

Abstract
Papillary thyroid carcinoma (PTC) is the most common type of thyroid carcinoma, and its incidence has been on the increase in recent years. However, the molecular mechanism of PTC is unclear and misdiagnosis remains a major issue. Therefore, the present study aimed to investigate this mechanism, and to identify key prognostic biomarkers. Integrated analysis was used to explore differentially expressed genes (DEGs) between PTC and healthy thyroid tissue. To investigate the functions and pathways associated with DEGs, Gene Ontology, pathway and protein-protein interaction (PPI) network analyses were performed. The predictive accuracy of DEGs was evaluated using the receiver operating characteristic (ROC) curve. Based on the four microarray datasets obtained from the Gene Expression Omnibus database, namely GSE33630, GSE27155, GSE3467 and GSE3678, a total of 153 DEGs were identified, including 66 upregulated and 87 downregulated DEGs in PTC compared with controls. These DEGs were significantly enriched in cancer-related pathways and the phosphoinositide 3-kinase-AKT signaling pathway. PPI network analysis screened out key genes, including acetyl-CoA carboxylase beta, cyclin D1, BCL2, and serpin peptidase inhibitor clade A member 1, which may serve important roles in PTC pathogenesis. ROC analysis revealed that these DEGs had excellent predictive performance, thus verifying their potential for clinical diagnosis. Taken together, the findings of the present study suggest that these genes and related pathways are involved in key events of PTC progression and facilitate the identification of prognostic biomarkers.
Keywords: papillary thyroid carcinoma, microarray, integrated analysis, key genes
Introduction
Thyroid carcinoma is the most common malignancy of the head and neck, and accounts for 91.5% of all endocrine malignancies (1). Its prevalence increased from 4.8 to 15.0 per 100,000 between 1975 and 2014 (2). In the thyroid carcinomas, 70–80% of them are pathologically diagnosed as papillary thyroid carcinomas (PTCs) (3). The incidence rate of PTC has been increasing rapidly worldwide over the past three decades (4). Although PTC has a favorable prognosis, certain cases exhibit aggressive clinical characteristics, including invasion, metastasis, recurrence or drug resistance (5). However, the clinical symptoms of early-stage PTC are usually absent or nonspecific. Various methods are currently used for PTC diagnosis, including sonography, CT, MRI, fine-needle aspiration and cytological examination, though diagnostic tests require improvement to achieve better accuracy (6). Recently, molecular biomarkers have been considered as promising approaches for PTC diagnosis, and understanding the molecular mechanism of this type of cancer may also aid in determining its pathogenesis, thus improving disease management (7).
Several molecular events and biological processes have been identified to serve critical roles in PTC tumorigenesis, progression and metastasis, including genetic or epigenetic alterations, non-coding RNAs, apoptosis, autophagy and the epithelial-to-mesenchymal transition (EMT) (8). For instance, C-X-C motif chemokine ligand 12 contributes to the development of PTC, syndecan-4 gene silencing leads to cell apoptosis and cadherin 6 (CDH6) has been reported to promote PTC metastasis through autophagy suppression (9–11). However, despite notable research progress, the mechanisms underlying PTC tumorigenesis remain elusive. A more detailed understanding of the gene changes, molecular impacts, protein interactions and signaling pathways involved in PTC may aid in the development of novel therapeutic approaches.
In recent years, high-throughput technology, including DNA microarray and RNA sequencing, has been used in cancer research to monitor thousands of targets simultaneously (12). A number of independent studies have applied microarrays to identify gene alterations in PTC (2,13,14), though, to the best of our knowledge, few integrated analyses have been conducted to screen for common gene alterations. Therefore, in the present study, the key genes associated with PTC were screened using bioinformatics analysis of four publicly available datasets. In addition, in order to understand disease pathogenesis and identify potential biomarkers, Gene Ontology (GO) and pathway analyses were performed. Furthermore, a protein-protein interaction (PPI) network was constructed in order to screen out central genes, and a receiver operating characteristic (ROC) curve analysis was performed to evaluate the predictive accuracy of differentially expressed genes (DEGs).
Materials and methods
Dataset selection and description
In order to identify DEGs in PTC, several datasets were analyzed in the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) (15). GSE33630 (dataset 1) (16) included 49 PTC and 45 healthy tissue samples, GSE27155 (dataset 2) (17) contained 51 PTC and four healthy samples, GSE3467 (dataset 3) (18) contained nine PTC and nine healthy samples and GSE3678 (dataset 4) (19) comprised of seven PTC and matched healthy tissues. The HG-U133 Plus 2 platform (Affymetrix Human Genome U133 Plus 2.0 Array) was used for profiling these four datasets. The selection of these four datasets was based on the following inclusion criteria: i) Use of the same detection platform; ii) inclusion of both PTC and normal tissues; iii) accessibility to the original data. The exclusion criteria were: i) The use of different detection platforms; and ii) analysis of cell line samples and not tissues. Therefore, the four aforementioned datasets were ultimately enrolled to screen key genes in PTC.
Data preprocessing and differential expression analyses
The gene screening strategy is presented in Fig. 1A. GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r), an online tool, was used separately for each dataset to identify genes that were differentially expressed in PTC. After obtaining four profiles of differentially expressed Affymetrix gene IDs, Venn diagram production online software (https://bioinfogp.cnb.csic.es/tools/venny/index.html, VENNY 2.1) was used to intersect DEGs. Differently colored areas represent different datasets, and cross areas represent key genes. Gene IDs were subsequently converted into gene symbols using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (15). Duplicated and uncharacterized genes were removed, and a final list of DEGs was generated. Since dataset 1 was identified to have a larger sample size of both PTC and healthy tissues, heat maps and volcano plots were further established from the gene expression profiles of this dataset in order to visualize DEGs using Morpheus (https://software.broadinstitute.org/morpheus, version 1.0) and Main software (BOA Bioinformatics, version 8.1). P<0.01 and log2 (fold-change) ≥1.5 were determined as significance cut-off levels.
Figure 1.

Gene screening strategy. (A) The four datasets of GSE33630 (dataset 1), GSE27155 (dataset 2), GSE3467 (dataset 3) and GSE3678 (dataset 4) were selected for the identification of DEGs. (B) Different colored areas represent different datasets. The crossed areas correspond to the common DEGs. DEGs, differentially expressed genes; GEO, Gene Expression Omnibus; PTC, papillary thyroid carcinoma; adj. P, adjusted P-value; DAVID, Database for Annotation, Visualization and Integrated Discovery.
GO and pathway enrichment analyses
In order to identify biological and functional attributes of DEGs, GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed using DAVID (https://david.ncifcrf.gov/) to explore comprehensive high-throughput gene functional annotation. P<0.05 was considered to indicate a statistically significant difference.
Integrated analysis of GO and pathway enrichment
ClueGO (version 2.5.4), a Cytoscape (http://www.cytoscape.org/) plug-in (version 3.7.1), was used to improve the biological interpretation of DEGs. ClueGO integrates GO terms and KEGG/Reactome pathways to generate a functional organized GO/pathway term network, which is then intuitively represented. The analysis parameters were set as follows: i) Right-sided hypergeometric test; ii) adjusted P<0.05 with Benjamini-Hochberg correction; iii) GO levels 6–14; and iv) a Kappa score threshold of 0.4.
PPI network construction
The Search Tool for the Retrieval of Interacting Genes (STRING, version 10.5) database (http//string-db.org) provides both experimental and predicted interaction information (20). A PPI network of DEGs was constructed with STRING to further evaluate the molecular mechanism of cellular processing. The PPI network was visualized in Cytoscape based on the gene expression values of dataset 1.
Statistical analysis
The ROC curve was used to evaluate classifiers in bioinformatic applications. To further assess the predictive accuracy of the DEGs, ROC analysis was performed to discriminate PTC from healthy tissues. ROC curves for genes with log2 (fold-change) ≥1.5 were generated using SPSS software (version 16.0; SPSS, Inc.) based on the obtained DEGs and their expression profile data from dataset 1. The area under the ROC curve (AUC) was calculated and used to compare the diagnostic value of these genes.
Results
Identification of DEGs in PTC
A total of four GEO datasets were selected for gene screening (Fig. 1). All data were acquired from microarray analyses of both PTC and healthy control tissues in the datasets. The screening strategy revealed a total of 153 DEGs among the four datasets, including 66 upregulated and 87 downregulated DEGs (Fig. 1; Table I).
Table I.
DEGs.
| DEGs | Gene name |
|---|---|
| Upregulated (66) | ABCC3, ADORA1, ALDH1A3, CAMK2N1, CCND1, CDH3, CDH6, CHI3L1, CITED1, COL13A1, DDB2, DPP4, DTX4, DUSP4, DVL1, EDEM1, ENTPD1, EPS8, ETV5, FN1, GALE, GALNT7, GGCT, GJB3, HEY2, HMGA2, HRH1, KCNJ2, KDELR3, KLHL2, KLK10, LAD1, LAMB3, LGALS3, LRP4, MCTP2, MET, MRC2, MSN, MVP, MYH10, NPC2, NRCAM, P2RX5-TAX1BP3, P4HA2, PDZK1IP1, PLXNC1, PROS1, PSD3, PTPRE, QPCT, RAB27A, RAD23B, RXRG, SCEL, SEL1L3, SERPINA1, SLC34A2, SPINT1, TBC1D2, TENM1, TGFA, TIAM1, TNIK, TUSC3TUSC3, UBE2A |
| Downregulated (87) | ACACB, ADAM22, ADGRA3, ADH1B, AGTR1, AOX1, ARHGAP24, ARHGEF28, ASXL3, ATP2C2, BCL2, CDH16, CDR2, CITED2, CLCNKA, CLMN, CRABP1, CSGALNACT1, CUX2, CWH43, DEPTOR, DGKI, DIRAS2, DPP6, ELMO1, EML1, EPHA3, ESRRG, FABP4, FAM234B, FCGBP, FGFR2, FHL1, GATM, GHR, GPM6A, HGD, HSBP1, ID4, IMPA2, IPCEF1, ITPR1, KIT, KIZ, KLHL3, LIFR, LRIG1, LRP1B, MAFB, MARC2, MATN2, MPC1, MPPED2, MROH7, MT1F, NETO2, OGDHL, PAPSS2, PEG3, PLA2R1, PLCH1, PPARGC1A, RAP1GAP, RGS16, RYR2, SDC2, SELENBP1, SHANK2, SLC4A4, SLITRK5, SORBS2, SPTBN1, STARD13, SYNE1, TBC1D4, TCF7L1, TFCP2L1, TFF3, TLE1, TMPRSS3, TNS3, TPO, TTC30A, WASF3, WFS1, WWOX, ZMAT4 |
DEGs, differentially expressed genes; ACACB, acetyl-CoA carboxylase β; CCND1, cyclin D1; SERPINA1, serpin peptidase inhibitor clade A member 1; CDH6, cadherin 6; COL13A1, collagen type XIII α 1 chain; ADH1B, dehydrogenase 1B (class I), β polypeptide; TBC1D4, TBC1 domain family member 4; TPO, thyroid peroxidase.
Validation and visualization of DEGs in microarray data
Using Morpheus software, the microarray data of dataset 1 were used in order to confirm the reliability and accuracy of DEGs in PTC. As presented in the cluster heat map of Fig. 2A, 153 DEGs demonstrated a distinct difference between PTC and healthy groups. The volcano plot (Fig. 2B) generated using Main software further verified that the majority of these DEGs had a log2 value (fold-change) >1.5 and P<0.05. These identified DEGs were used for the functional and pathway enrichment analyses.
Figure 2.

Validation and visualization of DEGs in dataset 1. (A) Heat maps were established based on the gene expression profile of dataset 1. Expression levels of the DEGs are represented by the different colors Red, up-regulated; black, normal expression; and green, down-regulated. (B) Volcano plot demonstrating the expression of DEGs with log2 value (fold-change)>1.5 and P<0.05. Blue dotted lines represent cut-off levels, red dots indicate high gene expression, black dots represent normal expression and green dots indicate low gene expression. DEGs, differentially expressed genes; PTC, papillary thyroid carcinoma.
GO analysis
DEGs were classified into the three functional groups of GO analysis: Biological processes, molecular function and cellular component (Fig. 3A; Table II). GO analysis showed that DEGs were primarily enriched in ‘postsynaptic density’, ‘melanocyte differentiation’, ‘dendrite morphogenesis’ and ‘positive regulation of cell proliferation’ and ‘cell migration’; ‘RNA polymerase II transcription coactivator activity’ and ‘molybdopterin cofactor binding’ were significantly enriched in molecular function; and in the cellular component group, DEGs were mainly enriched in ‘extracellular exosomes’, ‘dendritic spine’ and ‘receptor complex’.
Figure 3.

GO and pathway enrichment analyses of DEGs in PTC. (A) Significantly enriched GO terms of DEGs in PTC classified based on their biological functions. (B) Signaling pathway enrichment analysis of DEGs using KEGG. GO, Gene Ontology; DEGs, differentially expressed genes; PTC, papillary thyroid carcinoma; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Table II.
GΟ enrichment analysis differentially expressed genes in papillary thyroid carcinoma.
| Category | Term | Description | Count | P-value |
|---|---|---|---|---|
| Molecular function | GO:0001105 | Rna polymerase II transcription coactivator activity | 3 | 3.06×10−2 |
| GO:0043546 | Molybdopterin cofactor binding | 2 | 4.65×10−2 | |
| Cellular component | GO:0070062 | Extracellular exosome | 34 | 2.03×10−3 |
| GO:0043197 | Dendritic spine | 4 | 3.12×10−3 | |
| GO:0032045 | Guanyl-nucleotide exchange factor complex | 2 | 3.52×10−2 | |
| GO:0043235 | Receptor complex | 4 | 4.66×10−2 | |
| GO:0014069 | Postsynaptic density | 5 | 7.41×10−4 | |
| Biological process | GO:0030318 | Melanocyte differentiation | 4 | 7.71×10−4 |
| GO:0071340 | Skeletal muscle acetylcholine-gated channel clustering | 3 | 1.23×10−3 | |
| GO:0048813 | Dendrite morphogenesis | 4 | 2.82×10−3 | |
| GO:0016601 | Rac protein signal transduction | 3 | 5.21×10−3 | |
| GO:0008284 | Positive regulation of cell proliferation | 8 | 7.44×10−3 | |
| GO:0008584 | Male gonad development | 4 | 1.34×10−2 | |
| GO:0016477 | Cell migration | 5 | 1.59×10−2 | |
| GO:0070741 | Response to interleukin−6 | 2 | 1.83×10−2 | |
| GO:0035987 | Endodermal cell differentiation | 3 | 1.87×10−2 | |
| GO:0030178 | Negative regulation of wnt signaling pathway | 3 | 2.02×10−2 | |
| GO:0061156 | Pulmonary artery morphogenesis | 2 | 4.51×10−2 | |
| GO:0031532 | Actin cytoskeleton reorganization | 3 | 4.53×10−2 | |
| GO:0048538 | Thymus development | 3 | 4.98×10−2 |
GO, Gene Ontology.
Pathway enrichment analysis
Signaling pathway enrichment analysis of DEGs was conducted using DAVID. DEGs were found to be significantly enriched in nine classical pathways, including the phosphoinositide 3-kinase (PI3K)-AKT signaling pathway, and pathways associated with thyroid, prostate and small cell lung cancer (Fig. 3B; Table III). Signaling pathway analysis confirmed that DEGs were mainly involved in cancer-related pathways.
Table III.
Pathway enrichment analysis differentially expressed genes in papillary thyroid carcinoma.
| Pathway term | Description | Count | P-value | Genes |
|---|---|---|---|---|
| ptr00350 | Tyrosine metabolism | 5 | 4.51×10−4 | ALDH1A3, AOX1, HGD, ADH1B, TPO |
| ptr05200 | Pathways in cancer | 12 | 2.42×10−3 | FGFR2, AGTR1, LAMB3, CCND1, BCL2, MET, RXRG, TGFA, KIT, TCF7L1, DVL1, FN1 |
| ptr05215 | Prostate cancer | 5 | 1.37×10−2 | FGFR2, CCND1, BCL2, TGFA, TCF7L1 |
| ptr05222 | Small cell lung cancer | 5 | 1.48×10−2 | LAMB3, CCND1, BCL2, RXRG, FN1 |
| ptr05205 | Proteoglycans in cancer | 7 | 1.86×10−2 | CCND1, TIAM1, MET, MSN, SDC2, ITPR1, FN1 |
| ptr04924 | Renin secretion | 4 | 3.36×10−2 | AGTR1, KCNJ2, ADORA1, ITPR1 |
| ptr05216 | Thyroid cancer | 3 | 3.89×10−2 | CCND1, RXRG, TCF7L1 |
| ptr04151 | PI3K-AKT signaling pathway | 8 | 4.25×10−2 | FGFR2, LAMB3, CCND1, BCL2, MET, KIT, GHR, FN1 |
| ptr04141 | Protein processing in endoplasmic reticulum | 5 | 4.98×10−2 | RAD23B, TUSC3, WFS1, BCL2, EDEM1 |
CCND1, cyclin D1; ADH1B, dehydrogenase 1B (class I), β polypeptide; FN1, fibronectin 1; TPO, thyroid peroxidase; PI3K, phosphoinositide 3-kinase.
Integrated analysis of GO and pathway enrichment
To comprehensively investigate the biological processes associated with DEGs, integrated analysis of GO and pathway enrichment was conducted using Cytoscape. DEGs were found to be mainly involved in transcriptional regulation by the AP-2 (TFAP2) family of transcription factors, signaling by MET, regulation of lipase activity, dendrite morphogenesis, and postsynaptic density (Fig. 4), suggesting that these processes may constitute the underlying molecular mechanism of PTC.
Figure 4.

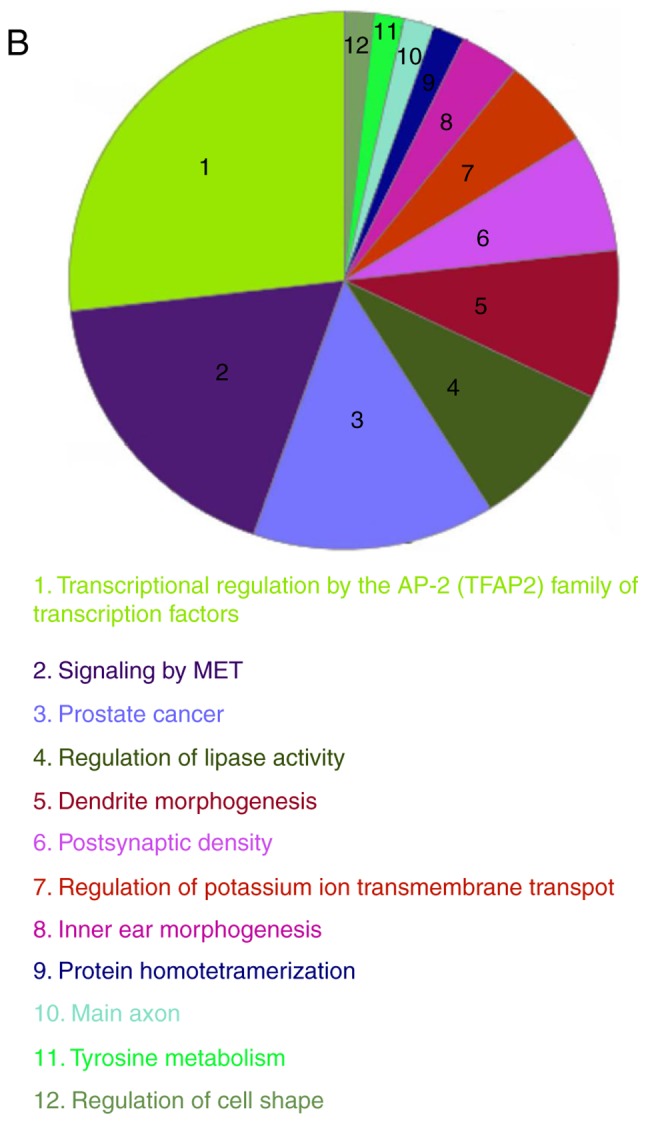
Integrated GO and pathway enrichment analyses. (A) Functionally grouped networks presented as colored circles. Circle size represents the enrichment significance of each term. Functionally associated groups were observed to partially overlap. (B) Overview chart illustrating the functional groups of DEGs. GO, Gene Ontology; DEGs, differentially expressed genes; CCND1, cyclin D1; ACACB, acetyl-CoA carboxylase β; TPO, thyroid peroxidase; FN1, fibronectin 1; ADH1B, dehydrogenase 1B (class I), β polypeptide.
PPI network
The PPI network included 152 nodes and 87 edges, representative of proteins and protein-protein interactions, respectively (Fig. 5). Moreover, acetyl-CoA carboxylase beta (ACACB) (nine edges), cyclin D1 (CCND1) (nine edges), BCL2 (seven edges) and serpin peptidase inhibitor clade A member 1 (SERPINA1) (five edges) were identified as central genes.
Figure 5.

PPI network of DEGs. PPI network was constructed based on the gene expression values of dataset 1. ACACB (9 edges), CCND1 (9 edges), BCL2 (7 edges) and SERPINA1 (5 edges) were identified as the central genes. Expression levels of the DEGs are represented by the different colors: Red denotes upregulated and green denotes downregulated expression. PPI, protein-protein interaction; DEGs, differentially expressed genes; FC, fold-change; CCND1, cyclin D1; ACACB, acetyl-CoA carboxylase beta; FN1, fibronectin 1; SERPINA1, serpin peptidase inhibitor clade A member 1.
Identification of key diagnostic genes by ROC analysis
The ROC curve was used to assess the predictive accuracy of DEGs with log2 (fold-change) ≥1.5. AUC was calculated and used to select the most appropriate cut-off gene expression levels. ROC curves and AUC values are presented in Fig. 6 and Table SI, respectively. All AUC values exceeded 0.8, while the upregulated genes CDH6 and collagen type XIII α1 chain (COL13A1), and downregulated genes alcohol dehydrogenase 1B (class I), β polypeptide (ADH1B) and TBC1 domain family member 4 (TBC1D4) had AUC values >0.98. The ROC analysis revealed that the expression levels of these genes had excellent predictive performance and were able to discriminate PTC from healthy tissues.
Figure 6.

ROC curve analysis presenting the sensitivity and specificity of DEGs in PTC diagnosis. (A) ROC curves of upregulated genes (B) ROC curves of downregulated genes. PTC, papillary thyroid carcinoma; ROC, receiver operating characteristic; SERPINA1, serpin peptidase inhibitor clade A member 1; FN1, fibronectin 1; PROS1, protein S; TPO, thyroid peroxidase; ADH1B, dehydrogenase 1B (class I), β polypeptide; CDH6, cadherin 6.
Discussion
PTC is a type of endocrine neoplasm, in which the incidence rate is gradually increasing in many parts of the world, such as the USA, Italy and Australia (21). Its transition to a well-differentiated type of tumor is considered a multifactorial and multistage process, and certain cases exhibit aggressive clinical characteristics (22). In the present study, it was identified that 153 genes were differentially expressed in four publicly available datasets, indicating that these genes and the associated pathways may regulate key events in PTC. Further ROC curve analysis revealed that their expression levels had excellent predictive performance in PTC diagnosis.
Microarray technology has been widely applied to investigate gene expression in PTC (13,20,22), although the common gene alterations in these studies remain poorly understood. Previous bioinformatics analyses have revealed the molecular mechanisms of various tumors, including hepatocellular carcinoma, anaplastic thyroid carcinoma, cutaneous squamous cell carcinoma and nasopharyngeal carcinoma (23–26). A total of four datasets were selected to screen DEGs from a total of 181 samples, including 116 tumor and 65 healthy tissues. The overlapping DEGs increased the reliability of the results.
Although PTC pathogenesis has previously been reported to be associated with various biological processes (27,28), the underlying molecular mechanisms remain unclear. GO and pathway enrichment analyses of the identified DEGs demonstrated that they were primarily associated with the RNA polymerase II (Pol II) transcription coactivator, and positive regulation of cell proliferation, cell migration and extracellular exosomes. Consistent with these findings, a previous study also reported that PTC was involved in the positive regulation of Pol II promoter transcription (20). Additionally, PTC progression was found to be associated with cell proliferation, invasion and apoptosis (27,29,30). Exosomes have been proposed as novel regulators in tumor initiation, while overexpressed microRNAs in exosomes were reported to alter cell proliferation in PTC (31).
In the present study, pathway enrichment analysis revealed that the DEGs were primarily involved in the PI3K-AKT signaling pathway and pathways associated with thyroid cancer and cancer proteoglycans. The PI3K-AKT signaling pathway regulates a wide range of cellular processes including survival, proliferation, metabolism, angiogenesis and metastasis; changes in this pathway are common in PTC (32–34). For instance, tumor protein p53-inducible protein 3 and C-X-C chemokine receptor type 7 serve an oncogenic role in PTC cell proliferation and metastasis by regulating the PI3K/AKT/PTEN pathway (2,35). In line with the present study, a previous network-based analysis also identified key genes as being enriched in proteoglycans in PTC (36).
The PPI network in the present study identified CCND1, ACACB, BCL2 and SERPINA1 as central genes. CCND1 is a gatekeeper that regulates the cell cycle transition from the G1 to S phase and is typically upregulated in various types of human cancer (37). Several studies on PTC have reported that CCND1 expression is associated with aggressive clinicopathological features, and that it can be used as a diagnostic and predictive biomarker (38,39). Furthermore, CCND1 is also involved in the PI3K-AKT signaling pathway and cancer proteoglycans, highlighting its importance in PTC initiation and progression. FN1 was revealed to serve an important role in several pathways described in the present study, and it was observed to be upregulated in numerous types of cancer, including colorectal cancer, oral squamous cell carcinoma and breast cancer (40–42). FN1 upregulation is thought to contribute to PTC development by regulating EMT, and it has been reported to act as a potential biomarker for PTC treatment (5).
Gene expression has promising diagnostic potential in a number of types of cancer, including colorectal, ovarian, and breast cancer (43–45). In the present study, the calculated AUC values ranged from 0.7–0.9, which is considered an excellent discriminatory power (46), while the AUC values for CDH6, COL13A1, ADH1B and TBC1D4 were increased. Consistent with the results of the present study, FN1 and the protein S gene have been proven to be associated with the diagnosis of PTC, and COL13A1 has been identified as a diagnostic marker that is able to clearly distinguish PTC from control samples (47,48). The combined use of gene expression values, including TACSTD2 (tumor-associated calcium signal transducer 2), subtilisin-like serine protease-2 and neural cell adhesion molecule 1, has been previously reported to be an effective diagnostic method for PTC (49). Furthermore, the thyroid peroxidase gene combined with other genes (including TIMP3, RARB2, SERPINB5, RASSF1, TPO and TSHR) demonstrated high diagnostic sensitivity (91%) and specificity (81%) in differentiating PTC from healthy thyroid tissue (50). The excellent predictive performance of the genes demonstrated in the present study suggests that all identified DEGs are potential biomarkers for PTC diagnosis.
The present study had a number of limitations. First, it lacked correlation analysis between DEGs and clinicopathological data in the publicly available datasets. Secondly, the DEGs were not validated using quantitative PCR in patients with PTC. Thirdly, western blot analysis or immunohistochemical staining was not performed to evaluate protein expression. Therefore, further studies that focus on the biological and molecular mechanisms of PTC are required in order to assess the accuracy of these genes in diagnosing this type of cancer.
An integrated analysis based on four public datasets was performed that identified 153 DEGs in PTC. Bioinformatics analysis results revealed that these DEGs may contribute to the pathogenesis of PTC via the PI3K-AKT signaling pathway and cancer-associated pathways. Diagnostic analysis further verified that expression levels of these DEGs had excellent predictive performance, suggesting their use as potential biomarkers for PTC diagnosis or targeted drug development.
Supplementary Material
Acknowledgements
The authors would like to thank Dr Sarah Williams (University of Oxford), for editing the original draft of this manuscript.
Funding
The present study was funded by the National Natural Science Foundation of China (grant no. 81702463 and 81702787) and the Beijing Health System Top Level Technical Personnel Training Plan (grant no. 20153079).
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Authors' contributions
YG and YBY designed the study. YL, HY, PC, SH and JL contributed to the data analysis. SG, YJ and YRY interpreted the results from the point of basic research, while clinical interpretation was made by JT, SW and XN. YL and SG wrote the initial draft of the manuscript. YG, XN and YBY revised the paper. All authors approved the final version manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Brennan K, Holsinger C, Dosiou C, Sunwoo JB, Akatsu H, Haile R, Gevaert O. Development of prognostic signatures for intermediate-risk papillary thyroid cancer. BMC Cancer. 2016;16:736. doi: 10.1186/s12885-016-2771-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang H, Teng X, Liu Z, Zhang L, Liu Z. Gene expression profile analyze the molecular mechanism of CXCR7 regulating papillary thyroid carcinoma growth and metastasis. J Exp Clin Cancer Res. 2015;34:16. doi: 10.1186/s13046-015-0132-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carling T, Udelsman R. Thyroid cancer. Annu Rev Med. 2014;65:125–137. doi: 10.1146/annurev-med-061512-105739. [DOI] [PubMed] [Google Scholar]
- 5.Qu T, Li YP, Li XH, Chen Y. Identification of potential biomarkers and drugs for papillary thyroid cancer based on gene expression profile analysis. Mol Med Rep. 2016;14:5041–5048. doi: 10.3892/mmr.2016.5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prasad NB, Somervell H, Tufano RP, Dackiw AP, Marohn MR, Califano JA, Wang Y, Westra WH, Clark DP, Umbricht CB, et al. Identification of genes differentially expressed in benign versus malignant thyroid tumors. Clin Cancer Res. 2008;14:3327–3337. doi: 10.1158/1078-0432.CCR-07-4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu X, He M, Hou Y, Liang B, Zhao L, Ma S, Yu Y, Liu X. Expression profiles of microRNAs and their target genes in papillary thyroid carcinoma. Oncol Rep. 2013;29:1415–1420. doi: 10.3892/or.2013.2263. [DOI] [PubMed] [Google Scholar]
- 8.Hu Y, Wang H, Chen E, Xu Z, Chen B, Lu G. Candidate microRNAs as biomarkers of thyroid carcinoma: A systematic review, meta-analysis, and experimental validation. Cancer Med. 2016;5:2602–2614. doi: 10.1002/cam4.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang S, Wang Y, Chen M, Sun L, Han J, Elena VK, Qiao H. CXCL12 methylation-mediated epigenetic regulation of gene expression in papillary thyroid carcinoma. Sci Rep. 2017;7:44033. doi: 10.1038/srep44033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen LL, Gao GX, Shen FX, Chen X, Gong XH, Wu WJ. SDC4 gene silencing favors human papillary thyroid carcinoma cell apoptosis and inhibits epithelial mesenchymal transition via Wnt/β-catenin pathway. Mol Cells. 2018;41:853–867. doi: 10.14348/molcells.2018.0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gugnoni M, Sancisi V, Gandolfi G, Manzotti G, Ragazzi M, Giordano D, Tamagnini I, Tigano M, Frasoldati A, Piana S, Ciarrocchi A. Cadherin-6 promotes EMT and cancer metastasis by restraining autophagy. Oncogene. 2017;36:667–677. doi: 10.1038/onc.2016.237. [DOI] [PubMed] [Google Scholar]
- 12.Reiner A, Yekutieli D, Benjamini Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics. 2003;19:368–375. doi: 10.1093/bioinformatics/btf877. [DOI] [PubMed] [Google Scholar]
- 13.Min XS, Huang P, Liu X, Dong C, Jiang XL, Yuan ZT, Mao LF, Chang S. Bioinformatics analyses of significant prognostic risk markers for thyroid papillary carcinoma. Tumour Biol. 2015;36:7457–7463. doi: 10.1007/s13277-015-3410-6. [DOI] [PubMed] [Google Scholar]
- 14.Zhu W, Li C, Ai Z. Candidate agents for papillary thyroid cancer identified by gene expression analysis. Pathol Oncol Res. 2013;19:597–604. doi: 10.1007/s12253-013-9625-1. [DOI] [PubMed] [Google Scholar]
- 15.Huang Da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 16.Dom G, Tarabichi M, Unger K, Thomas G, Oczko-Wojciechowska M, Bogdanova T, Jarzab B, Dumont JE, Detours V, Maenhaut C. A gene expression signature distinguishes normal tissues of sporadic and radiation-induced papillary thyroid carcinomas. Br J Cancer. 2012;107:994–1000. doi: 10.1038/bjc.2012.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giordano TJ, Kuick R, Thomas DG, Misek DE, Vinco M, Sanders D, Zhu Z, Ciampi R, Roh M, Shedden K, et al. Molecular classification of papillary thyroid carcinoma: Distinct BRAF, RAS, and RET/PTC mutation-specific gene expression profiles discovered by DNA microarray analysis. Oncogene. 2005;24:6646–6656. doi: 10.1038/sj.onc.1208822. [DOI] [PubMed] [Google Scholar]
- 18.He H, Jazdzewski K, Li W, Liyanarachchi S, Nagy R, Volinia S, Calin GA, Liu CG, Franssila K, Suster S, et al. The role of microRNA genes in papillary thyroid carcinoma. Proc Natl Acad Sci USA. 2005;102:19075–19080. doi: 10.1073/pnas.0509603102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu X, Yao J, Tian W. Microarray technology to investigate genes associated with papillary thyroid carcinoma. Mol Med Rep. 2015;11:3729–3733. doi: 10.3892/mmr.2015.3180. [DOI] [PubMed] [Google Scholar]
- 20.Tu Y, Fan G, Xi H, Zeng T, Sun H, Cai X, Kong W. Identification of candidate aberrantly methylated and differentially expressed genes in thyroid cancer. J Cell Biochem. 2018;11:8797–8806. doi: 10.1002/jcb.27129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu Z, Sheng J, Zhang Y, Deng J, Li Y, Lu A, Zhang J, Yu H, Zhang M, Xiong Z, et al. Clonality analysis of multifocal papillary thyroid carcinoma by using genetic profiles. J Pathol. 2016;239:72–83. doi: 10.1002/path.4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu J, Mai W, Cui Y, Kong L. Key genes and pathways predicted in papillary thyroid carcinoma based on bioinformatics analysis. J Endocrinol Invest. 2016;39:1285–1293. doi: 10.1007/s40618-016-0491-z. [DOI] [PubMed] [Google Scholar]
- 23.Shi KQ, Lin Z, Chen XJ, Song M, Wang YQ, Cai YJ, Yang NB, Zheng MH, Dong JZ, Zhang L, et al. Hepatocellular carcinoma associated microRNA expression signature: Integrated bioinformatics analysis, experimental validation and clinical significance. Oncotarget. 2015;6:25093–25108. doi: 10.18632/oncotarget.4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pan Z, Fang Q, Zhang Y, Li L, Huang P. Identification of key pathways and drug repurposing for anaplastic thyroid carcinoma by integrated bioinformatics analysis. Zhejiang Da Xue Xue Bao Yi Xue Ban. 2018;47:187–193. doi: 10.3785/j.issn.1008-9292.2018.04.13. (In Chinese) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei W, Chen Y, Xu J, Zhou Y, Bai X, Yang M, Zhu J. Identification of biomarker for cutaneous squamous cell carcinoma using microarray data analysis. J Cancer. 2018;9:400–406. doi: 10.7150/jca.21381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen F, Shen C, Wang X, Wang H, Liu Y, Yu C, Lv J, He J, Wen Z. Identification of genes and pathways in nasopharyngeal carcinoma by bioinformatics analysis. Oncotarget. 2017;8:63738–63749. doi: 10.18632/oncotarget.19478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yan R, Yang T, Zhai H, Zhou Z, Gao L, Li Y. MicroRNA- 150-5p affects cell proliferation, apoptosis, and EMT by regulation of the BRAFV600E mutation in papillary thyroid cancer cells. J Cell Biochem. 2018;119:8763–8772. doi: 10.1002/jcb.27108. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H, Yang L, Liu Z, Liu C, Teng X, Zhang L, Yin B, Liu Z. iTRAQ-coupled 2D LC/MS-MS analysis of CXCR7-transfected papillary thyroid carcinoma cells: A new insight into CXCR7 regulation of papillary thyroid carcinoma progression and identification of potential biomarkers. Oncol Lett. 2017;14:3734–3740. doi: 10.3892/ol.2017.6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diao Y, Fu H, Wang Q. MiR-221 exacerbate cell proliferation and invasion by targeting TIMP3 in papillary thyroid carcinoma. Am J Ther. 2017;24:e317–e328. doi: 10.1097/MJT.0000000000000420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Z, Zhang J, Gao J, Li Y. MicroRNA-4728 mediated regulation of MAPK oncogenic signaling in papillary thyroid carcinoma. Saudi J Biol Sci. 2018;25:986–990. doi: 10.1016/j.sjbs.2018.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee JC, Zhao JT, Gundara J, Serpell J, Bach LA, Sidhu S. Papillary thyroid cancer-derived exosomes contain miRNA-146b and miRNA-222. J Surg Res. 2015;196:39–48. doi: 10.1016/j.jss.2015.02.027. [DOI] [PubMed] [Google Scholar]
- 32.Han M, Chen L, Wang Y. miR-218 overexpression suppresses tumorigenesis of papillary thyroid cancer via inactivation of PTEN/PI3K/AKT pathway by targeting Runx2. Onco Targets Ther. 2018;11:6305–6316. doi: 10.2147/OTT.S172152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng Z, Zhou X, Cai Y, Chen E, Zhang X, Wang O, Wang Q, Liu H. TEKT4 promotes papillary thyroid cancer cell proliferation, colony formation, and metastasis through activating PI3K/Akt pathway. Endocr Pathol. 2018;29:310–316. doi: 10.1007/s12022-018-9549-0. [DOI] [PubMed] [Google Scholar]
- 34.Yu ST, Zhong Q, Chen RH, Han P, Li SB, Zhang H, Yuan L, Xia TL, Zeng MS, Huang XM. CRLF1 promotes malignant phenotypes of papillary thyroid carcinoma by activating the MAPK/ERK and PI3K/AKT pathways. Cell Death Dis. 2018;9:371. doi: 10.1038/s41419-018-0352-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu J, Cai J, Jin X, Yang J, Shen Q, Ding X, Liang Y. PIG3 plays an oncogenic role in papillary thyroid cancer by activating the PI3K/AKT/PTEN pathway. Oncol Rep. 2015;34:1424–1430. doi: 10.3892/or.2015.4096. [DOI] [PubMed] [Google Scholar]
- 36.Zhao H, Li H. Network-based meta-analysis in the identification of biomarkers for papillary thyroid cancer. Gene. 2018;661:160–168. doi: 10.1016/j.gene.2018.03.096. [DOI] [PubMed] [Google Scholar]
- 37.Alao JP. The regulation of cyclin D1 degradation: Roles in cancer development and the potential for therapeutic invention. Mol Cancer. 2007;6:24. doi: 10.1186/1476-4598-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeon S, Kim Y, Jeong YM, Bae JS, Jung CK. CCND1 splice variant as a novel diagnostic and predictive biomarker for thyroid cancer. Cancers (Basel) 2018;10 doi: 10.3390/cancers10110437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin Y, Hong S, Yu S, Huang Y, Chen S, Liu Y, Zhang Q, Li Y, Xiao H. MiR-195 inhibits tumor growth and metastasis in papillary thyroid carcinoma cell lines by targeting CCND1 and FGF2. Int J Endocrinol. 2017;2017:6180425. doi: 10.1155/2017/6180425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou Y, Lu L, Jiang G, Chen Z, Li J, An P, Chen L, Du J, Wang H. Targeting CDK7 increases the stability of Snail to promote the dissemination of colorectal cancer. Cell Death Differ. 2019;26:1442–1452. doi: 10.1038/s41418-018-0222-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin T, Zhang B, He H. Identification of genes correlated with oral squamous cell carcinoma. J Cancer Res Ther. 2018;14:S675–S679. doi: 10.4103/0973-1482.202893. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Xu H, Zhu B, Qiu Z, Lin Z. Systematic identification of the key candidate genes in breast cancer stroma. Cell Mol Biol Lett. 2018;23:44. doi: 10.1186/s11658-018-0110-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shou X, Li Y, Hu W, Ye T, Wang G, Xu F, Sui M, Xu Y. A six-gene Assay as a new biomarker in the blood of patients with colorectal cancer: Establishment and clinical validation. Mol Oncol. 2019;13:781–791. doi: 10.1002/1878-0261.12427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li L, Cai S, Liu S, Feng H, Zhang J. Bioinformatics analysis to screen the key prognostic genes in ovarian cancer. J Ovarian Res. 2017;10:27. doi: 10.1186/s13048-017-0323-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jasbi P, Wang D, Cheng SL, Fei Q, Cui JY, Liu L, Wei Y, Raftery D, Gu H. Breast cancer detection using targeted plasma metabolomics. J Chromatogr B Analyt Technol Biomed Life Sci. 2019;1105:26–37. doi: 10.1016/j.jchromb.2018.11.029. [DOI] [PubMed] [Google Scholar]
- 46.Johann DJ, Jr, McGuigan MD, Patel AR, Tomov S, Ross S, Conrads TP, Veenstra TD, Fishman DA, Whiteley GR, Petricoin EF, III, Liotta LA. Clinical proteomics and biomarker discovery. Ann NY Acad Sci. 2004;1022:295–305. doi: 10.1196/annals.1318.045. [DOI] [PubMed] [Google Scholar]
- 47.Port M, Boltze C, Wang Y, Röper B, Meineke V, Abend M. A radiation-induced gene signature distinguishes post-Chernobyl from sporadic papillary thyroid cancers. Radiat Res. 2007;168:639–649. doi: 10.1667/RR0968.1. [DOI] [PubMed] [Google Scholar]
- 48.Zhang K, Liu J, Li C, Peng X, Li H, Li Z. Identification and validation of potential target genes in papillary thyroid cancer. Eur J Pharmacol. 2019;843:217–225. doi: 10.1016/j.ejphar.2018.11.026. [DOI] [PubMed] [Google Scholar]
- 49.Yang X, Hu Y, Shi H, Zhang C, Wang Z, Liu X, Chen H, Zhang L, Cui D. The diagnostic value of TROP-2, SLP-2 and CD56 expression in papillary thyroid carcinoma. Eur Arch Otorhinolaryngol. 2018;275:2127–2134. doi: 10.1007/s00405-018-5045-x. [DOI] [PubMed] [Google Scholar]
- 50.Stephen JK, Chen KM, Merritt J, Chitale D, Divine G, Worsham MJ. Methylation markers differentiate thyroid cancer from benign nodules. J Endocrinol Invest. 2018;41:163–170. doi: 10.1007/s40618-017-0702-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.