

Abstract
Spreading depolarization is observed as a large negative shift of the direct current potential, swelling of neuronal somas, and dendritic beading in the brain’s gray matter and represents a state of a potentially reversible mass injury. Its hallmark is the abrupt, massive ion translocation between intraneuronal and extracellular compartment that causes water uptake (= cytotoxic edema) and massive glutamate release. Dependent on the tissue’s energy status, spreading depolarization can co-occur with different depression or silencing patterns of spontaneous activity. In adequately supplied tissue, spreading depolarization induces spreading depression of activity. In severely ischemic tissue, nonspreading depression of activity precedes spreading depolarization. The depression pattern determines the neurological deficit which is either spreading such as in migraine aura or migraine stroke or nonspreading such as in transient ischemic attack or typical stroke. Although a clinical distinction between spreading and nonspreading focal neurological deficits is useful because they are associated with different probabilities of permanent damage, it is important to note that spreading depolarization, the neuronal injury potential, occurs in all of these conditions. Here, we first review the scientific basis of the continuum of spreading depolarizations. Second, we highlight the transition zone of the continuum from reversibility to irreversibility using clinical cases of aneurysmal subarachnoid hemorrhage and cerebral amyloid angiopathy. These illustrate how modern neuroimaging and neuromonitoring technologies increasingly bridge the gap between basic sciences and clinic. For example, we provide direct electrophysiological evidence for the first time that spreading depolarization-induced spreading depression is the pathophysiological correlate of the migraine aura.
Keywords: Spreading depression, Migraine aura, Cerebral amyloid angiopathy, Subarachnoid hemorrhage, Delayed cerebral ischemia
Introduction
Globally, ischemic stroke, hemorrhagic stroke, and traumatic brain injury (TBI) are leading causes of death and disability, imposing a huge burden on patients, their families, and society. Stroke is the second leading cause of death and third leading cause of disability worldwide (Murray et al. 2012). In the USA, the American Heart Association and American Stroke Association project that the total cost of stroke, which includes both direct and indirect spending, will increase from $105 billion in 2012 to $241 billion by 2030 (Ovbiagele et al. 2013). Aging is the strongest nonmodifiable risk factor for stroke—most strokes occur in people older than 65 years (Benjamin et al. 2017; Feigin et al. 2003). Aged patients with stroke have higher mortality and poorer quality of life after stroke compared with younger patients (Arboix et al. 2000; Dennis et al. 1993; Di Carlo et al. 1999; Kammersgaard et al. 2004; Pohjasvaara et al. 1997; Rojas et al. 2007).
More than 50 million TBIs occur worldwide each year (Maas et al. 2017). The incidence of TBI in developed countries has increased in aged individuals to a greater extent than expected from demographic ageing (Brazinova et al. 2018; Kleiven et al. 2003; Koskinen and Alaranta 2008). Age is also among the strongest outcome predictors in TBI, with mortality and unfavorable outcome rising continuously with age (Hukkelhoven et al. 2003; Mushkudiani et al. 2007). However, therapeutic nihilism in the elderly is not justified as the poor outcome resulting from delayed neuroimaging, lower likelihood of transfer to specialist facilities, care by more junior staff, and less aggressive treatment might lead to a self-fulfilling prophecy of unfavorable prognosis (Stocchetti et al. 2017). For example, if patients with severe TBI between age 60 and 69 are treated promptly and competently in the intensive care unit, about 40% have a good prognosis (Stocchetti et al. 2012).
The general value of neurocritical care and emergency medicine is supported by a decline in mortality of TBI at a rate of 9% per decade from 1970 to 1990 (Stein et al. 2010). Similarly, the case fatality rate, e.g., for aneurysmal subarachnoid hemorrhage (aSAH), decreased by 8% per decade between 1960 and 1992 (Hop et al. 1997). In parallel, the proportion of patients who recovered independent function increased. Aneurysmal SAH is caused by the rupture of a basal cerebral arterial aneurysm, is the second most common hemorrhagic stroke, and often requires treatment in the intensive care unit because it is a particularly devastating form of stroke (Macdonald and Schweizer 2017). Within the direct health costs, intensive care medicine is particularly expensive. Within the costs for intensive care medicine, the high cost group accounts for half of the total costs and aSAH is among the top 4 causes of high costs (Reardon et al. 2018). Yet, the rate of improvement per decade has generally decreased for TBI and aSAH since 1990. To counter this trend by developing targeted treatments, it is mandatory to better understand the pathophysiology of acute neuronal mass injury, which contains surprising paradoxes complicating intuitive understanding at first glance. One of these paradoxes is the scientific connection between the harmless migraine aura and the central pathomechanism that leads to severe permanent brain damage in stroke, TBI, cardiac arrest, and related conditions.
The uniqueness of acute neuronal mass injury
Acute neuronal mass injury to the brain’s gray matter shows commonalities, yet also unique features compared with acute injuries of other somatic cells of the periphery. Its key features are that thousands if not millions or billions of neurons are affected by an abrupt release of approximately 90% of the Gibbs free energy contained in the ion concentration gradients across the neuronal membranes (Dreier et al. 2013a), that this release of free energy occurs at a given recording point in the tissue within a few seconds, and usually but not always spreads in the tissue at a rate between 2 and 9 mm/min (Aitken et al. 1998; Bogdanov et al. 2016; Dreier 2011; Farkas et al. 2010; Jarvis et al. 2001; Lauritzen et al. 2011; Lauritzen and Hansen 1992; Marshall 1959; Woitzik et al. 2013). The near-complete breakdown of the ion gradients is associated with a near-complete sustained neuronal membrane depolarization. Therefore, this process is termed spreading depolarization. The released energy is converted into heat, which has been measured with a sensitive thermal detector in the isolated retinae of the bullfrog and toad as a wave of brain temperature increase by about 5 to 30 mK (Tasaki and Byrne 1991).
The propagation of the neuronal mass injury seems to follow a reaction/diffusion mechanism in which neurons release neuroactive substances which diffuse to adjacent neurons, where they trigger a self-propagating regenerative process (Milakara et al. 2017; Zandt et al. 2013). However, the exact nature of the neuroactive substances has not yet been fully clarified (Dreier and Reiffurth 2015). It has been proposed that glutamate could be the neuroactive substance driving the mass injury (van Harreveld 1959), but the massive glutamate release only follows the membrane depolarization and near-complete breakdown of the ion gradients (Enger et al. 2015). Potassium is another candidate (Grafstein 1956). The large increase of extracellular potassium precedes the large increase of extracellular glutamate, but potassium is not released before the depolarization in naïve tissue, as it should if it was the neuroactive substance driving the propagation (Hansen and Zeuthen 1981; Herreras and Somjen 1993; Lehmenkuhler 1990; Somjen 2001). The almost complete collapse of the ion gradients in concert with the release of not only excitatory neurotransmitters such as glutamate but also inhibitory neurotransmitters such as GABA, which promotes the influx of chloride ions, leads to massive uptake of water by the neurons and to the sustainment of the depolarized state (Aiba and Shuttleworth 2012; Allen et al. 2004; Dreier et al. 2018a; Fabricius et al. 1993; Hinzman et al. 2015; Reinhart and Shuttleworth 2018; Revah et al. 2016; van Harreveld 1959).
Recovery from neuronal mass injury largely depends on proper function of the energy dependent sodium pump (LaManna and Rosenthal 1975; Major et al. 2017; Reiffurth et al. 2019). This enzyme consumes about 50% of the brain energy even under normal resting conditions (Astrup et al. 1981; Tidow et al. 2010). When an event of neuronal mass injury migrates through the tissue, activation of this and other energy-dependent pumps causes a fall in the tissue ATP concentration to at least 50% of its normal value (Mies and Paschen 1984; Selman et al. 2004). Because of the great importance of the sodium pump for the recovery from neuronal mass injury, the transition from neuronal mass injury to cell death induced by pharmacological sodium pump inhibition is widely considered as a model for the transition from injury to cell death under anoxia or aglycemia, although the tissue remains normoxic and normoglycemic in the presence of a sodium pump inhibitor (Balestrino et al. 1999; Basarsky et al. 1998; Jarvis et al. 2001).
Spreading depolarization is the injury potential of the brain’s gray matter
Acute neuronal mass injury to the brain’s gray matter is observed as a large, abrupt, negative shift of its steady (direct current (DC)) potential (frequency range < 0.05 Hz) (Dreier et al. 2017). Using animal models of electrical injury, asphyxia, and global ischemia, Aristides Leão and Anthoni van Harreveld were the first to unequivocally demonstrate the usefulness of the electrophysiological modality to record acute injury to the central nervous system (CNS) in the 1940s (Leão 1947; van Harreveld 1946). They were also the first to clearly relate the large negative DC shift of injury to the depolarization of neurons. On this basis, the term spreading depolarization is used today. However, it should be added that Jan Bures mentioned in 1957 that steady potential shifts in the context of cellular injury had already “been thoroughly investigated for over a century” (author’s emphasis) (Bures 1957). In the same article, Bures also pointed out the fundamental basis of the large negative DC shift characteristic of spreading depolarization when he explained (i) that the cortical surface of the physiological mammalian brain is steadily positive to other parts of the nervous system (sciatic nerve, cerebral ventricles) by approximately 10–20 mV and (ii) that this potential difference abruptly breaks down under various stressors such as, for example, cerebral anoxia, resulting in a negative shift of the steady potential. In the 1950s, the physiological positivity of the cerebral cortex had already been measured in various species, including rat, rabbit, cat, dog, monkey, and man (Burr and Harmann 1939; Goldensohn et al. 1951; Goldring and O’Leary 1951; Goldring et al. 1950; Kempinsky 1954). Also the large negative DC shift characteristic of spreading depolarization had been recorded in many species (Marshall 1959). Today, we know that spreading depolarization is not only the characteristic injury potential of the gray matter of mammals but also occurs in analogous neural structures of fish, insects, reptiles, and birds (Kraig and Nicholson 1978; Lauritzen et al. 1988; Martins-Ferreira et al. 1974; Robertson et al. 2017; Spong et al. 2017). The fact that this large negative DC shift is also found in brain slices that lack an intact blood-brain barrier (BBB) is one of the many arguments that it is not generated at the BBB (Kang et al. 2013; Somjen 2001). Its generation rather shows commonalities to that of postsynaptic potentials (Makarova et al. 2010). Generally speaking, its main component appears to result from a net transfer of positive charges from the extracellular to the intraneuronal compartment, although there may also be a smaller net transfer of positive charges to the intraastroglial compartment (Dreier et al. 2013a; Makarova et al. 2010).
After the discovery in animals, it took several more decades until the negative DC shift characteristic of spreading depolarization was also recorded unequivocally in patients in vivo and human neocortical brain slices using DC-coupled amplifiers (Avoli et al. 1991; Dreier et al. 2009; Mayevsky et al. 1996; Sramka et al. 1977). The occurrence in connection with acute damage to the human brain underlines once again that the large negative DC shift is an injury potential by its nature, even if it is not always followed by cellular death. Thus, the large negative DC shift marks the onset of toxic cellular changes that eventually lead to cell death, but is not a marker of cell death per se, since the depolarization is reversible—up to a point—with restoration of the physiological neuronal environment. In order to emphasize that spreading depolarization is an injury potential of the brain’s gray matter, we will use the terms spreading depolarization and injury depolarization synonymously in this manuscript.
Surrogate measures of acute neuronal mass injury to the brain’s gray matter
Over the last decade, DC potential recordings with subdural electrodes have increasingly become a routine procedure in specialized neurocritical care units (Carlson et al. 2018b; Dreier et al. 2018b; Dreier et al. 2019; Drenckhahn et al. 2012; Hartings et al. 2011b; Hartings et al. 2017b; Luckl et al. 2018; Oliveira-Ferreira et al. 2010). The continuum of injury depolarizations describes the spectrum from transient events with negative DC shifts of intermediate to short duration in less ischemic or adequately supplied tissue to terminal events in severely ischemic tissue characterized by long-lasting DC shifts and transition of the neurons from the state of injury to cell death (Dreier 2011; Dreier and Reiffurth 2015; Hartings et al. 2017a). Transient injury depolarizations have been detected in 60% of patients with severe TBI requiring neurosurgical intervention to alleviate their primary symptoms (Fabricius et al. 2006; Hartings et al. 2011b; Strong et al. 2002), 70% of patients with spontaneous intracerebral hemorrhage (ICH) (Fabricius et al. 2006; Helbok et al. 2017), 70–80% of patients with severe aSAH (Bosche et al. 2010; Dreier et al. 2009; Dreier et al. 2006; Sakowitz et al. 2013), and 90–100% of patients with malignant hemispheric stroke (MHS) (Dohmen et al. 2008; Pinczolits et al. 2017; Woitzik et al. 2013). Terminal injury depolarizations were recently recorded in patients who either died in the wake of circulatory arrest (Fig. 1a) (Dreier et al. 2018b) or suffered brain death despite sustained circulatory function (Carlson et al. 2018b; Dreier et al. 2019) or developed neuroimaging-proven early or delayed ischemic strokes after aSAH (Luckl et al. 2018). Prior to ischemic strokes after aSAH, clustered, increasingly prolonged injury depolarizations are often recorded (Fig. 1b) (Dreier et al. 2009; Dreier et al. 2006; Drenckhahn et al. 2012; Hartings et al. 2011b; Luckl et al. 2018; Oliveira-Ferreira et al. 2010). In addition, during the development of ischemic strokes after aSAH, electrodes typically show clustered injury depolarizations when neighboring electrodes show terminal events. Injury depolarizations propagate widely from metabolically stressed zones, thereby affording even remote detection of newly developing strokes which cannot be detected otherwise in real time. Accordingly, early transient injury depolarizations have recently shown high negative and positive predictive values for neuroimaging-proven early focal necrosis after aSAH (Eriksen et al. 2019; Hartings et al. 2017b).
Fig. 1.

Terminal spreading depolarization (SD) (= injury depolarization) during the dying process of the human brain after circulatory arrest (a) and a cluster of injury depolarizations during the development of a focal ischemic stroke after aneurysmal subarachnoid hemorrhage (aSAH) (b). a The two top traces show raw direct current (DC) (f 0–45 Hz) recordings from two subdural electrodes (6-contact Wyler recording strip, Ad-Tech Medical, Racine, WI, USA; BrainAmp amplifier, BrainProducts GmbH, Munich, Germany), the two middle traces show the changes in spontaneous brain activity at the same electrodes (f 0.5–45 Hz), and the three bottom traces show the changes in regional cerebral blood flow (rCBF) (opto-electrode strip and Periflux 4001 for laser Doppler flowmetry, Perimed AB, Järfälla, Sweden (Dreier et al. 2009; Drenckhahn et al. 2016)), tissue partial pressure of oxygen (Licox CC1P1, Integra Lifesciences Corporation, Plainsboro, NJ, USA) (Bosche et al. 2010; Dreier et al. 2009; Stuart et al. 2010)), and arterial blood pressure (radial artery catheter). The case has been previously published (patient 2 in Dreier et al. (2018b)). During the dying process in the wake of circulatory arrest, the first DC change was a very slow, homogeneous DC positivity that coincided with the progressive decline in arterial pressure, rCBF, and tissue partial pressure of oxygen. It has been recently suggested that this DC positivity results from interferences of chemicals with the platinum/iridium electrodes (Dreier et al. 2019). Nonspreading depression of spontaneous brain activity (middle traces, cf. blue asterisks) rendered the brain isoelectric within ~ 340 s after onset of the decline in mean arterial pressure or, respectively, 220 s after rCBF had fallen to 20% of baseline. The terminal injury depolarization (top traces) started 53 s after the spontaneous brain activity had fully ceased. In the figure, the terminal event is first seen at electrode 3 (transition from dark blue to red DC traces) from where it spread to electrode 6. The initial component of the terminal event is the injury depolarization component, whereas the late component is called the negative ultraslow potential (NUP). The negative ultraslow potential is experimentally defined by three properties: (i) that it is preceded by the initial injury depolarization component, (ii) that the ion shifts and cell edema do not fully recover during this extremely long DC negativity, and (iii) the death of neurons (Dreier 2011; Luckl et al. 2018). b A cluster of increasingly long-lasting injury depolarizations in a patient with aSAH who developed a delayed ischemic stroke (Luckl et al. 2018). The traces are similar to a. The case has been previously published (patient 4 in Luckl et al. (2018)). The initial injury depolarization had a short-lasting DC shift (top traces) at the three recording sites and caused a spreading depression of spontaneous activity (middle traces, red asterisks). Thereafter, the activity remained completely and persistently depressed, i.e., isoelectric. Development of isoelectricity occurs progressively as a consequence of injury depolarizations, as evidenced by DC shifts propagating between the different electrodes (red arrows). Injury depolarizations in electrically inactive tissue are termed isoelectric spreading depolarizations. Isoelectric spreading depolarizations indicate that there is energy deprivation in the tissue at or near the recording site (Dreier et al. 2017; Hartings et al. 2017a). All injury depolarizations following the first one are isoelectric spreading depolarizations in this figure. Note that injury depolarizations of decreasing amplitude occurred superimposed on a negative ultraslow potential, best seen at electrodes 3 and 5. The negative ultraslow potential suggests that neurons are dying. It thus marks the penumbra core conversion in ischemic tissue (Higuchi et al. 2002). Accordingly, the patient developed a delayed ischemic infarct at the recording site (Luckl et al. 2018). Each injury depolarization is associated with a sharp decrease of the tissue partial pressure of oxygen. This results (i) from the injury depolarization-induced increase in the cerebral metabolic rate of oxygen due to sodium pump activation (Haglund and Schwartzkroin 1990; LaManna and Rosenthal 1975; Piilgaard and Lauritzen 2009) in combination with (ii) an rCBF decrease due to the inverse hemodynamic response to the injury depolarization (Dreier 2011; Dreier et al. 2009)
Negative DC shift recording is the oldest, simplest, and most cost-effective technique for observing the continuum of neuronal mass injury to the brain’s gray matter (Leão 1947; van Harreveld 1946). Similar DC recording technology as in patients can also be used in organotypical cultures, acute brain slices, and animals in vivo (Dreier et al. 1998; Dreier et al. 2001a; Kunkler and Kraig 1998; Maslarova et al. 2011; Pomper et al. 2006; Somjen 2001). However, various other modalities are also suitable for recording neuronal mass injury to the brain’s gray matter, and changes in these modalities occur practically simultaneously with the injury depolarization with only minor variations (Makarova et al. 2010; Risher et al. 2010; Somjen 2001). For example, the water uptake into neurons, the so-called cytotoxic edema, leads to abrupt dendritic beading and swelling of neuronal somas. The beading can be visualized using two-photon microscopy or electron microscopy (Murphy et al. 2008; Risher et al. 2010; Steffensen et al. 2015; Takano et al. 2007; Van Harreveld and Khattab 1967). The beaded morphology allows the cells to contain a larger volume of fluid within the same surface area (Budde and Frank 2010). In normal dendrites, water molecules diffusing along the main axis of the dendrite encounter few barriers on the timescale of diffusion-weighted magnetic resonance imaging (DW-MRI). By contrast, the beaded morphology, characterized by the alternation between dendrite constriction and dilatation, causes reduction in the mobility of intracellular water along the main axis of each dendrite. This intracellular diffusion restriction is then seen as a hyperintensity with DW-MRI and as a hypointensity with maps of the apparent diffusion coefficient (ADC) (Beaulieu et al. 2000; Busch et al. 1998; Busch et al. 1996; Cain et al. 2017; de Crespigny et al. 1998; Dreier and Reiffurth 2017; Rother et al. 1996; Takano et al. 1996). Whereas two-photon microscopy can only be used in brain slices or in animals in vivo, DW-MRI and ADC maps are also routinely used in patients to detect neuronal mass injury (Lublinsky et al. 2019; Luckl et al. 2018). It is important to note that, similar to injury depolarizations, MRI-proven diffusion restrictions are initially reversible in patients as well (Fiehler et al. 2004).
A fourth comparatively simple method for detecting neuronal mass injury is the so-called intrinsic optical signal (IOS), which can be recorded in the isolated retina, in brain slices, and in vivo (Fernandes de Lima et al. 1994; Gouras 1958; Martins-Ferreira and de Castro 1966; Maslarova et al. 2011; Reiffurth et al. 2019; Reiffurth et al. 2012). The IOS represents alterations of optical features of unstained tissue which can be measured as a change of either light transmittance or reflectance with a camera mounted on a microscope. Measurement of the IOS does not necessitate any tissue manipulation other than illumination of the preparation. The injury depolarization in brain slices is characterized by a sustained IOS change of large amplitude over a wide range of wavelengths (Major et al. 2017; Mane and Muller 2012; Petzold et al. 2005). Unfortunately, the IOS in vivo is not only a marker for the injury depolarization but also strongly influenced by the cerebral blood volume. This renders the interpretation of the IOS in vivo difficult, but it can still be used for the assessment of the spatial expansion of the injury depolarization because the depolarization typically triggers a neurovascular response, as explained below, that secondarily affects the cerebral blood volume (Oliveira-Ferreira et al. 2019; Santos et al. 2017; Scholl et al. 2017).
Microdialyis is yet another modality to measure neuronal mass injury as it can detect the massive glutamate release in response to the injury depolarization or, for example, the extracellular changes in lactate and pyruvate (Fabricius et al. 1993; Sakowitz et al. 2013; Sakowitz et al. 2001; van Harreveld 1959). Microdialysis can be used both experimentally and in patients. While standard microdialysis is relatively slow, rapid sampling microdialysis offers the possibility to investigate the temporal dynamics of the molecules (Feuerstein et al. 2010; Hashemi et al. 2009). In brain slices and animals, an even better temporal resolution and more accurate estimation of the true concentration changes can be achieved using microelectrodes for extracellular metabolites (Balanca et al. 2017; Hinzman et al. 2015). Microelectrodes also allow extracellular measurements of various ion concentration alterations during the injury depolarization such as the increase in potassium concentration from 3 to 60 mM, the falls in sodium concentration from 150 to 60 mM, calcium concentration from 1.3 to < 0.1 mM, chloride concentration from 145 to 95 mM, pH from 7.4 to 6.7, or the decrease in the size of the extracellular space by approximately 70% (Dreier et al. 2002a; Hansen and Zeuthen 1981; Kraig and Nicholson 1978; Lauritzen 1994; Mazel et al. 2002; Oliveira-Ferreira et al. 2010; Perez-Pinzon et al. 1995; Petzold et al. 2005; Vyskocil et al. 1972; Windmuller et al. 2005). The large intracellular ion concentration changes during the injury depolarization such as the more than thousand-fold increase in the intraneuronal calcium concentration can be detected with ion-sensitive dyes using microscopy (Aiba and Shuttleworth 2012; Dietz et al. 2008; Menyhart et al. 2018). However, since the changes are so pronounced, their quantification is challenging, as, for example, the usual calcium dyes go into saturation during the injury depolarization (Dreier and Reiffurth 2015; Revah et al. 2016). Also the changes in holding current and input resistance of patch-clamped neurons represent general characteristics of injury depolarizations and are basically stable with minor variations, regardless of whether the neuronal mass injury is short-lasting and harmless or will later develop into cell death (Somjen 2001).
Further important methods for the detection of neuronal mass injury are histology and immunohistochemistry (Dreier et al. 2007; Dreier et al. 2018a; Dreier et al. 2013b; Klatzo 1967; Klatzo 1987). A problem with these methods is that they only allow measurements at one point in time and that in the first hours after a damaging event, it is often difficult to decide whether a cell is still injured or has already died (Dreier et al. 2018a).
Ultimately, it can be assumed that the concentration gradients of all small molecules in the intracellular and extracellular space change during an injury depolarization and that millions of different measuring methods are conceivable in order to fully capture the phenomenon. Depending on the applied methods to detect the injury depolarization, other terms are often used instead of the term injury depolarization. Thus, the pathologist often speaks of cytotoxic edema, the neuroradiologist of diffusion restriction, the microscopist of dendritic beading or neuronal swelling, and the electrophysiologist of spreading depolarization, but it is important to recognize that all these names refer to the same fundamental process of acute neuronal mass injury, which is initially always reversible and only becomes irreversible from a certain point in time. This point is termed the commitment point (Dreier 2011; Dreier et al. 2018a; Hartings et al. 2017a; Luckl et al. 2018; Somjen 2004). Whether the commitment point is reached depends on whether the cause of the mass injury is persistent and thus prevents the neuronal recovery. Importantly, the commitment point is not a universal value but is modified by additional factors, such as the level of the remaining perfusion and the local temperature (Dreier et al. 2013b). Moreover, it differs between different types of neurons involved (Heiss and Rosner 1983; Pulsinelli 1985).
Spreading depression and nonspreading depression of spontaneous activity
The continuum of injury depolarizations is also reflected in its consequences on spontaneous brain activity. In electrically active tissue, the injury depolarization causes spreading depression of activity, measured in the alternating current (AC)-ECoG range (> 0.5 Hz) (Figs. 1b and 2a, b) (Dreier et al. 2017). The depression is short-lasting in otherwise normal tissue but progressively prolonged in metabolically impaired areas (Dreier et al. 2006; Fabricius et al. 2006). In and immediately around metabolically impaired areas, the injury depolarization can cause even persistent electrical inactivity (= isoelectricity) (Fig. 1b) (Luckl et al. 2018; Oliveira-Ferreira et al. 2010; Winkler et al. 2017). Thereafter, injury depolarizations cannot induce further spreading depressions. Under this condition, injury depolarizations are often termed isoelectric spreading depolarizations (Fig. 1b). Isoelectric spreading depolarizations were associated with poor outcome after TBI (Hartings et al. 2011a). Transient injury depolarizations with prolonged activity depressions were linked to worse outcome after aSAH (Dreier et al. 2012; Winkler et al. 2017).
Fig. 2.

Normal and inverse hemodynamic responses to spreading depolarizations (SD) (= injury depolarizations) after aneurysmal subarachnoid hemorrhage (aSAH). a The two top traces show raw direct current (DC) (f 0–45 Hz) recordings from two subdural electrodes, traces 3 and 4 show the changes in spontaneous brain activity at the same electrodes (f 0.5–45 Hz), traces 5 and 6 show the changes in regional cerebral blood flow (rCBF) (laser-Doppler flowmetry), and traces 7 and 8 show the changes in low-frequency vascular fluctuations (= rCBF fluctuations in the frequency range between f 0.05–0.1 Hz). Two injury depolarizations are observed in the DC recordings as large negative DC shifts. The injury depolarizations cause spreading depression of the spontaneous activity (red asterisks in traces 3 and 4). They propagate in a through otherwise relatively normal tissue. Accordingly, they lead to predominantly hyperemic responses (= normal hemodynamic response to the injury depolarization, red asterisks in traces 5 and 6). Low-frequency vascular fluctuations, detectable by laser Doppler flowmetry and functional MRI, are determined by the brain’s resting neuronal activity. Injury depolarizations are associated with a depolarization block of the resting activity, recorded electrophysiologically as spreading depression of spontaneous electrocorticographic (ECoG) activity. Accordingly, injury depolarizations not only lead to spreading depression of the spontaneous activity but also to spreading suppression of low-frequency vascular fluctuations (Dreier et al. 2009) (red asterisks in traces 7 and 8). b The traces are similar to a, but more electrodes and optodes are shown. Two injury depolarizations are observed in the DC recordings as large negative DC shifts. When the neurovascular coupling process is disturbed, an injury depolarization can induce severe vasoconstriction instead of vasodilatation leading to a local perfusion deficit (= spreading ischemia). This delays the neuronal and astroglial repolarization and perpetuates the injury (= inverse hemodynamic response to the injury depolarization, red asterisks in traces 9-12) (Dreier et al. 1998; Dreier et al. 2009). The repolarization phases of the negative DC shifts are only slightly less steep in b than in a. However, the injury depolarization-induced spreading depressions of spontaneous activity (red asterisks in traces 5-8) and spreading suppressions of low-frequency vascular fluctuations (red asterisks in traces 13-16) are clearly longer in b than in a indicating that the tissue requires more time for recovery when the injury depolarizations trigger spreading ischemias
Interestingly, not only the injury depolarization but also the depression of activity can be recorded using very different measuring modalities. Thus, low-frequency vascular fluctuations are determined by the brain’s resting neuronal activity, as ultra-slow modulation of high-frequency neuronal activity can entrain vasomotion (Chan and Murphy 2017; Fox and Raichle 2007; Mateo et al. 2017). Accordingly, injury depolarizations not only induce spreading depressions of activity but also spreading suppressions of low-frequency vascular fluctuations (Dreier et al. 2009). The latter can be detected by laser Doppler flowmetry (Fig. 2a, b). At least theoretically, it should also be possible to visualize these using functional MRI.
Injury depolarization-induced spreading depression must be distinguished from nonspreading depression of activity, which precedes the injury depolarization in conditions of severe hypoxia or ischemia (Fig. 1a) (Dreier 2011). Complete ischemia causes this simultaneous isoelectricity of affected gray matter within approximately 20–40 s both in animals and humans (Dreier et al. 2018b; Leão 1947). Time intervals until arrest of evoked potentials are typically somewhat longer than those until arrest of the spontaneous activity (Pana et al. 2016). Nonspreading depression has been interpreted as an austerity program to curb neuronal energy usage by the shutdown of nonessential cell functions before the ATP pool is depleted and prospects of tissue recovery vanish (Hochachka et al. 1996; Somjen 2004). In contrast to spreading depression, which results from neuronal depolarization, neurons are at least initially in a hyperpolarized state during nonspreading depression (Revah et al. 2016; Tanaka et al. 1997). The possible mechanisms underlying nonspreading depression have been reviewed previously (Dreier and Reiffurth 2015). Despite this austerity program, sodium and calcium always leak into cells and potassium leaks out. Eventually, ATP is depleted and the sodium pump fails to restore the leaking ions. At a threshold level of failure, usually reached 1–5 min after nonspreading depression has made the ischemic tissue isoelectric, neurons then abruptly undergo the injury depolarization. Because the tissue is already isoelectric, the injury depolarization will not induce further spreading depression of activity under this condition.
Neurons can also die without an abrupt negative shift of the steady potential
This is a relevant caveat not only for cell cultures which lack the normal tissue assembly and thus the prerequisite for the occurrence of a neuronal mass injury as a sudden, expanding network event but may also apply to special conditions in vivo.
For example, injury depolarizations do not occur in neonatal brains (Bures 1957; Schade 1959), although neonatal neurons can certainly die. Somjen suggested that the lack of injury depolarizations could protect the brain at birth, which is the point in the life cycle with the highest statistical risk for hypoxia (Somjen 2004). In rodents, susceptibility to injury depolarizations then rapidly reaches its maximum at 16–30 days of age and slowly decreases again thereafter (Hablitz and Heinemann 1989; Hertelendy et al. 2019; Maslarova et al. 2011; Menyhart et al. 2017).
Another example is the cerebellar cortex that contains highly vulnerable neurons to ischemia, the Purkinje cells, but is in general less susceptible to injury depolarizations than, for example, the neocortex (Somjen 2001). Possibly, this is due to the extracellular volume fraction of the superficial molecular layer which is significantly higher than that of neocortex (Sykova and Nicholson 2008). The larger extracellular space should dilute ions, metabolites, and neuroactive substances released from the cells and should thus protect against neuronal mass injury (Lehmenkuhler et al. 1993). Accordingly, the application of the vasoconstrictor endothelin-1 (ET-1) to the cerebellum (Oliveira-Ferreira et al. 2019) led to a similar decrease in blood flow as in previous studies on the neocortex (Dreier et al. 2002a; Kleeberg et al. 2004; Oliveira-Ferreira et al. 2010; Petzold et al. 2003). However, in contrast to the neocortex, injury depolarizations, as assessed by DC-ECoG, IOS, and regional cerebral blood flow (rCBF) recordings, often did not occur. Yet, quantitative cell counting found that the proportion of necrotic Purkinje cells was significantly higher in ET-1-treated rats than sham controls even if ET-1 had not caused large negative DC shifts. This means that Purkinje cells can enter the state of injury relatively isolated from each other, depolarize isolated from each other, and die. Large injury depolarization as a network event however occurs in the cerebellum when ischemia is more severe. Accordingly, the characteristic sequence of a terminal large negative DC shift in the wake of circulatory arrest does not differ significantly between cerebellum and neocortex (Fifkova et al. 1961; Kraig et al. 1983; Nicholson et al. 1977; Oliveira-Ferreira et al. 2019).
The debate whether the reduced susceptibility of the cerebellum to mass injury depolarizations has a protective effect on Purkinje cells, which is required because Purkinje cells are particularly vulnerable, or vice versa, is a factor contributing to the high vulnerability of Purkinje cells, is complex, and also conducted for other brain structures. This has been reviewed previously (Dreier 2011; Dreier and Reiffurth 2015; Hartings et al. 2017a). Our current view is that injury depolarizations facilitate death in tissue with disturbed metabolism and disturbed neurovascular coupling (Dreier 2011), while they could be beneficial as a signal to surrounding adequately supplied tissue where they may upregulate growth factors, stress response proteins and inflammatory mediators (Jander et al. 2001; Kunkler et al. 2004; Richter et al. 2017; Sharp et al. 2000; Urbach et al. 2006), have preconditioning actions (Matsushima et al. 1996), enhance plasticity (Berger et al. 2008) and regeneration (Urbach et al. 2017; Yanamoto et al. 2005), and reduce the vascular steal effect on ischemic zones through the physiological oligemia (cf. below) (Oliveira-Ferreira et al. 2010). However, it should be added that the evidence that injury depolarizations can also have beneficial effects has so far been controversially disputed. Most of these considerations are of a more theoretical nature, and research efforts should be intensified in this area (Dreier 2011; Shuttleworth et al. 2019).
Inhibiting neuronal mass injury as a target of neuroprotective treatment
Due to the potentially positive and negative effects of injury depolarizations on the one hand and on the other, it is also a controversial issue whether direct inhibition of injury depolarizations, for example, using N-methyl-D-aspartate receptor (NMDAR) antagonists (Hertle et al. 2012; Lauritzen and Hansen 1992; Marrannes et al. 1988; Sakowitz et al. 2009), would have a net beneficial effect (Reinhart and Shuttleworth 2018). This discussion is further complicated by the fact that NMDAR antagonists increasingly lose efficacy against injury depolarizations where they are increasingly harmful (Hernandez-Caceres et al. 1987; Koroleva et al. 1998; Lauritzen and Hansen 1992; Madry et al. 2010; Muller and Somjen 1998). Thus, only a bathing medium containing high concentrations of, e.g., DNQX/NBQX to block all AMPA/kainate receptors, the combination of MK-801 and APV to potently block NMDARs, and bicuculline methiodide to block GABAA receptors succeeded in preventing the signature of injury depolarization on the single neuron level in severely ischemic (Allen et al. 2004; Madry et al. 2010) or anoxic brain slices (Revah et al. 2016). Nevertheless, high doses of NMDAR antagonists such as MK-801 may block at least a fraction of injury depolarizations in moderately ischemic tissue. Thus, high doses of MK-801 were clearly neuroprotective in animal models of focal cerebral ischemia (Dezsi et al. 1992; Dirnagl et al. 1990; Dreier et al. 2002a; Gill et al. 1992; Iijima et al. 1992; Ozyurt et al. 1988; Park et al. 1988a; Park et al. 1988b; Roussel et al. 1992; Yao et al. 1994; Yi et al. 2019). By contrast, NMDAR antagonists were exclusively administered at low doses in clinical studies of stroke and TBI, as drug developers wanted to avoid psychotropic side effects (Kemp and McKernan 2002). Consistent with the notion that NMDAR antagonists are insufficient to inhibit injury depolarizations in the ischemic penumbra at low doses (Klass et al. 2018), the clinical trials showed neither significant benefit nor harm from the NMDAR antagonists (Muir and Lees 2003). In recent years, however, the NMDAR antagonist ketamine has found its way at high doses into everyday clinical routine of neurocritical care medicine to sedate patients with acute cerebral injuries because it shows beneficial cardiocirculatory and bronchodilatory effects (Von der Brelie et al. 2017). Ketamine acts at the same binding site of NMDARs as MK-801 (Rashidy-Pour et al. 1995). Thus, at higher doses applied for sedation, ketamine may also block injury depolarizations in the penumbra and there are moves afoot to investigate this more systematically in neuromonitoring-guided multicenter trials (Carlson et al. 2018a; Hartings et al. 2018; Hertle et al. 2012; Sakowitz et al. 2009).
A further question is whether unfavorable properties of injury depolarizations causing their local perpetuation could be therapeutically targeted. For example, the inverse hemodynamic response to injury depolarization could be a worthwhile target in this context. Thus, in otherwise normal tissue, the intense neuronal and astrocytic depolarization acts as a potent stimulus to increase rCBF (= spreading hyperemia) (Fig. 2a). After the neuronal and astroglial repolarization, this increase is typically followed by a physiological, prolonged, moderate hypoperfusion (= oligemia) (Ayata and Lauritzen 2015; Fordsmann et al. 2013; Lauritzen 1994). Under these conditions, there is practically always a complete recovery from the neuronal mass injury (Nedergaard and Hansen 1988). However, in other more pathological situations, the injury depolarization can induce severe vasoconstriction instead of vasodilatation leading to a long-lasting local perfusion deficit that prevents the neuronal and astroglial repolarization and perpetuates the injury (Bere et al. 2014a; Bere et al. 2014b; Dreier et al. 1998; Dreier et al. 2001a; Dreier et al. 2002c; Dreier et al. 2002d; Feuerstein et al. 2014; Petzold et al. 2003; Petzold et al. 2008; Petzold et al. 2005; Shin et al. 2006; Sonn and Mayevsky 2000; Strong et al. 2007; Sukhotinsky et al. 2008). The term inverse hemodynamic response has been used to describe this process in which neuronal depolarization leads to spreading ischemia and neuronal cell death (Fig. 2b) (Dreier et al. 2000). Experimentally, spreading ischemia could be the sole cause of widespread infarcts (Dreier et al. 2000). The continuum from spreading hyperemia to spreading ischemia was not only recorded in animals but also in patients with aSAH, TBI, and MHS (Dreier 2011; Dreier et al. 2009; Hinzman et al. 2014; Woitzik et al. 2013). In aSAH patients, long-lasting spreading ischemia was associated with imaging-proven lesion progression (Luckl et al. 2018). Interestingly, the tendency to inverse neurovascular coupling seems to increase with age, suggesting that this is an interesting therapeutic target especially in the elderly (Csipo et al. 2019; Menyhart et al. 2015).
Clinical symptoms in the context of neuronal mass injury
Medical history and physical examination inform the neurologist about the shadows that pathological processes cast on body functions by their effects on brain activity. Among the most characteristic shadows of neurology are the spreading neurological deficit of a migraine aura or the sudden, nonspreading deficit of stroke characterized by the simultaneous involvement of various modalities such as speech, motor, sensory, or visual function. By contrast, epileptic seizures typically do not appear as shadows but as an involuntary excess of perceptions or body functions, e.g., as extreme stiffness of the limbs and subsequent jerking movements during a tonic-clonic seizure. The nature of these shadows or excesses, termed negative or positive symptoms, forms the basis for the neurologist’s clinical diagnosis. Their underlying pathophysiological mechanisms are in principle known today (Dreier and Reiffurth 2015).
Thus, it is now increasingly recognized that the sudden, nonspreading neurological deficit typical of stroke results from nonspreading depression of activity (Dreier and Reiffurth 2015; Dreier et al. 2015). By contrast, migraine aura is assumed to result from injury depolarization-induced spreading depression of spontaneous activity when (i) this propagates in an eloquent brain region, which is not metabolically impaired before arrival of the wave and thus not subject to any changes of the spontaneous brain activity before arrival of the wave, and (ii) the patient is in a state of conscious awareness that permits the perception, memorization, recall, and report of the symptoms (Dreier and Reiffurth 2015; Dreier et al. 2015; Leão and Morison 1945). However, (i) since the continuum of injury depolarizations can affect various cortical and subcortical representation fields, (ii) since they can exhibit a broad spectrum of depression patterns of spontaneous activity, (iii) since they can occur in the form of occasional or clustered events (Dreier et al. 2006; Sakowitz et al. 2013), (iv) since depressions in one brain region may affect other brain regions depending on connectivity (Gold and Lauritzen 2002), and (v) since epileptic activities may alternate with injury depolarizations (Fabricius et al. 2008; Revankar et al. 2017; Winkler et al. 2012) or (vi) may occur superimposed on periods of injury depolarization-induced activity depression (Dreier et al. 2012), this results in a very broad spectrum of possible clinical symptoms and provides a plausible explanation for the wide variety of global and focal presentations of numerous pathological conditions of the CNS (Dreier and Reiffurth 2015).
The typical clinical presentation of a migraine aura
The most common form of migraine aura concerns the visual system. A visual aura is typically characterized by a slowly growing, kidney-shaped scotoma, which usually begins in the visual field center, runs to the periphery within 10–20 min, and is often surrounded by a narrow, scintillating rim (Lashley 1941). Auras that affect the somatosensory system, motor function, or speech spread in a similar fashion across the corresponding cortical representation fields, and symptoms then migrate slowly, for example, along the arm of the patient in the case of a somatosensory aura, etc. In general, a migraine aura often shows a small area of increased excitation in the front that precedes the loss of neurological function (Dreier and Reiffurth 2015). For example, in the case of a somatosensory aura, tingling sensations often occur before numbness develops. If different modalities such as visual, somatosensory, motor, or speech systems are affected, the symptoms usually spread from one modality to the next. According to the International Headache Society, a migraine aura must last longer than 5 min. However, the caveat is added that this is an arbitrary threshold. In humans, there are not only plausibility considerations but also clear measurements that the injury depolarization (i) can also occur in spatially very limited fields and (ii) can spread in the cortical tissue at a wide range of propagation speeds between ~ 2 and 9 mm/min (Dahlem and Hadjikhani 2009; Woitzik et al. 2013).
Indirect evidence that neuronal mass injury-induced spreading depression of activity is the pathophysiological correlate of the migraine aura was previously provided by the planar intracarotideal 133Xenon method and single-photon emission computed tomography (SPECT) in patients undergoing migraine aura (Lauritzen et al. 1983; Olesen et al. 1981). This evidence was based on the similarity of the recorded rCBF changes during migraine aura with the normal hemodynamic response to injury depolarization in animal experiments (Lauritzen 1994). The large number of recorded migraine auras in these studies resulted from the procedure of catheterizing and injecting the carotid artery, which provoked visual aura in more than 50% of migraineurs (Lassen and Friberg 1991). Today, it is widely believed that the mechanism that led to these auras was cerebral microembolisms that caused brief ischemic episodes (Dreier et al. 2002a; Dreier and Reiffurth 2015; Nozari et al. 2010). In addition, a positron emission tomography and a functional MRI study strongly supported the concept that injury depolarization-induced spreading depression is the pathophysiological correlate of the migraine aura (Hadjikhani et al. 2001; Woods et al. 1994). Consistent evidence for this was also provided by magnetoencephalography (Bowyer et al. 2001).
As a rule, a migraine aura passes without lasting damage. However, a proportion of migraineurs showed white matter abnormalities associated with small vessel disease in MRI studies (Kurth et al. 2012). Since injury depolarizations are limited to gray matter, these abnormalities are unlikely to result from injury depolarizations. Yet, the small vessel disease may not only damage white matter tracts but also the cortex. This could lead to injury depolarizations manifesting as migraine with aura (Dichgans et al. 1998). For a more comprehensive account of the relationship between migraine aura and ischemic stroke, we refer the reader to the following review (Dreier and Reiffurth 2015).
Nonspreading and spreading focal neurological deficits in hemorrhagic stroke
Although a clinical distinction between spreading and nonspreading focal neurological deficits is useful because they are associated with different probabilities of permanent damage, it is nevertheless important to note that these events have a common pathophysiological basis, occur along a continuum, and do not show a black and white contrast. Thus, the sudden, nonspreading deficit in various modalities is the typical presentation of an ischemic stroke, but an ischemic stroke can also present with symptoms of a migraine aura in rare cases (Dreier and Reiffurth 2015). Accordingly, the typical presentation of a hemorrhagic stroke is a sudden, nonspreading deficit, yet migraine aura may be the initial symptom in rare cases. This has been observed, for example, for aSAH (Dreier et al. 2001b; Evans and Davidoff 2001), sulcal hemorrhages due to cerebral amyloid angiopathy (Izenberg et al. 2009; Kleinig et al. 2008; Samanci et al. 2016; Stanton et al. 2019), or bleeding due to an arteriovenous malformation (Jeon et al. 2016) or a venous cavernoma (Ulrich et al. 2016). In order to illustrate this, we will present an aSAH patient in the following in whom subdural DC/AC-ECoG recorded at least two events of injury depolarization-induced spreading depression of activity while the patient underwent a migraine aura with aphasic/dysarthric and motor symptoms. Second, we will discuss an analogous patient with repeated migraine auras who had a sulcal hemorrhage due to a mild trauma on the basis of a probable cerebral amyloid angiopathy. Written informed consent was obtained from both patients. Research was conducted in accordance with the Declaration of Helsinki.
Illustrative case 1
A 56-year-old woman suffered from aSAH due to a left middle cerebral artery aneurysm. Initially, she presented with sudden bilateral headache, aphasia, and decreased consciousness (World Federation of Neurosurgical Societies (WFNS) score 4, modified Fisher scale 3) (Fig. 3a). She had no history of migraine with or without aura. Upon admission, the patient was included in the Co-Operative Studies of Brain Injury Depolarizations (COSBID). Prospective inclusion and exclusion criteria for COSBID have been described previously (Winkler et al. 2017). A standard collinear subdural electrode strip with six platinum contacts was implanted over the left frontal cortex directly after surgical clip ligation of the aneurysm (Fig. 4b). The patient was extubated in the neurocritical care unit (NCU) on the following day and remained without sedation until the end of the neuromonitoring period. Until day 11, the patient had 160 transient injury depolarizations. Most of those spread from the frontal pole in occipital direction. The occurrence of focal neurological deficits was not noted during this period. However, during the routine restart of the recording system (performed by SM) on day 11, the patient was talking to the NCU nurse while SM was present. Suddenly, the patient developed progressive difficulties to speak and the nurse called the intensivists in charge. A few minutes later, the patient developed a facial paresis on the right side, followed by a paresis of the right arm again a few minutes later. The intensivists induced hypertension with intravenous noradrenaline to treat the suspected episode of delayed cerebral ischemia. Proximal vasospasm of the left middle cerebral artery (MCA) was ruled out using transcranial Doppler sonography (mean velocity < 100 cm/s) (Vora et al. 1999). During this episode characteristic of a migraine aura, an injury depolarization-induced spreading depression of activity was recorded (Fig. 4). Subsequently, the symptoms fully resolved within approximately 30 min. Three hours later, another similar event of injury depolarization-induced spreading depression of activity was recorded and a similar episode of migraine aura with speech difficulties and facial paresis was documented. The electrode strip was removed on day 14. In total, the patient had 189 transient injury depolarizations. Cerebral MRI performed on day 15 revealed discrete signs of cytotoxic edema in the cortex of the left MCA territory, especially in areas neighboring the subarachnoid blood clot (Fig. 3b). In the follow-up MRI performed 7 months later, signs of cortical atrophy were seen in this region (Fig. 3c). At this time, the patient had no focal neurological deficit, but neuropsychological testing revealed a mild cognitive dysfunction of the dysexecutive type (Glasgow coma score extended (GOSE): upper moderate disability = score 6/8).
Fig. 3.

Neuroimaging of illustrative case 1 (migraine aura after aneurysmal subarachnoid hemorrhage (aSAH)). a Initial computed tomography (CT) scan showing an aSAH with blood accumulation in the Sylvian fissure due to rupture of a left middle cerebral artery aneurysm. b Diffusion-weighted magnetic resonance imaging (DW-MRI) on day 15 after aSAH demonstrating diffusion restrictions typical of delayed cerebral ischemia at the cortical surface and around the Sylvian fissure (Dreier et al. 2002b; Weidauer et al. 2008) (arrows). The assessment is slightly reduced by motion artifacts. c Fluid-attenuated inversion recovery (FLAIR)-weighted MRI 7 months after aSAH with cortical atrophy in the area of the left Sylvian fissure
Fig. 4.

Spreading depolarization (= injury depolarization)-induced spreading depression of activity during a migraine aura on day 11 after aneurysmal subarachnoid hemorrhage (aSAH) (illustrative case 1). a The five top traces show raw direct current (DC) (f 0–45 Hz) recordings from subdural electrodes 2–6, traces 6–10 show the changes in spontaneous brain activity at the same electrodes (f 0.5–45 Hz), trace 11 shows the tissue partial pressure of oxygen, and trace 12 the intracranial pressure. During the routine restart of the recording system on day 11, the patient developed typical symptoms of a motor migraine aura. In parallel, an injury depolarization-induced spreading depression of activity was recorded in more frontal areas of the cortex. It is likely that electrode 6 would have recorded a negative DC shift similar to electrodes 3–5, if the recording had restarted earlier because the activity was depressed at electrode 6 at the onset of the recording and gradually recovered thereafter. Moreover, practically all other injury depolarizations in this patient involved electrode 6. b Projection of electrodes 1–6 of the subdural, collinear recording strip on the left brain cortex of the patient. The electrodes were obtained from a postoperative computed tomography (CT) scan by simple thresholding as previously described (Milakara et al. 2017). The surface of the isolated electrodes was geometrically reconstructed by MATLAB’s (MathWorks Inc., Natick, MA, USA) isosurface function. The cortical reconstruction was performed with FreeSurfer (Martinos Center for Biomedical Imaging, Charlestown, MA, USA, http://surfer.nmr.mgh.harvard.edu/) from the magnetic resonance imaging (MRI) scan on day 15. To superimpose the electrodes on the geometric mesh representing the cortical surface, the CT scan including the electrodes was rigidly co-registered to the MRI scan reoriented and resampled with FreeSurfer. The arrows indicate the direction of the spread of the injury depolarization. c The primary motor cortex is located in the precentral gyrus and handles signals coming from the premotor area of the frontal lobes. The motor homunculus represents a map of areas in the primary motor cortex dedicated to motor processing for different anatomical divisions of the body. The subdural electrode strip that recorded the injury depolarization-induced spreading depression of brain activity was not located directly on the motor cortex affected by the migraine aura, but it is plausible that it spread not only towards the frontal pole along the recording strip but also upwards along the primary motor cortex (arrow)
Illustrative case 2
A 72-year-old woman with a history of smoking, arterial hypertension, and hypothyroidism but no history of migraine with or without aura suffered a fall on the back of her head while climbing on a chair (day 0). This caused a skull laceration and a marked external hematoma. She did not report loss of consciousness or dizziness, but the fall was followed by moderate to severe headache over the following days. On the day of accident, she presented to the emergency room of another hospital where the skull laceration was treated. Thereafter, the patient was discharged. On day 2, the patient was admitted to the stroke unit of the Charité University Hospital because of an episode of paresis of the right arm and face followed by dysarthria as well as tingling and numbness of the right hand. The symptoms fully resolved within 10 to 15 min. On the stroke unit, we saw a patient without any focal neurological deficit. However, the symptoms typical of a complicated migraine aura then repeatedly occurred on our unit during the next 10 days (Fig. 5d). The highest frequency was observed on day 4 with seven events. Overall, the episodes were stereotypical but decreased in severity over time. While the face was initially included, the paresis remained restricted to the right hand during the later episodes. Then, the motor symptoms fully disappeared, and the patient only noted somatosensory symptoms. Eventually, only the fingers of her right hand were involved. The last episode occurred on day 12. On day 14, the patient was discharged without any permanent deficit or additional medication.
Fig. 5.
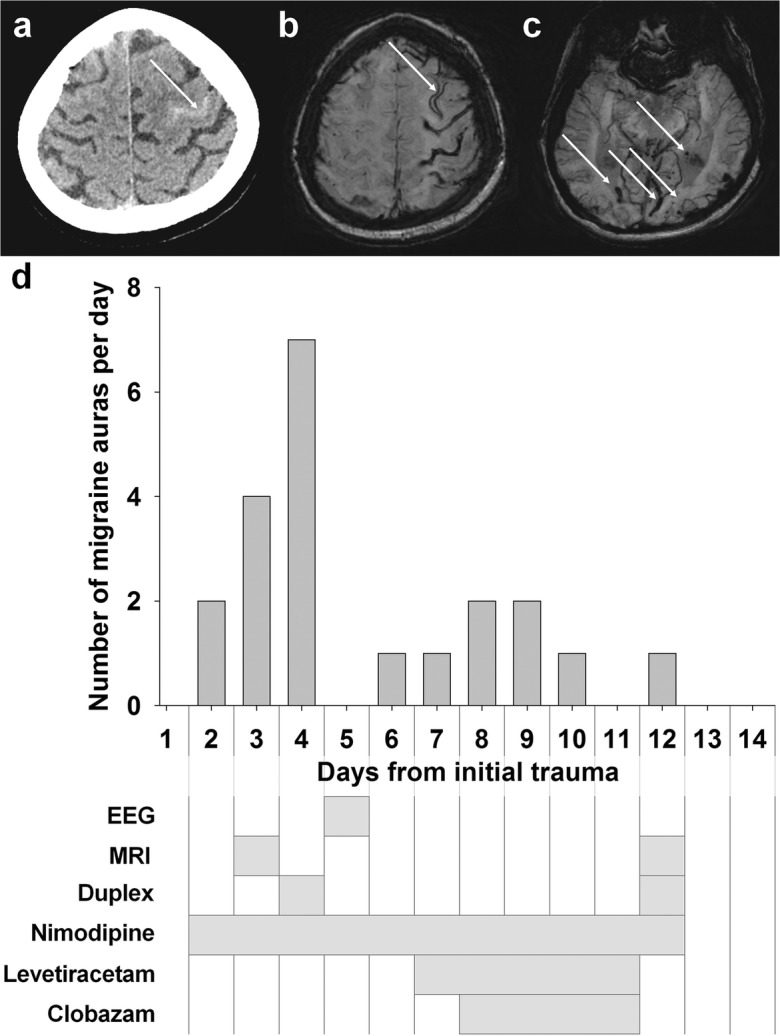
Repeated migraine auras after traumatic subarachnoid hemorrhage (tSAH) in the left central sulcus in a patient with probable cerebral amyloid angiopathy (illustrative case 2). a Initial computed tomography (CT) on day 2 showing localized acute cortical tSAH within the central sulcus adjacent to the hand knob (arrow). b The susceptibility-weighted magnetic resonance imaging (MRI) on day 12 demonstrates widespread left frontoparietal superficial hemosiderosis. No visible signal changes were seen in the parenchyma adjacent to the tSAH (not shown). c The susceptibility-weighted minimum intensity projection reveals numerous juxtacortical microbleeds and smaller lobar hemorrhagic residua typical of cerebral amyloid angiopathy. d Time course of the occurrence of migraine auras and the medical treatment as well as time points of the diagnostic tests after the sulcal hemorrhage
The initial computed tomography (CT) (Fig. 5a) and computed tomography angiography (CTA) on day 2 revealed a sulcal SAH in the left central sulcus without evidence for a bleeding source such as an arteriovenous fistula, malformation, or aneurysm. MRI scans were conducted on day 3 and day 12 showing a widespread left frontoparietal superficial hemosiderosis and numerous juxtacortical microbleeds, as well as smaller lobar hemorrhagic residua (Fig. 5b, c). In view of the radiological signs, the diagnosis of a probable cerebral amyloid angiopathy according to the Boston criteria was made (Greenberg and Charidimou 2018). Extracranial and transcranial Duplex sonography on days 4 and 12 revealed minor atherosclerosis without stenosis or signs of proximal vasospasm. Scalp electroencephalography (EEG) was performed on day 5 without evidence of epileptic discharges.
A prophylaxis for delayed cerebral ischemia with oral nimodipine (4 × 60 mg) was initiated on day 2 and discontinued on day 13 under blood pressure monitoring with a target mean arterial pressure of 85 mmHg. Additionally, antiepileptic treatment with levetiracetam (2 × 500 mg) and clobazam (2 × 5 mg) was given for 5 and 4 days, respectively (Fig. 5d). The antiepileptic treatment did not result in any obvious improvement of the frequency of the migraine auras. Accordingly, the differential diagnosis of focal epileptic seizures was deemed unlikely, and the antiepileptic treatment was discontinued again.
Conclusion
In the past, the continuum of injury depolarizations was much easier to comprehend in experimental research than in the clinic because the technologies required to this end had only been available to experimental research (Dijkhuizen et al. 1999; Dreier and Reiffurth 2015; Hartings et al. 2017a; Leão 1947; Leão and Morison 1945; Marshall 1959). However, modern neuromonitoring technologies in the neurocritical care unit have now come much closer to the recording methods used in animal experiments. Thus, neuromonitoring increasingly succeeds in closing the gap between basic research and clinical medicine. For example, such a gap has been the lack of direct electrophysiological proof for the concept that injury depolarization-induced spreading depression of activity is the pathophysiological correlate of the migraine aura. In case 1, this evidence has now been found for the first time. The subdural electrode strip was not located directly on the affected motor cortex, but it is likely that the two injury depolarization-induced spreading depressions, recorded simultaneously with the two migraine auras, spread not only along the recording strip but also in the motor cortex as further explained in Fig. 4.
It may be added that the percepts and behavioral correlates associated with injury depolarizations may well include a wider spectrum than just migraine aura or sudden nonspreading focal deficits typical of stroke. For example, a case of aSAH with repeated episodes of freezing behavior and mutism associated with brief clusters of injury depolarizations has been previously reported (Dreier et al. 2006). In rodents, the occurrence of injury depolarization-induced spreading depression was associated with episodes of freezing, grooming, wet dog shake and head shake behavior, or no symptoms (Akcali et al. 2010; Filiz et al. 2019; Oitzl and Huston 1984; Shibata 1978).
The depression pattern of spontaneous activity and the resulting clinical symptoms are related to the injury in a complex fashion, so that the nature and severity of the clinical symptoms allow predictions of prognosis within certain limits. Thus, clinical experience has shown that a focal neurological deficit that occurs in the form of a migraine aura is predominantly, however, not always associated with a good prognosis (Dreier and Reiffurth 2015). Although the significance of our illustrative case 2 is somewhat limited, as injury depolarizations have not been unequivocally confirmed with electrophysiology, both cases seem to confirm this. In case 2, there was no permanent damage. In case 1, the diffusion restriction of the cortex in the early MRI and the atrophy in the late MRI show that there was a permanent damage in some regions, but this was relatively mild compared with other cases of delayed cerebral ischemia. Overall, the clinical outcome was also good in case 1 with only mild cognitive dysfunction and no clear permanent focal neurological deficit.
The two cases as well as the experimental literature suggest that the underlying diseases such as brain ischemia or hemorrhage, TBI, a brain tumor, or an encephalitis (Dreier and Reiffurth 2015; Ellis et al. 2017; Evans et al. 2015; Klingebiel et al. 2008; Olesen et al. 1993; Vosoughi et al. 2018) play the decisive role for the occurrence of symptomatic migraine auras, but not whether the patient ever had a migraine in the past. Therefore, it would probably be more appropriate (i) to simply speak of spreading focal neurological deficit instead of migraine aura, (ii) to distinguish this from nonspreading focal neurological deficit, (iii) to be aware that a spreading focal neurological deficit results from spreading depression of activity and a nonspreading focal neurological deficit from nonspreading depression of activity (Dreier 2011), (iv) that both spreading and nonspreading depressions occur with injury depolarization, and (v) that the injury depolarization rather than the depression pattern determines whether the tissue will survive or die.
Funding information
This study is supported by grants of the Deutsche Forschungsgemeinschaft DFG DR 323/5-1 to Dr. Dreier and Dr. Woitzik and DFG DR 323/10-1, FP7 no. 602150 CENTER-TBI and Era-Net Neuron EBio2 to Dr. Dreier.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Aiba I, Shuttleworth CW. Sustained NMDA receptor activation by spreading depolarizations can initiate excitotoxic injury in metabolically compromised neurons. J Physiol. 2012;590:5877–5893. doi: 10.1113/jphysiol.2012.234476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken PG, Tombaugh GC, Turner DA, Somjen GG. Similar propagation of SD and hypoxic SD-like depolarization in rat hippocampus recorded optically and electrically. J Neurophysiol. 1998;80:1514–1521. doi: 10.1152/jn.1998.80.3.1514. [DOI] [PubMed] [Google Scholar]
- Akcali D, Sayin A, Sara Y, Bolay H. Does single cortical spreading depression elicit pain behaviour in freely moving rats? Cephalalgia. 2010;30:1195–1206. doi: 10.1177/0333102409360828. [DOI] [PubMed] [Google Scholar]
- Allen NJ, Rossi DJ, Attwell D. Sequential release of GABA by exocytosis and reversed uptake leads to neuronal swelling in simulated ischemia of hippocampal slices. J Neurosci. 2004;24:3837–3849. doi: 10.1523/JNEUROSCI.5539-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arboix A, Garcia-Eroles L, Massons J, Oliveres M, Targa C. Acute stroke in very old people: clinical features and predictors of in-hospital mortality. J Am Geriatr Soc. 2000;48:36–41. doi: 10.1111/j.1532-5415.2000.tb03026.x. [DOI] [PubMed] [Google Scholar]
- Astrup J, Sorensen PM, Sorensen HR. Oxygen and glucose consumption related to Na+-K+ transport in canine brain. Stroke. 1981;12:726–730. doi: 10.1161/01.str.12.6.726. [DOI] [PubMed] [Google Scholar]
- Avoli M, Drapeau C, Louvel J, Pumain R, Olivier A, Villemure JG. Epileptiform activity induced by low extracellular magnesium in the human cortex maintained in vitro. Ann Neurol. 1991;30:589–596. doi: 10.1002/ana.410300412. [DOI] [PubMed] [Google Scholar]
- Ayata C, Lauritzen M. Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev. 2015;95:953–993. doi: 10.1152/physrev.00027.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balanca B, Meiller A, Bezin L, Dreier JP, Marinesco S, Lieutaud T. Altered hypermetabolic response to cortical spreading depolarizations after traumatic brain injury in rats. J Cereb Blood Flow Metab. 2017;37:1670–1686. doi: 10.1177/0271678X16657571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balestrino M, Young J, Aitken P. Block of (Na+,K+)ATPase with ouabain induces spreading depression-like depolarization in hippocampal slices. Brain Res. 1999;838:37–44. doi: 10.1016/s0006-8993(99)01674-1. [DOI] [PubMed] [Google Scholar]
- Basarsky TA, Duffy SN, Andrew RD, MacVicar BA. Imaging spreading depression and associated intracellular calcium waves in brain slices. J Neurosci. 1998;18:7189–7199. doi: 10.1523/JNEUROSCI.18-18-07189.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu C, Busch E, de Crespigny A, Moseley ME. Spreading waves of transient and prolonged decreases in water diffusion after subarachnoid hemorrhage in rats. Magn Reson Med. 2000;44:110–116. doi: 10.1002/1522-2594(200007)44:1<110::aid-mrm16>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Benjamin EJ, et al. Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bere Z, Obrenovitch TP, Bari F, Farkas E. Ischemia-induced depolarizations and associated hemodynamic responses in incomplete global forebrain ischemia in rats. Neuroscience. 2014;260:217–226. doi: 10.1016/j.neuroscience.2013.12.032. [DOI] [PubMed] [Google Scholar]
- Bere Z, Obrenovitch TP, Kozak G, Bari F, Farkas E. Imaging reveals the focal area of spreading depolarizations and a variety of hemodynamic responses in a rat microembolic stroke model. J Cereb Blood Flow Metab. 2014;34:1695–1705. doi: 10.1038/jcbfm.2014.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger M, Speckmann EJ, Pape HC, Gorji A. Spreading depression enhances human neocortical excitability in vitro. Cephalalgia. 2008;28:558–562. doi: 10.1111/j.1468-2982.2008.01556.x. [DOI] [PubMed] [Google Scholar]
- Bogdanov VB, et al. Susceptibility of primary sensory cortex to spreading depolarizations. J Neurosci. 2016;36:4733–4743. doi: 10.1523/JNEUROSCI.3694-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosche B, et al. Recurrent spreading depolarizations after subarachnoid hemorrhage decreases oxygen availability in human cerebral cortex. Ann Neurol. 2010;67:607–617. doi: 10.1002/ana.21943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowyer SM, Aurora KS, Moran JE, Tepley N, Welch KM. Magnetoencephalographic fields from patients with spontaneous and induced migraine aura. Ann Neurol. 2001;50:582–587. doi: 10.1002/ana.1293. [DOI] [PubMed] [Google Scholar]
- Brazinova A et al (2018) Epidemiology of traumatic brain injury in Europe: a living systematic review. J Neurotrauma. 10.1089/neu.2015.4126 [DOI] [PMC free article] [PubMed]
- Budde MD, Frank JA. Neurite beading is sufficient to decrease the apparent diffusion coefficient after ischemic stroke. Proc Natl Acad Sci U S A. 2010;107:14472–14477. doi: 10.1073/pnas.1004841107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bures J. The ontogenetic development of steady potential differences in the cerebral cortex in animals. Electroencephalogr Clin Neurophysiol. 1957;9:121–130. doi: 10.1016/0013-4694(57)90116-5. [DOI] [PubMed] [Google Scholar]
- Burr HS, Harmann PJJ. Voltage gradients in the nervous system. Trans Amer Neurol Ass. 1939;65:11–14. [Google Scholar]
- Busch E, Gyngell ML, Eis M, Hoehn-Berlage M, Hossmann KA. Potassium-induced cortical spreading depressions during focal cerebral ischemia in rats: contribution to lesion growth assessed by diffusion-weighted NMR and biochemical imaging. J Cereb Blood Flow Metab. 1996;16:1090–1099. doi: 10.1097/00004647-199611000-00002. [DOI] [PubMed] [Google Scholar]
- Busch E, Beaulieu C, de Crespigny A, Moseley ME. Diffusion MR imaging during acute subarachnoid hemorrhage in rats. Stroke. 1998;29:2155–2161. doi: 10.1161/01.str.29.10.2155. [DOI] [PubMed] [Google Scholar]
- Cain SM et al (2017) In vivo imaging reveals that pregabalin inhibits cortical spreading depression and propagation to subcortical brain structures. Proc Natl Acad Sci U S A in press [DOI] [PMC free article] [PubMed]
- Carlson Andrew P., Abbas Mohammad, Alunday Robert L., Qeadan Fares, Shuttleworth C. William. Spreading depolarization in acute brain injury inhibited by ketamine: a prospective, randomized, multiple crossover trial. Journal of Neurosurgery. 2019;130(5):1513–1519. doi: 10.3171/2017.12.JNS171665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson Andrew P., Shuttleworth C. William, Major Sebastian, Lemale Coline L., Dreier Jens P., Hartings Jed A. Terminal spreading depolarizations causing electrocortical silencing prior to clinical brain death: case report. Journal of Neurosurgery. 2019;131(6):1773–1779. doi: 10.3171/2018.7.JNS181478. [DOI] [PubMed] [Google Scholar]
- Chan AW, Murphy TH. Good vibrations: resting-state functional connectivity reflects entrainment of vasomotion. Neuron. 2017;96:716–717. doi: 10.1016/j.neuron.2017.10.035. [DOI] [PubMed] [Google Scholar]
- Csipo Tamas, Mukli Peter, Lipecz Agnes, Tarantini Stefano, Bahadli Dhay, Abdulhussein Osamah, Owens Cameron, Kiss Tamas, Balasubramanian Priya, Nyúl-Tóth Ádám, Hand Rachel A., Yabluchanska Valeriya, Sorond Farzaneh A., Csiszar Anna, Ungvari Zoltan, Yabluchanskiy Andriy. Assessment of age-related decline of neurovascular coupling responses by functional near-infrared spectroscopy (fNIRS) in humans. GeroScience. 2019;41(5):495–509. doi: 10.1007/s11357-019-00122-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlem MA, Hadjikhani N. Migraine aura: retracting particle-like waves in weakly susceptible cortex. PLoS One. 2009;4:e5007. doi: 10.1371/journal.pone.0005007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Crespigny A, Rother J, van Bruggen N, Beaulieu C, Moseley ME. Magnetic resonance imaging assessment of cerebral hemodynamics during spreading depression in rats. J Cereb Blood Flow Metab. 1998;18:1008–1017. doi: 10.1097/00004647-199809000-00010. [DOI] [PubMed] [Google Scholar]
- Dennis MS, Burn JP, Sandercock PA, Bamford JM, Wade DT, Warlow CP. Long-term survival after first-ever stroke: the Oxfordshire Community Stroke Project. Stroke. 1993;24:796–800. doi: 10.1161/01.str.24.6.796. [DOI] [PubMed] [Google Scholar]
- Dezsi L, Greenberg JH, Hamar J, Sladky J, Karp A, Reivich M. Acute improvement in histological outcome by MK-801 following focal cerebral ischemia and reperfusion in the cat independent of blood flow changes. J Cereb Blood Flow Metab. 1992;12:390–399. doi: 10.1038/jcbfm.1992.56. [DOI] [PubMed] [Google Scholar]
- Di Carlo A, et al. Stroke in the very old : clinical presentation and determinants of 3-month functional outcome: a European perspective. European BIOMED Study of Stroke Care Group. Stroke. 1999;30:2313–2319. doi: 10.1161/01.str.30.11.2313. [DOI] [PubMed] [Google Scholar]
- Dichgans M, et al. The phenotypic spectrum of CADASIL: clinical findings in 102 cases. Ann Neurol. 1998;44:731–739. doi: 10.1002/ana.410440506. [DOI] [PubMed] [Google Scholar]
- Dietz RM, Weiss JH, Shuttleworth CW. Zn2+ influx is critical for some forms of spreading depression in brain slices. J Neurosci. 2008;28:8014–8024. doi: 10.1523/JNEUROSCI.0765-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkhuizen RM, Beekwilder JP, van der Worp HB, van der Sprenkel JW B, Tulleken KA, Nicolay K. Correlation between tissue depolarizations and damage in focal ischemic rat brain. Brain Res. 1999;840:194–205. doi: 10.1016/s0006-8993(99)01769-2. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Tanabe J, Pulsinelli W. Pre- and post-treatment with MK-801 but not pretreatment alone reduces neocortical damage after focal cerebral ischemia in the rat. Brain Res. 1990;527:62–68. doi: 10.1016/0006-8993(90)91060-t. [DOI] [PubMed] [Google Scholar]
- Dohmen C, et al. Spreading depolarizations occur in human ischemic stroke with high incidence. Ann Neurol. 2008;63:720–728. doi: 10.1002/ana.21390. [DOI] [PubMed] [Google Scholar]
- Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med. 2011;17:439–447. doi: 10.1038/nm.2333. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Reiffurth C. The stroke-migraine depolarization continuum. Neuron. 2015;86:902–922. doi: 10.1016/j.neuron.2015.04.004. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Reiffurth C. Exploitation of the spreading depolarization-induced cytotoxic edema for high-resolution, 3D mapping of its heterogeneous propagation paths. Proc Natl Acad Sci U S A. 2017;114:2112–2114. doi: 10.1073/pnas.1700760114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier JP, et al. Nitric oxide scavenging by hemoglobin or nitric oxide synthase inhibition by N-nitro-L-arginine induces cortical spreading ischemia when K+ is increased in the subarachnoid space. J Cereb Blood Flow Metab. 1998;18:978–990. doi: 10.1097/00004647-199809000-00007. [DOI] [PubMed] [Google Scholar]
- Dreier JP, et al. Products of hemolysis in the subarachnoid space inducing spreading ischemia in the cortex and focal necrosis in rats: a model for delayed ischemic neurological deficits after subarachnoid hemorrhage? J Neurosurg. 2000;93:658–666. doi: 10.3171/jns.2000.93.4.0658. [DOI] [PubMed] [Google Scholar]
- Dreier JP, et al. Ischaemia triggered by spreading neuronal activation is inhibited by vasodilators in rats. J Physiol. 2001;531:515–526. doi: 10.1111/j.1469-7793.2001.0515i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier JP, Sakowitz OW, Unterberg AW, Benndorf G, Einhaupl KM, Valdueza JM. Migrainous aura starting several minutes after the onset of subarachnoid hemorrhage. Neurology. 2001;57:1344–1345. doi: 10.1212/wnl.57.7.1344. [DOI] [PubMed] [Google Scholar]
- Dreier JP, et al. Endothelin-1 potently induces Leao’s cortical spreading depression in vivo in the rat: a model for an endothelial trigger of migrainous aura? Brain. 2002;125:102–112. doi: 10.1093/brain/awf007. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Sakowitz OW, Harder A, Zimmer C, Dirnagl U, Valdueza JM, Unterberg AW. Focal laminar cortical MR signal abnormalities after subarachnoid hemorrhage. Ann Neurol. 2002;52:825–829. doi: 10.1002/ana.10383. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Windmuller O, Petzold G, Lindauer U, Einhaupl KM, Dirnagl U. Ischemia caused by inverse coupling between neuronal activation and cerebral blood flow in rats. In: Tomita M, Kanno I, Hamel E, editors. Brain activation and CBF control. Amsterdam: Elsevier; 2002. pp. 487–492. [Google Scholar]
- Dreier JP, Windmuller O, Petzold G, Lindauer U, Einhaupl KM, Dirnagl U. Ischemia triggered by red blood cell products in the subarachnoid space is inhibited by nimodipine administration or moderate volume expansion/hemodilution in rats. Neurosurgery. 2002;51:1457–1465. [PubMed] [Google Scholar]
- Dreier JP, et al. Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain. 2006;129:3224–3237. doi: 10.1093/brain/awl297. [DOI] [PubMed] [Google Scholar]
- Dreier JP, et al. Endothelin-1-induced spreading depression in rats is associated with a microarea of selective neuronal necrosis. Exp Biol Med (Maywood) 2007;232:204–213. [PubMed] [Google Scholar]
- Dreier JP, et al. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain. 2009;132:1866–1881. doi: 10.1093/brain/awp102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier JP, et al. Spreading convulsions, spreading depolarization and epileptogenesis in human cerebral cortex. Brain. 2012;135:259–275. doi: 10.1093/brain/awr303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier JP, Isele T, Reiffurth C, Offenhauser N, Kirov SA, Dahlem MA, Herreras O. Is spreading depolarization characterized by an abrupt, massive release of gibbs free energy from the human brain cortex? Neuroscientist. 2013;19:25–42. doi: 10.1177/1073858412453340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier JP, et al. Electrochemical failure of the brain cortex is more deleterious when it is accompanied by low perfusion. Stroke. 2013;44:490–496. doi: 10.1161/STROKEAHA.112.660589. [DOI] [PubMed] [Google Scholar]
- Dreier JP, et al. How spreading depolarization can be the pathophysiological correlate of both migraine aura and stroke. Acta Neurochir Suppl. 2015;120:137–140. doi: 10.1007/978-3-319-04981-6_23. [DOI] [PubMed] [Google Scholar]
- Dreier JP, et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: review and recommendations of the COSBID research group. J Cereb Blood Flow Metab. 2017;37:1595–1625. doi: 10.1177/0271678X16654496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier JP, Lemale CL, Kola V, Friedman A, Schoknecht K. Spreading depolarization is not an epiphenomenon but the principal mechanism of the cytotoxic edema in various gray matter structures of the brain during stroke. Neuropharmacology. 2018;134:189–207. doi: 10.1016/j.neuropharm.2017.09.027. [DOI] [PubMed] [Google Scholar]
- Dreier JP, et al. Terminal spreading depolarization and electrical silence in death of human cerebral cortex. Ann Neurol. 2018;83:295–310. doi: 10.1002/ana.25147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier JP, et al. Correlates of spreading depolarization, spreading depression, and negative ultraslow potential in epidural versus subdural electrocorticography. Front Neurosci. 2019;13:373. doi: 10.3389/fnins.2019.00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenckhahn C, et al. Correlates of spreading depolarization in human scalp electroencephalography. Brain. 2012;135:853–868. doi: 10.1093/brain/aws010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenckhahn C, et al. Complications in aneurysmal subarachnoid hemorrhage patients with and without subdural electrode strip for electrocorticography. J Clin Neurophysiol. 2016;33:250–259. doi: 10.1097/WNP.0000000000000274. [DOI] [PubMed] [Google Scholar]
- Ellis MJ, Cordingley D, Girardin R, Ritchie L, Johnston J. Migraine with aura or sports-related concussion: case report, pathophysiology, and multidisciplinary approach to management. Curr Sports Med Rep. 2017;16:14–18. doi: 10.1249/JSR.0000000000000323. [DOI] [PubMed] [Google Scholar]
- Enger R, et al. Dynamics of ionic shifts in cortical spreading depression. Cereb Cortex. 2015;25:4469–4476. doi: 10.1093/cercor/bhv054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen N, et al. Early focal brain injury after subarachnoid hemorrhage correlates with spreading depolarizations. Neurology. 2019;92:e326–e341. doi: 10.1212/WNL.0000000000006814. [DOI] [PubMed] [Google Scholar]
- Evans RW, Davidoff RA. Subarachnoid hemorrhage or migraine? Headache. 2001;41:99–101. doi: 10.1046/j.1526-4610.2001.111006099.x. [DOI] [PubMed] [Google Scholar]
- Evans Randolph W., Timm Josefine S., Baskin David S. A Left Frontal Secretory Meningioma Can Mimic Transformed Migraine With and Without Aura. Headache: The Journal of Head and Face Pain. 2015;55(6):849–852. doi: 10.1111/head.12580. [DOI] [PubMed] [Google Scholar]
- Fabricius M, Jensen LH, Lauritzen M. Microdialysis of interstitial amino acids during spreading depression and anoxic depolarization in rat neocortex. Brain Res. 1993;612:61–69. doi: 10.1016/0006-8993(93)91644-8. [DOI] [PubMed] [Google Scholar]
- Fabricius M, Fuhr S, Bhatia R, Boutelle M, Hashemi P, Strong AJ, Lauritzen M. Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain. 2006;129:778–790. doi: 10.1093/brain/awh716. [DOI] [PubMed] [Google Scholar]
- Fabricius M, et al. Association of seizures with cortical spreading depression and peri-infarct depolarisations in the acutely injured human brain. Clin Neurophysiol. 2008;119:1973–1984. doi: 10.1016/j.clinph.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas E, Bari F, Obrenovitch TP. Multi-modal imaging of anoxic depolarization and hemodynamic changes induced by cardiac arrest in the rat cerebral cortex. Neuroimage. 2010;51:734–742. doi: 10.1016/j.neuroimage.2010.02.055. [DOI] [PubMed] [Google Scholar]
- Feigin VL, Lawes CM, Bennett DA, Anderson CS. Stroke epidemiology: a review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. Lancet Neurol. 2003;2:43–53. doi: 10.1016/s1474-4422(03)00266-7. [DOI] [PubMed] [Google Scholar]
- Fernandes de Lima VM, Goldermann M, Hanke WR. Calcium waves in gray matter are due to voltage-sensitive glial membrane channels. Brain Res. 1994;663:77–83. doi: 10.1016/0006-8993(94)90464-2. [DOI] [PubMed] [Google Scholar]
- Feuerstein D, et al. Dynamic metabolic response to multiple spreading depolarizations in patients with acute brain injury: an online microdialysis study. J Cereb Blood Flow Metab. 2010;30:1343–1355. doi: 10.1038/jcbfm.2010.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerstein D, et al. Detecting tissue deterioration after brain injury: regional blood flow level versus capacity to raise blood flow. J Cereb Blood Flow Metab. 2014;34:1117–1127. doi: 10.1038/jcbfm.2014.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiehler J, et al. Predictors of apparent diffusion coefficient normalization in stroke patients. Stroke. 2004;35:514–519. doi: 10.1161/01.STR.0000114873.28023.C2. [DOI] [PubMed] [Google Scholar]
- Fifkova E, Bures J, Koshtoyants OK, Krivanek J, Weiss T. Leao’s spreading depression in the cerebellum of rat. Experientia. 1961;17:572–573. doi: 10.1007/BF02156433. [DOI] [PubMed] [Google Scholar]
- Filiz A, Tepe N, Eftekhari S, Boran HE, Dilekoz E, Edvinsson L, Bolay H. CGRP receptor antagonist MK-8825 attenuates cortical spreading depression induced pain behavior. Cephalalgia. 2019;39:354–365. doi: 10.1177/0333102417735845. [DOI] [PubMed] [Google Scholar]
- Fordsmann JC, et al. Increased 20-HETE synthesis explains reduced cerebral blood flow but not impaired neurovascular coupling after cortical spreading depression in rat cerebral cortex. J Neurosci. 2013;33:2562–2570. doi: 10.1523/JNEUROSCI.2308-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox MD, Raichle ME. Spontaneous fluctuations in brain activity observed with functional magnetic resonance imaging. Nat Rev Neurosci. 2007;8:700–711. doi: 10.1038/nrn2201. [DOI] [PubMed] [Google Scholar]
- Gill R, Andine P, Hillered L, Persson L, Hagberg H. The effect of MK-801 on cortical spreading depression in the penumbral zone following focal ischaemia in the rat. J Cereb Blood Flow Metab. 1992;12:371–379. doi: 10.1038/jcbfm.1992.54. [DOI] [PubMed] [Google Scholar]
- Gold L, Lauritzen M. Neuronal deactivation explains decreased cerebellar blood flow in response to focal cerebral ischemia or suppressed neocortical function. Proc Natl Acad Sci U S A. 2002;99:7699–7704. doi: 10.1073/pnas.112012499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldensohn ES, Schoenfeld RL, Hoefer PF. The slowly changing voltage of the brain and the electrocorticogram Electroencephalogr. Clin Neurophysiol. 1951;3:231–236. doi: 10.1016/0013-4694(51)90016-8. [DOI] [PubMed] [Google Scholar]
- Goldring S, O’Leary JL. Experimentally derived correlates between ECG and steady cortical potential. J Neurophysiol. 1951;14:275–288. doi: 10.1152/jn.1951.14.4.275. [DOI] [PubMed] [Google Scholar]
- Goldring S, Ulett G, O’Leary J, Greditzer A. Initial survey of slow potential changes obtained under resting conditions and incident to convulsive therapy. Electroencephalogr Clin Neurophysiol. 1950;2:297–308. doi: 10.1016/0013-4694(50)90061-7. [DOI] [PubMed] [Google Scholar]
- Gouras P. Spreading depression of activity in amphibian retina. Am J Physiol. 1958;195:28–32. doi: 10.1152/ajplegacy.1958.195.1.28. [DOI] [PubMed] [Google Scholar]
- Grafstein B. Mechanism of spreading cortical depression. J Neurophysiol. 1956;19:154–171. doi: 10.1152/jn.1956.19.2.154. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Charidimou A. Diagnosis of cerebral amyloid angiopathy: evolution of the Boston Criteria. Stroke. 2018;49:491–497. doi: 10.1161/STROKEAHA.117.016990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hablitz JJ, Heinemann U. Alterations in the microenvironment during spreading depression associated with epileptiform activity in the immature neocortex. Brain Res Dev Brain Res. 1989;46:243–252. doi: 10.1016/0165-3806(89)90288-5. [DOI] [PubMed] [Google Scholar]
- Hadjikhani N, et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A. 2001;98:4687–4692. doi: 10.1073/pnas.071582498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haglund MM, Schwartzkroin PA. Role of Na-K pump potassium regulation and IPSPs in seizures and spreading depression in immature rabbit hippocampal slices. J Neurophysiol. 1990;63:225–239. doi: 10.1152/jn.1990.63.2.225. [DOI] [PubMed] [Google Scholar]
- Hansen AJ, Zeuthen T. Extracellular ion concentrations during spreading depression and ischemia in the rat brain cortex. Acta Physiol Scand. 1981;113:437–445. doi: 10.1111/j.1748-1716.1981.tb06920.x. [DOI] [PubMed] [Google Scholar]
- Hartings JA, et al. Spreading depolarisations and outcome after traumatic brain injury: a prospective observational study. Lancet Neurol. 2011;10:1058–1064. doi: 10.1016/S1474-4422(11)70243-5. [DOI] [PubMed] [Google Scholar]
- Hartings JA, et al. Spreading depolarizations have prolonged direct current shifts and are associated with poor outcome in brain trauma. Brain. 2011;134:1529–1540. doi: 10.1093/brain/awr048. [DOI] [PubMed] [Google Scholar]
- Hartings JA, et al. The continuum of spreading depolarizations in acute cortical lesion development: examining Leao’s legacy. J Cereb Blood Flow Metab. 2017;37:1571–1594. doi: 10.1177/0271678X16654495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartings JA, et al. Subarachnoid blood acutely induces spreading depolarizations and early cortical infarction. Brain. 2017;140:2673–2690. doi: 10.1093/brain/awx214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartings Jed A., Ngwenya Laura B., Carroll Christopher P., Foreman Brandon. Letter to the Editor. Ketamine sedation for the suppression of spreading depolarizations. Journal of Neurosurgery. 2019;130(6):2095–2097. doi: 10.3171/2018.6.JNS18235. [DOI] [PubMed] [Google Scholar]
- Hashemi P, Bhatia R, Nakamura H, Dreier JP, Graf R, Strong AJ, Boutelle MG. Persisting depletion of brain glucose following cortical spreading depression, despite apparent hyperaemia: evidence for risk of an adverse effect of Leao’s spreading depression. J Cereb Blood Flow Metab. 2009;29:166–175. doi: 10.1038/jcbfm.2008.108. [DOI] [PubMed] [Google Scholar]
- Heiss WD, Rosner G. Functional recovery of cortical neurons as related to degree and duration of ischemia. Ann Neurol. 1983;14:294–301. doi: 10.1002/ana.410140307. [DOI] [PubMed] [Google Scholar]
- Helbok R, et al. Spreading depolarizations in patients with spontaneous intracerebral hemorrhage: association with perihematomal edema progression. J Cereb Blood Flow Metab. 2017;37:1871–1882. doi: 10.1177/0271678X16651269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Caceres J, Macias-Gonzalez R, Brozek G, Bures J. Systemic ketamine blocks cortical spreading depression but does not delay the onset of terminal anoxic depolarization in rats. Brain Res. 1987;437:360–364. doi: 10.1016/0006-8993(87)91652-0. [DOI] [PubMed] [Google Scholar]
- Herreras O, Somjen GG. Analysis of potential shifts associated with recurrent spreading depression and prolonged unstable spreading depression induced by microdialysis of elevated K+ in hippocampus of anesthetized rats. Brain Res. 1993;610:283–294. doi: 10.1016/0006-8993(93)91412-l. [DOI] [PubMed] [Google Scholar]
- Hertelendy P, Varga DP, Menyhart A, Bari F, Farkas E. Susceptibility of the cerebral cortex to spreading depolarization in neurological disease states: the impact of aging. Neurochem Int. 2019;127:125–136. doi: 10.1016/j.neuint.2018.10.010. [DOI] [PubMed] [Google Scholar]
- Hertle DN, et al. Effect of analgesics and sedatives on the occurrence of spreading depolarizations accompanying acute brain injury. Brain. 2012;135:2390–2398. doi: 10.1093/brain/aws152. [DOI] [PubMed] [Google Scholar]
- Higuchi T, Takeda Y, Hashimoto M, Nagano O, Hirakawa M. Dynamic changes in cortical NADH fluorescence and direct current potential in rat focal ischemia: relationship between propagation of recurrent depolarization and growth of the ischemic core. J Cereb Blood Flow Metab. 2002;22:71–79. doi: 10.1097/00004647-200201000-00009. [DOI] [PubMed] [Google Scholar]
- Hinzman JM, et al. Inverse neurovascular coupling to cortical spreading depolarizations in severe brain trauma. Brain. 2014;137:2960–2972. doi: 10.1093/brain/awu241. [DOI] [PubMed] [Google Scholar]
- Hinzman JM, DiNapoli VA, Mahoney EJ, Gerhardt GA, Hartings JA. Spreading depolarizations mediate excitotoxicity in the development of acute cortical lesions. Exp Neurol. 2015;267:243–253. doi: 10.1016/j.expneurol.2015.03.014. [DOI] [PubMed] [Google Scholar]
- Hochachka PW, Buck LT, Doll CJ, Land SC. Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci U S A. 1996;93:9493–9498. doi: 10.1073/pnas.93.18.9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hop JW, Rinkel GJ, Algra A, van Gijn J. Case-fatality rates and functional outcome after subarachnoid hemorrhage: a systematic review. Stroke. 1997;28:660–664. doi: 10.1161/01.str.28.3.660. [DOI] [PubMed] [Google Scholar]
- Hukkelhoven CW, et al. Patient age and outcome following severe traumatic brain injury: an analysis of 5600 patients. J Neurosurg. 2003;99:666–673. doi: 10.3171/jns.2003.99.4.0666. [DOI] [PubMed] [Google Scholar]
- Iijima T, Mies G, Hossmann KA. Repeated negative DC deflections in rat cortex following middle cerebral artery occlusion are abolished by MK-801: effect on volume of ischemic injury. J Cereb Blood Flow Metab. 1992;12:727–733. doi: 10.1038/jcbfm.1992.103. [DOI] [PubMed] [Google Scholar]
- Izenberg A, Aviv RI, Demaerschalk BM, Dodick DW, Hopyan J, Black SE, Gladstone DJ. Crescendo transient aura attacks: a transient ischemic attack mimic caused by focal subarachnoid hemorrhage. Stroke. 2009;40:3725–3729. doi: 10.1161/STROKEAHA.109.557009. [DOI] [PubMed] [Google Scholar]
- Jander S, Schroeter M, Peters O, Witte OW, Stoll G. Cortical spreading depression induces proinflammatory cytokine gene expression in the rat brain. J Cereb Blood Flow Metab. 2001;21:218–225. doi: 10.1097/00004647-200103000-00005. [DOI] [PubMed] [Google Scholar]
- Jarvis CR, Anderson TR, Andrew RD. Anoxic depolarization mediates acute damage independent of glutamate in neocortical brain slices. Cereb Cortex. 2001;11:249–259. doi: 10.1093/cercor/11.3.249. [DOI] [PubMed] [Google Scholar]
- Jeon JP, et al. Long-term treatment outcome of venous-predominant arteriovenous malformation. J Neurosurg. 2016;124:1100–1106. doi: 10.3171/2015.4.JNS142475. [DOI] [PubMed] [Google Scholar]
- Kammersgaard LP, Jorgensen HS, Reith J, Nakayama H, Pedersen PM, Olsen TS, Copenhagen Stroke S. Short- and long-term prognosis for very old stroke patients. The Copenhagen Stroke Study. Age Ageing. 2004;33:149–154. doi: 10.1093/ageing/afh052. [DOI] [PubMed] [Google Scholar]
- Kang EJ, Major S, Jorks D, Reiffurth C, Offenhauser N, Friedman A, Dreier JP. Blood-brain barrier opening to large molecules does not imply blood-brain barrier opening to small ions. Neurobiol Dis. 2013;52:204–218. doi: 10.1016/j.nbd.2012.12.007. [DOI] [PubMed] [Google Scholar]
- Kemp JA, McKernan RM. NMDA receptor pathways as drug targets. Nat Neurosci. 2002;5(Suppl):1039–1042. doi: 10.1038/nn936. [DOI] [PubMed] [Google Scholar]
- Kempinsky WH. Steady potential gradients in experimental cerebral vascular occlusion. Electroencephalogr Clin Neurophysiol. 1954;6:375–388. doi: 10.1016/0013-4694(54)90052-8. [DOI] [PubMed] [Google Scholar]
- Klass A, Sanchez-Porras R, Santos E. Systematic review of the pharmacological agents that have been tested against spreading depolarizations. J Cereb Blood Flow Metab. 2018;38:1149–1179. doi: 10.1177/0271678X18771440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klatzo I. Presidental address. Neuropathological aspects of brain edema. J Neuropathol Exp Neurol. 1967;26:1–14. doi: 10.1097/00005072-196701000-00001. [DOI] [PubMed] [Google Scholar]
- Klatzo I. Pathophysiological aspects of brain edema. Acta Neuropathol. 1987;72:236–239. doi: 10.1007/BF00691095. [DOI] [PubMed] [Google Scholar]
- Kleeberg J, Petzold GC, Major S, Dirnagl U, Dreier JP. ET-1 induces cortical spreading depression via activation of the ETA receptor/phospholipase C pathway in vivo. Am J Physiol Heart Circ Physiol. 2004;286:H1339–H1346. doi: 10.1152/ajpheart.00227.2003. [DOI] [PubMed] [Google Scholar]
- Kleinig TJ, Kiley M, Thompson PD. Acute convexity subarachnoid haemorrhage: a cause of aura-like symptoms in the elderly. Cephalalgia. 2008;28:658–663. doi: 10.1111/j.1468-2982.2008.01570.x. [DOI] [PubMed] [Google Scholar]
- Kleiven S, Peloso PM, von Holst H. The epidemiology of head injuries in Sweden from 1987 to 2000. Inj Control Saf Promot. 2003;10:173–180. doi: 10.1076/icsp.10.3.173.14552. [DOI] [PubMed] [Google Scholar]
- Klingebiel R, Friedman A, Shelef I, Dreier JP. Clearance of a status aurae migraenalis in response to thrombendarterectomy in a patient with high grade internal carotid artery stenosis. J Neurol Neurosurg Psychiatry. 2008;79:89–90. doi: 10.1136/jnnp.2007.119230. [DOI] [PubMed] [Google Scholar]
- Koroleva VI, Korolev OS, Loseva E, Bures J. The effect of MK-801 and of brain-derived polypeptides on the development of ischemic lesion induced by photothrombotic occlusion of the distal middle cerebral artery in rats. Brain Res. 1998;786:104–114. doi: 10.1016/s0006-8993(97)01448-0. [DOI] [PubMed] [Google Scholar]
- Koskinen S, Alaranta H. Traumatic brain injury in Finland 1991-2005: a nationwide register study of hospitalized and fatal TBI. Brain Inj. 2008;22:205–214. doi: 10.1080/02699050801938975. [DOI] [PubMed] [Google Scholar]
- Kraig RP, Nicholson C. Extracellular ionic variations during spreading depression. Neuroscience. 1978;3:1045–1059. doi: 10.1016/0306-4522(78)90122-7. [DOI] [PubMed] [Google Scholar]
- Kraig RP, Ferreira-Filho CR, Nicholson C. Alkaline and acid transients in cerebellar microenvironment. J Neurophysiol. 1983;49:831–850. doi: 10.1152/jn.1983.49.3.831. [DOI] [PubMed] [Google Scholar]
- Kunkler PE, Kraig RP. Calcium waves precede electrophysiological changes of spreading depression in hippocampal organ cultures. J Neurosci. 1998;18:3416–3425. doi: 10.1523/JNEUROSCI.18-09-03416.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkler PE, Hulse RE, Kraig RP. Multiplexed cytokine protein expression profiles from spreading depression in hippocampal organotypic cultures. J Cereb Blood Flow Metab. 2004;24:829–839. doi: 10.1097/01.WCB.0000126566.34753.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurth T, Chabriat H, Bousser MG. Migraine and stroke: a complex association with clinical implications. Lancet Neurol. 2012;11:92–100. doi: 10.1016/S1474-4422(11)70266-6. [DOI] [PubMed] [Google Scholar]
- LaManna JC, Rosenthal M. Effect of ouabain and phenobarbital on oxidative metabolic activity associated with spreading cortical depression in cats. Brain Res. 1975;88:145–149. doi: 10.1016/0006-8993(75)90963-4. [DOI] [PubMed] [Google Scholar]
- Lashley KS. Patterns of cerebral integration indicated by the scotomas of migraine. Arch Neurol Psychiatry. 1941;46:331–339. [Google Scholar]
- Lassen NA, Friberg L. Cerebral blood flow measured by xenon 133 using the intraarterial injection method or inhalation combined with SPECT in migraine research. In: Olesen J, editor. Migraine and other headaches. The vascular mechanisms. New York: Raven Press; 1991. pp. 5–13. [Google Scholar]
- Lauritzen M. Pathophysiology of the migraine aura. The spreading depression theory. Brain. 1994;117(Pt 1):199–210. doi: 10.1093/brain/117.1.199. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Hansen AJ. The effect of glutamate receptor blockade on anoxic depolarization and cortical spreading depression. J Cereb Blood Flow Metab. 1992;12:223–229. doi: 10.1038/jcbfm.1992.32. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Skyhoj Olsen T, Lassen NA, Paulson OB. Changes in regional cerebral blood flow during the course of classic migraine attacks. Ann Neurol. 1983;13:633–641. doi: 10.1002/ana.410130609. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Rice ME, Okada Y, Nicholson C. Quisqualate, kainate and NMDA can initiate spreading depression in the turtle cerebellum. Brain Res. 1988;475:317–327. doi: 10.1016/0006-8993(88)90620-8. [DOI] [PubMed] [Google Scholar]
- Lauritzen M, Dreier JP, Fabricius M, Hartings JA, Graf R, Strong AJ. Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab. 2011;31:17–35. doi: 10.1038/jcbfm.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leão AAP. Further observations on the spreading depression of activity in the cerebral cortex. J Neurophysiol. 1947;10:409–414. doi: 10.1152/jn.1947.10.6.409. [DOI] [PubMed] [Google Scholar]
- Leão AAP, Morison RS. Propagation of spreading cortical depression. J Neurophysiol. 1945;8:33–45. [Google Scholar]
- Lehmenkuhler A (1990) [Spreading depression—cortical reactions: disorders of the extracellular microenvironment] EEG-EMG Z Elektroenzephalographie. Elektromyogr Verwandte Geb 21:1–6 [PubMed]
- Lehmenkuhler A, Sykova E, Svoboda J, Zilles K, Nicholson C. Extracellular space parameters in the rat neocortex and subcortical white matter during postnatal development determined by diffusion analysis. Neuroscience. 1993;55:339–351. doi: 10.1016/0306-4522(93)90503-8. [DOI] [PubMed] [Google Scholar]
- Lublinsky S, et al. Early blood-brain barrier dysfunction predicts neurological outcome following aneurysmal subarachnoid hemorrhage. EBioMedicine. 2019;43:460–472. doi: 10.1016/j.ebiom.2019.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckl J, et al. The negative ultraslow potential, electrophysiological correlate of infarction in the human cortex. Brain. 2018;141:1734–1752. doi: 10.1093/brain/awy102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas AIR, et al. Traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017;16:987–1048. doi: 10.1016/S1474-4422(17)30371-X. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Schweizer TA. Spontaneous subarachnoid haemorrhage. Lancet. 2017;389:655–666. doi: 10.1016/S0140-6736(16)30668-7. [DOI] [PubMed] [Google Scholar]
- Madry C, Haglerod C, Attwell D. The role of pannexin hemichannels in the anoxic depolarization of hippocampal pyramidal cells. Brain. 2010;133:3755–3763. doi: 10.1093/brain/awq284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major S, et al. A role of the sodium pump in spreading ischemia in rats. J Cereb Blood Flow Metab. 2017;37:1687–1705. doi: 10.1177/0271678X16639059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova J, Makarov VA, Herreras O. Generation of sustained field potentials by gradients of polarization within single neurons: a macroscopic model of spreading depression. J Neurophysiol. 2010;103:2446–2457. doi: 10.1152/jn.01045.2009. [DOI] [PubMed] [Google Scholar]
- Mane M, Muller M. Temporo-spectral imaging of intrinsic optical signals during hypoxia-induced spreading depression-like depolarization. PLoS One. 2012;7:e43981. doi: 10.1371/journal.pone.0043981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrannes R, Willems R, De Prins E, Wauquier A. Evidence for a role of the N-methyl-D-aspartate (NMDA) receptor in cortical spreading depression in the rat. Brain Res. 1988;457:226–240. doi: 10.1016/0006-8993(88)90690-7. [DOI] [PubMed] [Google Scholar]
- Marshall WH. Spreading cortical depression of Leao. Physiol Rev. 1959;39:239–279. doi: 10.1152/physrev.1959.39.2.239. [DOI] [PubMed] [Google Scholar]
- Martins-Ferreira H, de Castro GO. Light-scattering changes accompanying spreading depression in isolated retina. J Neurophysiol. 1966;29:715–726. doi: 10.1152/jn.1966.29.4.715. [DOI] [PubMed] [Google Scholar]
- Martins-Ferreira H, De Oliveira CG, Struchiner CJ, Rodrigues PS. Circling spreading depression in isolated chick retina. J Neurophysiol. 1974;37:773–784. doi: 10.1152/jn.1974.37.4.773. [DOI] [PubMed] [Google Scholar]
- Maslarova A, Alam M, Reiffurth C, Lapilover E, Gorji A, Dreier JP. Chronically epileptic human and rat neocortex display a similar resistance against spreading depolarization in vitro. Stroke. 2011;42:2917–2922. doi: 10.1161/STROKEAHA.111.621581. [DOI] [PubMed] [Google Scholar]
- Mateo C, Knutsen PM, Tsai PS, Shih AY, Kleinfeld D. Entrainment of arteriole vasomotor fluctuations by neural activity is a basis of blood-oxygenation-level-dependent “resting-state” connectivity. Neuron. 2017;96:936-948 e933. doi: 10.1016/j.neuron.2017.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima K, Hogan MJ, Hakim AM. Cortical spreading depression protects against subsequent focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1996;16:221–226. doi: 10.1097/00004647-199603000-00006. [DOI] [PubMed] [Google Scholar]
- Mayevsky A, Doron A, Manor T, Meilin S, Zarchin N, Ouaknine GE. Cortical spreading depression recorded from the human brain using a multiparametric monitoring system. Brain Res. 1996;740:268–274. doi: 10.1016/s0006-8993(96)00874-8. [DOI] [PubMed] [Google Scholar]
- Mazel T, Richter F, Vargova L, Sykova E. Changes in extracellular space volume and geometry induced by cortical spreading depression in immature and adult rats. Physiol Res. 2002;51(Suppl 1):S85–S93. [PubMed] [Google Scholar]
- Menyhart A, Makra P, Szepes BE, Toth OM, Hertelendy P, Bari F, Farkas E. High incidence of adverse cerebral blood flow responses to spreading depolarization in the aged ischemic rat brain. Neurobiol Aging. 2015;36:3269–3277. doi: 10.1016/j.neurobiolaging.2015.08.014. [DOI] [PubMed] [Google Scholar]
- Menyhart A, et al. Spreading depolarization remarkably exacerbates ischemia-induced tissue acidosis in the young and aged rat brain. Sci Rep. 2017;7:1154. doi: 10.1038/s41598-017-01284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menyhart A, et al. Large-conductance Ca(2+)-activated potassium channels are potently involved in the inverse neurovascular response to spreading depolarization. Neurobiol Dis. 2018;119:41–52. doi: 10.1016/j.nbd.2018.07.026. [DOI] [PubMed] [Google Scholar]
- Mies G, Paschen W. Regional changes of blood flow, glucose, and ATP content determined on brain sections during a single passage of spreading depression in rat brain cortex. Exp Neurol. 1984;84:249–258. doi: 10.1016/0014-4886(84)90222-x. [DOI] [PubMed] [Google Scholar]
- Milakara D, et al. Simulation of spreading depolarization trajectories in cerebral cortex: correlation of velocity and susceptibility in patients with aneurysmal subarachnoid hemorrhage. NeuroImage Clinical. 2017;16:524–538. doi: 10.1016/j.nicl.2017.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir KW, Lees KR (2003) Excitatory amino acid antagonists for acute stroke Cochrane Database Syst Rev CD001244. 10.1002/14651858.CD001244 [DOI] [PMC free article] [PubMed]
- Muller M, Somjen GG. Inhibition of major cationic inward currents prevents spreading depression-like hypoxic depolarization in rat hippocampal tissue slices. Brain Res. 1998;812:1–13. doi: 10.1016/s0006-8993(98)00812-9. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Li P, Betts K, Liu R. Two-photon imaging of stroke onset in vivo reveals that NMDA-receptor independent ischemic depolarization is the major cause of rapid reversible damage to dendrites and spines. J Neurosci. 2008;28:1756–1772. doi: 10.1523/JNEUROSCI.5128-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CJ, et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2197–2223. doi: 10.1016/S0140-6736(12)61689-4. [DOI] [PubMed] [Google Scholar]
- Mushkudiani NA, et al. Prognostic value of demographic characteristics in traumatic brain injury: results from the IMPACT study. J Neurotrauma. 2007;24:259–269. doi: 10.1089/neu.2006.0028. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Hansen AJ. Spreading depression is not associated with neuronal injury in the normal brain. Brain Res. 1988;449:395–398. doi: 10.1016/0006-8993(88)91062-1. [DOI] [PubMed] [Google Scholar]
- Nicholson C, Bruggencate GT, Steinberg R, Stockle H. Calcium modulation in brain extracellular microenvironment demonstrated with ion-selective micropipette. Proc Natl Acad Sci U S A. 1977;74:1287–1290. doi: 10.1073/pnas.74.3.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozari A, et al. Microemboli may link spreading depression, migraine aura, and patent foramen ovale. Ann Neurol. 2010;67:221–229. doi: 10.1002/ana.21871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oitzl MS, Huston JP. Electroencephalographic spreading depression and concomitant behavioral changes induced by intrahippocampal injections of ACTH1-24 and D-Ala2-Met-enkephalinamide in the rat. Brain Res. 1984;308:33–42. doi: 10.1016/0006-8993(84)90914-4. [DOI] [PubMed] [Google Scholar]
- Olesen J, Larsen B, Lauritzen M. Focal hyperemia followed by spreading oligemia and impaired activation of rCBF in classic migraine. Ann Neurol. 1981;9:344–352. doi: 10.1002/ana.410090406. [DOI] [PubMed] [Google Scholar]
- Olesen J, Friberg L, Olsen TS, Andersen AR, Lassen NA, Hansen PE, Karle A. Ischaemia-induced (symptomatic) migraine attacks may be more frequent than migraine-induced ischaemic insults. Brain. 1993;116(Pt 1):187–202. doi: 10.1093/brain/116.1.187. [DOI] [PubMed] [Google Scholar]
- Oliveira-Ferreira AI, et al. Experimental and preliminary clinical evidence of an ischemic zone with prolonged negative DC shifts surrounded by a normally perfused tissue belt with persistent electrocorticographic depression. J Cereb Blood Flow Metab. 2010;30:1504–1519. doi: 10.1038/jcbfm.2010.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira-Ferreira AI, Major S, Przesdzing I, Kang EJ, Dreier JP (2019) Spreading depolarizations in the rat endothelin-1 model of focal cerebellar ischemia J Cereb Blood Flow Metab in press [DOI] [PMC free article] [PubMed]
- Ovbiagele B, et al. Forecasting the future of stroke in the United States: a policy statement from the American Heart Association and American Stroke Association. Stroke. 2013;44:2361–2375. doi: 10.1161/STR.0b013e31829734f2. [DOI] [PubMed] [Google Scholar]
- Ozyurt E, Graham DI, Woodruff GN, McCulloch J. Protective effect of the glutamate antagonist, MK-801 in focal cerebral ischemia in the cat. J Cereb Blood Flow Metab. 1988;8:138–143. doi: 10.1038/jcbfm.1988.18. [DOI] [PubMed] [Google Scholar]
- Pana R, Hornby L, Shemie SD, Dhanani S, Teitelbaum J. Time to loss of brain function and activity during circulatory arrest. J Crit Care. 2016;34:77–83. doi: 10.1016/j.jcrc.2016.04.001. [DOI] [PubMed] [Google Scholar]
- Park CK, Nehls DG, Graham DI, Teasdale GM, McCulloch J. Focal cerebral ischaemia in the cat: treatment with the glutamate antagonist MK-801 after induction of ischaemia. J Cereb Blood Flow Metab. 1988;8:757–762. doi: 10.1038/jcbfm.1988.124. [DOI] [PubMed] [Google Scholar]
- Park CK, Nehls DG, Graham DI, Teasdale GM, McCulloch J. The glutamate antagonist MK-801 reduces focal ischemic brain damage in the rat. Ann Neurol. 1988;24:543–551. doi: 10.1002/ana.410240411. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Tao L, Nicholson C. Extracellular potassium, volume fraction, and tortuosity in rat hippocampal CA1, CA3, and cortical slices during ischemia. J Neurophysiol. 1995;74:565–573. doi: 10.1152/jn.1995.74.2.565. [DOI] [PubMed] [Google Scholar]
- Petzold GC, Einhaupl KM, Dirnagl U, Dreier JP. Ischemia triggered by spreading neuronal activation is induced by endothelin-1 and hemoglobin in the subarachnoid space. Ann Neurol. 2003;54:591–598. doi: 10.1002/ana.10723. [DOI] [PubMed] [Google Scholar]
- Petzold GC, et al. Increased extracellular K+ concentration reduces the efficacy of N-methyl-D-aspartate receptor antagonists to block spreading depression-like depolarizations and spreading ischemia. Stroke. 2005;36:1270–1277. doi: 10.1161/01.STR.0000166023.51307.e0. [DOI] [PubMed] [Google Scholar]
- Petzold GC, et al. Nitric oxide modulates spreading depolarization threshold in the human and rodent cortex. Stroke. 2008;39:1292–1299. doi: 10.1161/STROKEAHA.107.500710. [DOI] [PubMed] [Google Scholar]
- Piilgaard H, Lauritzen M. Persistent increase in oxygen consumption and impaired neurovascular coupling after spreading depression in rat neocortex. J Cereb Blood Flow Metab. 2009;29:1517–1527. doi: 10.1038/jcbfm.2009.73. [DOI] [PubMed] [Google Scholar]
- Pinczolits A, et al. Standard-sampling microdialysis and spreading depolarizations in patients with malignant hemispheric stroke. J Cereb Blood Flow Metab. 2017;37:1896–1905. doi: 10.1177/0271678X17699629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohjasvaara T, Erkinjuntti T, Vataja R, Kaste M. Comparison of stroke features and disability in daily life in patients with ischemic stroke aged 55 to 70 and 71 to 85 years. Stroke. 1997;28:729–735. doi: 10.1161/01.str.28.4.729. [DOI] [PubMed] [Google Scholar]
- Pomper JK, Haack S, Petzold GC, Buchheim K, Gabriel S, Hoffmann U, Heinemann U. Repetitive spreading depression-like events result in cell damage in juvenile hippocampal slice cultures maintained in normoxia. J Neurophysiol. 2006;95:355–368. doi: 10.1152/jn.00186.2005. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA. Selective neuronal vulnerability: morphological and molecular characteristics. Prog Brain Res. 1985;63:29–37. doi: 10.1016/S0079-6123(08)61973-1. [DOI] [PubMed] [Google Scholar]
- Rashidy-Pour A, Motaghed-Larijani Z, Bures J. Tolerance to ketamine-induced blockade of cortical spreading depression transfers to MK-801 but not to AP5 in rats. Brain Res. 1995;693:64–69. doi: 10.1016/0006-8993(95)00692-j. [DOI] [PubMed] [Google Scholar]
- Reardon PM, et al. Characteristics, outcomes, and cost patterns of high-cost patients in the intensive care unit crit. Care Res Pract. 2018;2018:5452683. doi: 10.1155/2018/5452683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiffurth C, Kirov SA, Dreier JP. Spreading depolarization. In: Chen J, Xu XM, Xu ZC, Zhang JH, editors. Animal models of acute neurological injuries II, Injury and mechanistic assessments. New York: Humana Press; 2012. pp. 339–352. [Google Scholar]
- Reiffurth C, Alam M, Zahedi-Khorasani M, Major S, Dreier JP (2019) Na+/K+-ATPase α isoform deficiency results in distinct spreading depolarization phenotypes J Cereb Blood Flow Metab in press [DOI] [PMC free article] [PubMed]
- Reinhart KM, Shuttleworth CW. Ketamine reduces deleterious consequences of spreading depolarizations. Exp Neurol. 2018;305:121–128. doi: 10.1016/j.expneurol.2018.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revah O, Lasser-Katz E, Fleidervish IA, Gutnick MJ. The earliest neuronal responses to hypoxia in the neocortical circuit are glutamate-dependent. Neurobiol Dis. 2016;95:158–167. doi: 10.1016/j.nbd.2016.07.019. [DOI] [PubMed] [Google Scholar]
- Revankar GS, et al. Spreading depolarizations and seizures in clinical subdural electrocorticographic recordings. In: Varelas PN, Claassen J, et al., editors. Seizures in Critical Care. A Guide to Diagnosis and Therapeutics. New York: Springer; 2017. pp. 77–90. [Google Scholar]
- Richter F, Eitner A, Leuchtweis J, Bauer R, Lehmenkuhler A, Schaible HG. Effects of interleukin-1ss on cortical spreading depolarization and cerebral vasculature. J Cereb Blood Flow Metab. 2017;37:1791–1802. doi: 10.1177/0271678X16641127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risher WC, Ard D, Yuan J, Kirov SA. Recurrent spontaneous spreading depolarizations facilitate acute dendritic injury in the ischemic penumbra. J Neurosci. 2010;30:9859–9868. doi: 10.1523/JNEUROSCI.1917-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson RM, Spong KE, Srithiphaphirom P. Chill coma in the locust, Locusta migratoria, is initiated by spreading depolarization in the central nervous system. Sci Rep. 2017;7:10297. doi: 10.1038/s41598-017-10586-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas JI, Zurru MC, Romano M, Patrucco L, Cristiano E. Acute ischemic stroke and transient ischemic attack in the very old risk factor profile and stroke subtype between patients older than 80 years and patients aged less than 80 years. Eur J Neurol. 2007;14:895–899. doi: 10.1111/j.1468-1331.2007.01841.x. [DOI] [PubMed] [Google Scholar]
- Rother J, de Crespigny AJ, D’Arceuil H, Mosley ME. MR detection of cortical spreading depression immediately after focal ischemia in the rat. J Cereb Blood Flow Metab. 1996;16:214–220. doi: 10.1097/00004647-199603000-00005. [DOI] [PubMed] [Google Scholar]
- Roussel S, Pinard E, Seylaz J. Effect of MK-801 on focal brain infarction in normotensive and hypertensive rats. Hypertension. 1992;19:40–46. doi: 10.1161/01.hyp.19.1.40. [DOI] [PubMed] [Google Scholar]
- Sakowitz OW, et al. Relation of cerebral energy metabolism and extracellular nitrite and nitrate concentrations in patients after aneurysmal subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2001;21:1067–1076. doi: 10.1097/00004647-200109000-00004. [DOI] [PubMed] [Google Scholar]
- Sakowitz OW, et al. Preliminary evidence that ketamine inhibits spreading depolarizations in acute human brain injury. Stroke. 2009;40:e519–e522. doi: 10.1161/STROKEAHA.109.549303. [DOI] [PubMed] [Google Scholar]
- Sakowitz OW, et al. Clusters of spreading depolarizations are associated with disturbed cerebral metabolism in patients with aneurysmal subarachnoid hemorrhage. Stroke. 2013;44:220–223. doi: 10.1161/STROKEAHA.112.672352. [DOI] [PubMed] [Google Scholar]
- Samanci B, Coban O, Baykan B. Late onset aura may herald cerebral amyloid angiopathy: a case report. Cephalalgia. 2016;36:998–1001. doi: 10.1177/0333102415620253. [DOI] [PubMed] [Google Scholar]
- Santos Edgar, Sánchez-Porras Renán, Sakowitz Oliver W, Dreier Jens P, Dahlem Markus A. Heterogeneous propagation of spreading depolarizations in the lissencephalic and gyrencephalic brain. Journal of Cerebral Blood Flow & Metabolism. 2017;37(7):2639–2643. doi: 10.1177/0271678X16689801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schade JP. Maturational aspects of EEG and of spreading depression in rabbit. J Neurophysiol. 1959;22:245–257. doi: 10.1152/jn.1959.22.3.245. [DOI] [PubMed] [Google Scholar]
- Scholl MJ, et al. Large field-of-view movement-compensated intrinsic optical signal imaging for the characterization of the haemodynamic response to spreading depolarizations in large gyrencephalic brains. J Cereb Blood Flow Metab. 2017;37:1706–1719. doi: 10.1177/0271678X16668988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman WR, Lust WD, Pundik S, Zhou Y, Ratcheson RA. Compromised metabolic recovery following spontaneous spreading depression in the penumbra. Brain Res. 2004;999:167–174. doi: 10.1016/j.brainres.2003.11.016. [DOI] [PubMed] [Google Scholar]
- Sharp FR, Lu A, Tang Y, Millhorn DE. Multiple molecular penumbras after focal cerebral ischemia. J Cereb Blood Flow Metab. 2000;20:1011–1032. doi: 10.1097/00004647-200007000-00001. [DOI] [PubMed] [Google Scholar]
- Shibata M. Unilateral cortical spreading depression in the rat: effects on feeding, drinking and other behaviors. Brain research bulletin. 1978;3:395–400. doi: 10.1016/0361-9230(78)90108-9. [DOI] [PubMed] [Google Scholar]
- Shin HK, Dunn AK, Jones PB, Boas DA, Moskowitz MA, Ayata C. Vasoconstrictive neurovascular coupling during focal ischemic depolarizations. J Cereb Blood Flow Metab. 2006;26:1018–1030. doi: 10.1038/sj.jcbfm.9600252. [DOI] [PubMed] [Google Scholar]
- Shuttleworth C. William, Andrew R. David, Akbari Yama, Ayata Cenk, Balu Ramani, Brennan K. C., Boutelle Martyn, Carlson Andrew P., Dreier Jens P., Fabricius Martin, Farkas Eszter, Foreman Brandon, Helbok Raimund, Henninger Nils, Jewell Sharon L., Jones Stephen C., Kirov Sergei A., Lindquist Britta E., Maciel Carolina B., Okonkwo David, Reinhart Katelyn M., Robertson R. Meldrum, Rosenthal Eric S., Watanabe Tomas, Hartings Jed A. Which Spreading Depolarizations Are Deleterious To Brain Tissue? Neurocritical Care. 2019;32(1):317–322. doi: 10.1007/s12028-019-00776-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev. 2001;81:1065–1096. doi: 10.1152/physrev.2001.81.3.1065. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Irreversible hypoxic (ischemic) neuron injury. In: Somjen GG, editor. Ions in the brain. New York: Oxford University Press; 2004. pp. 338–372. [Google Scholar]
- Sonn J, Mayevsky A. Effects of brain oxygenation on metabolic, hemodynamic, ionic and electrical responses to spreading depression in the rat. Brain Res. 2000;882:212–216. doi: 10.1016/s0006-8993(00)02827-4. [DOI] [PubMed] [Google Scholar]
- Spong Kristin E., Dreier Jens P., Robertson R. Meldrum. A new direction for spreading depolarization: Investigation in the fly brain. Channels. 2016;11(2):97–98. doi: 10.1080/19336950.2016.1239898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sramka M, Brozek G, Bures J, Nadvornik P. Functional ablation by spreading depression: possible use in human stereotactic neurosurgery. Appl Neurophysiol. 1977;40:48–61. [PubMed] [Google Scholar]
- Stanton Joel Elliot Dane, Chandratheva Arvind, Wilson Duncan, Hostettler Isabel Charlotte, Islam Saiful, Werring David John. Clinical features distinguish cerebral amyloid angiopathy-associated convexity subarachnoid haemorrhage from suspected TIA. Journal of Neurology. 2019;267(1):133–137. doi: 10.1007/s00415-019-09558-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffensen AB, Sword J, Croom D, Kirov SA, MacAulay N. Chloride cotransporters as a molecular mechanism underlying spreading depolarization-induced dendritic beading. J Neurosci. 2015;35:12172–12187. doi: 10.1523/JNEUROSCI.0400-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein SC, Georgoff P, Meghan S, Mizra K, Sonnad SS. 150 years of treating severe traumatic brain injury: a systematic review of progress in mortality. J Neurotrauma. 2010;27:1343–1353. doi: 10.1089/neu.2009.1206. [DOI] [PubMed] [Google Scholar]
- Stocchetti N, Paterno R, Citerio G, Beretta L, Colombo A. Traumatic brain injury in an aging population. J Neurotrauma. 2012;29:1119–1125. doi: 10.1089/neu.2011.1995. [DOI] [PubMed] [Google Scholar]
- Stocchetti N, et al. Severe traumatic brain injury: targeted management in the intensive care unit. Lancet Neurol. 2017;16:452–464. doi: 10.1016/S1474-4422(17)30118-7. [DOI] [PubMed] [Google Scholar]
- Strong AJ, et al. Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke. 2002;33:2738–2743. doi: 10.1161/01.str.0000043073.69602.09. [DOI] [PubMed] [Google Scholar]
- Strong AJ, et al. Peri-infarct depolarizations lead to loss of perfusion in ischaemic gyrencephalic cerebral cortex. Brain. 2007;130:995–1008. doi: 10.1093/brain/awl392. [DOI] [PubMed] [Google Scholar]
- Stuart RM, et al. Intracranial multimodal monitoring for acute brain injury: a single institution review of current practices. Neurocritical care. 2010;12:188–198. doi: 10.1007/s12028-010-9330-9. [DOI] [PubMed] [Google Scholar]
- Sukhotinsky I, Dilekoz E, Moskowitz MA, Ayata C. Hypoxia and hypotension transform the blood flow response to cortical spreading depression from hyperemia into hypoperfusion in the rat. J Cereb Blood Flow Metab. 2008;28:1369–1376. doi: 10.1038/jcbfm.2008.35. [DOI] [PubMed] [Google Scholar]
- Sykova E, Nicholson C. Diffusion in brain extracellular space. Physiol Rev. 2008;88:1277–1340. doi: 10.1152/physrev.00027.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano K, et al. The role of spreading depression in focal ischemia evaluated by diffusion mapping. Ann Neurol. 1996;39:308–318. doi: 10.1002/ana.410390307. [DOI] [PubMed] [Google Scholar]
- Takano T, et al. Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci. 2007;10:754–762. doi: 10.1038/nn1902. [DOI] [PubMed] [Google Scholar]
- Tanaka E, Yamamoto S, Kudo Y, Mihara S, Higashi H. Mechanisms underlying the rapid depolarization produced by deprivation of oxygen and glucose in rat hippocampal CA1 neurons in vitro. J Neurophysiol. 1997;78:891–902. doi: 10.1152/jn.1997.78.2.891. [DOI] [PubMed] [Google Scholar]
- Tasaki I, Byrne PM. Demonstration of heat production associated with spreading depression in the amphibian retina. Biochem Biophys Res Commun. 1991;174:293–297. doi: 10.1016/0006-291x(91)90519-d. [DOI] [PubMed] [Google Scholar]
- Tidow Henning, Aperia Anita, Nissen Poul. How are ion pumps and agrin signaling integrated? Trends in Biochemical Sciences. 2010;35(12):653–659. doi: 10.1016/j.tibs.2010.05.004. [DOI] [PubMed] [Google Scholar]
- Ulrich ND, Lapeyre ER, Moore RC. Hemorrhagic stroke resulting from venous malformation at 20 weeks of pregnancy. Ochsner J. 2016;16:542–544. [PMC free article] [PubMed] [Google Scholar]
- Urbach A, Bruehl C, Witte OW. Microarray-based long-term detection of genes differentially expressed after cortical spreading depression. Eur J Neurosci. 2006;24:841–856. doi: 10.1111/j.1460-9568.2006.04862.x. [DOI] [PubMed] [Google Scholar]
- Urbach A, Baum E, Braun F, Witte OW. Cortical spreading depolarization increases adult neurogenesis, and alters behavior and hippocampus-dependent memory in mice. J Cereb Blood Flow Metab. 2017;37:1776–1790. doi: 10.1177/0271678X16643736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Harreveld A. Depolarisation in the spinal cord caused by asphyxiation. Am J Physiol. 1946;147:669–684. doi: 10.1152/ajplegacy.1946.147.4.669. [DOI] [PubMed] [Google Scholar]
- van Harreveld A. Compounds in brain extracts causing spreading depression of cerebral cortical activity and contraction of crustacean muscle. J Neurochem. 1959;3:300–315. doi: 10.1111/j.1471-4159.1959.tb12636.x. [DOI] [PubMed] [Google Scholar]
- Van Harreveld A, Khattab FI. Changes in cortical extracellular space during spreading depression investigated with the electron microscope. J Neurophysiol. 1967;30:911–929. doi: 10.1152/jn.1967.30.4.911. [DOI] [PubMed] [Google Scholar]
- Von der Brelie C, Seifert M, Rot S, Tittel A, Sanft C, Meier U, Lemcke J. Sedation of patients with acute aneurysmal subarachnoid hemorrhage with ketamine is safe and might influence the occurrence of cerebral infarctions associated with delayed cerebral ischemia world. Neurosurg. 2017;97:374–382. doi: 10.1016/j.wneu.2016.09.121. [DOI] [PubMed] [Google Scholar]
- Vora YY, Suarez-Almazor M, Steinke DE, Martin ML, Findlay JM. Role of transcranial Doppler monitoring in the diagnosis of cerebral vasospasm after subarachnoid hemorrhage. Neurosurgery. 1999;44:1237–1247. [PubMed] [Google Scholar]
- Vosoughi R, Walkty A, Drebot MA, Kadkhoda K. Jamestown Canyon virus meningoencephalitis mimicking migraine with aura in a resident of Manitoba. CMAJ. 2018;190:E262–E264. doi: 10.1503/cmaj.170940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyskocil F, Kritz N, Bures J. Potassium-selective microelectrodes used for measuring the extracellular brain potassium during spreading depression and anoxic depolarization in rats. Brain Res. 1972;39:255–259. doi: 10.1016/0006-8993(72)90802-5. [DOI] [PubMed] [Google Scholar]
- Weidauer S, Vatter H, Beck J, Raabe A, Lanfermann H, Seifert V, Zanella F. Focal laminar cortical infarcts following aneurysmal subarachnoid haemorrhage. Neuroradiology. 2008;50:1–8. doi: 10.1007/s00234-007-0294-1. [DOI] [PubMed] [Google Scholar]
- Windmuller O, Lindauer U, Foddis M, Einhaupl KM, Dirnagl U, Heinemann U, Dreier JP. Ion changes in spreading ischaemia induce rat middle cerebral artery constriction in the absence of NO. Brain. 2005;128:2042–2051. doi: 10.1093/brain/awh545. [DOI] [PubMed] [Google Scholar]
- Winkler MK, et al. Impaired neurovascular coupling to ictal epileptic activity and spreading depolarization in a patient with subarachnoid hemorrhage: possible link to blood-brain barrier dysfunction. Epilepsia. 2012;53(Suppl 6):22–30. doi: 10.1111/j.1528-1167.2012.03699.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler MK, et al. Oxygen availability and spreading depolarizations provide complementary prognostic information in neuromonitoring of aneurysmal subarachnoid hemorrhage patients. J Cereb Blood Flow Metab. 2017;37:1841–1856. doi: 10.1177/0271678X16641424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woitzik J, et al. Propagation of cortical spreading depolarization in the human cortex after malignant stroke. Neurology. 2013;80:1095–1102. doi: 10.1212/WNL.0b013e3182886932. [DOI] [PubMed] [Google Scholar]
- Woods RP, Iacoboni M, Mazziotta JC. Brief report: bilateral spreading cerebral hypoperfusion during spontaneous migraine headache. N Engl J Med. 1994;331:1689–1692. doi: 10.1056/NEJM199412223312505. [DOI] [PubMed] [Google Scholar]
- Yanamoto H, et al. Induced spreading depression activates persistent neurogenesis in the subventricular zone, generating cells with markers for divided and early committed neurons in the caudate putamen and cortex. Stroke. 2005;36:1544–1550. doi: 10.1161/01.STR.0000169903.09253.c7. [DOI] [PubMed] [Google Scholar]
- Yao H, Markgraf CG, Dietrich WD, Prado R, Watson BD, Ginsberg MD. Glutamate antagonist MK-801 attenuates incomplete but not complete infarction in thrombotic distal middle cerebral artery occlusion in Wistar rats. Brain Res. 1994;642:117–122. doi: 10.1016/0006-8993(94)90912-1. [DOI] [PubMed] [Google Scholar]
- Yi NX, et al. MK-801 attenuates lesion expansion following acute brain injury in rats: a meta-analysis. Neural Regen Res. 2019;14:1919–1931. doi: 10.4103/1673-5374.259619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandt BJ, ten Haken B, van Putten MJ. Diffusing substances during spreading depolarization: analytical expressions for propagation speed, triggering, and concentration time courses. J Neurosci. 2013;33:5915–5923. doi: 10.1523/JNEUROSCI.5115-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]