J., Vincze T., Posfai J., Macelis D.. REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2015; 43:D298–D299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Murray I.A., Morgan R.D., Luyten Y., Fomenkov A., Correa I.R. Jr, Dai N., Allaw M.B., Zhang X., Cheng X., Roberts R.J.. The non-specific adenine DNA methyltransferase M.EcoGII. Nucleic Acids Res. 2018; 46:840–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu S.Y., Zhu Z., Zhang P., Chan S.H., Samuelson J.C., Xiao J., Ingalls D., Wilson G.G.. Discovery of natural nicking endonucleases Nb.BsrDI and Nb.BtsI and engineering of top-strand nicking variants from BsrDI and BtsI. Nucleic Acids Res. 2007; 35:4608–4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Crooks G.E., Hon G., Chandonia J.M., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu S.Y., Boitano M., Clark T.A., Vincze T., Fomenkov A., Kumar S., Too P.H., Gonchar D., Degtyarev S.K., Roberts R.J.. Complete Genome Sequence Analysis of Bacillussubtilis T30. Genome Announcements. 2015; 3:e00395-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Flusberg B.A., Webster D.R., Lee J.H., Travers K.J., Olivares E.C., Clark T.A., Korlach J., Turner S.W.. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods. 2010; 7:461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marchler-Bauer A., Derbyshire M.K., Gonzales N.R., Lu S., Chitsaz F., Geer L.Y., Geer R.C., He J., Gwadz M., Hurwitz D.I. et al.. CDD: NCBI's conserved domain database. Nucleic Acids Res. 2015; 43:D222–D226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J.. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015; 10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Holm L., Laakso L.M.. Dali server update. Nucleic Acids Res. 2016; 44:W351–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Frickey T., Lupas A.. CLANS: a Java application for visualizing protein families based on pairwise similarity. Bioinformatics. 2004; 20:3702–3704. [DOI] [PubMed] [Google Scholar]
- 34. Ashkenazy H., Abadi S., Martz E., Chay O., Mayrose I., Pupko T., Ben-Tal N.. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016; 44:W344–W350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pei J., Kim B.H., Grishin N.V.. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008; 36:2295–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wilson G.G., Young K.Y., Edlin G.J., Konigsberg W.. High-frequency generalised transduction by bacteriophage T4. Nature. 1979; 280:80–82. [DOI] [PubMed] [Google Scholar]
- 37. Georgopoulos C.P., Revel H.R.. Studies with glucosyl transferase mutants of the T-even bacteriophages. Virology. 1971; 44:271–285. [DOI] [PubMed] [Google Scholar]
- 38. Pingoud A., Wilson G.G., Wende W.. Type II restriction endonucleases - a historical perspective and more. Nucleic Acids Res. 2016; 44:8011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kisiala M., Copelas A., Czapinska H., Xu S.Y., Bochtler M.. Crystal structure of the modification-dependent SRA-HNH endonuclease TagI. Nucleic Acids Res. 2018; 46:10489–10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moodley S., Maxwell K.L., Kanelis V.. The protein gp74 from the bacteriophage HK97 functions as a HNH endonuclease. Protein Sci. 2012; 21:809–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xu S.Y. Sequence-specific DNA nicking endonucleases. Biomol. Concepts. 2015; 6:253–267. [DOI] [PubMed] [Google Scholar]
- 42. Julien O., Wells J.A.. Caspases and their substrates. Cell Death Differ. 2017; 24:1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hoskisson P.A., Sumby P., Smith M.C.. The phage growth limitation system in Streptomycescoelicolor A(3)2 is a toxin/antitoxin system, comprising enzymes with DNA methyltransferase, protein kinase and ATPase activity. Virology. 2015; 477:100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gonzalez-Ceron G., Miranda-Olivares O.J., Servin-Gonzalez L.. Characterization of the methyl-specific restriction system of Streptomycescoelicolor A3(2) and of the role played by laterally acquired nucleases. FEMS Microbiol. Lett. 2009; 301:35–43. [DOI] [PubMed] [Google Scholar]
- 45. Doron S., Melamed S., Ofir G., Leavitt A., Lopatina A., Keren M., Amitai G., Sorek R.. Systematic discovery of antiphage defense systems in the microbial pangenome. Science. 2018; 359:eaar4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Flodman K., Tsai R., Xu M.Y., Correa I.R. Jr, Copelas A., Lee Y.J., Xu M.Q., Weigele P., Xu S.Y.. Type II Restriction of Bacteriophage DNA With 5hmdU-Derived Base Modifications. Front. Microbiol. 2019; 10:584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tsai R., Correa I.R., Xu M.Y., Xu S.Y.. Restriction and modification of deoxyarchaeosine (dG+)-containing phage 9 g DNA. Sci. Rep. 2017; 7:8348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mulligan E.A., Hatchwell E., McCorkle S.R., Dunn J.J.. Differential binding of Escherichiacoli McrA protein to DNA sequences that contain the dinucleotide m5CpG. Nucleic Acids Res. 2010; 38:1997–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Czapinska H., Kowalska M., Zagorskaite E., Manakova E., Slyvka A., Xu S.Y., Siksnys V., Sasnauskas G., Bochtler M.. Activity and structure of EcoKMcrA. Nucleic Acids Res. 2018; 46:9829–9841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Murai J., Tang S.W., Leo E., Baechler S.A., Redon C.E., Zhang H., Al Abo M., Rajapakse V.N., Nakamura E., Jenkins L.M.M. et al.. SLFN11 blocks stressed replication forks independently of ATR. Mol. Cell. 2018; 69:371–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Horton J.R., Mabuchi M.Y., Cohen-Karni D., Zhang X., Griggs R.M., Samaranayake M., Roberts R.J., Zheng Y., Cheng X.. Structure and cleavage activity of the tetrameric MspJI DNA modification-dependent restriction endonuclease. Nucleic Acids Res. 2012; 40:9763–9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kazrani A.A., Kowalska M., Czapinska H., Bochtler M.. Crystal structure of the 5hmC specific endonuclease PvuRts1I. Nucleic Acids Res. 2014; 42:5929–5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Duan J.Q., Li L., Lu J., Wang W., Ye K.Q.. Structural mechanism of substrate RNA recruitment in H/ACA RNA-Guided pseudouridine synthase. Mol. Cell. 2009; 34:427–439. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.