

Abstract
Background
Interstitial pulmonary fibrosis (IPF) is harmful for patients’ life and health. The effective treatment of IPF is lacking because of unclear pathogenesis. Necrostatin-1 has protective effects on lung injury and can suppress the fibrosis development. I this study we investigated whether necrostatin-1 could decrease the proliferation of pulmonary fibroblasts, pulmonary fibrosis and expression of extracellular matrix (ECM) in IPF.
Material/Methods
The IPF mice model was conducted by intra-tracheal injection of bleomycin (BLM) (2 mg/kg) for C57BL/6N mice. Necrostatin-1 treatment was performed with 1 mg/kg necrostatin-1 by an intravenous injection for C57BL/6N mice. Lung tissue structures and collagen deposition were observed by hematoxylin and eosin staining and Masson staining. IPF in vitro model was constructed by MRC-5 cells induced by transforming growth factor beta 1 (TGF-β1). And, 20 μM necrostatin-1 was used to treat the TGF-β1 induced MRC-5 cells. Cell Counting Kit-8 (CCK-8) assay detected the viability of MRC-5 cells. The expression of receptor-interacting protein kinase-1 and -3 (RIPK1 and RIPK3), α smooth muscle actin (α-SMA), collagen IV, collagen I, fibronectin (FN), and transforming growth factor-β (TGF-β) in lung tissues and MRC-5 cells was measured by western blot analysis. The α-SMA expression in lung tissues was also analyzed by immunohistochemistry.
Results
The expression of RIPK1 and RIPK3 in lung tissues of BLM induced mice was increased. The degree of pulmonary fibrosis and expression of α-SMA, collagen IV, collagen I, FN, and TGF-β in lung tissues of BLM induced mice was enhanced. The proliferation of MRC-5 cells was increased when MRC-5 cells were induced by TGF-β. The expression of RIPK1, RIPK3, α-SMA, collagen IV, collagen I, and FN was increased in TGF-β induced MRC-5 cells. And, necrostatin-1 could effectively reverse the changes of pulmonary fibrosis, RIPK1, RIPK3, and ECM in vivo and in vitro experiments.
Conclusions
Necrostatin-1 attenuated pulmonary fibrosis in lung tissues of BLM induced mice and inhibited the fibroblast proliferation. And, necrostatin-1 also decreased the expression of RIPK1, RIPK3, and ECM in lung tissues of BLM induced mice and TGF-β induced fibroblasts. Necrostatin-1 could be a new effective drug for the treatment of IPF.
MeSH Keywords: Bleomycin, Extracellular Matrix, Pulmonary Fibrosis
Background
Interstitial pulmonary fibrosis (IPF), also known as interstitial lung disease (ILD), is a collection of diseases with diffuse exudation, infiltration and fibrosis [1]. Up to now, western treatments for IPF mainly include glucocorticoid immunosuppressant, anti-fibrosis drug, antacid drug, oxygen therapy, mechanical ventilation, pulmonary rehabilitation, and lung transplantation [2,3]. The official guidelines of 2015 reported that pirfenidone and nintedanib were recommended for use under certain conditions, but their clinical application was limited due to unclear potential benefits, heavy economic burden and obvious adverse reactions [4]. Therefore, the treatment of IPF remains to be further studied.
The receptor-interacting protein (RIP) kinase family includes seven members, each of which having a homologous kinase domain and various functional domains [5]. Previous researches have showed that RIP kinase family is related to many biological processes, such as tumorigenesis [6], cell death [7], necrosis [8], and inflammation [9].
Receptor-interacting protein kinase-1 (RIPK1) is the first member of the RIP family, which functions in the transformation between apoptosis and necroptosis [10,11]. Moreover, it plays an important regulatory role in signaling pathways between inflammatory response, apoptosis and necroptosis [12]. RIPK3 is a typical serine/threonine protein kinase that is activated by RIPKl to exert broad downstream effects [13]. Recent study has showed that the excessive necroptosis of alveolar epithelial cells is closely related to the development of pulmonary fibrosis, and receptor-interacting protein kinase-1 and -3 (RIPK1/3) can regulate the initiation of necroptosis. Therefore, it can be seen that RIPK1/3 are related to the pulmonary fibrosis [14]. Necrostatin-1 inhibits kinase activity and prevents the mutual phosphorylation of RIP1 and RIP3 by acting on the kinase parts of RIP1 and RIP3, thereby inhibiting the formation of RIP1-RIP3 complexes to restrain the occurrence of necrosis [15,16]. Necrostatin-1 is a RIP inhibitor, which is studied in the fibrosis development in wild-type mice [14]. Literature has shown that necrostatin-1 often plays a protective role in peripheral nerve injury [17], osteoarthritis [18], and acute kidney injury [19]. In addition, necrostatin-1 can reduce lipopolysaccharide (LPS)-induced lung injury and acute respiratory distress syndrome by inhibiting necroptosis and inflammation [20,21]. However, its role in IPF is not clear, especially its role in alleviating the pulmonary fibrosis and expression of extracellular matrix (ECM).
Therefore, we studied the effects of necrostatin-1 on proliferation of pulmonary fibroblasts, pulmonary fibrosis and expression of RIPKl, RIPK3, and ECM in the bleomycin (BLM)-induced interstitial pulmonary fibrosis.
Material and Methods
Animal model
Forty male C57BL/6N mice (6 to 8 weeks old, weighing 20 g) were obtained from Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). Mice were fed with standard water and food in an environmentally controlled room (22±2°C, 12-hour light-12-hour dark cycle). Forty male C57BL/6N mice were divided into 4 groups (n=10) including control group, BLM-induced group, BLM-induced+dimethylsulfoxide (DMSO) group and BLM-induced+necrostatin-1 group. Mice in the BLM-induced group were induced by injecting 2 mg/kg BLM (Nippon Kayaku Co., Tokyo, Japan) into their tracheas with a single dose. One milligram of necrostatin-1 (Selleckchem, Houston, USA) was dissolved in 20 mL of 2% DMSO (Sigma, Saint Louis, MO, USA) as described in a previous study [22]. In the BLM-induced+necrostatin-1 group, mice were pretreated with intravenous injection of 1 mg/kg necrostatin-1 and then induced by BLM. In the BLM-induced+DMSO group, mice were pretreated with intravenous injection of equal volume of DMSO. The mice were euthanized with their lungs collected. Finally, all groups were sacrificed on day 28.
Cell culture and cell induction
MRC-5 cells purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) high-sugar medium containing 100 mL/L fetal bovine serum (FBS), 100 IU/mL penicillin, and 100 μg/mL streptomycin at 37°C in the 50 mL/L CO2 incubator. The cells grew adherently and were passaged 1 time every 2 days. The MRC-5 cells grew to logarithmic stage were selected for the experiment. MRC-5 cells were exposed to 10 ng/mL TGF-β1 (transforming growth factor beta 1) for 48 hours and then treated with 20 μM necrostatin-1 for 24 hours, 48 hours, and 72 hours.
Quantitative real-time PCR (RT-qPCR) analysis
The TRIzol method (Invitrogen) was used to extract the total RNA in lung tissues, and the RNA quality was then identified. The amount of RNA was used to synthesize the cDNA with SYBR Premix Ex TaqTM II (Takara, Dalian, China). RT-qPCR was performed with the synthesized RIPK1 and RIPK3 mRNA primers using SYBRON Premix ExTaq™ kit (Invitrogen). The primer sequences for qPCR were as follows:
GAPDH forward, 5′-GAAGGTGAAGGTCGGAGTC-3′, and
reverse, 5′-GAAGATGGTGATGGGATTTC-3′;
RIPK1 forward, 5′-TCAGCTCCTTGCCACCAACA-3′, and
reverse, 5′-TCGTCCCACCAATCTCCATA-3′;
RIPK3 forward, 5′-CCTCTCAGTCCACACTCCGA-3′, and
reverse, 5′-TGACGCACCAGTAGGCCAT-3′.
The mRNA expression of RIPK1 and RIPK3 was detected with the 2−ΔΔCt method. The expression level was normalized to GAPDH.
Western blot analysis
Western blot analysis was conducted according to previous studies [23,24]. The primary antibodies used were anti-RIPK1 (Abnova, Taiwan, China; MAB0836), anti-RIPK3 (Proteintech, Chicago, IL, USA; 17563-1-AP), anti-α smooth muscle actin (α-SMA) (Abcam, UK; ab32575), anti-collagen IV (Abcam, UK; ab214417), anti-collagen I (Abcam, UK; ab34710), anti-fibronectin (FN) (Abcam, UK; ab32419), anti- transforming growth factor-β (TGF-β) (Abcam, UK; ab92486). The denatured protein samples were subjected to 12% SDS-polyacrylamide gel electrophoresis (PAGE), and then transferred to polyvinylidene fluoride (PVDF) membrane. After the treatment of membranes with 5% non-fat dry milk overnight at 4°C, secondary antibodies were added to the membranes for another 2 hours at 25°C. GAPDH (Abcam, UK; ab181602) was applied to detect the relative protein expression.
Hematoxylin and eosin staining (H&E staining)
The lung tissues of mice were fixed with 4% paraformaldehyde for 24 hours, then conventional paraffin embedding sections were performed. The lung sections of mice were stained with hematoxylin solution, counterstained with eosin solution, differentiated with hydrochloric acid alcohol, washed with running tap water, dehydrated with graded ethanol, vitrified by dimethylbenzene and sealed with neutral gum. Finally, the structure changes of lung tissues were observed by an inverted fluorescence microscope (MF53; Micro-shot Technology Co., Ltd., Guangzhou, China).
Masson staining
The collagen deposition was assessed by Masson staining. The lung sections of mice were stained with Masson dye according to the instructions. The collagen deposition in interstitial fibrous tissues was observed by an inverted fluorescence microscope (MF53; Micro-shot Technology Co., Ltd., Guangzhou).
Immunohistochemistry (IHC)
Immumohistochemical staining was performed to detect the positive expression of α-SMA in the lung tissues. The specific operation was conducted strictly in accordance with the introductions of α-SMA IHC kit (Shanghai Yansheng Industrial Co., Ltd., China). After immunohistochemical staining, the positive results were brown-yellow particles, which were observed by Image-Pro Plus6.0 analysis system.
Cell Counting Kit-8 (CCK-8) assay
After induced by TGF-β1 and treated with necrostatin-1, MRC-5 cells were seeded in a 96-well plate, with each well containing 1×104 cells. Five compound holes were set for each group. Each well of the 96-well plate was added with 10 μL CCK-8 solution, followed by another 4 hours of incubation. Finally, the OD450 value was determined by a Synergy™ 2 Multi-function Microplate Reader. The CCK-8 solution contained WST-8, which was reduced to a highly water-soluble yellow formazan dye by dehydrogenase in the cell mitochondria, and the amount of formazan was proportional to the number of living cells.
Statistical analysis
SPSS 21.0 software was used for the analysis of experimental data and measurement data were expressed as the mean±standard deviation. One-way analysis of variance compared the differences among multiple groups with SNK-q test. The difference of experimental data was statistically significant when P<0.05.
Results
Necrostatin-1 reduced the expression of RIPK1 and RIPK3 in BLM-induced lung tissues
The protein expression and mRNA expression of RIPK1 and RIPK3 in lung tissues were shown in Figure 1A and 1B. BLM induction promoted the expression of RIPK1 and RIPK3 in lung tissues of BLM induced mice. However, the expression of RIPK1 and RIPK3 in lung tissues of BLM induced mice treated by necrostatin-1 was obviously decreased compared with BLM induced group and DMSO treatment group.
Figure 1.

Necrostatin-1 reduces the expression of RIPK1 and RIPK3 in BLM-induced lung tissues. (A) The protein expression of RIPK1 and RIPK3 in lung tissues detected by western blot analysis. ** P<0.01 and *** P<0.001 versus control group. # P<0.05 versus BML-induced group. @ P<0.05 versus BML-induced+DMSO group. (B) The mRNA expression of RIPK1 and RIPK3 in lung tissues detected by RT-qPCR analysis. * P<0.05, ** P<0.01 and *** P<0.001 versus control group. ### P<0.001 versus BML-induced group. @@@ P<0.001 versus BML-induced+DMSO group. RIPK1 – receptor-interacting protein kinase-1; RIPK3 – receptor-interacting protein kinase-3; BLM – bleomycin; DMSO – dimethylsulfoxide; RT-qPCR – real-time quantitative polymerase chain reaction.
Necrostatin-1 alleviated pathological changes and collagen deposition in lung tissues induced by BLM
The pathological changes of lung tissue are shown in Figure 2A. The lung tissues in the control group showed that the structure of lung tissue was clear, and the alveolar septum was not thickened. Also, there was no inflammation and edema in lung tissue. But, the lung tissues in the BLM-induced model group showed that the alveolar structure was destroyed with thickened alveolar septum and exfoliated epithelial cells, inflammatory exudation and pulmonary interstitial congestion was observed in the alveolar cavities. The condition of collagen deposition in lung tissues was shown in Figure 2B. The lung tissues in the control group showed no or very few thin filamentous fiber deposition, while lung tissues in the BLM-induced model group showed large amount of blue fiber deposition. However, the lung tissues in necrostatin-1 treated model group showed that the degree of pulmonary interstitial congestion, inflammatory cell infiltration, and collagen deposition was significantly decreased.
Figure 2.

Necrostatin-1 alleviates pathological changes and collagen deposition in lung tissues induced by BLM. (A) The pathological changes of lung tissue observed after H&E staining. (B) The lung organization fibrosis degree observed after Masson staining. BLM – bleomycin; H&E – hematoxylin and eosin.
Necrostatin-1 decreased the ECM expression in lung tissues induced by BLM
The ECM expression in lung tissues was determined by immunohistochemistry and western blot analysis. BLM expression was represented as the brown-yellow particles in IHC and the result indicated that BLM induction promoted the expression of α-SMA in mice lung tissues and necrostatin-1 effectively suppressed the expression of α-SMA in BLM induced mice (Figure 3A). As shown in Figure 3B, the expression of ECM (α-SMA, collagen IV, collagen I, FN, and TGF-β) was increased in BLM-induced lung tissues and necrostatin-1 obviously decreased the ECM expression. The ECM expression was closely related to the fibrosis.
Figure 3.

Necrostatin-1 decreases the ECM expression in lung tissues induced by BLM. (A) The expression of α-SMA in lung tissues detected by immunohistochemistry. (B) The expression of α-SMA, collagen IV, collagen I, FN, and TGF-β in lung tissues detected by western blot analysis. ** P<0.01 and *** P<0.001 versus control group. # P<0.05 and ## P<0.01 versus BML-induced group. @ P<0.05 and @@ P<0.01 versus BML-induced+DMSO group. ECM – extracellular matrix; BLM – bleomycin; α-SMA – smooth muscle actin; FN – fibronectin; TGF-β1 – transforming growth factor beta 1.
Necrostatin-1 decreased the ECM expression in pulmonary fibroblasts induced by TGF-β1
The expression of collagen and ECM is shown in Figure 4. Necrostatin-1 decreased the expression of α-SMA, collagen IV, collagen I, and FN in MRC-5 cells while TGF-β1 promoted the aforementioned proteins expression in MRC-5 cells. After MRC-5 cells induced by TGF-β1, necrostatin-1 treatment could effectively downregulate the aforementioned proteins expression in TGF-β1 induced MRC-5 cells, but the expression level of the aforementioned proteins was still higher than that in the control group. Therefore, necrostatin-1 decreases the ECM expression in MRC-5 cells induced by TGF-β1.
Figure 4.

Necrostatin-1 decreases the ECM expression in pulmonary fibroblasts induced by TGF-β1. The expression of α-SMA, collagen IV, collagen I, and FN in lung tissues detected by western blot analysis. * P<0.05, ** P<0.01 and *** P<0.001 versus control group. ### P<0.001 versus necrostatin-1 group. @ P<0.05 versus TGF-β1 group. ECM – extracellular matrix; TGF-β1 – transforming growth factor beta 1; α-SMA – smooth muscle actin; FN – fibronectin.
Necrostatin-1 inhibited the proliferation of pulmonary fibroblasts induced by TGF-β1
The viability of MRC-5 cells was decreased when MRC-5 cells were treated with necrostatin-1, while increased when MRC-5 cells were treated with TGF-β1. After MRC-5 cells induced by TGF-β1, necrostatin-1 could effectively suppress the viability of TGF-β1 induced MRC-5 cells but the cell viability was still lower than that in the control group (Figure 5). Therefore, necrostatin-1 inhibits the proliferation of MRC-5 cells induced by TGF-β1.
Figure 5.
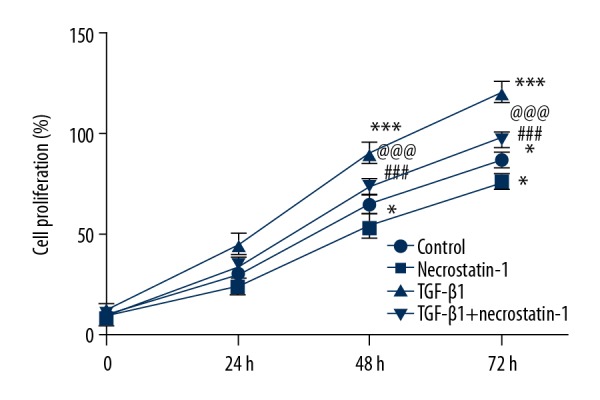
Necrostatin-1 inhibits the proliferation of pulmonary fibroblasts induced by TGF-β1. The cell viability detected by CCK-8 assay. * P<0.05 versus control group. ### P<0.001 versus necrostatin-1 group. @@@ P<0.001 versus TGF-β1 group. TGF-β1 – transforming growth factor beta 1; CCK-8 – Cell Counting Kit-8.
Necrostatin-1 decreased the expression of RIPK1 and RIPK3 in pulmonary fibroblasts induced by TGF-β1
As shown in Figure 6, the expression of RIPK1 and RIPK3 was obviously decreased in MRC-5 cells treated with necrostatin-1 while TGF-β1 could upregulate the aforementioned proteins expression in MRC-5 cells. After MRC-5 cells induced by TGF-β1, necrostatin-1 effectively down-regulate the expression of RIPK1 and RIPK3 in TGF-β1 induced MRC-5 cells.
Figure 6.

Necrostatin-1 decreases the expression of RIPK1 and RIPK3 in pulmonary fibroblasts induced by TGF-β1. The expression of RIPK1 and RIPK3 in lung tissues detected by western blot analysis. ** P<0.01 and *** P<0.001 versus control group. ### P<0.001 versus necrostatin-1 group. @ P<0.05 and @@ P<0.01 versus TGF-β1 group. RIPK1 – receptor-interacting protein kinase-1; RIPK3 – receptor-interacting protein kinase-3; TGF-β1 – transforming growth factor beta 1.
Discussion
IPF has been a serious threat to human health. In recent years, studies have found that the incidence of pulmonary fibrosis is increasing [25]. The IPF model for this study was performed by transtracheal injection of BLM for mice. And, MRC-5 cells induced by TGF-β1 simulated the IPF model in vitro. This study indicated that necrostatin-1 could effectively reduce the expression of RIPK and RIPK3 in lung tissues, improve the lung structure, decrease collagen deposition and ECM expression in IPF animal model. And, necrostatin-1 also decreased cell viability and suppressed the expression of RIPK, RIPK3, α-SMA, collagen IV, collagen I, and FN in TGF-β induced MRC-5 cells.
BLM is currently recognized as a pulmonary fibrosis drug in the world. Experiments showed that BLM could damage the pulmonary tissue of rats, widen alveolar septum and cause a large number of inflammatory cells infiltration, fibroblasts proliferation and obviously increased expression of collagen-I and α-SMA [26,27]. Necrostatin-1 has anti-fibrosis effect. Previous study indicated that necrostatin-1 could significantly inhibit myocardial fibrosis response and improve cardiac function in rats with chronic myocardial ischemia and showed good effects on myocardial protection [28]. Necrostatin-1 could also protect against the lung injury by reducing the inflammation [20,21,29]. RIPK1 and RIPK3 are the necrosis markers which increased in inflammation related diseases and necrosis death [30]. RIPK1 activates the necroptotic kinase cascade by RIPK3 phosphorylation [31]. RIPK1 kinase activity is vital to the activation of necroptosis and necrostatin-1 can inhibit the necroptosis [16]. Study demonstrated that irregular necroptosis could lead to tissue fibrosis [32]. In the present study, the expression of RIPK1 and RIPK3 was increased in lung tissues of BLM-induced mice. Therefore, necrostatin-1, as RIP1 inhibitor, suppressed the expression of RIPK1 and RIPK3 in lung tissues and effectively decrease the inflammatory cells infiltration and collagen deposition and improve lung fibrosis.
The typical characteristics of IPF are that a large number of fibroblasts are accumulated in lung tissues and the ECM is deposited with inflammatory cell infiltration. Excessive proliferation of fibroblasts is the main cause of ECM aggregation [33]. EMT is related to injury healing, tissue regeneration and organ fibrosis [34]. If the inflammation continuously stimulates the injury, epithelial mesenchymal transition (EMT) will exist persistently to produce excessive extracellular matrix (ECM), thereby leading to organ fibrosis [35]. Hepatocytes promote the occurrence of liver fibrosis through EMT [36–38]. EMT in hepatocytes is manifested by decreased expression of epithelial markers such as e-cadherin and increased expression of interstitial markers such as α-SMA [39]. TGF-β is the most important cytokine in the development of pulmonary fibrosis [40]. Shen et al. [41] demonstrated that myocardial fibrosis in rats was related to the inhibition of TGF-β signaling pathway. TGF-β could promote ECM-producing cells to express α-SMA and make ECM-producing cells converted to myofibroblast (MFB) [42–44]. As one of the most important fibrogenic cytokines, TGF-β1 could increase the mRNA expression of collagen I in cardiac fibroblast to upregulate the expression of collagen I [45,46]. The late changes in EMC of IPF presented that collagen IV expression was increased on alveolar epithelium and capillary basement membrane and distributed in the lung interstitium full of collagen, which indicating that collagen IV was closely related to the IPF development [47]. FN is not only the component of ECM protein, but also a scaffold for deposition of other extracellular proteins. It is also a chemotactic factor of fibroblasts. Reducing the synthesis of FN and increasing the degradation of FN are of importance for preventing the formation of renal tubule-interstitial fibrosis [48]. In the interstitial fibrosis, it was found that the deposition and increase of FN. The FN expression was not obvious in the kidney, but it increased in renal interstitial lesions [49]. This shows that α-SMA, collagen IV, collagen I, FN, and TGF-β are functioned in the development of organ fibrosis. In this study, the expression of α-SMA, collagen IV, collagen I, FN, and TGF-β was increased in lung tissues of BLM induced mice and the expression of α-SMA, collagen IV, collagen I and FN was increased in TGF-β1 induced MRC-5 cells. Necrostatin-1 alleviated the fibrosis in the lung tissues, inhibited the proliferation of MRC-5 cells and decreased the expression of RIPK1, RIPK3, α-SMA, collagen IV, collagen I, FN, and TGF-β.
Conclusions
In conclusion, we demonstrated that the degree of pulmonary fibrosis and ECM expression was reduced by necrostatin-1 in BLM-induced mice and necrostatin-1 could also inhibit the proliferation of MRC-5 cells induced by TGF-β1. Therefore, necrostatin-1 could be a new effective drug for the treatment of IPF. However, this study had some limitations. The effective dose of necrostatin-1 was taken as only one value. In further studies, the gradient values of necrostatin-1 should be used for the treatment of BLM-induced mice. Thus, the dose of necrostatin-1 that can produce side effects on BLM-induced mice should be investigated.
Footnotes
Source of support: Departmental sources
Conflicts of interest
None.
References
- 1.Mari PV, Jones MG, Richeldi L. Contemporary concise review 2018: Interstitial lung disease. Respirology. 2019;24:809–16. doi: 10.1111/resp.13572. [DOI] [PubMed] [Google Scholar]
- 2.Chinese Society of Respiratory Diseases. Draft Guidelines for the diagnosis and treatment of idiopathic pulmonary (interstitial) fibrosis. Chin J Tuberc Respir Dis. 2002;25:387–89. [Google Scholar]
- 3.Interstitial Pulmonary Disease Committee RS, Chinese Medical Association. Chinese expert consensus on diagnosis and treatment of idiopathic pulmonary fibrosis. Chin J Tuberc Respir Dis. 2016;39:427–32. [Google Scholar]
- 4.Raghu G, Rochwerg B, Zhang Y, et al. An Official ATS/ERS/JRS/ALAT Clinical practice guideline: Treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015;192:e3–19. doi: 10.1164/rccm.201506-1063ST. [DOI] [PubMed] [Google Scholar]
- 5.Zhang D, Lin J, Han J. Receptor-interacting protein (RIP) kinase family. Cell Mol Immunol. 2010;7:243–49. doi: 10.1038/cmi.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pachiyappan K, Alhazzazi TY, Theodora D, et al. Receptor-interacting protein (RIP) and Sirtuin-3 (SIRT3) are on opposite sides of anoikis and tumorigenesis. Cancer. 2012;118:5800–10. doi: 10.1002/cncr.27655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun J, Yu X, Wang C, et al. RIP-1/c-FLIPL induce hepatic cancer cell apoptosis through regulating tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) Med Sci Monit. 2017;23:1190–99. doi: 10.12659/MSM.899727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang C, Luo Y, Zhao J, et al. Shikonin kills glioma cells through necroptosis mediated by RIP-1. PLoS One. 2013;8:e66326. doi: 10.1371/journal.pone.0066326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moriwaki K, Chan FKM. Necrosis-dependent and independent signaling of the RIP kinases in inflammation. Cytokine Growth Factor Rev. 2014;25:167–74. doi: 10.1016/j.cytogfr.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blériot C, Lecuit M. RIPK1, a key survival factor for hepatocytes. J Hepatol. 2017;66:1118–19. doi: 10.1016/j.jhep.2017.02.009. [DOI] [PubMed] [Google Scholar]
- 11.Stanger BZ, Leder P, Lee TH, et al. RIP: A novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell. 1998;81:513–23. doi: 10.1016/0092-8674(95)90072-1. [DOI] [PubMed] [Google Scholar]
- 12.Manolis P, Peter V. Necroptosis and its role in inflammation. Nature. 2015;517:311–20. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 13.Tom VB, Andreas L, Sandrine JL, et al. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–47. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 14.Lee JM, Yoshida M, Kim MS, et al. Involvement of alveolar epithelial cell necroptosis in IPF pathogenesis. Am J Respir Cell Mol Biol. 2018;59:215–24. doi: 10.1165/rcmb.2017-0034OC. [DOI] [PubMed] [Google Scholar]
- 15.Cammas S, Renard I, Girault JP, Guérin P. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–23. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Degterev A, Hitomi J, Germscheid M, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–21. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang YX, Wang NN, Zhang ZY, et al. Necrostatin-1 ameliorates peripheral nerve injury-induced neuropathic pain by inhibiting the RIP1/RIP3 pathway. Front Cell Neurosci. 2019;13:211. doi: 10.3389/fncel.2019.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liang S, Lv ZT, Zhang JM, et al. Necrostatin-1 attenuates trauma-induced mouse osteoarthritis and IL-1 beta induced apoptosis via HMGB1/TLR4/SDF-1 in primary mouse chondrocytes. Front Pharmacol. 2018;9:1378. doi: 10.3389/fphar.2018.01378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dong W, Li ZL, Chen YH, et al. Necrostatin-1 attenuates sepsis-associated acute kidney injury by promoting autophagosome elimination in renal tubular epithelial cells. Mol Med Rep. 2018;17:3194–99. doi: 10.3892/mmr.2017.8214. [DOI] [PubMed] [Google Scholar]
- 20.Pan L, Yao DC, Yu YZ, et al. Necrostatin-1 protects against oleic acid-induced acute respiratory distress syndrome in rats. Biochem Biophys Res Commun. 2016;478:1602–8. doi: 10.1016/j.bbrc.2016.08.163. [DOI] [PubMed] [Google Scholar]
- 21.Guan E, Wang Y, Wang C, et al. Necrostatin-1 attenuates lipopolysaccharide-induced acute lung injury in mice. Exp Lung Res. 2017;43:378–87. doi: 10.1080/01902148.2017.1384083. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L, Cui Y, Wang B, et al. Protective effect of necrostatin-1 on the liver of rats with trauma induced hemorrhagic shock. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2014;26:17–22. [in Chinese] [PubMed] [Google Scholar]
- 23.Geng J, Huang X, Li Y, et al. Down-regulation of USP13 mediates phenotype transformation of fibroblasts in idiopathic pulmonary fibrosis. Respir Res. 2015;16:124. doi: 10.1186/s12931-015-0286-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu X, Wan X, Geng J, et al. Rapamycin regulates connective tissue growth factor expression of lung epithelial cells via phosphoinositide 3-kinase. Exp Biol Med (Maywood, NJ) 2013;238:1082–94. doi: 10.1177/1535370213498976. [DOI] [PubMed] [Google Scholar]
- 25.Ganesh R, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu PF, Yao YK, Zhou JY. Particulate matter with a diameter of ≤2.5 mu m induces and enhances bleomycin-induced pulmonary fibrosis by stimulating endoplasmic reticulum stress in rat. Biochem Cell Biol. 2019;97:357–63. doi: 10.1139/bcb-2018-0053. [DOI] [PubMed] [Google Scholar]
- 27.Suryadevara V, Huang LS, Kim SJ, et al. Role of phospholipase D in bleomycin-induced mitochondrial reactive oxygen species generation, mitochondrial DNA damage, and pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2019;317:L175–87. doi: 10.1152/ajplung.00320.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang W, Xiao-Liang W, Jie Z. [Effect of necrostatin-1 in chronically myocardial ischemia]. Chinese Journal of Cardiovascular Review. 2008;6:542–44. [in Chinese] [Google Scholar]
- 29.Pan L, Yao DC, Yu YZ, et al. Necrostatin-1 protects against oleic acid-induced acute respiratory distress syndrome in rats. Biochem Biophys Res Commun. 2016;478:1602–8. doi: 10.1016/j.bbrc.2016.08.163. [DOI] [PubMed] [Google Scholar]
- 30.Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. 2016;18:127–36. doi: 10.1038/nrm.2016.149. [DOI] [PubMed] [Google Scholar]
- 31.Sun L, Wang H, Wang Z, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–27. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 32.O’Donnell JA, Lehman J, Roderick JE, et al. Dendritic cell RIPK1 maintains immune homeostasis by preventing inflammation and autoimmunity. J Immunol. 2018;200:737–48. doi: 10.4049/jimmunol.1701229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.King TE, Jr, Annie P, Moisés S. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–61. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 34.Raghu K, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2015;119:1420–28. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen LCG, Chen YW, Chen Y, Li D. [Research progress on the regulatory mechanism of epithelial-mesenchymal transformation in liver fibrosis]. International Journal of Digestive Diseases. 2010;30:222–25. [in Chinese] [Google Scholar]
- 36.Zeisberg M, Yang C, Martino M, et al. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007;82:23337–47. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]
- 37.Magness ST, Ramón B, Liu Y, Brenner DA. A dual reporter gene transgenic mouse demonstrates heterogeneity in hepatic fibrogenic cell populations. Hepatology. 2010;40:1151–59. doi: 10.1002/hep.20427. [DOI] [PubMed] [Google Scholar]
- 38.Syn WK, Jung Y, Omenetti A, et al. Hedgehog-mediated epithelial-to-mesenchymal transition and fibrogenic repair in nonalcoholic fatty liver disease. Gastroenterology. 2009;137:1478–88.e1478. doi: 10.1053/j.gastro.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429–37. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hye-Ryun K, Soo Jung C, Chun Geun L, et al. Transforming growth factor (TGF)-beta1 stimulates pulmonary fibrosis and inflammation via a Bax-dependent, bid-activated pathway that involves matrix metalloproteinase-12. J Biol Chem. 2007;282:7723–32. doi: 10.1074/jbc.M610764200. [DOI] [PubMed] [Google Scholar]
- 41.Xiang-Chun S, Yu-Ping Y, Ting-Ting X, et al. Protective effect of oxymatrine on myocardial fibrosis induced by acute myocardial infarction in rats involved in TGF-β1-Smads signal pathway. J Asian Nat Prod Res. 2011;13:215–24. doi: 10.1080/10286020.2010.550883. [DOI] [PubMed] [Google Scholar]
- 42.Marcin D, Wei C, Frangogiannis NG. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J Mol Cell Cardiol. 2011;51:600–6. doi: 10.1016/j.yjmcc.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kamato D, Burch ML, Piva TJ, et al. Transforming growth factor-β signalling: Role and consequences of Smad linker region phosphorylation. Cell Signal. 2013;25:2017–24. doi: 10.1016/j.cellsig.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 44.Colwell AS, Krummel TM, Longaker MT, Lorenz HP. Fetal and adult fibroblasts have similar TGF-beta-mediated, Smad-dependent signaling pathways. Plast Reconstr Surg. 2006;117:2277–83. doi: 10.1097/01.prs.0000224299.16523.76. [DOI] [PubMed] [Google Scholar]
- 45.Lijnen PJ, Petrov VV, Fagard RH. Induction of cardiac fibrosis by transforming growth factor-β 1. Mol Genet Metabol. 2000;71:418–35. doi: 10.1006/mgme.2000.3032. [DOI] [PubMed] [Google Scholar]
- 46.Nakajima H, Nakajima HO, Salcher O, et al. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-beta(1) transgene in the heart. Circ Res. 2000;86:571–79. doi: 10.1161/01.res.86.5.571. [DOI] [PubMed] [Google Scholar]
- 47.Su YL, Gu HY, Weng D, et al. Association of serum levels of laminin, type IV collagen, procollagen III N-terminal peptide, and hyaluronic acid with the progression of interstitial lung disease. Medicine (Baltimore) 2017;96:e6617. doi: 10.1097/MD.0000000000006617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fu P, Feng M, Zhang Z. [Experimental study on effect of matrine in alleviating renal tubulointerstitial fibrosis in unilateral ureteral obstruction model]. Zhongguo Zhong Xi Yi Jie He Za Zhi. 2006;26(2):140–43. [in Chinese] [PubMed] [Google Scholar]
- 49.Satoh M, Kashihara N, Yamasaki Y, et al. Renal interstitial fibrosis is reduced in angiotensin II type 1a receptor-deficient mice. J Am Soc Nephrol. 2001;12:317–25. doi: 10.1681/ASN.V122317. [DOI] [PubMed] [Google Scholar]