

Abstract
Alcohol-associated liver disease is accompanied by dysregulation of bile acid metabolism and gut barrier dysfunction. Peroxisome proliferator-activated receptor-delta (PPARδ) agonists are key metabolic regulators and have anti-inflammatory properties. Here, we evaluated the effect of the selective PPAR-delta agonist seladelpar (MBX–8025) on gut barrier function and bile acid metabolism in a mouse model of ethanol-induced liver disease. Wild type C57BL/6 mice were fed LieberDe-Carli diet containing 0%–36% ethanol (caloric) for 8 weeks followed by a single binge of ethanol (5 g/kg). Pair fed mice received an isocaloric liquid diet as control. MBX-8025 (10 mg/kg/d) or vehicle were added to the liquid diet during the entire feeding period (prevention), or during the last 4 weeks of Lieber DeCarli diet feeding (intervention). In both prevention and intervention trials, MBX-8025 protected mice from ethanol-induced liver disease, characterized by lower serum alanine aminotransferase (ALT) levels, hepatic triglycerides, and inflammation. Chronic ethanol intake disrupted bile acid metabolism by increasing the total bile acid pool and serum bile acids. MBX-8025 reduced serum total and secondary bile acids, and the total bile acid pool as compared with vehicle treatment in both prevention and intervention trials. MBX-8025 restored ethanol-induced gut dysbiosis and gut barrier dysfunction. Data from this study demonstrates that seladelpar prevents and treats ethanol-induced liver damage in mice by direct PPARδ agonism in both the liver and the intestine.
Keywords: Gut barrier, Enterohepatic circulation, Microbiome
INTRODUCTION
Alcohol-related liver disease is one of the most prevalent liver diseases worldwide.1 Alcohol-related liver disease can develop gradually from hepatic steatosis to alcoholic steatohepatitis, alcoholic cirrhosis, and hepatocellular carcinoma,2 contributing to the increasing rates of liver transplantation and liver-related mortality.3,4 Many efforts have been made to evaluate the pathogenesis of alcohol-related liver disease, which involves direct hepatotoxic effects of ethanol, alcohol-associated gut barrier dysfunction, alterations of the gut microbiota and epigenetic changes.5–7
Peroxisome proliferator-activated receptors (PPARs) are nuclear hormone receptors that regulate gene transcription.8 Peroxisome proliferator-activated receptor-delta (PPARδ; also referred to as PPARβ) is emerging as a key therapeutic target due to its ubiquitous expression in tissues9 and its favorable effects on multiple diseases including diabetes,10 atherogenic dyslipidemia,11 nonalcoholic steatohepatitis,12 and liver fibrosis.13 PPARδ agonists attenuate ethanol-induced liver disease via reducing insulin resistance and endoplasmic reticulum (ER) stress.14,15 Due to its expression in the intestine, PPARδ activation can increase the proliferation of epithelial cells in small intestine16 and suppress macrophage-derived inflammation.17 Whether PPARδ activation reduces alcohol-associated liver disease by stabilizing gut barrier function and reducing intestinal dysbiosis is not known. In the present study, we addressed the effects of a specific PPARδ agonist MBX–8025 (seladelpar) on ethanol-induced gut barrier dysfunction, dysbiosis, and liver disease.
METHODS
Mice.
C57BL/6 mice were purchased from Charles River. Female mice (age 9–10 weeks) were subjected to a chronic Lieber-DeCarli diet model for 8 weeks as previously described18 followed by 1 binge of ethanol.19 The caloric intake from ethanol was 0 on day 1, 10% of total calories on days 2 and 3, 20% on days 4 and 5, 30% from day 6 until the end of 6 weeks, and 36% for the last 2 weeks. Then, mice were gavaged with 1 dose of ethanol (5 g/kg body weight) in the morning and sacrificed 9 hours later. Control mice were fed with an isocaloric amount of isomaltose instead of ethanol.
To evaluate the effects of MBX-8025 on liver disease, C57BL/6 female mice (control diet: n = 10/group; ethanol diet: n = 19–24/group) were treated with vehicle (H2O) or MBX-8025 daily (10 mg/kg/d)12 by adding MBX-8025 to the liquid diet during the entire feeding period (prevention) or during the last 4 weeks of ethanol feeding (intervention).
All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee of the University of California, San Diego.
MBX-8025 and bile acid measurements.
Total bile acids were measured using a mouse total bile acid kit (Crystal Chem). To estimate the total bile acid pool, total liver bile acids, total gallbladder bile acids, and total bile acids from the intestinal contents were measured and calculated per 100 g of body weight. Individual serum bile acids were analyzed by liquid chromatography–mass spectrometry (LC-MS) as described.20
MBX-8025 and C4 levels were analyzed using a tandem LC-MS at Quintara Discovery (Hayward, CA). Both methods used an Exion UHPLC, with the column equilibrated at 40°C, and a Sciex Qtrap 6500+ detector. Samples were prepared from plasma by spiking the samples with the respective internal standard prior to acetonitrile precipitation. The precipitated samples were centrifuged and aliquots of the supernatant were analyzed. Standards were prepared in undiluted blank mouse plasma and subjected to the same extraction methods. MBX-8025 was resolved using a Thermo Hypersil Gold C18 50 × 2.1 mm, 5 μm column with a mobile phase of 5 mM ammonium acetate in water (A) and acetonitrile (B) with the gradient (B% [t(min)]: 10 [0], 10[0.3], 95[1.2], 100[2.9], 10[3]), using warfarin as an internal standard. C4 was resolved using a Phenomenex Luna C18(2) 100 × 2 mm, 5 μm column and a mobile phase of water (A) and methanol (B), both with 0.1% acetic acid with the gradient (B% [t (min)]: 50[0], 50[0.2], 95[0.5], 100[5.5], 50[5.6]), using d7-C4 as an internal standard.
Bacterial DNA isolation and 16S rDNA sequencing
DNA from mouse feces was extracted using QIAamp Fast DNA Stool kit (Qiagen) and further concentrated using the Zymo gDNA Clean and Concentrate kit (Zymo). Then, PCR reactions were performed to amplify the V3-V4 hypervariable segment of the 16S rDNA gene, using PCR forward primer 5′-TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG CCT ACG GGN GGC WGC AG-3′ and reverse primer 5′-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GGA CTA CHV GGG TAT CTA ATC C-3′.21 The following PCR conditions were used: initial denaturation at 95°C for 3 minutes, followed by 35 cycles of denaturation (95°C for 30 seconds), annealing at 55°C for 30 seconds, extension at 72°C for 30 seconds, and a final extension step at 72°C for 5 minutes.
Amplified products were prepared for sequencing using the Nextera DNA Library Prep XT kit (Illumina, Inc) according to manufacturer instructions. PCR products were purified with Ampure XP beads (Beckman-Coulter) and visualized using a High Sensitivity DNA Kit on a Bioanalyzer (Agilent Technologies). Samples were finally sequenced on the Illumina MiSeq platform (Illumina) yielding 250 bp paired-end reads. Data were analyzed using QIIME 2 pipeline.22 Representative OTUs from each set were chosen at a minimum sequence identity of 97% which uses the Greengenes database.23 Metabolic function of gut microbiota was analyzed using PIC-RUSt.24 Sequence data were registered at NCBI under BioProject PRJNA597350. Sequence reads are available at NCBI under the following consecutive BioSample IDs: SAMN13673358-SAMN13673442.
RNA isolation and RNA sequencing.
Total RNA from mouse liver and ileum were extracted using RNeasy Mini Kit (Qiagen). RNA-Seq libraries were sequenced (Illumina NovaSeq 6000) to a depth of ≈41 × 106 reads, and aligned to the mouse genome (NCBI GRCm38.p6) using Salmon version 0.14.1.25 Gene expression profiling was performed using DESeq2 (false discovery rate calculations).26 Gene expression fold change ≥2 and P value <0.05 were set as the threshold values. Pathway analysis was performed with Database for Annotation, Visualization and Integrated Discovery online tool.27 Pathway enrichment was determined using the Kyoto Encyclopedia of Genes and Genomes Pathway annotation showing top 10 biological processes according to −log(P value). Sequence data were registered at NCBI under BioProject PRJNA597350. Sequence reads are available at NCBI under the following consecutive BioSample IDs: SAMN13673201-SAMN13673357.
Biochemical assays.
Serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured using the Infinity ALT kit (Thermo Scientific) and Infinity AST kit (Thermo Scientific). Hepatic triglyceride and cholesterol levels were measured using the Triglyceride Liquid Reagents Kit (Pointe Scientific) and Cholesterol (Liquid) Reagent Kit (Pointe Scientific), respectively. Serum levels of ethanol were measured using the Ethanol Assay Kit (BioVision). Fecal albumin and serum LPS were determined by ELISA kits (Bethyl Labs and Lifeome Biolabs, respectively).
Staining procedures.
Formalin-fixed tissue samples for liver were embedded in paraffin (Paraplast plus, McCornick). Sections were cut at 5 μm thickness and stained with hematoxylin-eosin (Leica Biosystems). To determine lipid accumulation, liver sections were embedded in OCT compound. Frozen sections were then cut at 8 μm thickness and stained with Oil Red O (Sigma-Aldrich).
Western blot.
Liver samples were collected from the central lobule for protein analysis. Tissues were lysed in ice-cold RIPA lysis buffer. After centrifugation of the lysates at 16,000 g for 15 minutes at 4°C, the supernatants were collected and stored at −80°C until analysis. Protein was loaded into Mini-PROTEAN TGX Precast Protein Gels (BIO-RAD), and transferred to PVDF membranes. Membranes were subsequently probed using antibodies specific for Cyp7a1 (1:1000 dilution, ab65596, Abcam) or α-tubulin (1:5000 dilution, Santa Cruz). Bands were quantitated using NIH ImageJ analysis.
Statistical analysis.
All data is expressed as mean ± s.e.m. unless otherwise specified. For comparisons of 2 groups within one experimental setting, Mann-Whitney-Wilcoxon test was used. For comparisons of >2 groups within 1 experimental setting, 1-way analysis of variance with Tukey’s post hoc test or Kruskal-Wallis test with Dunn’s post hoc test were used. Unweighted UniFrac distances were used for principal coordinate analysis and P values were determined by permutational multivariate analysis of variance (PERMANOVA) followed by false discovery rate (FDR) procedures to correct for multiple comparisons.
Statistical analyses were performed using the statistical software R (V.3.5.1, 2018 the R Foundation for Statistical Computing) and GraphPad Prism (V.7.04). A P value <0.05 was considered significant.
RESULTS
PPARδ agonist MBX-8025 reduces ethanol-induced liver injury.
To evaluate the effects of MBX-8025 on ethanol-induced liver disease, mice were subjected to chronic plus binge ethanol feeding for 8 weeks.18,19 Mice were treated with MBX-8025 during the entire feeding period (prevention), or 4 weeks after ethanol feeding was started (intervention). Serum MBX-8025 was detected in mice treated with MBX-8025, but not in vehicle treated mice (Fig 1A). MBX-8025 reduced ethanol-induced liver injury (as demonstrated by lower plasma ALT and AST levels) (Fig 1B, supplementary Fig 1A), steatosis (Fig 1C–E, supplementary Fig 1B) and inflammation (Fig 1F), both in the prevention and intervention trial. After absorption in the gastrointestinal tract, ethanol is metabolized in the liver via alcohol-dehydrogenase 1 (Adh1) and cytochrome P450 family 2 subfamily E polypeptide 1 (Cyp2e1). Although mice treated with MBX-8025 showed increased expression of hepatic Adh1 following chronic ethanol administration, serum ethanol levels were comparable between groups (supplementary Fig 1C–D). Hepatic Cyp2e1 was not significantly different after treatment with MBX-8025 (supplementary Fig 1E). In addition, although body weight decreased in ethanol diet-fed mice compared with control diet-fed mice, there was no change of body weight after MBX-8025 treatment (supplementary Fig 1F). Liver weight and the ratio of liver weight to body weight increased in both MBX-8025 intervention and prevention groups compared with vehicle treated mice after ethanol administration (supplementary Fig 1G–H). Taken together, MBX-8025 reduces ethanol-induced liver disease in mice.
Fig 1.
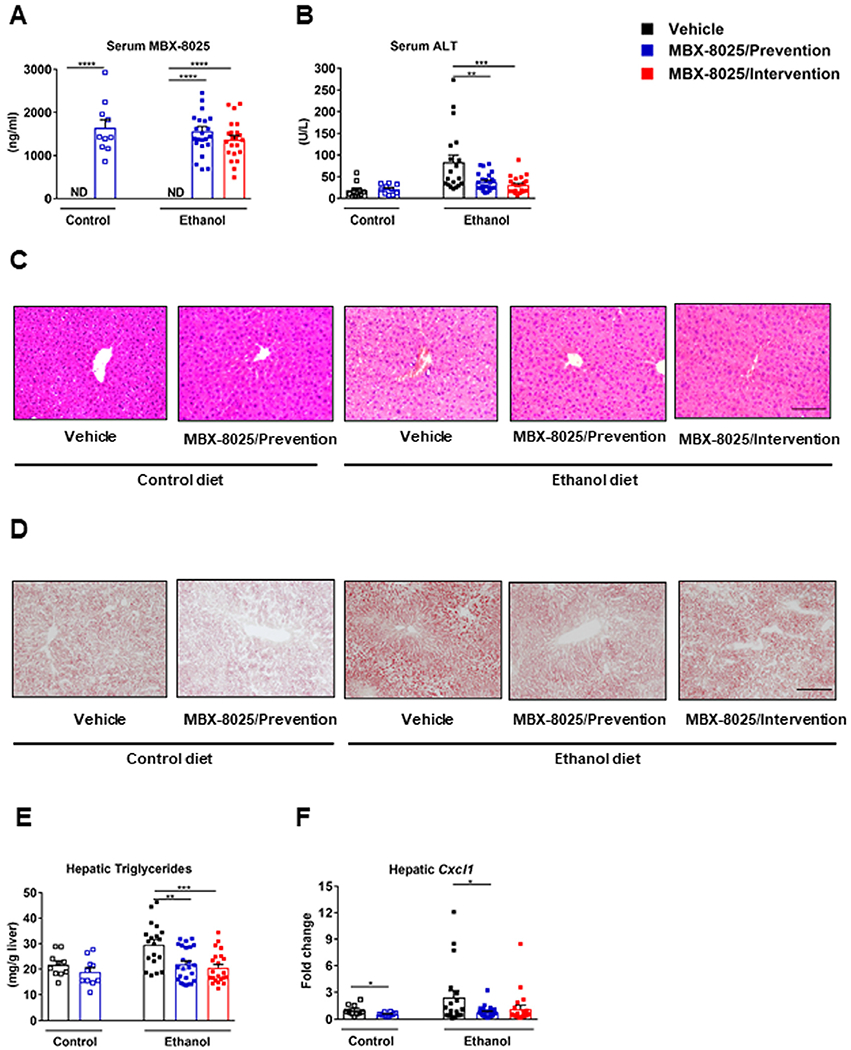
MBX-8025 protects from ethanol-induced liver disease. C57BL/6 mice were fed an oral isocaloric (control) diet (1–2 technical replicates) or a chronic Lieber-DeCarli diet for 8 weeks followed by 1 binge of ethanol (3–4 technical replicates), and treated with vehicle, or MBX-8025 (10 mg/kg/d) daily by adding MBX-8025 in the liquid diet during the entire feeding period (prevention) or during the last 4 weeks of ethanol feeding (intervention). (A) Serum levels of MBX-8025. (B) Serum levels of alanine aminotransferase (ALT). (C) Representative images of H&E stained liver tissue. (D) Representative images of Oil Red O stained liver tissue. (E) Hepatic triglyceride levels. (F) Hepatic levels of Cxcl1 mRNA. Control diet: Vehicle, n = 8–10; MBX-8025/ Prevention, n= 10; Ethanol diet: Vehicle, n= 15–19; MBX-8025/Prevention, n = 23–24; MBX-8025/Intervention, n = 22. Scale bar = 50 μm (C). Scale bar= 100 μm (D). Results are expressed as mean § s.e.m. (A, B, E, F). P values were determined using Mann-Whitney-Wilcoxon test for control diet-fed mouse groups (A, B, E, F), and Kruskal-Wallis test with Dunn’s post hoc test for ethanol-fed mouse groups (A) and one-way ANOVA with Tukey’s post hoc test for ethanol-fed mouse groups (B, E, F). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
MBX-8025 regulates liver inflammation and lipid metabolism.
To better understand the hepatoprotective effect of MBX-8025 on the molecular level, hepatic RNA was extracted and gene expression profile was analyzed by RNA-sequencing. Consistent with our results above, MBX-8025 regulated hepatic inflammatory and lipid metabolism pathways in both prevention and intervention trials (Fig 2A and B).
Fig 2.
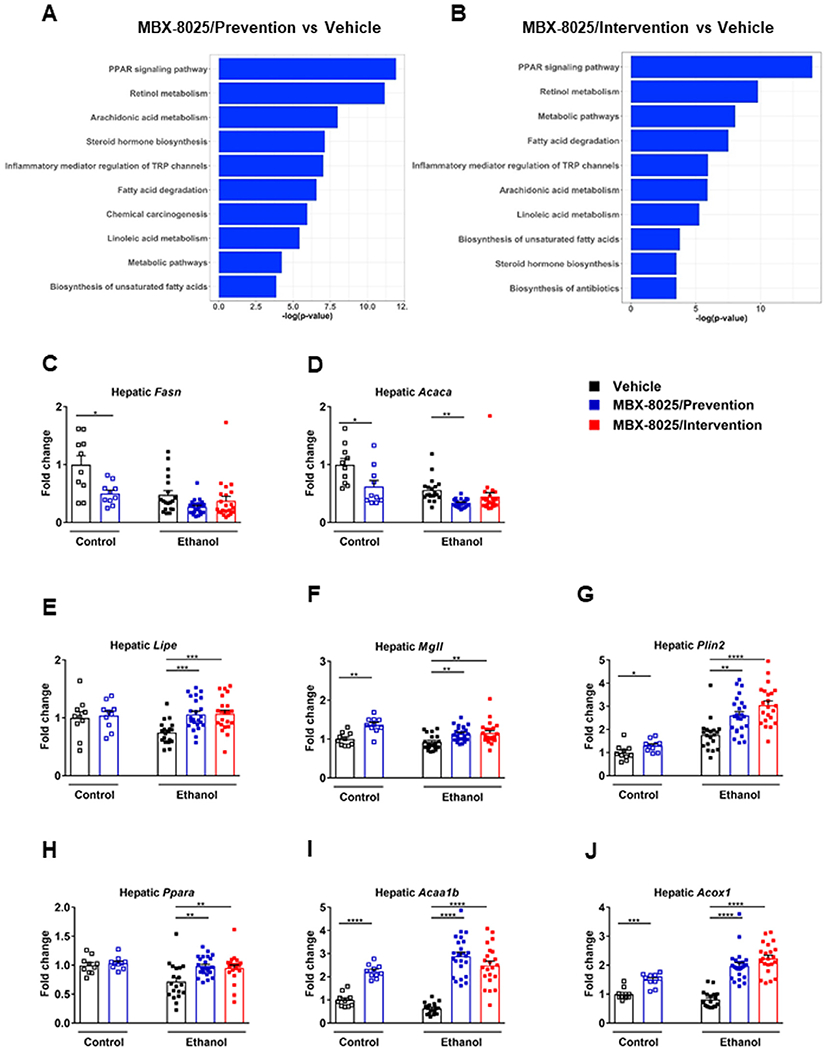
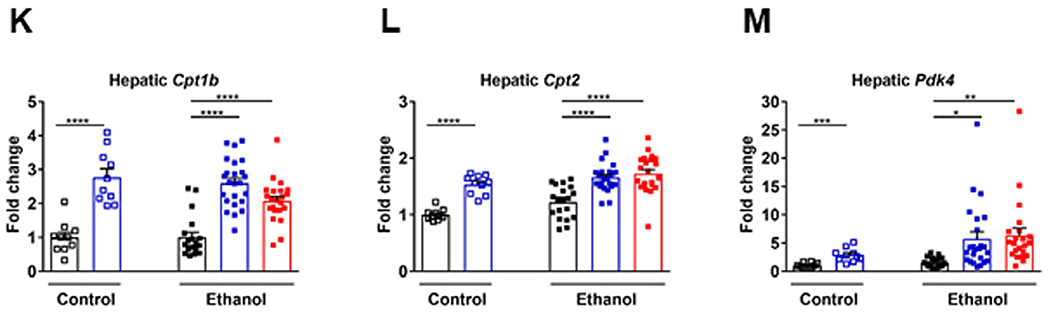
Effect of MBX-8025 on hepatic gene expression. C57BL/6 mice were fed an oral isocaloric (control) diet (1-2 technical replicates) or a chronic Lieber-DeCarli diet for 8 weeks followed by 1 binge of ethanol (3–4 technical replicates), and treated with vehicle, or MBX-8025 (10 mg/kg/d) daily by adding MBX-8025 in the liquid diet during the entire feeding period (prevention) or during the last 4 weeks of ethanol feeding (intervention). (A) Ranking of top 10 biological processes by −log(P value) in the liver of MBX-8025 treated (prevention group) compared with vehicle treated, ethanol-fed mice. (B) Ranking of top 10 biological processes by −log(P value) in the liver of MBX-8025 treated (intervention group) compared with vehicle treated, ethanol-fed mice. (C) Hepatic levels of Fasn mRNA. (D) Hepatic levels of Acaca mRNA. (E) Hepatic levels of Lipe mRNA. (F) Hepatic levels of Mgll mRNA. (G) Hepatic levels of Plin2 mRNA. (H) Hepatic levels of Ppara mRNA. (I) Hepatic levels of Acaa1b mRNA. (J) Hepatic levels of Acox1 mRNA. (K) Hepatic levels of Cpt1b mRNA. (L) Hepatic levels of Cpt2 mRNA. (M) Hepatic levels of Pdk4 mRNA. Control diet: Vehicle, n = 10; MBX-8025/Prevention, n = 10; Ethanol diet: Vehicle, n = 19; MBX-8025/Prevention, n = 24; MBX-8025/Intervention, n = 22. Results are expressed as mean § s.e.m. (C-M). P values were determined using one-way ANOVA with Tukey’s post hoc test for ethanol-fed mouse groups and Mann-Whitney-Wilcoxon test for control diet-fed mouse groups (C-M). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Changes in gene expression related to lipid metabolism were further analyzed. Fatty acid synthesis (FASN) and acetyl CoA carboxylase (ACACA) regulate lipogenesis. MBX-8025 reduced hepatic Fasn mRNA level in the prevention group of control diet-fed mice (Fig 2C), and reduced hepatic Acaca mRNA level in the prevention groups of control and ethanol diet-fed mice (Fig 2D). Both genes were reduced in the MBX-8025 intervention trial although this was not significantly different (Fig 2C and D). Besides, hepatic mRNA level of genes involved in lipolysis, such as lipase E (Lipe), monoacylglycerol lipase (Mgll), and perilipin 2 (Plin2), fatty acid oxidation such as Ppara, acetyl-Coenzyme A acyltransferase 1B (Acaa1b), acyl-CoA oxidase 1 (Acox1), carnitine palmitoyltransferase 1B (Cpt1b) and Cpt2, and mitochondrial metabolism such as pyruvate dehydrogenase kinase 4 (Pdk4) were increased in ethanol-fed mice treated with MBX-8025 in the prevention and intervention approach (Fig 2E–M). In line with published results,28 hepatic Ppard mRNA level was lower in the MBX-8025 intervention group compared to vehicle group following chronic ethanol administration (supplementary Fig 2A), while there was no difference in hepatic Pparg mRNA level among groups in ethanol-fed mice (supplementary Fig 2B).
Intestinal farnesoid X receptor (FXR) /ceramide axis mediates hepatic lipid accumulation in experimental high fat diet-induced fatty liver disease.29 We found no differences in the expression of FXR target genes small heterodimer partner (Shp) and fibroblast growth factor 15 (Fgf15) in ileum among groups in ethanol-fed mice (supplementary Fig 2C and D). In addition, intestinal mRNAs encoded by de novo ceramide synthesis–related genes were not uniformly changed in ethanol-fed mice. Serine palmitoyltransferase, long-chain base subunit 2(Sptlc2) increased (supplementary Fig 2E), while ceramide synthase 4 (Cers4) decreased in both MBX-8025 prevention and intervention groups, while Cers6 decreased in the MBX-8025 intervention group after chronic ethanol feeding (supplementary Fig 2F and G). There was no change in expression of genes involved in ceramide catabolism after MBX-8025 treatment (data not shown).
These results confirm that MBX-8025 has beneficial effects on hepatic lipid metabolism by decreasing lipogenesis and increasing lipolysis and fatty acid oxidation, which likely contributes to its beneficial effect on ethanol-induced liver disease.
MBX-8025 restores bile acid homeostasis during chronic ethanol feeding.
Chronic ethanol administration disrupts bile acid homeostasis, which is characterized by increased systemic levels of bile acids and increased secondary bile acids.20 To assess the effects of MBX-8025 on bile acid metabolism, total bile acids were quantified using a colorimetric assay and bile acid composition was assessed using LC-MS. In line with our previous study,20 ethanol-fed mice showed increased serum total bile acids and an altered composition with an increase in secondary bile acids (Fig 3A–C). In addition, the total bile acid pool (Fig 3D), bile acids in small intestine (Fig 3E) were increased, whereas bile acids in colon were decreased following chronic ethanol administration (Fig 3F). Importantly, MBX-8025 reversed these changes in the prevention and intervention groups following chronic ethanol administration (Fig 3A–F). In addition, total liver bile acids were lower in control diet-fed mice treated with MBX-8025 (prevention) and in ethanol-fed mice treated with MBX-8025 (intervention; Fig 3G).
Fig 3.
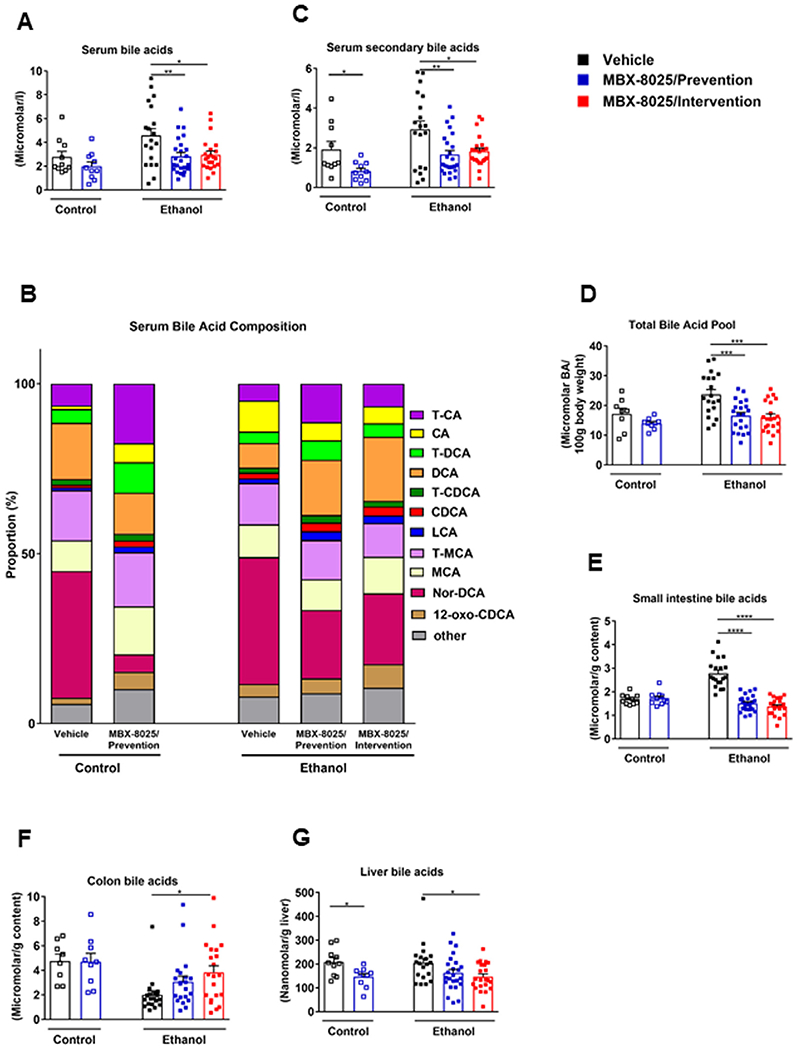
Effect of MBX-8025 on bile acid metabolism. C57BL/6 mice were fed an oral isocaloric (control) diet (1–2 technical replicates) or a chronic Lieber-DeCarli diet for 8 weeks followed by 1 binge of ethanol (3–4 technical replicates), and treated with vehicle, or MBX-8025 (10 mg/kg/d) daily by adding MBX-8025 in the liquid diet during the entire feeding period (prevention) or during the last 4 weeks of ethanol feeding (intervention). (A) Bile acid concentration in serum. (B) Serum bile acid composition ratio. (C) Secondary bile acid concentration in serum. (D) Total bile acid pool. (E) Bile acid concentration in small intestinal contents. (F) Bile acid concentration in colon contents. (G) Bile acid concentration in liver. Control diet: Vehicle, n = 8–10; MBX-8025/ Prevention trial, n = 9–10; Ethanol diet: Vehicle, n = 19; MBX-8025/Prevention, n = 21–24; MBX-8025/Intervention, n = 21–22. Results are expressed as mean ± s.e.m. (A, C-G). P values were determined using one-way ANOVA with Tukey’s post hoc test for ethanol-fed mouse groups and Mann-Whitney-Wilcoxon test for control diet-fed mouse groups (A, C-G). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. CA, cholic acid; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; LCA, lithocholic acid; MCA, muricholic acid; T-CA, taurocholic acid; T-CDCA, taurochenodeoxycholic acid; T-DCA, taurodeoxycholic acid; T-MCA, tauromuricholic acid.
To better understand the effect of MBX-8025 on bile acid metabolism, molecules involved in bile acid synthesis, transport and enterohepatic circulation were analyzed. Expression of enzymes mediating bile acid synthesis, such as hepatic Cyp7a1 protein, Cyp8b1 and Cyp27a1 mRNA were not affected by MBX-8025 (supplementary Fig 3A–C). The bile acid synthesis intermediate C4, which serves as a marker for de novo bile acid synthesis, was reduced in control diet-fed mice treated with MBX-8025 (prevention), but was not significantly different in ethanol-fed mice treated with vehicle or MBX-8025 (supplementary Fig 3D), indicating that a decreased bile acid pool cannot be explained by suppressed bile acid synthesis, but rather through increased intestinal loss of bile acids in MBX-8025 treated mice following chronic ethanol administration (Fig 3D). ABCB11 (also known as bile salt export pump or BSEP) mediates bile acid efflux from hepatocytes into bile.30 Abcb11 mRNA showed no significant difference following MBX-8025 treatment in mice subjected to chronic ethanol administration (data not shown).
SLC10A2 (also known as the apical sodium–bile acid transporter) mediates uptake of luminal bile acids into enterocytes of the terminal ileum. Slc10a2 mRNA showed no significant difference following MBX-8025 treatment in mice subjected to chronic ethanol administration (supplementary Fig 3E), indicating that MBX-8025 did not result in decreased absorption of bile acids in the terminal ileum. Fibroblast growth factor 15 (FGF15) is secreted from enterocytes in the terminal ileum, reaches the liver via the portal vein, and inhibits CYP7A1 and bile acid synthesis.30 Ileal Fgf15 mRNA showed no significant difference following MBX-8025 treatment in mice subjected to chronic ethanol administration (supplementary Fig 2C).
Taken together, MBX-8025 restored bile acid homeostasis during chronic ethanol administration through decreasing the total bile acid pool and secondary bile acids and increasing intestinal excretion of bile acids.
MBX-8025 modulates ethanol-associated dysbiosis.
Gut microbiota is metabolizing primary into secondary bile acids, which determines intestinal absorption of bile acids. Chronic administration of ethanol is associated with intestinal bacterial overgrowth and gut dysbiosis.31 To determine whether MBX-8025 regulates the composition of the intestinal microbiota in ethanol fed mice, the expression of Ppara that is linked to production of antibacterial peptides and changes in microbiota composition was analyzed in the terminal ileum by RNA-sequencing. There was no significant difference in Ppara mRNA in ethanol-fed mice treated with vehicle or MBX-8025 (supplementary Fig 4A). Fecal microbiota was analyzed by 16S ribosomal RNA sequencing. In line with our previous study,31 the main principal components clustered significantly different between ethanol diet-fed and control diet-fed mice (Fig 4A), and between MBX-8025/intervention and vehicle-treated mice following chronic ethanol feeding (Fig 4A). Changes in fecal microbiota composition were noted in MBX-8025 (prevention and intervention) as compared with vehicle treated mice following chronic ethanol administration (Fig 4B). We further analyzed the metabolic function of microbiota using PICRUSt analysis24 and found that the principal components clustered significantly different between MBX-8025/intervention and vehicle-treated mice following chronic ethanol feeding (supplementary Fig 4B). In addition, MBX-8025/intervention mice had decreased levels of 3-dehydro-bile acid Delta4,6-reductase, an enzyme that takes part in the synthesis of secondary bile acid,32 as compared with vehicle-treated mice (supplementary Fig 4C). These results indicate that some of the microbiota changes associated with MBX-8025 administration contribute to reduced metabolism of primary into secondary bile acids that we have observed.
Fig 4.
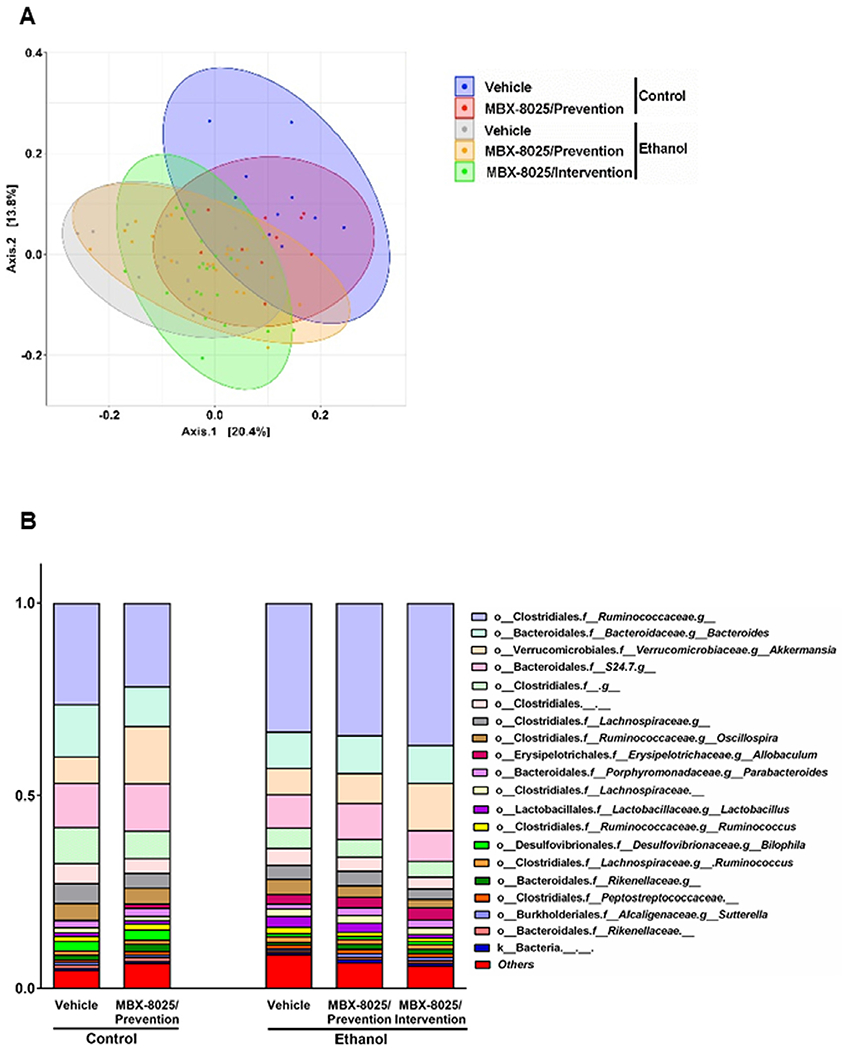
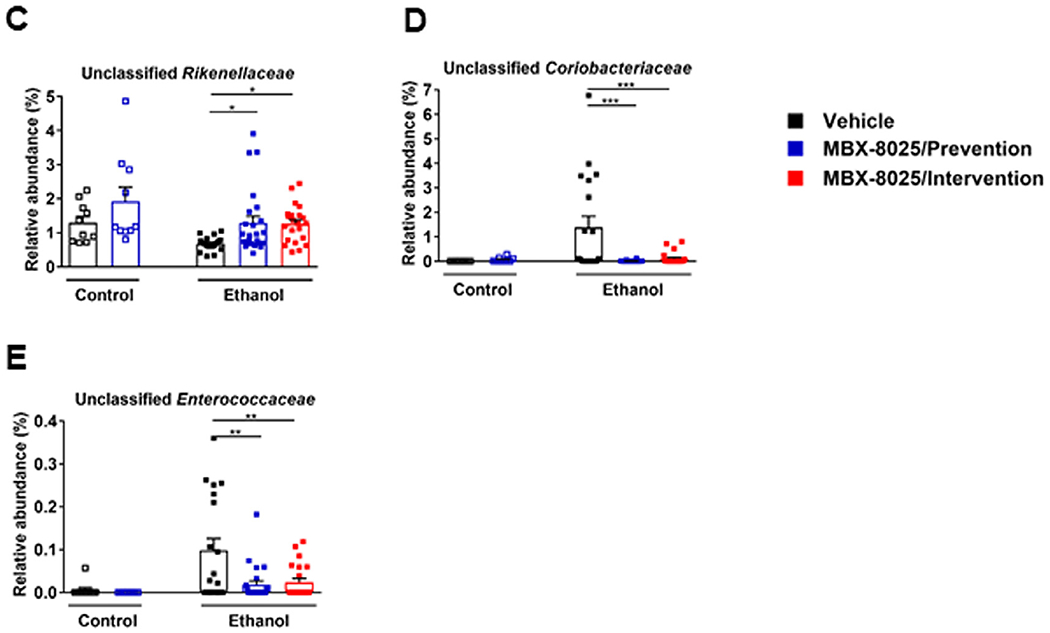
MBX-8025 modulates gut microbiota.C57BL/6 mice were fed an oral isocaloric (control) diet (1–2 technical replicates) or a chronic Lieber-DeCarli diet for 8 weeks followed by 1 binge of ethanol (3–4 technical replicates), and treated with vehicle, or MBX-8025 (10 mg/kg/d) daily by adding MBX-8025 in the liquid diet during the entire feeding period (prevention) or during the last 4 weeks of ethanol feeding (intervention). (A) Principal coordinate analysis (PCoA) based on Unweighted UniFrac distances was used to show-diversity among groups, at the genus level. The axes are the 2 most discriminating axes, based on binary metric; 20.4% of the variances were explained by axis 1 and 13.8% by axis 2. (B) The mean relative abundance of sequence reads in each genus for each group. 0–1 corresponds to 0%–100% abundance. (C) Relative abundance of unclassified Rikenellaceae. (D) Relative abundance of unclassified Coriobacteriaceae. (E) Relative abundance of unclassified Enterococcaceae. Control diet: Vehicle, n = 10; MBX-8025/Prevention, n = 10; Ethanol diet: Vehicle, n = 19; MBX-8025/Prevention, n = 24; MBX-8025/Intervention, n = 22. Results are expressed as mean ± s.e.m. (C–E). P values were determined using one-way ANOVA with Tukey’spost hoc test for ethanol-fed mouse groups and Mann-Whitney-Wilcoxon test for control diet-fed mouse groups (C–E). *P < 0.05, **P < 0.01, ***P < 0.001.
Further analyses showed that some specific taxa that are related to tissue or cell injury and lipid absorption were modified by MBX-8025. Rikenellaceae is one of the hydrogen–producing bacteria, and hydrogen protects against tissue and cell injuries as a unique antioxidant.33 The relative abundance of unclassified Rikenellaceae was decreased in ethanol-fed mice compared to control diet-fed mice, which was reversed by MBX-8025 in both prevention and intervention trials (Fig 4C). Unclassified Coriobacteriaceae negatively impacts cholesterol homeostasis through increasing cholesterol absorption.34 The relative increase of pathogenic family Enterococcaceae is linked to liver failure and endotoxin plasma levels.35 Unclassified Coriobacteriaceae and unclassified Enterococcaceae were overrepresented in the vehicle treated mice fed ethanol diet but was markedly reduced by MBX-8025 in both, the prevention and intervention trials (Fig 4D and E). These results indicate that MBX-8025-associated changes in the gut microbiota might contribute to protection from ethanol-induced liver disease not only by affecting intestinal bile acid metabolism, but also through additional other effects.
MBX-8025 reduces ethanol-induced gut barrier dysfunction.
PPAR-δ is expressed in epithelial cells of small intestine in mice and the PPAR-δ agonist GW501516 can suppress epithelial apoptosis in a dose-dependent manner in vitro.16 PPARβ/δ reduces inflammation in macrophages.36–38 To determine whether MBX-8025 has effects on intestinal permeability in vivo, we functionally assessed the integrity of the gut barrier. Compared with vehicle, MBX-8025 treated mice had improved gut barrier function as shown by reduced fecal albumin level (Fig 5A) and serum lipopolysaccharide (LPS) level following chronic ethanol administration (Fig 5B).
Fig 5.
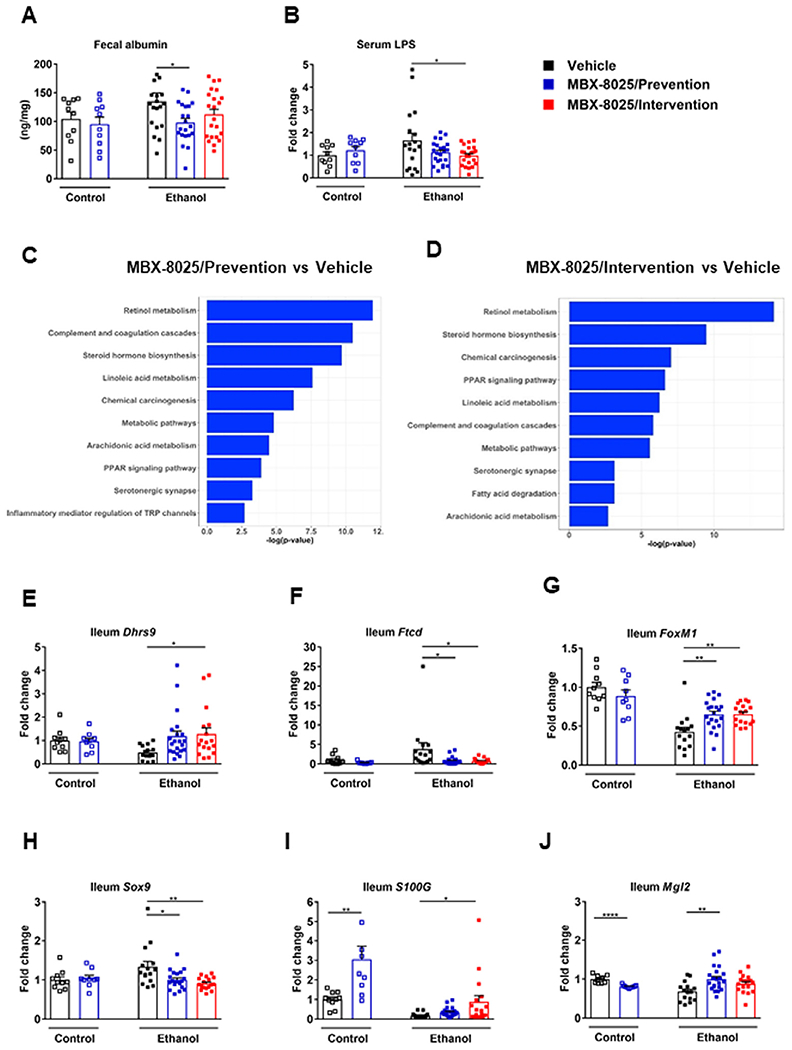
MBX-8025 restores gut barrier function during chronic ethanol feeding. C57BL/6 mice were fed an oral isocaloric (control) diet (1–2 technical replicates) or a chronic Lieber-DeCarli diet for 8 weeks followed by 1 binge of ethanol (3–4 technical replicates), and treated with vehicle, or MBX-8025 (10 mg/kg/d) daily by adding MBX-8025 in the liquid diet during the entire feeding period (prevention) or during the last 4 weeks of ethanol feeding (intervention). (A) Fecal albumin. (B) Serum LPS. (C) Ranking of top 10 biological processes by −log(P value) in the ileum of MBX-8025 treated (prevention group) compared with vehicle treated, ethanol-fed mice. (D) Ranking of top 10 biological processes by −log(P value) in the ileum of MBX-8025 treated (intervention group) compared with vehicle treated, ethanol-fed mice. (E) Ileal Dhrs9 mRNA level. (F) Ileal Ftcd mRNA level. (G) Ileal FoxMl mRNA level. (H) Ileal Sox9 mRNA level. (I) Ileal S100G mRNA level. (J) Ileal Mgl2 mRNA level. Control diet: Vehicle, n = 10; MBX-8025/Prevention, n = 9–10; Ethanol diet: Vehicle, n = 15–19; MBX-8025/Prevention trial, n = 21–23; MBX-8025/Intervention, n = 17–22. Results are expressed as mean ± s.e.m (A, B, E–J). P values were determined using one-way ANOVA with Tukey’s post hoc test for ethanol-fed mouse groups and Mann-Whitney-Wilcoxon test for control diet-fed mouse groups (A–C, F–K). *P < 0.05, **P < 0.01, ****P < 0.0001.
Gene expression in the terminal ileum was analyzed by RNA-sequencing. MBX-8025 activated multiple pathways that are related to the function of intestinal tight junctions, such as retinol metabolism and histidine metabolism (Fig 5C and D). Dehydrogenase/reductase 9 (DHRS9) participates in the conversion of retinol to retinal and is further involved in retinoic acid biosynthesis.39 Retinoic acid can regulate function of tight junctions in intestinal epithelial cells and promote barrier function.40,41 In the present study, ileal Dhrs9 mRNA level increased in MBX-8025 treated mice following chronic ethanol administration (Fig 5E). Formimidoyltransferase cyclodeaminase (FTCD) catalyzes two reactions in the histidine degradation pathway42 and depletion of Ftcd increases the levels of histidine,43 which protects against loss of intestinal tight junction proteins and increase of cytokine mRNA levels induced by copper in the intestine.44 Ileal Ftcd mRNA level was decreased in both MBX8025 prevention and intervention trials compared with vehicle following chronic ethanol administration (Fig 5F). MBX-8025 also regulated epithelial cell proliferation. Forkhead box M1 (FoxM1) is expressed in the intestinal mucosa and is involved in the proliferation of intestinal epithelial cells following ischemia/reperfusion injury.45 Ileal FoxM1 mRNA level was increased in both MBX-8025 prevention and intervention groups as compared with vehicle following chronic ethanol feeding (Fig 5G). SRY-box transcription factor 9 (Sox9) is a downstream target of Wnt signaling and represses caudal type homeobox 2 which is associated with epithelial cell differentiation.46 Sox9 mRNA level was decreased in MBX-8025 treated mice as compared with vehicle after ethanol feeding (Fig 5H). MBX-8025 also regulated pathways that are related to intestinal immunity and inflammation, such as complement and coagulation cascades, inflammatory mediator regulation of TRP channels, and cytokine-cytokine receptor interaction (Fig 5C and D). S100 calcium binding protein G (S100G) inhibits NF-κB activation and suppresses the production of monocyte chemotactic protein-1, suggesting its role as an important anti-inflammatory mediator in fibroblasts.47 Macrophage galactose (Gal)-type C-type lectin 2 (Mgl2) plays an important role in inhibiting inflammation.37 S100G mRNA and Mgl2 mRNA level were increased in MBX-8025 intervention trial and MBX-8025 prevention trial, respectively, in ethanol fed mice (Fig 5I and J).
These data indicated that functional improvement in gut barrier function by MBX-8025 likely contributes to protection from ethanol-induced liver disease.
DISCUSSION
PPARδ agonists attenuate ethanol-induced liver disease via reducing insulin resistance and endoplasmic reticulum (ER) stress in the liver.14,15 Since PPARδ is expressed in the intestine, we tested whether the specific PPARδ agonist MBX-8025 (seladelpar) affects gut barrier function and intestinal microbiota. We confirmed that MBX-8025 has a beneficial effect on hepatic lipid metabolism by decreasing lipid synthesis and increasing lipolysis and fatty acid oxidation. This is likely one of the main mechanisms for protection from ethanol-induced steatohepatitis. MBX-8025 showed several other effects that could mechanistically be involved in reducing ethanol-induced liver damage. First, MBX-8025 restores bile acid homeostasis, which is likely mediated by modulating the intestinal microbiota. MBX-8025 also improved gut barrier function during chronic ethanol administration. Finally, hepatic levels of Adh1 increased after MBX-8025 treatment, which might have contributed to protection from ethanol-induced liver disease. MBX-8025 may increase the level of Adh1 directly.
Our data indicates that MBX-8025 restored bile acid homeostasis by reducing bile acid pool and total serum bile acids. Reducing an increased bile acid pool associated with chronic ethanol feeding, is known to ameliorate ethanol-induced liver disease in mice.20 Patients with alcohol use disorder or alcoholic hepatitis have increased serum bile acid levels.48 The total bile acid pool is balanced by de novo bile acid synthesis in the liver and loss of bile acids via the feces. While de novo bile acid synthesis was largely unaffected following MBX-8025 treatment, intestinal excretion of bile acids was increased. This could be facilitated by MBX-8025-mediated functional changes in the gut microbiota, which is known to metabolize primary to secondary bile acids in the intestine. However, it is also possible that changes in intestinal bile acids might in turn modulate the microbiota. MBX-8025 may also maintain bile acid homeostasis by directly regulating and lowering hepatic cholesterol metabolism following chronic ethanol feeding. Cholesterol stimulates bile acid synthesis via liver X receptor (LXR).49,50 Thus, less hepatic cholesterol would not induce bile acid synthesis in MBX-8025 treated and ethanol-fed mice, thereby lowering the total bile acid pool. A cholesterol lowering effect of MBX-8025 has been observed clinically in the plasma or serum of patients with primary biliary cholangitis (PBC)51,52 and in liver cholesterol in a mouse model of nonalcoholic steatohepatitis.12
Stabilizing gut barrier function plays an important role in the treatment of ethanol-induced liver disease in mice.53 Our current study creates a link between improving gut barrier function and PPARδ. The regulation of gut barrier dysfunction by PPARβ/δ agonist can be caused by different mechanisms. PPARβ/δ activation in macrophages can inhibit the expression of monocyte chemotactic protein −1 and interleukin 1 beta (IL-1b) that can promote inflammation.36 PPARβ/δ activation also increases expression of anti-inflammatory genes including Mgl1, Mgl2, and Mannose Receptor C Type 2 (Mrc2),37 and upregulates expression of opsonin genes (complement C1q A Chain (C1qa), C1qb, C1qc, growth arrest-specific 6 (Gas6), milk fat globule-EGF factor 8, and thrombospondin 1) that regulate phagocytes to recognize and clear apoptotic bodies.38 Thus, PPARβ/δ activation might limit inflammation in macrophages. In addition, PPARβ/δ activation also can activate anti-apoptotic pathways and promote proliferation in intestinal epithelial cells.16 Our study confirmed that PPARδ agonist MBX-8025 inhibited the expression of Ftcd and Sox9 that are involved in reducing tight junction proteins, increasing inflammation, and inhibiting proliferation of epithelial cells, and upregulate the expression of Dhrs9, FoxMl, S100G, and Mgl2 that promote gut barrier function and have anti-inflammatory properties. This might contribute to the restoration of gut barrier function. Finally, reducing intestinal bacterial overgrowth and dysbiosis,53 or changing intestinal bile acid composition could also contribute to restored gut barrier function.20 Taken together, MBX-8025 might restore gut barrier integrity via different direct and indirect mechanisms.
Intervention with MBX-8025 appears to be more effective than the prevention treatment. The exact reason for this observation is not known, but it is possible that ethanol changes typical PPARδ-responsive genes when the timing of MBX-8025 is modified.
In summary, our study demonstrates that MBX-8025 reduced ethanol-induced liver disease by improving hepatic lipid metabolism, regulating bile acid metabolism, and attenuating gut barrier dysfunction and dysbiosis.
Supplementary Material
ACKNOWLEDGMENTS
Conflicts of Interest: B.S. is consulting for Ferring Research Institute. This study was supported by a laboratory service agreement from CymaBay to the University of California San Diego. E.C. is an employee from CymaBay Therapeutics (Newark, CA). All authors have read the journal’s authorship agreement and policy.
The study was also supported by the NIDDK-funded San Diego Digestive Diseases Research Center (P30 DK120515) and by the NIAAA-funded Southern California Research Center for ALPD and Cirrhosis (P50 AA011999).
Abbreviations:
- ABCB11
ATP-binding cassette, sub-family B member 11
- ASBT
apical sodium–bile acid transporter
- Acaa1b
acetyl-Coenzyme A acyltransferase 1B
- ACACA
acetyl CoA carboxylase
- Acox1
acyl-CoA oxidase 1
- Adh1
alcohol dehydrogenase 1
- ALT
alanine aminotransferase
- ANOVA
analysis of variance
- AST
aspartate aminotransferase
- BSEP
bile salt export pump
- C1qa
complement C1q A chain
- Cdx2
caudal type homeobox 2
- Cpt1b
carnitine palmitoyltransferase 1B
- Cxcl
chemokine (C-X-C motif) ligand
- Cyp2e1
cytochrome P450 family 2 subfamily E polypeptide 1
- DHRS9
Dehydrogenase/reductase 9
- ER
endoplasmic reticulum
- FASN
fatty acid synthesis
- FGF15
fibroblast growth factor 15
- FoxM1
forkhead box M1
- FTCD
formimidoyltransferase cyclodeaminase
- Gas6
growth arrest-specific 6
- IL-1β
interleukin 1 beta
- LC-MS
liquid chromatography-mass spectrometry
- Lipe
lipase E
- LPS
lipopolysaccharide
- LXR
liver X receptor
- MCP-1
monocyte chemotactic protein-1
- Mfge8
milk fat globule-EGF factor 8
- Mgl2
macrophage galactose(Gal)-type-C-type lectin 2
- Mgll
monoacylglycerol lipase
- Mrc2
Mannose Receptor C Type 2
- NASH
nonalcoholic steatohepatitis
- Pdk4
pyruvate dehydrogenase kinase 4
- Plin2
perilipin 2
- PPARs
peroxisome proliferator-activated receptors
- rRNA
ribosomal RNA
- S100G
S100 calcium binding protein G
- SLC10A2
solute carrier family 10 member 2
- Sox9
SRY-box transcription factor 9
- Thbs1
thrombospondin 1
Footnotes
SUPPLEMENTARY MATERIALS
Supplementary material associated with this article can be found in the online version at doi:10.1016/j.trsl.2020.06.006.
REFERENCES
- 1.Moon AM, Singal AG, Tapper EB. Contemporary epidemiology of chronic liver disease and cirrhosis. Clin Gastroenterol Hepatol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crabb DW, Im GY, Szabo G, Mellinger JL, Lucey MR. Diagnosis and treatment of alcohol-associated liver diseases: 2019 practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2020;71:306–33. [DOI] [PubMed] [Google Scholar]
- 3.Dang K, Hirode G, Singal AK, Sundaram V, Wong RJ. Alcoholic liver disease epidemiology in the United States: a retrospective analysis of 3 US databases. Am J Gastroenterol 2020;115:96–104. [DOI] [PubMed] [Google Scholar]
- 4.Kim D, Li AA, Perumpail RB, et al. Disparate trends in mortality of etiology-specific chronic liver diseases among hispanic sub-populations. Clin Gastroenterol Hepatol 2019;17:1607–15 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dunn W, Shah VH. Pathogenesis of alcoholic liver disease. Clin Liver Dis 2016;20:445–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 2011;141:1572–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osna NA, Donohue TM Jr., Kharbanda KK. Alcoholic liver disease: pathogenesis and current management. Alcohol Res 2017;38:147–61. [PMC free article] [PubMed] [Google Scholar]
- 8.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev 1999;20:649–88. [DOI] [PubMed] [Google Scholar]
- 9.Giordano Attianese GM, Desvergne B. Integrative and systemic approaches for evaluating PPARbeta/delta (PPARD) function. Nucl Recept Signal 2015;13:e001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tian XY, Wong WT, Wang N, et al. PPARdelta activation protects endothelial function in diabetic mice. Diabetes 2012;61:3285–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi YJ, Roberts BK, Wang X, et al. Effects of the PPAR-delta agonist MBX-8025 on atherogenic dyslipidemia. Atherosclerosis 2012;220:470–6. [DOI] [PubMed] [Google Scholar]
- 12.Haczeyni F, Wang H, Barn V, et al. The selective peroxi-some proliferator-activated receptor-delta agonist seladelpar reverses nonalcoholic steatohepatitis pathology by abrogating lipotoxicity in diabetic obese mice. Hepatol Commun 2017;1:663–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iwaisako K, Haimerl M, Paik YH, et al. Protection from liver fibrosis by a peroxisome proliferator-activated receptor delta agonist. Proc Natl Acad Sci U S A 2012;109:E1369–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramirez T, Tong M, Chen WC, Nguyen QG, Wands JR, de la Monte SM. Chronic alcohol-induced hepatic insulin resistance and endoplasmic reticulum stress ameliorated by peroxisome-proliferator activated receptor-delta agonist treatment. J Gastroenterol Hepatol 2013;28:179–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de la Monte SM, Pang M, Chaudhry R, et al. Peroxisome proliferator-activated receptor agonist treatment of alcohol-induced hepatic insulin resistance. Hepatol Res 2011;41:386–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gupta RA, Wang D, Katkuri S, Wang H, Dey SK, DuBois RN. Activation of nuclear hormone receptor peroxisome proliferator-activated receptor-delta accelerates intestinal adenoma growth. Nat Med 2004;10:245–7. [DOI] [PubMed] [Google Scholar]
- 17.Barish GD, Narkar VA, Evans RM. PPAR delta: a dagger in the heart of the metabolic syndrome. J Clin Invest 2006;116:590–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang AM, Inamine T, Hochrath K, et al. Intestinal fungi contribute to development of alcoholic liver disease. J Clin Invest 2017;127:2829–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu MJ, Cai Y, Wang H, et al. Fat-specific protein 27/CIDEC promotes development of alcoholic steatohepatitis in mice and humans. Gastroenterology 2015;149:1030–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartmann P, Hochrath K, Horvath A, et al. Modulation of the intestinal bile acid/farnesoid X receptor/fibroblast growth factor 15 axis improves alcoholic liver disease in mice. Hepatology 2018;67:2150–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jung CE, Chopyk J, Shin JH, et al. Benchmarking urine storage and collection conditions for evaluating the female urinary microbiome. Sci Rep 2019;9:13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010;7:335–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeSantis TZ, Hugenholtz P, Larsen N, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 2006;72:5069–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langille MG, Zaneveld J, Caporaso JG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 2013;31:814–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 2017;14:417–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang DW, Sherman BT, Tan Q, et al. The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol 2007;8:R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pang M, de la Monte SM, Longato L, et al. PPARdelta agonist attenuates alcohol-induced hepatic insulin resistance and improves liver injury and repair. J Hepatol 2009;50:1192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang C, Xie C, Li F, et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J Clin Invest 2015;125:386–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li T, Chiang JY. Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev 2014;66:948–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Llorente C, Jepsen P, Inamine T, et al. Gastric acid suppression promotes alcoholic liver disease by inducing overgrowth of intestinal Enterococcus. Nat Commun 2017;8:837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harris SC, Devendran S, Alves JMP, Mythen SM, Hylemon PB, Ridlon JM. Identification of a gene encoding a flavoprotein involved in bile acid metabolism by the human gut bacterium Clostridium scindens ATCC 35704. Biochim Biophys Acta Mol Cell Biol Lipids 2018;1863:276–83. [DOI] [PubMed] [Google Scholar]
- 33.Ohsawa I, Ishikawa M, Takahashi K, et al. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat Med 2007;13:688–94. [DOI] [PubMed] [Google Scholar]
- 34.Martinez I, Wallace G, Zhang C, et al. Diet-induced metabolic improvements in a hamster model of hypercholesterolemia are strongly linked to alterations of the gut microbiota. Appl Environ Microbiol 2009;75:4175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanduzzi Zamparelli M, Rocco A, Compare D, Nardone G. The gut microbiota: a new potential driving force in liver cirrhosis and hepatocellular carcinoma. United Eur Gastroenterol J 2017;5:944–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee CH, Chawla A, Urbiztondo N, et al. Transcriptional repression of atherogenic inflammation: modulation by PPARdelta. Science 2003;302:453–7. [DOI] [PubMed] [Google Scholar]
- 37.Kang K, Reilly SM, Karabacak V, et al. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab 2008;7:485–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mukundan L, Odegaard JI, Morel CR, et al. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med 2009;15:1266–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szatmari I, Pap A, Ruhl R, et al. PPARgamma controls CD1d expression by turning on retinoic acid synthesis in developing human dendritic cells. J Exp Med 2006;203:2351–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abdelhamid L, Luo XM. Retinoic acid, leaky gut, and autoimmune diseases. Nutrients 2018, 1016;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Osanai M, Nishikiori N, Murata M, Chiba H, Kojima T, Sawada N. Cellular retinoic acid bioavailability determines epithelial integrity: Role of retinoic acid receptor alpha agonists in colitis. Mol Pharmacol 2007;71:250–8. [DOI] [PubMed] [Google Scholar]
- 42.Mao Y, Vyas NK, Vyas MN, et al. Structure of the bifunctional and Golgi-associated formiminotransferase cyclodeaminase octamer. EMBO J 2004;23:2963–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kanarek N, Keys HR, Cantor JR, et al. Histidine catabolism is a major determinant of methotrexate sensitivity. Nature 2018;559:632–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang WD, Qu B, Feng L, et al. Histidine prevents cu-induced oxidative stress and the associated decreases in mRNA from encoding tight junction proteins in the intestine of grass carp (ctenopharyngodon idella). PLoS One 2016;11:e0157001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zu G, Guo J, Zhou T, et al. The transcription factor FoxM1 activates Nurr1 to promote intestinal regeneration after ischemia/reperfusion injury. Exp Mol Med 2019;51:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blache P, van de Wetering M, Duluc I, et al. SOX9 is an intestine crypt transcription factor, is regulated by the Wnt pathway, and represses the CDX2 and MUC2 genes. J Cell Biol 2004;166:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ishiguro K, Watanabe O, Nakamura M, et al. S100G expression and function in fibroblasts on colitis induction. Int Immunopharmacol 2016;39:92–6. [DOI] [PubMed] [Google Scholar]
- 48.Brandl K, Hartmann P, Jih LJ, et al. Dysregulation of serum bile acids and FGF19 in alcoholic hepatitis. J Hepatol 2018;69:396– 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peet DJ, Turley SD, Ma W, et al. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell 1998;93:693–704. [DOI] [PubMed] [Google Scholar]
- 50.Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 1996;383:728–31. [DOI] [PubMed] [Google Scholar]
- 51.Bays HE, Schwartz S, Littlejohn T, et al. MBX-8025, a novel peroxisome proliferator receptor-delta agonist: lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin. J Clin Endocrinol Metab 2011;96:2889–97. [DOI] [PubMed] [Google Scholar]
- 52.Jones D, Boudes PF, Swain MG, et al. Seladelpar (MBX-8025), a selective PPAR-delta agonist, in patients with primary biliary cholangitis with an inadequate response to ursodeoxycholic acid: a double-blind, randomised, placebo-controlled, phase 2, proof-of-concept study. Lancet Gastroenterol Hepatol 2017;2:716–26. [DOI] [PubMed] [Google Scholar]
- 53.Chen P, Starkel P, Turner JR, Ho SB, Schnabl B. Dysbiosis-induced intestinal inflammation activates tumor necrosis factor receptor I and mediates alcoholic liver disease in mice. Hepatology 2015;61:883–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.