

Supplemental Digital Content is available in the text.
Keywords: hypercholesterolemia, lipoproteins, mutations, proof of concept study, rare disease
Objective:
Homozygous familial hypercholesterolemia is a rare disease usually caused by LDLR (low-density lipoprotein receptor) mutations. Homozygous familial hypercholesterolemia is characterized by markedly elevated LDL-C (low-density lipoprotein cholesterol) levels and an extremely high risk of premature atherosclerotic cardiovascular disease. A phase 2, proof-of-concept study (NCT02265952) demonstrated that evinacumab, a fully human monoclonal antibody to ANGPTL3 (angiopoietin-like 3 protein), reduced LDL-C levels in 9 patients with genotypically confirmed homozygous familial hypercholesterolemia and was well tolerated. The aim of this study was to analyze the effects of evinacumab on LDLR activity in lymphocytes purified from patients in the proof-of-concept study.
Approach and Results:
LDLR activity was assessed in patient lymphocytes before and after treatment with evinacumab and versus lymphocytes carrying wild-type LDLR, and also in an LDLR-defective Chinese hamster ovary cell line (CHO-ldlA7) transfected with plasmids encoding the LDLR variants. Overall mean peak reduction in LDL-C with evinacumab was −58±18%, occurring between Week 4 and Week 12. Mutations identified in the 9 patients were shown to be pathogenic, with loss of LDLR activity versus wild type. Two of the LDLR variants, p.(Cys681*) and p.(Ala627Profs*38), were class 2 type mutations that are retained in the endoplasmic reticulum. Six variants were class 3 type mutations with impaired LDL-C binding activity: p.(Trp87Gly), occurring in 2 patients, p.(Gln254Pro), p.(Ser177Leu), p.(Gly335Val), and p.(Ser306Leu). Evinacumab had no effect on LDLR activity.
Conclusions:
These results suggest that evinacumab is effective for lowering LDL-C in patients with homozygous familial hypercholesterolemia, and the inhibition of ANGPTL3 in humans lowers LDL-C in a mechanism independent of the LDLR.
Highlights.
We analyzed the effect of ANGPTL3 (angiopoietin-like 3 protein) inhibition on LDLR (low-density lipoprotein receptor) activity in lymphocytes from homozygous familial hypercholesterolemia patients (n=9) who received evinacumab in a proof-of-concept study.
LDL-cholesterol was reduced with evinacumab in all patients, with mean peak reduction of −58±18% occurring between Weeks 4 and 12.
Mutations identified from the patients were shown to have loss of LDLR activity versus wild-type because of either defective endoplasmic reticulum transport or impaired LDL-cholesterol binding activity.
Evinacumab had no functional effect on LDLR activity.
ANGPTL3 inhibition lowers LDL-cholesterol independently of LDLR, suggesting that evinacumab may be effective for lowering LDL-cholesterol in patients with homozygous familial hypercholesterolemia who are otherwise difficult to treat.
Familial hypercholesterolemia (FH; MIM#143890) is an autosomal dominant disorder causing premature coronary heart disease that is characterized by increased plasma LDL-C (low-density lipoprotein cholesterol), tendon xanthomas, and deposits of cholesterol in peripheral tissues leading to accelerated atherosclerosis.1,2 In 95% of the cases, FH is because of mutations in the LDLR (low-density lipoprotein receptor; MIM# 606945) gene,3 which is responsible for the uptake of LDL particles into cells.4 Historically, FH was reported to have a homozygous frequency of 1 in 1 000 000,5 although the heterozygous frequency has recently been estimated as high as 1 in ≈200 (based on Dutch Lipid Clinic Network criteria) or 1 in 244 for FH based on molecular criteria in some northern European populations, and up to 1 in 80 in some founder populations such as French Canada,6 suggesting that the frequency of homozygous FH (HoFH) is in the region of 1:600 000 to 1:300 000.1 Although there is wide phenotypic variability among HoFH patients,7 LDL-C values in those who are untreated usually reach concentrations >500 mg/dL (13 mmol/L), leading to premature cardiovascular events1 and premature death.8
Currently, >2500 different variants have been described on the LDLR gene but not all of them are pathogenic (ie, FH-causing).9 The nature and location of the mutations within the LDLR gene determine the activity of the receptor and, therefore, mutations have been divided into 5 classes:10 Class 1: no detectable LDLR synthesis; Class 2: defective LDLR transport from the endoplasmic reticulum; Class 3: impaired LDL to LDLR binding; Class 4: no LDLR/LDL internalization because of defective clustering in clathrin-coated pits; and Class 5: no LDLR recycling. Recently, a sixth class has been proposed that includes mutants that are incorrectly inserted in the cell membrane.11,12 Among LDLR defective mutations, residual LDLR activity can vary in the range of 2% to 80%; therefore, the treatment strategy must be evaluated to achieve the highest effectiveness.
The currently available pharmacological treatments, which include statins, ezetimibe, mipomersen, lomitapide, and evolocumab, are insufficient to bring HoFH patients to optimal LDL-C levels.1,13,14 It has been shown that evolocumab, a PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibitor, promotes a 20% to 30% mean reduction in LDL-C in HoFH patients.15–17 However, the activity of the LDLR is reduced to variable degrees depending on class type mutation, and evolocumab does not lower LDL-C in HoFH patients with class 1 mutations or extreme LDLR mutations with low residual activity or binding capacity.15–19
Studies have shown that the ANGPTL3 (angiopoietin-like 3 protein) affects lipoprotein metabolism by inhibiting LPL (lipoprotein lipase), which hydrolyzes triglycerides from chylomicrons and low-density lipoproteins.20 Low plasma triglyceride and high-density lipoprotein cholesterol levels have been associated with ANGPTL3 loss-of-function variants because of a lack of LPL inhibition.21,22 In addition, ANGPTL3 inhibits endothelial lipase, which catalyzes the hydrolysis of high-density lipoprotein phospholipids and facilitates the clearance of high-density lipoprotein from the circulation.20 Recently, evinacumab, a fully human ANGPTL3-blocking antibody generated by VelocImmune technology, was found to lower cholesterol and triglyceride levels in healthy human volunteers.23 Furthermore, administration of evinacumab to 9 HoFH patients in an open-label, phase 2, proof-of-concept study resulted in further substantial reductions in LDL-C levels on top of those achieved with stable, aggressive lipid-lowering therapy.19
The aim of this study was to analyze the effects of evinacumab on the activity of LDLR in lymphocytes purified from the HoFH patients included in the proof-of-concept study, before and after treatment with the antibody. Enrolled patients had a documented history of HoFH diagnosis with mutation(s) in both LDLR alleles. LDLR activity was also assessed in a LDLR-defective Chinese hamster ovary (CHO) cell line (CHO-ldlA7) transfected with plasmids encoding the LDLR variants. From these analyses, we aimed to confirm that evinacumab has an LDLR-independent mechanism by assessing the level of LDLR expression, and LDL binding and uptake, in the LDLR variants.
Materials and Methods
Qualified researchers may request access to study documents (including the clinical study report, study protocol with any amendments, blank case report form, and statistical analysis plan) that support the methods and findings reported in this article. Individual anonymized participant data will be considered for sharing once the indication has been approved by a regulatory body, if there is legal authority to share the data and there is not a reasonable likelihood of participant reidentification. Submit requests to https://errs.regeneron.com/external.
Subject Selection and Treatment
Patient selection for the phase 2, proof-of-concept study (NCT02265952) was carried out by Regeneron Pharmaceuticals, Inc, details of which have been described previously.19 Briefly, male and female patients at least 18 years of age with an HoFH diagnosis by (1) documented FH-causing mutation(s) in both LDLR or APOB alleles, (2) documented presence of double heterozygous variants in LDLR, APOB, and PCSK9, or (3) documented skin fibroblast LDLR activity <20% of normal were eligible for inclusion in the study; however, only patients with HoFH and mutations in both LDLR alleles were enrolled. They were also required to be on stable, aggressive lipid-lowering therapy for at least 4 weeks (including statins, fibrates, ezetimibe, lomitapide, PCSK9 inhibitors, and portacaval shunt), and to have not undergone lipid apheresis within 4 weeks before the screening visit. All patients provided written informed consent.
Two control subjects, previously described,24,25 were used for internal method validation. The first was a heterozygous FH patient carrying a mutation in the LDLR gene that produces a defective binding protein because the mRNA contains an in-frame deletion of exons 3 and 4, c.(191-?_694+?del). The second was a heterozygous FH patient carrying the c.261G>A, p.(Trp87*) LDLR variant, a null allele mutant that does not produce LDLR.
Patients received evinacumab 250 mg administered subcutaneously in the abdominal area at baseline, and then a single 15 mg/kg intravenous dose 2 weeks later. Of the 9 patients included in the phase 2 study, 2 received 4 additional doses of evinacumab 450 mg subcutaneously at weeks 12 to 15.
Isolation and Culture of T-Lymphocytes
Patient blood samples were collected on the baseline visit, and peripheral blood lymphocytes from patient blood samples were isolated from Vacutainer CPT (BD Biosciences, CA) tubes following the manufacturer’s instructions. Cells were then cultured at 37°C in 5% CO2 at a concentration of 2×105 cells/mL in medium supplemented with lipoprotein-deficient serum, antibiotics, and antimycotics. Anti-CD3/CD28 beads (Dynal Biotech, ThermoFisher Scientific, Norway; 2×105 beads/mL) were also added to obtain suitable T-cell activation to upregulate the LDLR.
Lipoprotein Isolation
Lipoproteins were isolated from blood samples from normocholesterolemic controls in a 2-step centrifugation procedure. Briefly, blood was collected in ethylenediaminetetraacetic acid tubes and plasma was separated by centrifugation for 30 minutes, at 12 000×g at 4°C. Blood plasma LDL (1.019–1.050 g/mL) was isolated by density ultracentrifugation, adjusting the density of plasma to 1.21 g/mL by the addition of potassium bromide, and obtaining 2 phases with PBS buffer. The sample was centrifuged at 244 500×g for 19 hours 30 minutes at 4°C. The band corresponding to LDL was recovered carefully, stored at 4°C, and used within the next 4 to 5 days. Further analysis by complementary techniques of samples obtained with this procedure confirmed that the LDL purification was near purity.
Lipoprotein Labeling
LDL was labeled with fluorescein isothiocyanate (FITC), as previously described.10 Briefly, lipoproteins (1 mg/mL) in 0.1 M NaHCO3 (pH 9.0) were mixed with 10 µL/mL of FITC (2 mg/mL in dimethyl sulfoxide). The mixture was gently mixed by slow rocking at room temperature for 2 hours. The unreacted dye was removed by gel filtration on a Sephadex G-25 mounted in a PD-10 desalting column (GE Healthcare, IL) equilibrated with PBS ethylenediaminetetraacetic acid-free buffer. All fractions were assayed for protein content with BSA as standard (Pierce BCA protein assay, Pierce, ThermoFisher Scientific, MA).
Quantification of LDLR Activity and Expression by Flow Cytometry in Lymphocytes
LDLR cell surface expression and LDLR activity (including LDL binding capacity and LDL uptake) was determined by flow cytometry in lymphocytes. Lymphocytes were taken from patients before and after the patients were treated with evinacumab, and then the activity assays were performed. Lymphocytes were isolated using BD Vacutainer CPT Cell Preparation Tubes with Sodium Citrate (REF 362761, BD Biosciences, CA) following the manufacturer’s instructions. Lymphocytes (2×105 cells) were cultured for 72 hours in lipoprotein-deficient serum and stimulated with anti-CD3/CD28 beads to obtain a uniform fraction of lymphoblasts with a maximum upregulation of LDLR. Lymphoblasts are slightly bigger in size and can be more efficiently selected by flow cytometry for further analysis of LDLR activity. Cells were recovered and washed twice in PBS containing 1% BSA (PBS-1%BSA).
To determine LDLR expression, cells were fixed in 4% paraformaldehyde for 10 minutes, blocked with PBS-1%BSA-5% fetal bovine serum incubated with a mouse anti-LDLR primary antibody (1:100; 2.5 mg/L; Progen Biotechnik GmbH, Germany; reagent details are given in Major Resources Table in the online-only Data Supplement ) for 1 hour at room temperature, washed twice with PBS-1%BSA, and incubated with an Alexa Fluor 488-conjugated goat anti-mouse secondary immunoglobulin G antibody (1:100; Invitrogen, Molecular Probes, CA).
To determine LDLR activity, cells were incubated for 2 hours with 20 μg/mL FITC-LDL at either 37°C (to determine LDLR uptake) or 4°C (to determine LDL-LDLR binding). After incubation with FITC-LDL, lymphoblasts were washed twice in PBS-1%BSA, fixed in 4% paraformaldehyde for 10 minutes, and washed twice more with PBS-1%BSA.
To determine the amount of internalized LDL, Trypan blue solution (Sigma-Aldrich, Germany) was added directly to the same samples above to a final concentration of 0.2%, which quenches the extracellular signal because of the noninternalized LDLR-LDL complexes.26
Fluorescence intensities were measured by fluorescence-activated cell sorting in a FACSCalibur Flow cytometer (BD Biosciences, CA) according to the manufacturer’s instructions. Stimulated lymphoblasts could be separated from the unstimulated ones according to areas in flow cytometric analysis. For each sample, fluorescence of 10 000 events was acquired for data analysis and the results were expressed as the mean fluorescence of activated gated cells, selected in a forward- versus side-scatter window. All measurements were performed in triplicate. Heterozygous lymphocytes carrying c.(191-?_694+?del) LDLR variant that produces a defective binding receptor, and also heterozygous lymphocytes carrying c.261G>A, p.(Trp87*) LDLR variant, were used as internal controls of the assay.
Site-Directed Mutagenesis
Among the LDLR variants carried by the 9 HoFH patients, the expression of point mutation and frameshift LDLR variants was determined in vitro. The mutations were introduced into the human LDLR cDNA (NM_000527.4), in the mammalian expression vector pcDNA3 under control of a SV40 promoter by oligonucleotide site-directed mutagenesis using QuickChange Lightning mutagenesis kit (Agilent Technologies, CA), according to the manufacturer’s instructions. The oligonucleotides used to generate the plasmid carrying the LDLR variant under study were synthesized in vitro and subcloned using the restriction enzymes SacII and EcoRI. The presence of the desired nucleotide alteration was confirmed by polymerase chain reaction and restriction enzyme digestion of the appropriate fragments and the integrity of the remaining LDLR cDNA sequence of the construct was verified by direct sequence analysis.
CHO Cell Culture and Transfection
An LDLR-deficient CHO cell line ldlA7 (CHO-ldlA7) was cultured in Ham’s F-12 medium supplemented with 5% fetal bovine serum, 2 mmol/L L-glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin. CHO-ldlA7 cells were plated into 6- or 24-well culture plates and transfected with the plasmids carrying wild-type (wt) LDLR or c.259T>G, p.(Trp87Gly), c.2043C>A, p.(Cys681*), c.761A>C, p.(Gln254Pro), c.1878delA, p.(Ala627Profs*38), c.530C>T, p.(Ser177Leu), c.1004G>T, p.(Gly335Val), and c.917C>T, p.(Ser306Leu) LDLR variants using Lipofectamine LTX and Plus Reagent (Invitrogen, ThermoFisher Scientific, MA) according to the manufacturer’s instructions. Transfected cells were maintained in culture for 48 hours to achieve maximal LDLR expression.
Western Blot Analysis
LDLR expression in CHO-ldlA7 cells transfected with mutations of interest were analyzed by immunoblotting. Cell lysates were prepared, protein concentration determined, and fractionated by electrophoresis on 8.5% sodium dodecyl sulphate-polyacrylamide gel electrophoresis for semiquantitative immunoblotting. Membranes were immunostained with rabbit polyclonal anti-LDLR antibody (1:500; Cayman Chemical, MI) for 16 hours at 4°C and anti-GAPDH (GAPDH) antibody (1:1000) (Santa Cruz Biotechnology, Inc, CA) for 1 hour at room temperature (antibodies and washing buffer have 2 mmol/L CaCl2) and counterstained with a horseradish peroxidase-conjugated anti-rabbit antibody (GE Healthcare, Little Chalfont, United Kingdom). The signals were developed using SuperSignal West Dura Extended Substrate (Pierce Biotechnology, ThermoFisher Scientific, IL). ChemiDoc XRS (Bio-Rad, CA) was used to detect the signals, and Quantity One Basic 4.4.0 software (Bio-Rad) was used to quantify band intensities. The concentrations of the antibodies were optimized to achieve low background and a linear dose-dependent increase in signal intensity. The relative band intensity for the mature form of LDLR protein expressed for the different constructs was calculated as the ratio between the LDLR 160 kDa band to that of GAPDH. c.261G>A, p.(Trp87*), a variant that is not expressed, and c.1285G>C, p.(Val429Leu), a variant that is only expressed into its nonmature form, were used as internal controls of the assay.
Quantification of LDLR Activity and Expression by Flow Cytometry in Transfected CHO-ldlA7 Cells
LDLR activity (including LDLR cell surface expression, LDL binding capacity, and LDL uptake) was determined by flow cytometry in CHO-ldlA7 transfected cells with plasmids encoding the LDLR variants. Transfected CHO-ldlA7 cells were grown in 24-well culture plates. Forty-eight hours after transfection, cells were incubated for 2 hours with 20 µg/mL FITC-LDL at either 37°C or 4°C, as described above for lymphocytes.
To determine LDLR cell surface expression by fluorescence-activated cell sorting, transfected CHO-ldlA7 cells grown during 48 hours were incubated with a mouse anti-LDLR primary antibody (1:100; Cayman Chemical, cat no.: 10007665) for 1 hour at room temperature, then washed twice with PBS-1%BSA and incubated with secondary antibody Alexa Fluor 488-conjugated goat anti-mouse immunoglobulin G (1:100), as described above for lymphocytes.
Fluorescence intensities were measured in a FACSCalibur Flow cytometer as described above for lymphocytes, and all measurements were performed in triplicate.
Confocal Laser Scanning Microscopy
LDLR variants found in the HoFH patients were classified by mutation type using confocal laser scanning microscopy, based on analysis of LDLR cell surface expression, LDL-LDLR binding, and LDL uptake in LDLR-transfected CHO-ldlA7 cells. Briefly, cells were plated in coverslips and then transfected with the LDLR-containing plasmids and cultured for 48 hours at 37°C in 5% CO2. The medium was then removed and coverslips washed twice with PBS-1%BSA. To determine LDLR expression, cells were incubated for 16 hours at 4°C with a mouse primary antibody anti-LDLR, washed, and then incubated for 1 hour at room temperature with a secondary antibody Alexa Fluor 488-conjugated goat anti-mouse immunoglobulin G. To determine LDL-LDLR binding and LDL uptake, Dil-labeled lipoproteins (20 µg/mL LDL) were added and cells were incubated for an additional 4 hours at 4°C or 37°C, respectively. Cells were fixed with 4% paraformaldehyde for 10 minutes, washed 3× with PBS-1%BSA, and permeabilized with 1% Triton X-100 for 30 minutes at room temperature. Samples were then washed and blocked in PBS-10% fetal bovine serum for 1 hour and washed in PBS-1%BSA 3×. The samples were then incubated with the appropriate primary antibodies for 16 hours at 4°C followed by incubation with the appropriate fluorescent secondary antibodies. Subcellular colocalization of LDLR variants with the endoplasmic reticulum was determined by using a primary antibody that recognized the endoplasmic reticulum-specific marker calregulin and the appropriate fluorescent secondary antibody. Coverslips were mounted on a glass slide and samples were visualized using a confocal microscope (Olympus IX 81, Tokyo, Japan), with sequential excitation and capture image acquisition with a digital camera (Axiocam NRc5, Zeiss, Jena, Germany). Images were processed with Fluoview v.50 software. Image analysis to quantify the fluorescence intensities was accomplished using the public domain software ImageJ (available at http://rsb.info.nih.gov/ij) running on a standard PC. Cells transfected with the c.(191-?_694+?del) LDLR variant that produces a defective binding receptor were used as internal controls of the assay
Variant Classification
Variants were classified as recommended by the American College of Medical Genetics and Genomics,27 adapted for FH specifications.9
In Silico Prediction of the LDLR Variant Under Study
To predict the possible impact of amino acid substitutions on the structure and function of the LDLR, 4 different types of software were used: PhyloP (https://omictools.com/phylogenetic-p-values-tool), Polyphen-2 (http://genetics.bwh.harvard.edu/pph2), SIFT (http://sift.jcvi.org), and Mutation Taster (http://www.mutationtaster.org).
Statistical Analysis
All measurements were performed at least 3×, unless otherwise stated, and results are presented as mean±SD. To confirm that the data were normally distributed, a Shapiro-Wilk test was performed. The null hypothesis was verified, indicating that the data were normally distributed. As the intention was to compare wt with each mutant, that is, comparison of 2 variables, Student t-tests were employed for analysis. A 2-tailed Student t test with a significance level of 0.05 was used to test for differences in LDLR activity between wt patient samples and each patient’s samples before and after treatment. In addition, the same statistical method applies to the comparisons in LDLR activity between before and after treatment administration for each variant. All statistical analyses were performed with the SPSS 25 (SPSS, Inc, Chicago, IL).
Results
Patient Characteristics
Baseline characteristics, LDL-C levels, and genotypes of the 9 HoFH patients enrolled in the study are shown in the Table. There were 2 null homozygotes and 1 compound heterozygote with 2 null alleles.
Table.
Baseline Characteristics, LDLR Genotypes, and LDL-C Levels With Evinacumab Treatment of Individual Patients With HoFH

LDL-C Reduction in HoFH Patients After Evinacumab Treatment
Overall mean peak reduction in LDL-C with evinacumab was −58±18%, occurring between Week 4 and Week 12, showing that ANGPTL3 inhibition substantially reduced LDL-C in all participants (Table).
Effect of Evinacumab Treatment on LDLR Activity Determined in Lymphocytes From HoFH Patients
The LDLR activity was assessed in isolated lymphocytes from 9 HoFH patients before and after treatment with evinacumab, as described in the Materials and Methods section. As shown in Figure 1, lymphocytes from the HoFH patients had reduced LDLR activity compared with lymphocytes carrying wt LDLR. Evinacumab treatment did not modify LDLR activity, indicating no functional effect on LDLR activity with evinacumab treatment (Figure 1). As shown in Figure 1A, LDLR expression was highly heterogeneous, ranging from values of nearly 100% to 10%. However, both LDL binding and uptake values were reduced, ranging from ≈60% to 10% both before and after evinacumab treatment (Figure 1B and 1C).
Figure 1.

LDLR (low-density lipoprotein receptor) activity in lymphocytes from 9 homozygous familial hypercholesterolemia (HoFH) patients before and after evinacumab treatment. LDLR expression (A); LDL binding (B); and LDL uptake (C). LDLR activity and expression was quantified by flow cytometry as described in the Materials and Methods section. The values represent the mean of 3 independent experiments; error bars represent ±SD. wt=wild-type (non-HoFH) control; c.[191-?_694+?del] and p.(Trp87*)=LDLR defective heterozygous patient controls (see Methods). (1;2) represents the mean±SD of patients 1 and 2 which carry the same LDLR variants; the lymphocytes from these 2 patients were analyzed independently. The sex of the patients 1 to 9 from which the lymphocytes were sourced is indicated in the Table. HoFH indicates homozygous familial hypercholesterolemia; LDL, low-density lipoprotein; and LDLR, low-density lipoprotein receptor. *P<0.001 comparing wt with each variant before and after treatment. No statistical significance was found when after treatment vs before treatment LDLR activity data (expression, binding, and uptake) of each variant was compared.
Expression of LDLR Variants in CHO-ldlA7 Cells
CHO-ldlA7 cells were transfected with plasmids carrying wt LDLR or c.259T>G, p.(Trp87Gly); c.2043C>A, p.(Cys681*); c.761A>C, p.(Gln254Pro); c.1878del, p.(Ala627Profs*38); c.530C>T, p.(Ser177Leu); c.1004G>T, p.(Gly335Val); and c.917C>T, p.(Ser306Leu) LDLR variants. LDLR expression in cells with transfected mutants and wt LDLR was analyzed by immunoblotting as described in the Materials and Methods section. For internal controls of the assay, 2 mutants, c.261G>A, p.(Trp87*), a variant that is not expressed, and c.1285G>C, p.(Val429Leu), a variant that is only expressed in its nonmature form, were used. As shown in Figure 2A (upper panel), expression of p.(Trp87Gly), p.(Gln254Pro), p.(Ser306Leu), and p.(Gly335Val) LDLR variants is similar to the expression of wt LDLR. Equal loading of protein was confirmed by membrane stripping and analysis of cytosolic GAPDH protein (Figure 2A, lower). The extent of protein expression was determined by quantitative densitometric analysis (Figure 2B). On the contrary, and as expected due to the nature of the variants, p.(Cys681*) and p.(Ala627Profs*38) were expressed as nonmature proteins with a smaller size (Figure 2C, upper). Figure 2C also shows that the expression of the mature LDLR protein p.(Ser177Leu) is less efficient compared with the mature wt LDLR control. The extent of protein expression in p.(Cys681*), p.(Ala627Profs*38), and p.(Ser177Leu) was determined by quantitative densitometric analysis (Figure 2D).
Figure 2.
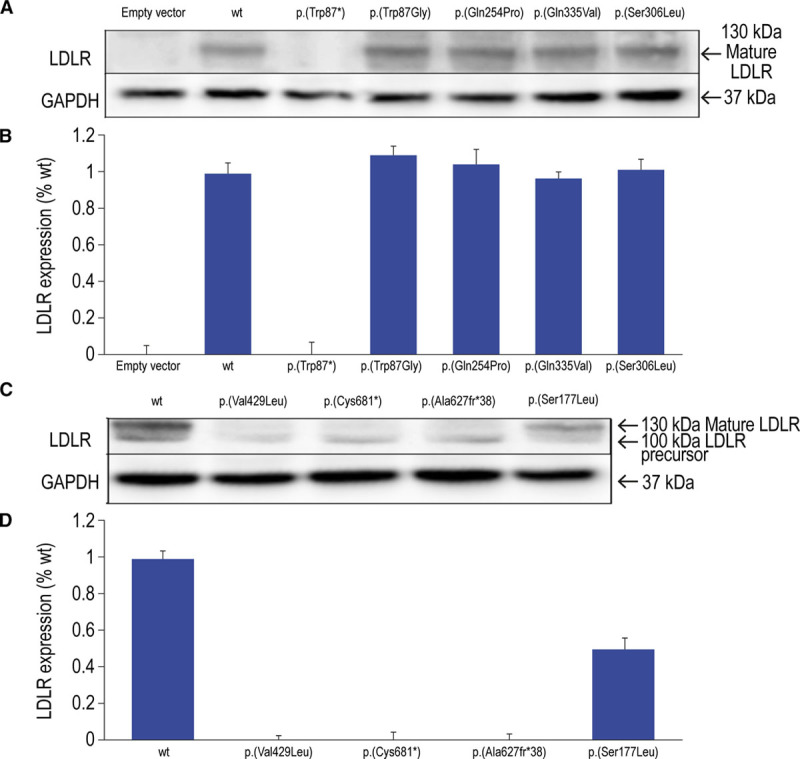
Expression of wild-type LDLR and LDLR variants in Chinese hamster ovary-ldlA7 transfected cells. LDLR (low-density lipoprotein receptor) expression (upper) and GAPDH expression (A and C; lower); LDLR to GAPDH densitometric analysis (B and D). Cells were transfected with the corresponding plasmids carrying the LDLR mutations of interest and immunoblot analysis was performed, as described in Materials and Methods. The relative band intensity of mature LDLR protein expression was calculated as the ratio of 160 kDa LDLR band intensity to that of GAPDH. A representative experiment from 3 independently performed assays is shown in A. Levels of significance were determined by a 2-tailed Student t test, and a CI of >95% (P<0.05) was used to establish statistical significance. No statistically significant differences were found among LDLR expression for variants analyzed in A and B, but differences were significant for variants analyzed in C and D. wt indicates wild-type.
Functional In vitro Characterization of the Point Mutations and Frameshift LDLR Variants Present in the HoFH Patients
Next, in vitro LDLR activity of p.(Trp87Gly), p.(Cys681*), p.(Gln254Pro), p.(Ala627Profs*38), p.(Ser177Leu), p.(Gly335Val), and p.(Ser306Leu) LDLR variants was determined by flow cytometry in CHO-ldlA7 cells as described in the Materials and Methods section. The p.(Trp87*) and c.191-?_694+?del LDLR variants were used as internal controls for the assay. As shown in Figure 3A, expression of p.(Trp87Gly), p.(Gln254Pro), p.(Gly335Val), and p.(Ser306Leu) was similar to wt LDLR. As with the results obtained by Western blot (Figure 2), expression of the p.(Ser177Leu) diminished by ≈40% compared with wt LDLR. As expected, expression of p.(Cys681*) and p.(Ala627Profs*38) at the cellular membrane surface was residual (Figure 3A).
Figure 3.

LDLR (low-density lipoprotein receptor) activity in Chinese hamster ovary-ldlA7-transfected cells. LDLR expression (A); LDL (low-density lipoprotein) binding (B); and LDL uptake (C). LDLR activity and expression was quantified by flow cytometry, as described in the Materials and Methods section. The values represent the mean of 3 independent experiments; error bars represent ±SD. wt indicates wild-type. *P<0.001 comparing wt with each variant.
As shown in Figure 3B, LDL-LDLR binding activities of p.(Trp87Gly), p.(Cys681*), p.(Gln254Pro), p.(Ala627Profs*38), and p.(Ser177Leu) were almost completely abolished as compared with wt. By contrast, the LDL-binding capacity of p.(Gly335Val) and p.(Ser306Leu) was partially impaired by ≈40% compared with LDLR wt control (Figure 3B).
As shown in Figure 3C and in agreement with LDL-LDLR binding results (Figure 3B), LDL internalization in CHO-ldlA7 cells expressing p.(Trp87Gly), p.(Cys681*), p.(Gln254Pro), p.(Ala627Profs*38), and p.(Ser177Leu) was highly diminished compared with wt. On the contrary, uptake activity of p.(Gly335Val) and p.(Ser306Leu) was significantly inhibited (by ≈40% compared with LDLR control) but to a lesser extent than the other variants studied (Figure 3C).
Analysis of LDLR Activity by Confocal Microscopy
LDLR variants found in the patients were classified by mutation type using confocal microscopy as described in Materials and Methods section. As shown in Figure 4A, the expressed LDLR variants p.(Cys681*) and p.(Ala627Profs*38) colocalize with calregulin (an endoplasmic reticulum-specific marker), and the receptors do not reach the cellular membrane. Consequently, and as shown in Figure 4B and 4C, respectively, there is no significant LDL binding or uptake. Therefore, p.(Cys681*) and p.(Ala627Profs*38) LDLR variants are class 2 type mutations.
Figure 4.

Class 2 type mutation assignment of variants found in homozygous familial hypercholesterolemia patients. LDLR (low-density lipoprotein receptor) expression and colocalization with calregulin in the endoplasmic reticulum (A); LDL (low-density lipoprotein)-LDLR binding activity (B); and LDL internalization activity (C). Confocal laser scanning microscopy was used to analyze LDLR activity, as described in the Materials and Methods section. wt indicates wild-type.
Next, we analyzed the class type mutation of p.(Trp87Gly), p.(Gln254Pro), p.(Ser177Leu), p.(Gly335Val), and p.(Ser306Leu) LDLR variants. As shown in Figure 5A, all are expressed in their mature form at the cellular surface, similar to the wt LDLR. By contrast, LDL binding and uptake are almost completely abolished in p.(Trp87Gly), p.(Gln254Pro), and p.(Ser177Leu) LDLR compared with wt LDLR (Figure 5B and 5C). Interestingly, and confirming the results obtained by fluorescence-activated cell sorting, LDL binding and uptake activities of p.(Gly335Val) and p.(Ser306Leu) LDLR variants are diminished compared with wt LDLR but functional to a significant extent (Figure 5B and 5C). According to these results, p.(Trp87Gly), p.(Gln254Pro), p.(Ser177Leu), p.(Gly335Val), and p.(Ser306Leu) LDLR variants can be classified as type 3 mutations.
Figure 5.

Class 3 type mutation assignment of variants found in homozygous familial hypercholesterolemia patients. LDLR (low-density lipoprotein receptor) expression at cellular membrane (A); LDL (low-density lipoprotein)-LDLR binding activity (B); and LDL internalization activity (C). Confocal laser scanning microscopy was used to analyze LDLR activity, as described in the Materials and Methods section. wt indicates wild-type.
Discussion
Results from the present study suggest that evinacumab treatment did not alter LDLR activity. LDLR variants studied in vitro had only residual expression and diminished LDL binding and uptake. Furthermore, LDL-C uptake was similarly low in lymphocytes taken before and after evinacumab treatment. In comparison, patients with HoFH and the corresponding LDLR variants had LDL-C reductions ranging 50.9% to 90.2% following evinacumab treatment. The functionality of the LDLR variants in vitro did not show a pattern in relation to the magnitude of LDL-C reductions observed in the patients with the corresponding LDLR variant. For example, 2 patients had the same Trp87Gly variant but had LDL-C reductions of 50.9% and 90.1%, respectively. The LDLR variants would be expected to have no relation to the LDL-C reduction if evinacumab has an LDLR-independent mechanism. The intrapatient variability in LDL-C reductions observed may be related to other factors that have not been investigated such as levels of ANGPTL3 and/or LPL. The mechanism leading to the large reduction in LDL-C with evinacumab is under investigation (Regeneron Pharmaceuticals, Inc). One possible mechanism may involve LPL as follows. ANGPTL3 inhibits LPL activity; inhibition of ANGPTL3 with evinacumab may lead to increased LPL-mediated hydrolysis of triglycerides associated with very-low density lipoprotein and chylomicrons. The observed LDL-C reductions may, at least in part, result from lower secretion of very-low density lipoprotein, the precursor to LDL.28,29
A potential limitation of these analyses that used cells from patients before and after they were treated with evinacumab is that the patient cells were maintained in culture for 48 hours, without addition of the evinacumab. It is possible that the antibody effect could be missed because cells are not in constant contact with the antibody. However, overall, the results indicate that the LDLR variants were either not processed or expressed or had low activity compared with wt, yet, the patients had LDL-C reductions. This supports the overall conclusion that evinacumab has an LDLR-independent mode of action.
In this study, we used the LDLR-deficient CHO cell line ldlA7 (CHO-ldlA7) for studying the LDLR variants instead of a hepatic cell line such as HepG2. CHO-ldlA7 have been previously used in several LDLR functional studies.30–32 An advantage of using CHO-ldlA7 instead of a hepatic cell line such as HepG2 is that the CHO-ldlA7 cells do not express LDLR, making them an excellent model to study the effect of LDLR variants as they mimic the homozygous situation. Hepatic cell lines express wt LDLR, thus transfection with an LDLR variant would result in a heterozygous-like phenotype, masking the effect of the variant under analysis. This makes it more difficult to interpret the real effect of the LDLR variant under study. In addition, using the CHO-ldlA7 cell line allows both characterization and classification of the LDLR mutation class type. In a previous study, we used a HepG2 cell line to characterize extracellular activity of PCSK9.33 In this case, we took advantage of the high LDLR expression in the HepG2 cells and added recombinant purified PCSK9 variants to the culture medium, then studied LDLR expression and LDL uptake. Also, we used HEK293 cells for transient transfection with PCSK9 variants because they do not express significant amounts of wt PCSK9.
Patients with HoFH who do not respond well to lipid-lowering therapy have a poor prognosis, as survival has been shown to be determined by on-treatment serum cholesterol level.34,35 Novel treatment approaches are therefore vital. Although the recent LDLR-independent agents mipomersen and lomitapide may be of benefit in some patients, particularly those with LDLR-negative HoFH,13 they are associated with potentially harmful effects such as hepatotoxicity (elevated transaminases) and hepatic steatosis in adults;36,37 there are, as of yet, no efficacy or safety data in children.14
The evinacumab proof-of-concept study demonstrated meaningful reductions in LDL-C on top of standard-of-care treatment in all 9 HoFH patients, including 3 null/null patients, and LDL-C reductions were sustained for up to 2 months following the intravenous injection at Week 2.19 Evinacumab was also well tolerated at the doses given, including the 450 mg subcutaneous dose given to 2 patients at Weeks 12 to 15. While all patients reported at least 1 adverse event (including nausea and back pain in 4 patients each), there were no treatment discontinuations because of adverse events; there was 1 serious adverse event (coronary artery disease because of underlying disease) in the main study period.19
Among the new treatments approved for lowering LDL-C are evolocumab and alirocumab, both monoclonal antibodies that prevent circulating PCSK9 from binding to the LDL receptor.38,39 Several lipid guidelines suggest that these PCSK9 inhibitor antibodies may be suitable for individuals with FH or secondary prevention in patients with dyslipidemia in whom statins provide insufficient LDL-C control, due to either resistance to statin therapy or insufficient therapeutic success with statin with/without ezetimibe.40–42 The addition of either alirocumab 150 mg every 2 weeks or evolocumab 140 mg monthly to statin therapy consistently resulted in an incremental decrease in LDL-C levels of up to 60% in HeFH compared with placebo.43–45 However, PCSK9 inhibitor treatment resulted in a less pronounced LDL-C lowering efficacy (mean LDL-C reductions of ≈20% were reported) in HoFH patients with LDLR Class 2 and 3 mutations and with patients with mutations in ApoB100, which fails in LDLR recognition. Importantly, there is heterogeneity in the response to PCSK9 inhibitors among LDLR defective HoFH patients, as illustrated in the present study where a HoFH patient with LDLR class 3 mutation had an LDL-C reduction of <2%, suggesting several factors including baseline LDL-bound or receptor-bound PCSK9 concentrations may affect the LDL-C lowering effect of PCSK9 inhibitor treatment in LDLR defective HoFH. By contrast, LDLR-negative patients were reported as having no or poor response to PCSK9 inhibitor treatment, as expected from the mechanism of action of this class of drugs.15–17 Thus, new therapeutic strategies such as administration of evinacumab could be beneficial, especially for LDLR-negative HoFH patients as shown in this study.
The functional and structural analysis reported here on these LDLR variants provides evidence that, in humans, inhibition of ANGPTL3 lowers LDL independently of LDLR, and is useful for a more targeted treatment of patients with significant defects in LDLR. The results also suggest that evinacumab may be effective in patients carrying Class 2 or 3 mutations with almost complete loss of LDL-binding capacity. This is important because the LDL-C-lowering capacity of other treatments, such as statins and PCSK9 inhibitor antibodies, is restricted in such cases.
Acknowledgments
We thank the patients, their families, and all investigators involved in this study. The LDLR-deficient CHO cell line ldlA7 (CHO-ldlA7) was kindly provided by Dr Monty Krieger, Massachusetts Institute of Technology, Cambridge, MA. Medical writing assistance and editorial support was provided by Grace Shim, PhD and Rob Campbell, PhD, of Prime (Knutsford, United Kingdom), funded by Regeneron Pharmaceuticals, Inc, according to Good Publication Practice guidelines (Link). The authors were responsible for all content and editorial decisions and received no honoraria related to the development of this publication.
Sources of Funding
This analysis was funded by Regeneron Pharmaceuticals, Inc.
Disclosures
P. Banerjee, K.-C. Chan, M. Tarabocchia, P.J. Skiba, and R. Pordy are stockholders (modest) and employees (significant) of Regeneron Pharmaceuticals, Inc. M. Bourbon has received projects grants from Alexion (modest) and a research grant from Regeneron Pharmaceuticals (modest). She has no financial interest in Regeneron Pharmaceuticals, Inc. D. Gaudet has received research grant support from FH Canada, Aegerion (Novelion Therapeutics), Amgen, Akcea, Esperion, Gemphire, Ionis, Regeneron Pharmaceuticals, Inc, and Sanofi and has served as a consultant for Amgen, Aegerion, Akcea, Ionis, Regeneron Pharmaceuticals, Inc, and Sanofi. He has no financial interest in Regeneron Pharmaceuticals, Inc. C. Martin has received a research grant from Regeneron Pharmaceuticals, Inc. (modest). He has no financial interest in Regeneron Pharmaceuticals, Inc. The other authors report no conflicts.
Supplementary Material
{kind=link}
Nonstandard Abbreviations and Acronyms
- ANGPTL3
- angiopoietin-like 3 protein
- CHO
- Chinese hamster ovary
- FITC
- fluorescein isothiocyanate
- FH
- familial hypercholesterolemia
- HoFH
- homozygous FH
- LDL-C
- low-density lipoprotein cholesterol
- LDLR
- low-density lipoprotein receptor
- LLT
- lipid-lowering therapy
- LPL
- lipoprotein lipase
- PCSK9
- proprotein convertase subtilisin/kexin type 9
- wt
- wild type
Dedication - We wish to dedicate this article to the memory of Daniel A. Gipe, MD, who died prematurely and unexpectedly after its submission, and to whom we are all profoundly indebted, not only for his contributions to this manuscript, but most importantly for his significant personal contribution to the recent advancements in the field that are here described and for his friendship.
For Sources of Funding and Disclosures, see page 2259.
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/ATVBAHA.119.313051.
References
- 1.Cuchel M, Bruckert E, Ginsberg HN, Raal FJ, Santos RD, Hegele RA, Kuivenhoven JA, Nordestgaard BG, Descamps OS, Steinhagen-Thiessen E, et al. European Atherosclerosis Society Consensus Panel on Familial Hypercholesterolaemia. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. 2014;35:2146–2157. doi: 10.1093/eurheartj/ehu274. doi: 10.1093/eurheartj/ehu274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marais AD. Familial hypercholesterolaemia. Clin Biochem Rev. 2004;25:49–68. [PMC free article] [PubMed] [Google Scholar]
- 3.Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR, Beaudet A, Sly WS, Valle D, editors. In: The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. pp. 2863–2913. [Google Scholar]
- 4.Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat. 1992;1:445–466. doi: 10.1002/humu.1380010602. doi: 10.1002/humu.1380010602. [DOI] [PubMed] [Google Scholar]
- 5.Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, et al. European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478–390a. doi: 10.1093/eurheartj/eht273. doi: 10.1093/eurheartj/eht273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gaudet D, Tremblay G, Perron P, Gagné C, Ouadahi Y, Moorjani S. [Familial hypercholesterolemia in eastern Quebec: a public health problem? The experience of the hyperlipidemia clinic of Chicoutimi]. Union Med Can. 1995;124:54–60. [PubMed] [Google Scholar]
- 7.Raal FJ, Sjouke B, Hovingh GK, Isaac BF. Phenotype diversity among patients with homozygous familial hypercholesterolemia: a cohort study. Atherosclerosis. 2016;248:238–244. doi: 10.1016/j.atherosclerosis.2016.03.009. doi: 10.1016/j.atherosclerosis.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 8.Raal FJ, Pilcher GJ, Panz VR, van Deventer HE, Brice BC, Blom DJ, Marais AD. Reduction in mortality in subjects with homozygous familial hypercholesterolemia associated with advances in lipid-lowering therapy. Circulation. 2011;124:2202–2207. doi: 10.1161/CIRCULATIONAHA.111.042523. doi: 10.1161/CIRCULATIONAHA.111.042523. [DOI] [PubMed] [Google Scholar]
- 9.Chora JR, Medeiros AM, Alves AC, Bourbon M. Analysis of publicly available LDLR, APOB, and PCSK9 variants associated with familial hypercholesterolemia: application of ACMG guidelines and implications for familial hypercholesterolemia diagnosis. Genet Med. 2018;20:591–598. doi: 10.1038/gim.2017.151. doi: 10.1038/gim.2017.151. [DOI] [PubMed] [Google Scholar]
- 10.Etxebarria A, Benito-Vicente A, Alves AC, Ostolaza H, Bourbon M, Martin C. Advantages and versatility of fluorescence-based methodology to characterize the functionality of LDLR and class mutation assignment. PLoS One. 2014;9:e112677. doi: 10.1371/journal.pone.0112677. doi: 10.1371/journal.pone.0112677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strøm TB, Laerdahl JK, Leren TP. Mutation p.L799R in the LDLR, which affects the transmembrane domain of the LDLR, prevents membrane insertion and causes secretion of the mutant LDLR. Hum Mol Genet. 2015;24:5836–5844. doi: 10.1093/hmg/ddv304. doi: 10.1093/hmg/ddv304. [DOI] [PubMed] [Google Scholar]
- 12.Strøm TB, Tveten K, Laerdahl JK, Leren TP. Mutation G805R in the transmembrane domain of the LDL receptor gene causes familial hypercholesterolemia by inducing ectodomain cleavage of the LDL receptor in the endoplasmic reticulum. FEBS Open Bio. 2014;4:321–327. doi: 10.1016/j.fob.2014.03.007. doi: 10.1016/j.fob.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zodda D, Giammona R, Schifilliti S.Treatment strategy for dyslipidemia in cardiovascular disease prevention: focus on old and new drugs. Pharmacy (Basel) 20186pii: E10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.France M. Homozygous familial hypercholesterolaemia: update on management. Paediatr Int Child Health. 2016;36:243–247. doi: 10.1080/20469047.2016.1246640. doi: 10.1080/20469047.2016.1246640. [DOI] [PubMed] [Google Scholar]
- 15.Raal FJ, Honarpour N, Blom DJ, Hovingh GK, Xu F, Scott R, Wasserman SM, Stein EA TESLA Investigators. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;385:341–350. doi: 10.1016/S0140-6736(14)61374-X. doi: 10.1016/S0140-6736(14)61374-X. [DOI] [PubMed] [Google Scholar]
- 16.Raal FJ, Hovingh GK, Blom D, Santos RD, Harada-Shiba M, Bruckert E, Couture P, Soran H, Watts GF, Kurtz C, et al. Long-term treatment with evolocumab added to conventional drug therapy, with or without apheresis, in patients with homozygous familial hypercholesterolaemia: an interim subset analysis of the open-label TAUSSIG study. Lancet Diabetes Endocrinol. 2017;5:280–290. doi: 10.1016/S2213-8587(17)30044-X. doi: 10.1016/S2213-8587(17)30044-X. [DOI] [PubMed] [Google Scholar]
- 17.Stein EA, Honarpour N, Wasserman SM, Xu F, Scott R, Raal FJ. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation. 2013;128:2113–2120. doi: 10.1161/CIRCULATIONAHA.113.004678. doi: 10.1161/CIRCULATIONAHA.113.004678. [DOI] [PubMed] [Google Scholar]
- 18.Thedrez A, Blom DJ, Ramin-Mangata S, Blanchard V, Croyal M, Chemello K, Nativel B, Pichelin M, Cariou B, Bourane S, et al. Homozygous Familial Hypercholesterolemia Patients With Identical Mutations Variably Express the LDLR (Low-Density Lipoprotein Receptor): Implications for the Efficacy of Evolocumab. Arterioscler Thromb Vasc Biol. 2018;38:592–598. doi: 10.1161/ATVBAHA.117.310217. doi: 10.1161/ATVBAHA.117.310217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaudet D, Gipe DA, Pordy R, Ahmad Z, Cuchel M, Shah PK, Chyu KY, Sasiela WJ, Chan KC, Brisson D, et al. ANGPTL3 Inhibition in Homozygous Familial Hypercholesterolemia. N Engl J Med. 2017;377:296–297. doi: 10.1056/NEJMc1705994. doi: 10.1056/NEJMc1705994. [DOI] [PubMed] [Google Scholar]
- 20.Hassan M. ANGPLT3: a novel modulator of lipid metabolism. Glob Cardiol Sci Pract. 2017;2017:e201706. doi: 10.21542/gcsp.2017.6. doi: 10.21542/gcsp.2017.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minicocci I, Santini S, Cantisani V, Stitziel N, Kathiresan S, Arroyo JA, Martí G, Pisciotta L, Noto D, Cefalù AB, et al. Clinical characteristics and plasma lipids in subjects with familial combined hypolipidemia: a pooled analysis. J Lipid Res. 2013;54:3481–3490. doi: 10.1194/jlr.P039875. doi: 10.1194/jlr.P039875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Romeo S, Yin W, Kozlitina J, Pennacchio LA, Boerwinkle E, Hobbs HH, Cohen JC. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J Clin Invest. 2009;119:70–79. doi: 10.1172/JCI37118. doi: 10.1172/JCI37118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dewey FE, Gusarova V, Dunbar RL, O’Dushlaine C, Schurmann C, Gottesman O, McCarthy S, Van Hout CV, Bruse S, Dansky HM, et al. Genetic and pharmacologic inactivation of ANGPTL3 and cardiovascular disease. N Engl J Med. 2017;377:211–221. doi: 10.1056/NEJMoa1612790. doi: 10.1056/NEJMoa1612790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Etxebarria A, Palacios L, Stef M, Tejedor D, Uribe KB, Oleaga A, Irigoyen L, Torres B, Ostolaza H, Martin C. Functional characterization of splicing and ligand-binding domain variants in the LDL receptor. Hum Mutat. 2012;33:232–243. doi: 10.1002/humu.21630. doi: 10.1002/humu.21630. [DOI] [PubMed] [Google Scholar]
- 25.Benito-Vicente A, Alves AC, Etxebarria A, Medeiros AM, Martin C, Bourbon M. The importance of an integrated analysis of clinical, molecular, and functional data for the genetic diagnosis of familial hypercholesterolemia. Genet Med. 2015;17:980–988. doi: 10.1038/gim.2015.14. doi: 10.1038/gim.2015.14. [DOI] [PubMed] [Google Scholar]
- 26.Melzer S, Nunes CS, Endringer DC, de Andrade TU, Tarnok A, Lenz D. Trypan blue as an affordable marker for automated live-dead cell analysis in image cytometry. Scanning. 2016;38:857–863. doi: 10.1002/sca.21335. doi: 10.1002/sca.21335. [DOI] [PubMed] [Google Scholar]
- 27.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lang W, Frishman WH. Angiopoietin-Like 3 protein inhibition: a new frontier in lipid-lowering treatment. Cardiol Rev. 2019;27:211–217. doi: 10.1097/CRD.0000000000000258. doi: 10.1097/CRD.0000000000000258. [DOI] [PubMed] [Google Scholar]
- 29.Tikka A, Jauhiainen M. The role of ANGPTL3 in controlling lipoprotein metabolism. Endocrine. 2016;52:187–193. doi: 10.1007/s12020-015-0838-9. doi: 10.1007/s12020-015-0838-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Etxebarria A, Benito-Vicente A, Palacios L, Stef M, Cenarro A, Civeira F, Ostolaza H, Martin C. Functional characterization and classification of frequent low-density lipoprotein receptor variants. Hum Mutat. 2015;36:129–141. doi: 10.1002/humu.22721. doi: 10.1002/humu.22721. [DOI] [PubMed] [Google Scholar]
- 31.Rodríguez-Jiménez C, Pernía O, Mostaza J, Rodríguez-Antolín C, de Dios García-Díaz J, Alonso-Cerezo C, García-Polo I, Blanco A, Lahoz C, Arrieta F, et al. Functional analysis of new variants at the low-density lipoprotein receptor associated with familial hypercholesterolemia. Hum Mutat. 2019;40:1181–1190. doi: 10.1002/humu.23801. doi: 10.1002/humu.23801. [DOI] [PubMed] [Google Scholar]
- 32.Silva S, Alves AC, Patel D, Malhó R, Soutar AK, Bourbon M. In vitro functional characterization of missense mutations in the LDLR gene. Atherosclerosis. 2012;225:128–134. doi: 10.1016/j.atherosclerosis.2012.08.017. doi: 10.1016/j.atherosclerosis.2012.08.017. [DOI] [PubMed] [Google Scholar]
- 33.Di Taranto MD, Benito-Vicente A, Giacobbe C, Uribe KB, Rubba P, Etxebarria A, Guardamagna O, Gentile M, Martín C, Fortunato G. Identification and in vitro characterization of two new PCSK9 Gain of Function variants found in patients with Familial Hypercholesterolemia. Sci Rep. 2017;7:15282. doi: 10.1038/s41598-017-15543-x. doi: 10.1038/s41598-017-15543-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moorjani S, Roy M, Torres A, Bétard C, Gagné C, Lambert M, Brun D, Davignon J, Lupien P. Mutations of low-density-lipoprotein-receptor gene, variation in plasma cholesterol, and expression of coronary heart disease in homozygous familial hypercholesterolaemia. Lancet. 1993;341:1303–1306. doi: 10.1016/0140-6736(93)90815-x. doi: 10.1016/0140-6736(93)90815-x. [DOI] [PubMed] [Google Scholar]
- 35.Thompson GR, Blom DJ, Marais AD, Seed M, Pilcher GJ, Raal FJ. Survival in homozygous familial hypercholesterolaemia is determined by the on-treatment level of serum cholesterol. Eur Heart J. 2018;39:1162–1168. doi: 10.1093/eurheartj/ehx317. doi: 10.1093/eurheartj/ehx317. [DOI] [PubMed] [Google Scholar]
- 36.Aegerion Pharmaceuticals Inc. JUXTAPID (lomitapide) capsules, for oral use. Highlights of prescribing information. 2017. Available at: http://www.juxtapid.com/prescribing-information. Accessed September 4, 2018.
- 37.Kastle Therapeutics. KYNAMRO (mipomersen sodium) Injection. Solution for Subcutaneous Injection. Highlights of Prescribing Information. 2016. Available at: http://www.kynamro.com/media/pdfs/Kynamro_Prescribing_information.pdf. Accessed September 4, 2018.
- 38.Amgen Inc. Repatha prescribing information. 2015. Available at: https://pi.amgen.com/~/media/amgen/repositorysites/pi-amgen-com/repatha/repatha_pi_hcp_english.ashx. Accessed July 2, 2018.
- 39.Sanofi-aventis U.S. LLC. Praluent prescribing information. 2015. Available at: http://products.sanofi.us/praluent/praluent.pdf. Accessed July 2, 2018.
- 40.Catapano AL, Graham I, De Backer G, Wiklund O, Chapman MJ, Drexel H, Hoes AW, Jennings CS, Landmesser U, Pedersen TR, et al. ESC Scientific Document Group. 2016 ESC/EAS guidelines for the management of dyslipidaemias. Eur Heart J. 2016;37:2999–3058. doi: 10.1093/eurheartj/ehw272. doi: 10.1093/eurheartj/ehw272. [DOI] [PubMed] [Google Scholar]
- 41.Landmesser U, Chapman MJ, Stock JK, Amarenco P, Belch JJF, Borén J, Farnier M, Ference BA, Gielen S, Graham I, et al. 2017 Update of ESC/EAS Task Force on practical clinical guidance for proprotein convertase subtilisin/kexin type 9 inhibition in patients with atherosclerotic cardiovascular disease or in familial hypercholesterolaemia. Eur Heart J. 2018;39:1131–1143. doi: 10.1093/eurheartj/ehx549. doi: 10.1093/eurheartj/ehx549. [DOI] [PubMed] [Google Scholar]
- 42.Lloyd-Jones DM, Morris PB, Ballantyne CM, Birtcher KK, Daly DD, Jr, DePalma SM, Minissian MB, Orringer CE, Smith SC., Jr. 2016 ACC expert consensus decision pathway on the role of non-statin therapies for LDL-cholesterol lowering in the management of atherosclerotic cardiovascular disease risk: a report of the American College of Cardiology Task Force on Clinical Expert Consensus Documents. J Am Coll Cardiol. 2016;68:92–125. doi: 10.1016/j.jacc.2016.03.519. [DOI] [PubMed] [Google Scholar]
- 43.Farnier M, Gaudet D, Valcheva V, Minini P, Miller K, Cariou B. Efficacy of alirocumab in high cardiovascular risk populations with or without heterozygous familial hypercholesterolemia: Pooled analysis of eight ODYSSEY Phase 3 clinical program trials. Int J Cardiol. 2016;223:750–757. doi: 10.1016/j.ijcard.2016.08.273. doi: 10.1016/j.ijcard.2016.08.273. [DOI] [PubMed] [Google Scholar]
- 44.Raal FJ, Stein EA, Dufour R, Turner T, Civeira F, Burgess L, Langslet G, Scott R, Olsson AG, Sullivan D, et al. RUTHERFORD-2 Investigators. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;385:331–340. doi: 10.1016/S0140-6736(14)61399-4. doi: 10.1016/S0140-6736(14)61399-4. [DOI] [PubMed] [Google Scholar]
- 45.Stein EA, Gipe D, Bergeron J, Gaudet D, Weiss R, Dufour R, Wu R, Pordy R. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet. 2012;380:29–36. doi: 10.1016/S0140-6736(12)60771-5. doi: 10.1016/S0140-6736(12)60771-5. [DOI] [PubMed] [Google Scholar]