


Abstract
Poly(ADP-ribosyl)ation regulates numerous cellular processes like genome maintenance and cell death, thus providing protective functions but also contributing to several pathological conditions. Poly(ADP-ribose) (PAR) molecules exhibit a remarkable heterogeneity in chain lengths and branching frequencies, but the biological significance of this is basically unknown. To unravel structure-specific functions of PAR, we used PARP1 mutants producing PAR of different qualities, i.e. short and hypobranched (PARP1\G972R), short and moderately hyperbranched (PARP1\Y986S), or strongly hyperbranched PAR (PARP1\Y986H). By reconstituting HeLa PARP1 knockout cells, we demonstrate that PARP1\G972R negatively affects cellular endpoints, such as viability, cell cycle progression and genotoxic stress resistance. In contrast, PARP1\Y986S elicits only mild effects, suggesting that PAR branching compensates for short polymer length. Interestingly, PARP1\Y986H exhibits moderate beneficial effects on cell physiology. Furthermore, different PARP1 mutants have distinct effects on molecular processes, such as gene expression and protein localization dynamics of PARP1 itself, and of its downstream factor XRCC1. Finally, the biological relevance of PAR branching is emphasized by the fact that branching frequencies vary considerably during different phases of the DNA damage-induced PARylation reaction and between different mouse tissues. Taken together, this study reveals that PAR branching and chain length essentially affect cellular functions, which further supports the notion of a ‘PAR code’.
INTRODUCTION
The post-translational modification poly(ADP-ribosyl)ation (PARylation) plays key roles in cellular physiology and stress response. The modification is catalyzed by members of the family of poly(ADP-ribose) polymerases (PARPs, also known as ARTDs) (1). It is involved in a host of cellular functions including genome maintenance, transcription, chromatin remodeling, stress response and regulation of cell death (2). Importantly, PARPs represent promising targets in cancer therapy, either as chemosensitizers in combination with classical DNA damaging chemotherapeutics or as stand-alone drugs for tumors deficient in homologous recombination, following the concept of synthetic lethality (3,4).
The PARP gene family consists of 17 homologs in humans, where at least four of the encoded enzymes produce poly(ADP-ribose) (PAR) by using NAD+ as a substrate, while the remaining members catalyze mono- and oligo(ADP-ribosyl)ation, or are catalytically inactive (5). Of those that produce PAR, PARP1 and PARP2 localize to the nucleus and can be activated by several forms of DNA damage, but in particular by DNA strand breaks. PARP1, which is responsible for ∼90% of genotoxic stress-induced PAR formation, produces PAR chains of up to 200 ADP-ribosyl units, which can be either linear or branched, and which are linked via α-O-glycosidic (1″-2′) ribose-ribose bonds or α-O-glycosidic (1″′-2″) ribose-ribose bonds, respectively (6). After PARP1 activation, target proteins are covalently modified with PAR at serine, glutamate, aspartate, lysine and tyrosine residues (7–12). One major target of PARylation is PARP1 itself (i.e. ‘PARP1 automodification’) (13), leading to changes in PARP1’s DNA binding ability and its catalytic activity (14,15). It has been reported that PARP1 can be active as a monomer or a dimer, resulting in PARylation in cis or in trans (16–22). Apart from covalent modification, proteins can also be regulated via non-covalent binding to PAR chains (23,24). Several different PAR binding motifs were identified, which mediate high-affinity PAR–protein interactions (25). Among those are the ‘classical’ PAR-binding motifs (PBMs) (26), as well as glycine- and arginine-rich domains (GAR) (27), PAR-binding zinc fingers (PBZ) (28), macrodomains (29) or WWE-domains (30). Like other post-translational modifications, PARylation is tightly regulated by catabolizing enzymes such as PARG and others, which hydrolyze PAR shortly after its synthesis (5,31). Thereby, physico-chemical properties, as well as spatio-temporal activities of hundreds of target proteins are dynamically regulated. PARP1-dependent PARylation is linked to pleiotropic cellular functions, yet in particular to the DNA damage response. Therefore, PARP1 was proposed as a caretaker of genomic stability (32,33). The recruitment of PARP1 to DNA lesions is one of the fastest cellular responses upon genotoxic stress and PARP1-mediated PARylation orchestrates many downstream DNA damage response processes (14,34,35). Furthermore, PARP1 is involved in chromatin regulation (33,36), telomere maintenance (37,38), replication (39), cell cycle control (40), inflammation and immunity (41,42) and cell death (43).
Biochemical data support the existence of a so-called ‘PAR code’, i.e. the hypothesis that PAR chains of specific lengths and branching frequencies exert specific functions (44–48). Thus, some proteins, such as histones, p53 or XPA, prefer binding to PAR chains of specific lengths (44,46,49,50). Moreover, PAR branching may serve as a recognition site for non-covalent protein binding and it has been reported that certain PAR-binding proteins, such as histones, show a preference for branched PAR (49). Intriguingly, a recent study revealed that the tandem PBZ domains of the histone chaperone APLF specifically recognize PAR branching points, which appears to be important for the removal of histone H3 upon DNA damage (51). Furthermore, the latter study also showed that PARP2 produces branched PAR chains at higher frequency compared to PARP1 (51), while on the contrary, TNKS1 was reported to produce only linear PAR chains (52). In addition to PARP isoform-specific effects, PAR branching and chain length may also be regulated by intracellular NAD+ levels (53) as well as post-translational modifications, which has not been evaluated so far. Moreover, not only protein binding properties appear to be influenced by PAR chain length and branching, but also the stability of the polymer is potentially determined by its structure. Thus, previous results suggested that long PAR molecules are degraded faster by PAR catabolizing enzymes than shorter ones (54). Furthermore, PAR branching may lead to stabilization of the PAR structure, since PARG appears to prefer linear to branched PAR for degradation (55,56). Despite these interesting initial findings, the cellular and organismic relevance of the heterogeneity in PAR chain length and branching is largely unexplored.
Here, we address the role of PAR heterogeneity on cellular physiology by combining a genetic approach with the analysis of select cellular and biochemical endpoints. To this end, we reconstituted HeLa PARP1 knock-out (KO) cells (34) with different PARP1 variants, which produce PAR of different qualities with respect to chain length and branching (57). We chose three different variants that were reported to synthesize either short PAR (PARP1\Y986S), hyperbranched PAR (PARP1\Y986H) or hypobranched PAR (PARP1\G972R) (57) and performed a comprehensive analysis of the biochemical properties of PAR in vitro and in cells, as well as of the resulting cellular consequences.
MATERIALS AND METHODS
Purification of recombinant PARP1 variants
Human recombinant (rec.) PARP1\WT, PARP1\G972R, PARP1\Y986H and PARP1\Y986S were purified as described previously (15,58), with some modifications. All steps were performed at 4°C. Bacterial pellets [Escherichia coli Rosetta 2 (DE3)] were resuspended in 25 mM HEPES (pH 8.0), 500 mM NaCl, 0.5 mM DTT and 10 mM benzamide and stored at –80°C. After thawing, the bacterial suspension was supplemented with 0.1% NP-40, Complete protease inhibitor (Roche), and 1 mg/ml lysozyme. This was followed by sonication (4 × 20 pulses), the addition of 10 μg/ml DNaseI and incubation for 1 h. Cellular debris was removed by centrifugation (2 h at 50 000 × g). The supernatant was filtered and injected into an ÄKTA FPLC system equipped with a 1-ml HisTrap HP column (GE Healthcare). This was followed by washing with 10 ml low-salt buffer [25 mM HEPES (pH 8.0), 500 mM NaCl], 10 ml high-salt buffer [25 mM HEPES (pH 8.0), 1 M NaCl] and again 10 ml low-salt buffer. Finally, PARP1 was eluted with 30 ml elution buffer [25 mM HEPES (pH 8.0), 500 mM NaCl, 500 mM imidazole]. In the next step, the eluted protein fractions were diluted with 10 ml no-salt heparin buffer [50 mM Na-phosphate (pH 7), 1 mM EDTA] and loaded onto a 1-ml heparin HP column (GE Healthcare). PARP1 was eluted with 30 ml NaCl using a gradient up to a concentration of 1 M. PARP1 containing fractions were further purified via size-exclusion chromatography (HiLoad 16/600 Superdex 200, GE Healthcare) in 50 mM Tris (pH 8.0), 150 mM NaCl and 0.5 mM DTT at a flow rate of 0.3 ml/min.
Differential scanning fluorimetry
Melting temperatures of rec. PARP1 variants were determined as described previously (59). Briefly, 0.1 mg/ml PARP1 was incubated for 10 min at room temperature (RT) in 50 mM Tris (pH 8.0), 150 mM NaCl and 0.5 mM DTT. To analyze the effect of DNA/NAD+ binding and/or auto-PARylation, the samples were supplemented with 5 mM MgCl2, 5 μM EcoRI double-stranded DNA linker oligonucleotide (TTCCGGAA) and/or 100 μM NAD+ and incubated for 20 min at RT. All samples were mixed with SYPRO Orange solution (ThermoFisher) and fluorescence signals were measured in a quantitative real-time CFX connect thermocycler (BioRad, TET channel) using a gradient from 20°C to 95°C at 1°C/min steps.
In-vitro PAR synthesis and purification
PAR of PARP1\WT and mutants was synthesized and purified as previously described (23,44). Rec. PARP1 (150 nM) was incubated in 100 mM Tris (pH 7.8), 10 mM MgCl2 and 1 mM DTT supplemented with 300 μg/ml histone H2a, 50 μg/ml EcoRI double-stranded DNA activator oligonucleotide (TTCCGGAA) and 1 mM NAD+ in a total volume of 5 ml at 37°C for 45 min to produce PAR. The reaction was stopped by the addition of 5 ml ice-cold 20% TCA, followed by incubation on ice for 15 min. PAR chains were separated from proteins by alkaline treatment (0.5 M KOH, 50 mM EDTA) for 10 min. Afterwards, pH was adjusted to 7.5–8.0 and DNA was digested by the addition of 50 mM MgCl2 and 50 μg/ml DNaseI and subsequent incubation at 37°C for 2 h. Proteins were digested via the addition of 1 mM CaCl2 and 50 μg/ml proteinase K and subsequent incubation at 37°C overnight. The next day, PAR was further purified via two steps of phenol/chloroform/isoamyl-alcohol (Roth) extraction and one step of chloroform extraction followed by ethanol precipitation. To remove residual phenol, buffer was exchanged via Vivacon 2 columns (2000 MWCO, Sartorius).
Mass spectrometric analysis of in-vitro generated PAR
Purified PAR was digested with alkaline phosphatase and phosphodiesterase to nucleosides. Digested PAR (2.5 pmol) was subjected to UPLC–MS/MS analysis and adenosine (Ado), ribosyl-adenosine (R-Ado) and diribosyladenosine (R2-Ado) were analyzed as described previously (60,61).
Analysis of chain length distribution of in-vitro generated PAR via HPLC
Purified PAR was separated via an Agilent 1100 HPLC system equipped with a DNA Pac PA-100 (ThermoFisher) analytical column. PAR (100 μl of a 100-μM solution) was loaded and eluted with increasing NaCl concentrations as published previously (23,44). Buffer A consisted of 25 mM Tris pH 9.0 and buffer B consisted of 25 mM Tris pH 9.0 and 1 M NaCl. The program was set as follows: 0 min (0% B), 3 min (20% B), 20 min (35% B), 40 min (42% B), 70 min (47% B), 110 min (53% B), 120 min (61% B), 131 min (70% B), 132 min (100% B), 152 min (100% B). Elution of PAR was detected by measuring the absorbance at 258 nm.
PARP1 automodification assay
Rec. PARP1 (1 pmol) was pre-incubated in 9 μl buffer [50 mM Tris (pH 8), 150 mM NaCl, 5 mM MgCl2, 0.5 mM DTT] in the presence or absence of 50 pmol EcoRI double-stranded DNA activator oligonucleotide (TTCCGGAA) for 10 min at 25°C. The PARylation reaction was started by the addition of 1 μl [TAMRA-labeled NAD+]/[NAD+] (1/10) at a final concentration of 100 μM (62). Reactions were stopped by the addition of SDS loading dye and subsequent heating at 95°C. Samples were separated on gradient gels (4–20%, Bio-Rad) and visualized on a Typhoon FLA 9000 bioimager (GE Healthcare).
PAR degradation assay
The degradation of PAR by PARG was analyzed using a previously published method (63). Briefly, 5 nmol PAR was pre-incubated for 5 min at 37°C in 55 μl PARG assay buffer [50 mM KCl, 50 mM KH2PO4 (pH 7.2)]. The reaction was started by the addition of 10 μl rec. human PARG (AdipoGen) to a final concentration of 60 pg/μl. Reactions were stopped simultaneously by the addition of 48 μl 2.5 M KOH and 48 μl 250 mM benzamidine and subsequent incubation at 110°C for 10 min. After cooling, samples were centrifuged for 5 min at 17 000 × g. A volume of 50 μl was loaded in triplicates into a 96-well half-area plate (Corning) and analyzed via a TECAN infinite F200 PRO plate reader (λex = 340 nm/λem = 440 nm). To quantify the generated ADP-ribose units, a calibration curve was included in each experiment. Therefore, 65 μl of ADP-ribose of different concentrations (0, 4, 8, 12, 20, 40, 60, 80 μM) was supplemented with 48 μl 2.5 M KOH and 48 μl 250 mM benzamide and treated the same way as the PAR samples.
Purification of cellular PAR for mass spectrometric analysis
HeLa WT and PARP1 KO cells were seeded in 6-well plates and transfected with different eGFP-N1::PARP1 constructs. Forty hours after transfection, cells were treated either with 500 μM H2O2 for 5 min or not treated. Cells were lysed by addition of ice-cold TCA, PAR was purified, digested to nucleosides, and R-Ado and R2-Ado were quantified as described previously (60,61).
Cell culture, transient transfection, and treatment with PARP inhibitor
HeLa WT and PARP1 KO cells were cultured at 37°C, 5% CO2 and 95% humidity in DMEM (Gibco) supplemented with 10% fetal bovine serum (Biochrom), 90 U/ml penicillin, 90 μg/ml streptomycin (Gibco) and 2 mM glutamine (Gibco). For transient transfections with the PARP1-eGFP constructs, Effectene transfection reagent (Qiagen) was used according to the manufacturer's instruction, except for the amount of Effectene, which was reduced by 50%. Treatment with 10 μM ABT888 (Selleckchem) was performed immediately before transfection and cells were incubated with ABT888 until harvesting. To determine transfection efficiencies, transfected HeLa PARP1 KO cells were harvested with trypsin-EDTA (Gibco), pelleted by centrifugation, and resuspended either in phenolred-free medium or in FACS-buffer [PBS, 0.5 mM EDTA, 1% (v/v) BSA]. Samples were analyzed using FACSCalibur (BD), FACSFortessa (BD) or FACSVerse (BD) instruments, depending on the experiment.
CRISPR/Cas9-mediated generation of a PARP1/XRCC1 double KO cell line
To delete functionality of the XRCC1 gene locus, gRNA oligonucleotides were designed to target exon 1 of XRCC1 and then assembled into a plasmid for transfection via the GeneArt CRISPR Nuclease Vector with OFP Reporter Kit (ThermoFisher). HeLa PARP1 KO cells were transfected with the targeting plasmids using Lipofectamine 2000 transfection reagent (ThermoScientific). Transfected cells were single-cell sorted using the FACSAria Cell Sorter (BD). Expanded cellular clones were then harvested, potential knockouts were screened via western blot analysis and subsequently validated via DNA sequencing.
Detection of PAR, XRCC1 and fibrillarin by fluorescence microscopy
HeLa WT and PARP1 KO cells were seeded on glass cover slips in 12-well plates. If required, cells were transiently transfected with the eGFP-N1::PARP1 constructs. Cells were treated with H2O2 either 24 or 40 h after transfection or not treated. Concentration and duration of treatment varied depending on the experiment as indicated. Treatment was followed by fixation of the cells with 4% paraformaldehyde in PBS (containing 10 μM ABT888 for staining with XRCC1 and fibrillarin) for 20 min. The reaction was stopped by washing with 100 mM glycine, followed by washing with PBS and permeabilization for 3 min with 0.4% Trition-X100 in PBS. The cells were washed for 5 min in PBS (30 min when stained for XRCC1 and fibrillarin), followed by blocking in PBS containing 20% (w/v) non-fat milk powder and 0.2% (v/v) Tween20 (PBSMT). Primary antibodies were diluted in PBSMT as follows: mouse-anti-PAR [10H, purified from culture supernatant of hybridoma cells, according to (64), 1:300], rabbit-anti-XRCC1 (Enzo Life Sciences, 1:1000) and mouse-anti-fibrillarin (Abcam, 1:1000). The cells were incubated with the respective primary antibody either for 1 h at 37°C or overnight at 4°C. The slides were washed three times in PBS for 10 min and incubated with secondary antibodies for 1 h at 37°C. The secondary antibodies were diluted as follows: Alexa546 (ThermoFisher, 1:400 or 1:600) and Alexa647 (ThermoFisher, 1:400) both in PBSMT. The slides were washed three times in PBS for 10 min followed by staining with 0.1 mg/μl Hoechst33342 in PBS for 5 min. This was followed by washing the cells three times for 10 min in PBS and mounting of the glass slides using Aqua Poly Mount (Polyscience). Microscopic images were acquired using either a Zeiss Axiovert 200M widefield microscope (for PAR detection) or a Zeiss LSM700 confocal microscope (for XRCC1 and fibrillarin detection). Image data was analyzed using automated KNIME workflows as described previously (34,65). Only cells with a GFP fluorescence signal >1.5-fold of the mean background fluorescence were considered as GFP-positive and thus analyzed for PAR, XRCC1 and fibrillarin.
NAD+ cycling assay
HeLa WT and PARP1 KO cells were seeded in 6-well plates and transfected with the eGFP-N1::PARP1 constructs as indicated. Forty hours after transfection, cells were either treated for 7 min with 500 μM H2O2 or not treated. After treatment, cells were washed once with PBS and detached with trypsin–EDTA (Gibco). The harvested cells were counted, and 2 × 106 cells were used for extraction of NAD+. The cells were pelleted and resuspended in pre-cooled PBS, lysed by addition of 24 μl 3.5 M perchloric acid and incubation for 15 min on ice. Cellular debris was removed via centrifugation for 10 min at 1500 × g and the supernatant was mixed with 350 μl phosphate buffer (0.33 M K2HPO4, 0.33 M KH2PO4, pH 7.5) followed by incubation for 15 min on ice. To clear precipitates, the samples were centrifuged twice for 10 min at 1500 × g with a 20-min incubation on ice in between. As reference for the NAD+ cycling assay, a standard curve with NAD+ concentrations up to 0.48 μM was prepared for each experiment. Each sample was measured in technical triplicates. To this end, either 200 μl of the standards or 40 μl of the samples diluted with 160 μl diluent (0.5 M H3PO4, 0.5 M NaOH) were distributed in a 96-well flat bottom plate (Corning). To each well, 100 μl reaction mix [0.48 M bicine (pH 8.0), 4 mg/ml BSA, 0.02 M EDTA, 2.4 M ethanol, 2 mM MTT, 0.96 mg alcohol dehydrogenase and 5.7 mM phenazine ethosulfate] were added. Absorption was measured at 550 nm after 30 min of incubation at 30°C using 690 nm as a reference wavelength. The intracellular NAD+ amount (n) was calculated according to the standard curve and was normalized to transfection efficiencies determined by FACS analysis. The following formula was used:
 |
Analysis of cell viability using Annexin V/PI staining
HeLa WT and PARP1 KO cells were seeded in 6-well plates and transfected with different eGFP-N1::PARP1 constructs as indicated. Twenty-four hours after transfection, the cells were either treated with various concentrations of camptothecin (CPT; Sigma-Aldrich) for 40 h or not treated. Afterwards, cells were harvested using trypsin-EDTA (Gibco) and 2.5 × 105 cells were used for analysis. The cells were pelleted and resuspended in 250 μl Annexin V-binding buffer [10 mM HEPES pH 7.4, 140 mM NaCl, 2.5 mM CaCl2]. A volume of 195 μl of the cell suspension was mixed with 2.5 μl Annexin V-APC (Enzo Life Sciences) and incubated for 15 min in the dark. A volume of 200 μl propidium iodide solution (10 μg/ml PI in Annexin V-binding buffer) was added and the samples were analyzed using a FACSCalibur instrument (BD). For each sample, 10 000 transfected cells were analyzed, and only GFP-positive cells were included in the analysis.
Analysis of cell proliferation
HeLa PARP1 KO cells were seeded in six-well plates and transfected with different eGFP-N1::PARP1 constructs. Forty hours after transfection, cells were harvested with trypsin-EDTA (Gibco) and incubated with CellTrace violet stain (Invitrogen) according to manufacturer's instructions. Three hundred thousand cells were seeded in 6-cm plates and allowed to grow for 5 days. Immediately after staining, the remaining cells, corresponding to the time point t0, were analyzed using a FACSFortessa instrument (BD). For exclusion of dead cells, SytoxRed dead cell stain (Invitrogen) was used in a 1:1000 dilution. For tracing proliferation, cells were harvested using trypsin-EDTA, stained with SytoxRed dead cell stain and analyzed every 24 h (time points t1–t5).
Colony formation assay
HeLa WT and PARP1 KO cells were seeded in six-well plates and transfected with various eGFP-N1::PARP1 constructs. Forty hours after transfection, cells were treated with different concentrations of H2O2 for 5 min and harvested with trypsin-EDTA (Gibco), pelleted, and resuspended in phenolred-free medium containing 1:1000 SytoxRed dead cell stain (Invitrogen). For each sample 1000 GFP-positive cells were sorted using a FACSAria instrument (BD) in each well of six-well plates. Plates were incubated for one week at 37°C at 5% CO2 before fixation with PFA and staining with crystal violet. All colonies larger than 100 cells were included in the analysis.
Cell cycle analysis
HeLa WT and PARP1 KO cells were seeded in six-well plates and transfected with various eGFP-N1::PARP1 constructs. Twenty-four hours after transfection, cells were treated with different concentrations of camptothecin (CPT, Sigma-Aldrich) and incubated for 40 h. For the PI staining, cells were harvested with trypsin-EDTA (Gibco), pelleted and resuspended in 300 μl ice-cold PBS. Ice-cold ethanol (700 μl) was added and samples were incubated for 20 min on ice. Then, cells were washed once with PBS and resuspended in 30 μl PBS. DNA extraction buffer [120 μl; 4 mM citric acid, 0.2 M Na2HPO4 pH 7.8] was added and samples were incubated for 20 min at room temperature on a shaker. Afterwards, cells were pelleted, resuspended in DNA staining buffer [PBS, 0.2 mg/ml RNase A (Thermo Scientific), 20 μg/ml propidium iodide (Sigma-Aldrich)], incubated for 30 min at RT in the dark and analyzed using a FACSCalibur instrument (BD). In case of transfection experiments, only GFP-positive cells were analyzed.
Western blot analysis of protein expression
All cell lines were seeded in six-well plates. HeLa PARP1 KO cells were transfected with various eGFP-N1::PARP1 constructs as indicated. Furthermore, as indicated, treatment with ABT888 was performed immediately before transfection and cells were incubated with ABT888 until harvesting. Cells were harvested with trypsin-EDTA (Gibco), pelleted and resuspended in PBS and SDS loading dye to a concentration of 1 × 106 cells per 100 μl. Subsequently, samples were heated at 95°C for 5 min and 20 μl (i.e. corresponding to 2 × 105 cell equivalents) were loaded onto SDS gels (10 or 12%). Western blots were performed using 0.45 μm nitrocellulose membranes (GE Healthcare), unspecific binding sites on membranes were blocked with 5% milk in TBS-T [150 mM NaCl, 10 mM Tris base (pH 8), 0.05% Tween20], and membranes were subsequently incubated with the respective primary antibodies either for 1 h at RT or overnight at 4°C: mouse-anti-PARP1 (CII10, purified from culture supernatant of hybridoma cells, 1:300), rabbit-anti-XRCC1 (Enzo Life Sciences, 1:1000) and mouse-anti-β-actin (Cell Signaling, 1:1000). Membranes were then incubated with respective secondary antibodies for 1 h at RT: goat-anti-mouse HRP-coupled or goat-anti-rabbit HRP-coupled (both Dako, 1:5000). Chemiluminescent signals were visualized on a ChemiDoc Imaging System (Bio-Rad). The detected signal for actin as a loading control was corrected according to the following formula to account for different transfection efficiencies (TFE):  . Subsequently, values detected for PARP1 were normalized to the
. Subsequently, values detected for PARP1 were normalized to the  value.
value.
Analysis of protein recruitment to laser-induced DNA damage
HeLa cells bearing a genomic knockout of PARP1 (PARP1 KO) or PARP1 and XRCC1 (PARP1/XRCC1 double KO, DKO) were seeded in μ-dishes (Ibidi). Approx. 18 h later, they were transfected with expression vectors encoding either WT or mutant versions of eGFP-N1::PARP1 alone, or in combination with mRFP-C1::XRCC1. Twenty-four to forty hours after transfection, medium was replaced by phenolred-free medium for microscopic analysis. Microirradiation and subsequent time-lapse imaging was performed using an inverted laser-scanning microscope (Zeiss LSM700) equipped with a multi-color femtosecond fiber laser setup (66). In-situ bandwidth-limited optical pulses with a center wavelength of 1035 nm were focused onto the sample using a 63× oil immersion objective lens (N.A. 1.4). The average optical power was between 16 and 18 mW. The laser was scanned along a 4.6 μm long linear path divided into 52 pixel positions, resulting in a pixel dwell time of 45 ms. In each experiment, at least 27 cells were subsequently irradiated. The experiments were performed at least three times on different days. Quantitative image data analysis of time-series was performed in FIJI with the ImageJ software macro BIC Macro Toolkit (BIC toolbox, University of Konstanz, Germany). The toolbox is available for download at https://www.biologie.uni-konstanz.de/bioimaging-centre/. To account for different expression levels and to ensure better comparability of the different PARP1 variants, only cells with comparable nuclear fluorescence intensity values were included in the evaluation.
RT qPCR
HeLa PARP1 KO cells were seeded in six-well plates and transfected with various eGFP-N1::PARP1 constructs. Sixty-four hours after transfection, cells were detached with trypsin–EDTA and 10% of the cells were used for determination of transfection efficiencies via flow cytometric analysis. The remaining cells were used for RNA isolation, followed by high throughput RT qPCR with Fluidigm dynamic arrays on the BioMark system as described previously (67). A complete list of all analyzed genes is shown in Supplementary Table S1. To take into account different transfection efficiencies (TFE), the ΔΔCq calculation of the respective genes was adjusted using gene expression values of PARP1 KO cells, according to the following formula:
 |
Statistical analyses
Statistical testing has been performed with GraphPad Prism 8.4 as indicated in individual figure legends, * P < 0.05, ** P < 0.01, *** P < 0.001, ‘n.s.’, non-significant.
RESULTS
Biochemical characterization of PARP1 variants synthesizing PAR of different chain length and branching
To obtain further insight into the role of the structural heterogeneity of PAR regarding chain length and branching in cellular (patho-)physiology, we used different PARP1 variants that have been previously reported to synthesize PAR of different qualities (57). We chose the variants PARP1\G972R (producing hypobranched PAR), PARP1\Y986H (producing hyperbranched PAR), and PARP1\Y986S (producing short PAR). For biochemical characterization and validation of the enzymatic properties, we purified the respective proteins using an E. coli expression system and analyzed biochemical properties of the PARP1 variants themselves, as well as of the PAR produced by them. To test if amino acid (aa) exchanges, which are all located within the catalytic domain of PARP1 (Figure 1A), affect the structural stabilities of the proteins, we performed differential scanning fluorimetry (DSF). PARP1\WT showed a melting temperature of 48°C (Supplementary Figure S1), which is consistent with previously published data (68). All three variants showed a slightly reduced thermal stability with PARP1\G972R and PARP1\Y986S being somewhat more affected than PARP1\Y986H (Supplementary Figure S1). This indicates that the introduced aa exchanges have slight destabilizing effects on the structure of PARP1—a finding which needs to be considered when interpreting the results of this study. Binding of PARP1 to DNA and the resulting destabilization of the HD domain are essential for efficient activation of PARP1 (16,17,69,70). To test for potential effects of the inserted mutations on PARP1 activation, we performed DSF experiments in the presence of DNA. As expected, we observed a slight reduction of the thermal stability of PARP1\WT in the presence of DNA, which is likely caused by the previously reported allosteric binding mechanism (Supplementary Figure S1B) (16,17,69,70). Interestingly, the observed destabilization effect in the presence of DNA was even enhanced for all three PARP1 variants. Therefore, DNA binding and resulting allosteric activation are probably not impaired by the inserted mutations. To test the enzymatic activities of the human recombinant (rec.) PARP1 variants, we performed an automodification assay using TAMRA-labeled NAD+ (Figure 1B and Supplementary Figure S2) (71). While PARP1\WT and PARP1\Y986H strongly modified themselves in a time-dependent manner, the automodification activities of PARP1\G972R and PARP1\Y986S were considerably reduced. Furthermore, PARP1\Y986S showed a reduced upwards shift in the gel, indicating the preferential formation of short PAR chains. Next, we used rec. PARP1 to synthesize PAR enzymatically, which was then analyzed regarding branching frequencies and chain length distributions. The amounts of PAR produced by the different PARP1 variants confirmed the differences in catalytic activities (Figure 1C). The highest amounts of PAR were obtained from PARP1\WT followed by PARP1\Y986H. Considerably lower amounts were produced by PARP1\G972R and PARP1\Y986S. Next, we analyzed branching frequencies of purified PAR. To this end, PAR was digested to the corresponding nucleosides and R2-Ado/R-Ado ratios were analyzed using a mass spectrometric approach (60,61) (Figure 1D). In accordance with results from Rolli et al. (57), PAR produced by PARP1\G972R exhibited a ∼30% reduced branching ratio in comparison to PAR produced by PARP1\WT, whereas PAR from PARP1\Y986H exhibited a 16-fold increase in branching ratio. A new finding was that PAR from PARP1\Y986S displayed a 1.7-fold increased branching frequency in comparison to PARP1\WT. Finally, we analyzed the chain length distribution of purified PAR (Figure 1E). For all three mutants, we observed a shift toward shorter chains in comparison to PARP1\WT. While PARP1\G972R produced PAR of medium size, especially PARP1\Y986S produced PAR of very short chain length. In summary, we generated and characterized three different non-natural PARP1 variants, which display distinct catalytic activities and synthesize PAR of different chain lengths and branching frequencies. Accordingly, these variants offer a suitable tool to unravel the significance of PAR heterogeneity in a cellular context.
Figure 1.

Biochemical characterization of rec. PARP1 variants. (A) Schematic representation of the domain organization of human PARP1 and crystal structure of the catalytic domain (PDB code: 5WRQ). Amino acids substituted in the PARP1 variants that are used in this study are highlighted in red, with the G972R substitution resulting in short and hypobranched, Y986S in short and moderately hyperbranched and Y986H in strongly hyperbranched PAR chains. The E988K substitution, which results in mono-ADP-ribosyl transferase activity, is highlighted in green. (B) Time-dependent automodification reaction of rec. PARP1 variants in the absence or presence of DNA. PARylation was started by the addition of 100 μM TAMRA-labelled NAD+ (NAD+:TAMRA-NAD+ = 10:1) and analyzed by SDS-PAGE. Full blots are shown in Supplementary Figure S2. (C) Amount of PAR purified from rec. PARP1 variants as determined by UV absorption at 258 nm. (D) Mass-spectrometric quantification of the [A.U.C. of R2-Ado]/[A.U.C. of R-Ado] ratio of PAR generated and purified from rec. PARP1 variants. Means of n=3. Statistical analysis was performed for the whole data set using 1-way ANOVA testing with Dunnett's post test (###P < 0.001), or separately between PARP1\WT and each variant using an unpaired t-test (**P < 0.01; ***P < 0.001). (E) Analysis of chain length distribution of PAR generated and purified from rec. PARP1 variants by HPLC. 10 nmol PAR was loaded respectively.
Cellular characterization of PARP1 variants synthesizing PAR of different chain length and branching
To test if the PARP1 variants produce differentially structured PAR also in a cellular environment, we transiently transfected HeLa PARP1 KO cells (34) with C-terminally eGFP-tagged versions of the different variants. To obtain sufficient amounts of cellular PAR for LC–MS/MS analysis, we stimulated cells by H2O2 treatment before PAR purification. Results of this analysis confirmed that all tested GFP-tagged PARP1 variants were catalytically active, when expressed in cells. As already seen in vitro, all three variants displayed a reduced activity, with the strongest reduction for PARP1\G972R (Figure 2A). The produced PAR was further analyzed with regards to branching frequencies by determining the R2-Ado/R-Ado ratio via LC-MS/MS. Comparable to the analysis of in vitro generated PAR, PARP1\G972R exhibited a 36% reduced branching frequency, whereas PARP1\Y986H and Y986S produced PAR with an ∼11-fold and ∼2-fold increased branching ratio, respectively (Figure 2B). These data were further supported by the analysis of intracellular NAD+ levels. After H2O2 treatment, the NAD+ levels decreased in cells transfected with all PARP1 variants, but after treatment with 50 μM H2O2 the decrease in NAD+ levels was slightly less pronounced for PARP1\G972R-expressing cells, confirming a reduced activity of this variant in a cellular environment (Figure 2C). To further analyze the catalytic activities of the PARP1 variants and PAR formation and degradation dynamics, we performed time-course immunofluorescence analyses using a PAR-specific antibody. We observed similar formation and degradation dynamics of PAR generated by the different PARP1 variants compared to PARP1\WT (Figure 2D and Supplementary Figure S3). Yet, PAR levels in PARP1\G972R-expressing cells decreased at a slower rate than in cells expressing PARP1\WT and the two other variants. Moreover, in accordance with the mass spectrometric data (Figure 2A), the maximal PAR signals were reduced for all PARP1 variants compared to PARP1\WT. In summary, the PARP1 variants that were chosen for the comparative analysis of PAR chain length- and branching-specific cellular effects, were all active in a cellular environment, and thus represent exquisite tools to analyze cellular consequences of potential PAR structure-specific effects.
Figure 2.

Characterization of PAR formation in HeLa PARP1 KO cells reconstituted with different PARP1-eGFP variants. (A) Mass-spectrometric quantification of PAR levels (R-Ado) after treatment with 500 μM H2O2 for 5 min. Means ± SEM of n = 3–8 independent experiments, each normalized to PARP1\WT. Statistical analysis was performed using one-way ANOVA testing with Dunnett's post-test. (B) Mass-spectrometric quantification of the [A.U.C. of R2-Ado] / [A.U.C. of R-Ado] ratio of purified PAR after treatment with 500 μM H2O2 for 5 min. Means ± SEM of n = 3–8 independent experiments. Statistical analysis was performed for the whole data set using one-way ANOVA testing with Dunnett's post-test (###P < 0.001), or separately between PARP1\WT and each variant using an unpaired t-test (*P < 0.05; ***P < 0.001). (C) Intracellular NAD+ levels with or without H2O2 treatment (concentrations as indicated) for 7 min, as measured by an enzymatic NAD+ cycling assay. Means ± SEM of n = 3–4 independent experiments. Statistical analysis was performed using two-way ANOVA testing with Sidak's post test. (D) Single-cell immuno-epifluorescence analysis of PAR-synthesis using the anti-PAR-specific 10H antibody. Densitometric quantification of epifluorescence imaging data using a KNIME workflow as described in material and methods. Means ± SEM of n = 3 independent experiments. Statistical analysis was performed using two-way ANOVA testing.
Impact of different PARP1 variants on cellular physiology
PARP1 is involved in diverse cellular processes like DNA replication, transcription, cell cycle regulation, and regulation of cell death. To determine potential PAR structure-specific roles in these processes, we reconstituted HeLa PARP1 KO cells with the different PARP1 variants and analyzed selected cellular endpoints, such as clonogenic survival, proliferation, cell cycle progression, and viability. As a general, yet very sensitive, read-out for potential alterations of cellular physiology, we performed clonogenic survival analyses, as this assay takes into account a spectrum of cellular processes like cell attachment, proliferation and viability (72). Intriguingly, we observed that cells transfected with PARP1\G972R—but not PARP1\Y986S—formed only half as many colonies as PARP1\WT-transfected cells. This suggests that hypobranched PAR negatively influences cell attachment, proliferation and/or viability. Remarkably, expression of the hyperbranching PARP1\Y986H variant led to more colonies compared to cells expressing PARP1\WT, suggesting a moderate positive effect of PAR branching on cell attachment and/or proliferation (Figure 3A). To analyze in detail, which processes are affected by the expression of the different PARP1 variants, we measured cell proliferation, using cell trace violet and flow cytometric analysis. These analyses revealed that cell growth is delayed in PARP1\G972R- and mildly affected in PARP1\Y986S-expressing cells. In contrast, cells expressing the PARP1\Y986H variant displayed normal cell growth compared to cells transfected with PARP1\WT (Figure 3B). This indicates that short and hypobranched PAR causes a delay in cell growth, whereas hyperbranched PAR does not influence cell proliferation under these conditions. The slower proliferation, as observed for PARP1\G972R- and PARP1\Y986S-expressing cells, points to a deregulated cell cycle progression. To test this hypothesis, we analyzed the cell cycle profile of HeLa PARP1 KO cells reconstituted with the different PARP1 variants by PI staining and subsequent flow cytometric analysis. The results demonstrate that PARP1\G972R-transfected cells — but not the PARP1\Y986S-expressing cells — indeed accumulated in G2 phase (Figure 3C). Again, hyperbranched PAR, as present in cells transfected with PARP1\Y986H, did not change the cell cycle profile. To test if cell death might also contribute to the strongly reduced colony formation of PARP1\G972R-expressing cells, we analyzed cell viability of transfected cells using Annexin V/PI staining and subsequent flow cytometric analysis. These experiments revealed that cells expressing the PARP1\G972R variant have a strongly reduced viability and enhanced levels of apoptosis and necrosis. In contrast, cells expressing the PARP1\WT, PARP1\Y986S or PARP1\Y986H variants showed normal viability and only very low levels of apoptosis and necrosis (Figure 3D). Importantly, the reduced viability observed for cells transfected with PARP1\G972R could be directly linked to the synthesis of short and hypobranched PAR chains, as inhibition of PAR formation by the PARP1-inhibitor ABT888 restored normal levels of apoptosis and necrosis (Figure 3D). To investigate the stability of the PARP1 variants in our cellular system, we analyzed protein expression levels in cells 24, 48 and 72 h after transfection and compared untreated cells to those treated with ABT888. Generally, PARP1 expression decreased over time and expression of the PARP1/G972R variant was lower than that of the other variants (Figure 4 and Supplementary Figure S4). We did, however, not find any significant differences in PARP expression levels between untreated cells and cells treated with ABT888. Importantly, this suggests that inhibitor binding does not stabilize PARP1 levels and is therefore not responsible for the observed rescue effect in cells transfected with PARP1\G972R. In summary, these results revealed that the PARP1 variants producing PAR of different chain lengths and branching frequencies indeed influence proliferation, cell cycle regulation, and viability. While cells expressing the hyperbranching PARP1\Y986H variant exhibited enhanced plating efficiencies, especially short and hypobranched PAR, as produced by the PARP1\G972R variant, negatively affected these processes leading to impaired cellular physiology.
Figure 3.

Analyses of different endpoints of cellular physiology of HeLa PARP1 KO cells reconstituted with PARP1-eGFP variants. In all cases, PARP1-reconstituted HeLa PARP1 KO cells were analyzed. (A) Clonogenic survival assay. After transfection, cells were sorted and 100 GFP-positive cells were plated and cultivated for 7 days prior to colony counting. Means ± SEM of n = 3 independent experiments. [N.B. this data set is part of the experiment shown in Figure 5A and replotted here for ease of reader friendliness]. (B) Cell proliferation assay via cell trace violet and subsequent flow cytometric analysis. n = 1. (C) Cell cycle analysis via PI staining and subsequent flow cytometric analysis. Means ± SEM of n = 4 independent experiments. Statistical analysis was performed using repeated measures one-way ANOVA testing with Dunnett's post-test. (D) Cytotoxicity analysis via Annexin V/PI staining and subsequent flow cytometric analysis of cells before and after treatment with 10 μM ABT888. Means ± SEM of n = 6 independent experiments for untreated (including n = 4 values from the experiment shown in Figure 5B) and of n = 2 independent experiments for ABT888 treated cells. Statistical analysis was performed using two-way ANOVA testing with Sidak's post-test.
Figure 4.

Expression analysis of PARP1-eGFP variants in reconstituted HeLa PARP1 KO cells compared to HeLa WT cells. (A–C) Densitometric analysis of western blot signal intensities of PARP1 protein levels in HeLa WT or PARP1 KO cells reconstituted with different PARP1 variants (A) 24 h, (B) 48 h or (C) 72 h after transfection. Cells were either untreated or treated with 10 μM ABT888 for the indicated duration. Signal intensities were normalized to transfection efficiencies, to β-actin as a loading control and to untreated HeLa WT cells for comparison. Means ± SEM of n = 2–3 independent experiments. Statistical analysis of ‘untreated’ versus ‘ABT888’ was performed using two-way ANOVA testing with Sidak's post-test. Representative blots are shown in Supplementary Figure S4.
Impact of different PARP1 variants on genotoxic stress response
To analyze, to which extent the different PARP1 variants mediate tolerance to genotoxic stress, we treated PARP1-reconstituted cells with H2O2 and performed clonogenic survival assays. Interestingly, the lowest dose of 1 μM H2O2 slightly increased plating efficiencies for all PARP1 variants. Above this concentration, a dose dependent decrease in colony formation was observed. Cells expressing PARP1\G972R were hardly capable of forming any colonies after treatment with ≥5 μM H2O2 (Figure 5A). In comparison, cells producing short but branched PAR by PARP1\Y986S formed colonies up to 50% compared to PARP1\WT-reconstituted cells, while cells transfected with PARP1\Y986H, producing short but hyperbranched PAR, formed up to ∼80% the number of colonies compared to PARP1\WT. Next, we were interested whether this effect was also evident after treatment with the topoisomerase I inhibitor camptothecin (CPT), which induces DNA strand breaks, protein–DNA adducts and replicative stress. Cells expressing PARP1\G972R were highly sensitized to CPT treatment, leading to a reduction to ∼11% viable cells already at the lowest applied concentration of 1 μM (Figure 5B), which was accompanied by strongly enhanced levels of apoptosis and necrosis. On the contrary, cells expressing PARP1\WT and PARP1\Y986H displayed 49% and 44% viable cells at 1 μM CPT, respectively. Likewise, PARP1\Y986S-reconstituted cells also showed reduced viability compared to PARP1\WT-reconstituted cells, but the effect was not as pronounced as in PARP1\G972R-reconstituted cells. Normalization of the results to the respective untreated controls revealed that not only was the reduced basal viability responsible for the strongly reduced viability of the PARP1\G972R-reconstituted cells after CPT treatment, but an additional sensitization to CPT was evident (Figure 5B). Of note, this effect could be completely rescued by pre-treatment with the PARP-inhibitor ABT888 (Supplementary Figure S5), which again clearly indicates that the catalytic activity of the PARP1\G972R variant is responsible for the observed effect. To test if impaired viability of PARP1\G972R-transfected cells is accompanied by changes in cell cycle distributions, we investigated cell cycle profiles after treatment of cells with nanomolar concentrations of CPT. For all different PARP1 variants, including PARP1\WT, cells underwent a G2 arrest upon CPT treatment (Figure 5C). Again, the effect was strongest for PARP1\G972R- and PARP1\Y986S-reconstituted cells, as already observed for colony forming capacities after H2O2 treatment. In summary, these results demonstrate that the PARP1 variants producing PAR of different chain length and branching significantly affect the cellular response and tolerance to genotoxic stress. Thus, the production of short and hypobranched PAR considerably impaired the cellular genotoxic stress response, in contrast to short and moderately or strongly hyperbranched PAR, which showed only mild or even beneficial effects.
Figure 5.

Genotoxic stress resistance of HeLa PARP1 KO cells reconstituted with PARP1-eGFP variants. In all cases, PARP1-reconstituted HeLa PARP1 KO cells were analyzed. (A) Clonogenic survival assay upon H2O2 treatment. After transfection and respective H2O2 treatment, cells were sorted and 100 GFP-positive cells were plated and cultivated for 7 days prior to colony counting. Means ± SEM of n = 3 independent experiments. (B) Cytotoxicity analysis after CPT treatment via Annexin V/PI staining and subsequent flow cytometric analysis. Normalization to DMSO controls is presented in the right panel. Means ± SEM of n = 4 independent experiments. (C) Effect of CPT treatment on cell cycle progression analyzed via PI staining and subsequent flow cytometry. Means ± SEM of n = 4 independent experiments. Statistical analysis was performed using two-way ANOVA testing with Sidak's post-test.
Subnuclear localization of PARP1 variants and XRCC1 upon induction of DNA damage
Since the expression of the PARP1\G972R and PARP1\Y986S variants influenced clonogenic survival and cell cycle progression after genotoxic stress, we analyzed the recruitment and dissociation behavior of PARP1 at DNA damage, as these localization dynamics represent one of the first steps of the PARP1-dependent DNA damage response (14,34). As expected, PARP1\WT showed a fast recruitment with maximum signal intensities at the irradiated site after 50 s, followed by relatively fast dissociation kinetics leading to its almost complete release 400 s post irradiation (Figure 6A, B). PARP1\Y986H displayed a behavior similar to PARP1\WT, but with slightly lower maximum recruitment levels. In contrast, dissociation of PARP1\Y986S, and especially PARP1\G972R, was strongly impaired and fluorescence signals did not return to the levels of the wildtype within the 400-s observation period. The differences in dissociation kinetics became more evident when the curves were normalized to the respective maximum intensity values of recruitment (Figure 6C). Additionally, PARP1\G972R showed slower recruitment kinetics, reaching maximum signal intensities at the irradiated site only after 100 s, compared to 40–60 s for all other variants. Therefore, recruitment of PARP1 to — and dissociation from – DNA damage appears to be directly connected to PAR quality, leading to longer retention in case of short and linear PAR chains. As a further cellular parameter for the functional relevance of PAR chain structure, we studied the recruitment of the PARP1 downstream factor XRCC1 to laser-irradiated sites. To eliminate any possible confounding effects due to the endogenously expressed protein, we generated HeLa PARP1/XRCC1 double KO (DKO) cells via CRISPR/Cas9-mediated genome editing (Supplementary Figure S6A). In line with the recruitment data obtained in reconstituted PARP1 KO cells (Figure 6), we observed a fast recruitment of PARP1\WT, as well as the Y986H and Y986S variants, with maximum signal intensities at the irradiated site after ∼50 s in PARP1/XRCC1 DKO cells. Instead, PARP1\G972R recruitment was again delayed to ∼100 s (Figure 7A, B). While the overall recruitment kinetics of the PARP1\G972R variant were similar in PARP1 KO and double-transfected PARP1\XRCC1 DKO cells, the relative amount of recruited PARP1\G972R was significantly higher in the DKO cells (Figure 7A and Supplementary Figure S6B). A further difference between experiments in PARP1 KO and PARP1\XRCC1 DKO cells was observed in the dissociation behavior of the PARP1\Y986H variant, which resembled PARP1\WT in PARP1 KO cells, while it entered steady-state levels above those of PARP1\WT at ∼270 s after irradiation in PARP1\XRCC1 DKO cells (Figure 7B). The level of recruitment of XRCC1 was significantly lower in all cells co-transfected with PARP1 mutants as compared to PARP1\WT (Figure 7C and Supplementary Figure S6B), yet recruitment and dissociation kinetics were similar in the presence of either PARP1 variant (Figure 7D). Thus, the highest signal of XRCC1 at irradiated sites was observed with PARP1/WT, while the lowest levels of XRCC1 recruitment was detected with PARP1\Y986S. The dissociation kinetics of XRCC1 were slightly, yet significantly, faster in case of reconstitution with the PARP1\G972R and Y986H variants (Figure 7D). To gain further insight into the influence of PAR quality on XRCC1 subnuclear dynamics, we investigated the translocation of XRCC1 from nucleoli to the nucleoplasm upon genotoxic stress in cells co-transfected with the different PARP1 variants by immunofluorescence staining and subsequent confocal microscopic analysis (Figure 8). In a previous study, we revealed that XRCC1 translocates from nucleoli to the nucleoplasm upon H2O2 treatment. In the presence of PARP1, XRCC1 is retained in the nucleoplasm, while in PARP1 KO cells, XRCC1 swiftly relocates back into nucleoli (65). In accordance with this, Figure 8 shows that in PARP1\WT-reconstituted cells, XRCC1 was released from nucleoli in response to H2O2-treatment. The same effect could be observed for both, PARP1\Y986H and PARP1\Y986S. For PARP1\G972R, however, the translocation was impaired. This resembles the effect observed for the previously tested mono-ADP-ribosylating PARP1\E988K variant, in which case translocation of XRCC1 was shown to be significantly inhibited, but not to the same extent as in PARP1 KO cells. This leads to the assumption that translocation of XRCC1 is at least partially dependent on the quality of the PAR chains produced. In summary, recruitment and dissociation kinetics of PARP1 at sites of laser-induced DNA damage, as well as recruitment/dissociation kinetics and nucleolar translocation dynamics of XRCC1, are significantly influenced by PARylation and PAR quality. Altered PARP1 automodification and impaired downstream signaling events could therefore at least in part explain the significantly reduced viability observed for PARP1\G972R-reconstituted cells.
Figure 6.

PARP1 recruitment and dissociation at sites of laser-induced DNA damage in HeLa PARP1 KO cells reconstituted with PARP1-eGFP variants. (A) Representative images. (B) Quantification of recruitment dynamics from image data as shown in (A). Means ± SEM of n = 3 independent experiments, ≥ 42 cells were analyzed per PARP1 variant. Statistical analysis was performed using two-way ANOVA testing. (C) Normalized PARP1 recruitment data from (A), with maximum intensity values of each curve set to 100%, respectively. Statistical analysis was performed using two-way ANOVA testing.
Figure 7.

PARP1 and XRCC1 recruitment in HeLa PARP1/XRCC1 double KO cells reconstituted with PARP1-eGFP variants and mRFP-XRCC1. (A) Quantification of PARP1 recruitment dynamics from image data as shown in Supplementary Figure S6. Means ± SEM of n = 4 independent experiments, ≥52 cells were analyzed per PARP1 variant. (B) Normalized PARP1 recruitment data from (A), with maximum intensity values of each curve set to 100%, respectively. (C) Quantification of XRCC1 recruitment dynamics from image data as shown in Supplementary Figure S6. Means ± SEM of n = 4 independent experiments, ≥52 cells were analyzed per PARP1 variant. (D) Normalized XRCC1 recruitment data from (C), with maximum intensity values for XRCC1 set to 100% for each condition, respectively. Statistical analysis was performed using two-way ANOVA testing.
Figure 8.
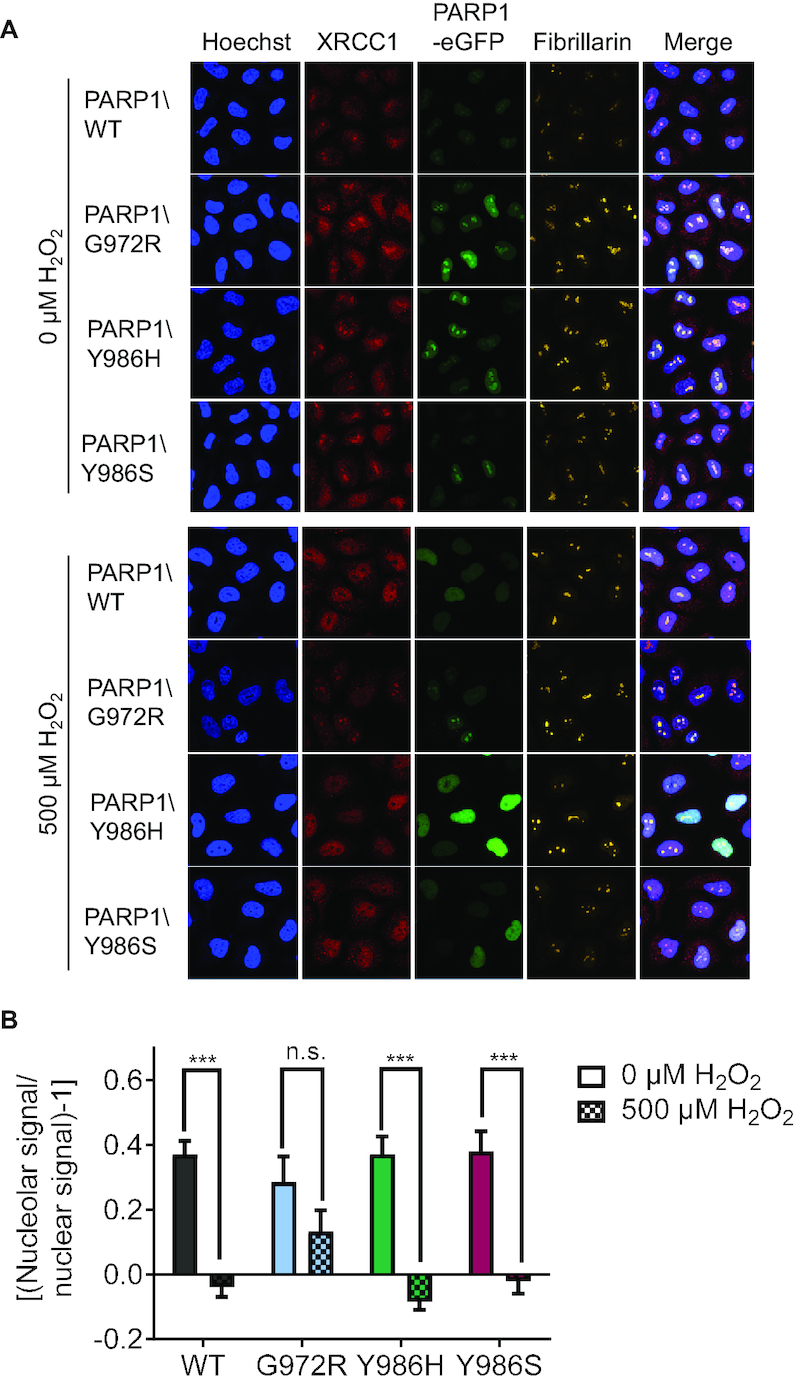
Nucleolar-nucleoplasmic translocation of endogenous XRCC1 in HeLa PARP1 KO cells reconstituted with PARP1-eGFP variants. (A) Representative images from single-cell immunofluorescence analysis of endogenous XRCC1 translocation from the nucleoli to the nucleoplasm. (B) Densitometric quantification of confocal imaging data from (A) analyzing XRCC1 translocation via a KNIME workflow. Means ± SEM of n = 4 independent experiments. Statistical analysis was performed using two-way ANOVA testing with Sidak's post-test.
Impact of PARP1 variants on gene expression profiles
To obtain insight into the downstream molecular mechanisms leading to the observed cellular phenotypes, we analyzed expression of a set of 80 genes involved in genome maintenance, cell death regulation and proliferation in cells reconstituted with the different PARP1 variants by using a very accurate and highly sensitive high-throughput qPCR approach, as described previously (23,67). We indeed observed subtle, yet significant, changes in the expression profiles for cells reconstituted with the different PARP1 variants in comparison to PARP1\WT-reconstituted cells (Figure 9). For cells producing hyperbranched PAR (i.e., PARP1\Y986H), most altered genes were slightly downregulated (fold regulation < 1.5). The most significant changes could be observed for growth factors like EGFR and VEGFA. In contrast, for cells reconstituted with PARP1\G972R, most altered genes were upregulated. Interestingly, especially the expression of genes regulating apoptosis like BCL2L1 and PMAIP1, cell cycle inhibitors like TP53 and CDKN1A (p21) and DNA damage repair genes like RAD51 was significantly increased. This is consistent with the observed cellular phenotype, showing a G2 arrest, increased rates of cell death, and higher sensitivity to genotoxic treatment in PARP1\G972R-expressing cells. In comparison, cells reconstituted with PARP1\Y986S showed hardly any changes in their expression profile, suggesting that the observed effects for PARP1\G972R-reconstituted cells are indeed caused by hypobranched PAR chains and not only by a reduced chain length, as PARP1\Y986S produces even shorter chains than PARP1\G972R. In summary, after reconstitution of HeLa PARP1 KO cells with the different PARP1 variants, the gene expression profile indeed changed, providing important indications how changes in PAR structure might be mechanistically connected to the observed cellular phenotypes. Especially for PARP1\G972R-reconstituted cells, which produce short, hypobranched PAR, the changes in gene expression profile directly mirror the observed cellular phenotype.
Figure 9.

Effects of PARP1 variants on expression profiles of an array of 80 genes-of-interest. In all cases, PARP1-eGFP-reconstituted HeLa PARP1 KO cells were analyzed by high throughput RT qPCR. The volcano plots show the magnitude (fold regulation, x-axis) and the significance (–log10P-value, y-axis) of gene expression changes between PARP1 KO cells reconstituted with the respective PARP1 variant compared to cells reconstituted with PARP1\WT. Horizontal lines indicate the statistical significance threshold (P ≤ 0.05, statistical analysis was performed by an independent samples t-test, n = 3 independent experiments); vertical lines indicate no change in gene expression. Genes that are differentially expressed at a significant level are labelled with the corresponding names. All genes included in the analysis are listed in Supplementary Table S1.
General significance of PAR branching in cellular and organismic physiology
As expression of the different non-natural PARP1 variants pointed to PAR branching-specific effects in cellular functions, we were interested whether PAR branching varies during genotoxic stress response in HeLa WT cells. To this end, PAR levels and branching frequencies were analyzed in HeLa cells at different time points after H2O2 treatment (Figure 10A). Interestingly, the R2-Ado/R-Ado ratios strongly increased between 5 and 7.5 min after treatment, which corresponds to the onset of the catabolic phase of PARylation dynamics. The increase in branching frequencies was then followed by a decrease until the basal branching ratio was reached 20 min post treatment. These results suggest that during the PARylation response, linear parts of PAR were degraded first, leading to an increase in the R2-Ado/R-Ado ratio. To follow up on this, we tested how PARG, which represents the main enzyme responsible for the degradation of cellular PAR, catabolizes PAR synthesized by the different non-natural PARP1 variants. Therefore, in-vitro produced PAR was digested with PARG for different periods of time and the resulting amounts of ADP-ribose were quantified (Figure 10B). Hypobranched PAR, produced by PARP1\G972R, as well as short PAR chains, produced by PARP1\Y986S, were degraded significantly faster than PAR produced by PARP1\WT and PARP1\Y986H. These results suggest that PARG preferentially degrades short and linear PAR chains, which would be consistent with the observed increase in PAR branching in HeLa cells at the early phase of PAR catabolism. In summary, we demonstrate that PAR branching indeed changes during the process of PAR degradation after genotoxic stress-induced PARylation, providing a potential mechanism for the temporal control of PAR structure-specific downstream processes in cells.
Figure 10.

Analysis of PAR branching during cellular genotoxic stress response and PARG activity on PAR produced by the different PARP1 variants in vitro. (A) Mass spectrometric quantification of R-Ado (black) and the [A.U.C. R2-Ado]/[A.U.C. R-Ado] ratio (orange) of PAR purified from HeLa WT cells after treatment with 500 μM H2O2 as indicated. Means ± SEM of n = 3 independent experiments. (B) Time-dependent degradation of PAR purified from rec. PARP1 variants by PARG. Generated ADP-ribose units were quantified via an ADP-ribose calibration curve as published previously (89). Background levels of ADP-ribose within the different PAR samples in the absence of PARG are subtracted. Means ± SEM of n = 3–4 independent experiments. Statistical analysis was performed using two-way ANOVA testing with Sidak's post-test.
To elucidate a potential in-vivo significance of PAR branching, we analyzed PAR levels and branching frequencies in 12 different mouse organs. Quantitative analysis by isotope dilution mass spectrometry revealed that PAR levels are strongly tissue-dependent. Especially skin, but also muscle, stomach, and brain exhibited high PAR levels. In contrast, spleen, lung, small intestine, testis, and colon revealed very low PAR amounts (Figure 11A). Strikingly, analysis of branching ratios revealed that PAR branching indeed differs significantly between different organs. Notably, colon and stomach had several fold higher R2-Ado/R-Ado ratios compared to all other organs (Figure 11B). Since high levels of PAR did not correlate with high levels of branching, it can be assumed that PAR branching does not just represent a side-product of extensive PAR formation, but is controlled by distinct underlying regulatory mechanisms in different cell types (Figure 11A and B). From these data, we can conclude that physiological PAR levels and PAR branching frequencies show considerable variability across different mammalian tissues, pointing towards a biological significance of PAR heterogeneity on an organismic level.
Figure 11.

Characterization of PAR levels and PAR branching in different mouse organs. (A) Mass-spectrometric analysis of PAR-amounts (R-Ado) purified from mouse tissues as indicated. Means ± SEM of n = 7 mice. Statistical analysis was performed using repeated measures one-way ANOVA testing with Dunnett's post-test. (B) Mass-spectrometric analysis of the [A.U.C. R2-Ado]/[A.U.C. R-Ado] ratio of purified PAR from mouse tissues as indicated. Means ± SEM of n = 6–7 mice. Statistical analysis was performed using mixed effects repeated measures analysis with Dunnett's post-test. ‘R’ refers to reference organ for statistical testing.
DISCUSSION
Although PARP1 and PARylation are generally well characterized and their importance, especially with regards to cancer and DNA repair, is widely accepted, the functions of the heterogeneity of PAR regarding chain length and branching are essentially unknown. Results from other post-translational modifications like the ubiquitin system indicate the existence of an ‘ubiquitin code’. Here, proteins can be modified with ubiquitin monomers as well as ubiquitin polymers, which may contain mixed linkages and/or two or more branches, showing distinct functions dependent on the structure (73,74). These findings indicate that the structure of a post-translational modification can determine its functions, supporting the notion of the existence of a ‘PAR-code’. To address this question, we have characterized the functions of PAR chain length and branching on the cellular level. To this end, we used a HeLa PARP1 KO cell system reconstituted with different non-natural PARP1 variants that synthesize PAR of different qualities, i.e., with regards to chain length and branching (34,57).
Our biochemical and cellular characterization of the PARP1 variants confirmed the formation of distinct PAR qualities with regards to chain length and branching. In addition to previous results from Rolli et al. (57), we demonstrate that PARP1\Y986S not only produces short, but also moderately (∼2-fold) hyperbranched PAR chains. Combined with the fact that PARP1\Y986H produces 16-fold higher branching ratios compared to PARP1\WT, our data indicate that the aa residue Y986 plays a central role in the branching reaction. This is in accordance with previous data, proposing that interactions of residue Y986 with the pyrophosphates of the distal ADP-ribose unit of the acceptor PAR chain stabilize the branching conformation (6,75). In contrast, the mechanism of aa substitution G972R, which is located at the surface of the catalytic domain, remains elusive. The observed destabilization effect on the structure of PARP1 suggests an indirectly mediated defect in elongation and branching. Since PARP1\G972R, PARP1\Y986S and PARP1\Y986H mainly produce short PAR chains of similar chain length, but with variable branching frequencies, those variants are very well suited to distinguish between the effect of PAR branching and the effect of PAR chain length when compared to PARP1\WT.
Interestingly, while PARP1\Y986S only mildly affected cellular physiology, PARP1\G972R displayed a strong phenotype affecting clonogenic survival, cell viability, proliferation, cell cycle progression as well as tolerance to genotoxic stress. Importantly, those effects could be directly linked to the formation of short and hypobranched PAR, as they could be rescued by PARP inhibitor treatment. We can assume that the observed rescue effect is indeed due to the lack of hypobranched PAR, rather than stabilization of the G972R variant through inhibitor binding, as expression levels did not change after inhibitor treatment. Notably, PARP1\E988K-reconstituted cells (only performing MARylation) displayed a similar phenotype, which could be rescued by PARP inhibitor treatment (34). Therefore, it can be assumed that not only the complete loss of PARylation ability of PARP1, but already defects in PAR elongation and/or branching capacity can result in severe cellular effects. Hanzlikova et al. recently showed that PARP1 functions as a sensor of unligated Okazaki fragments, as PAR was detected at sites of DNA replication during S phase (39). It would therefore be reasonable to assume that differentially structured PAR might also influence cellular physiology at the level of DNA replication, altering the repair of Okazaki fragments not processed by the canonical pathway. How it might thereby affect cell cycle progression and potentially manifest in a G2 phase arrest, as seen for the PARP1\G972R variant, is an intriguing topic for further studies. The significance of a correct PAR structure is further demonstrated by a recently described PARP1 knock-in mouse model carrying a point mutation within the catalytic domain of PARP1 (D993A) (76). Interestingly, the mutation resulted in significantly less branched PAR (in addition to slightly shorter chain length) compared to PARP1\WT. Of note, those mice were hypersensitive to alkylating agents and showed a similar phenotype to PARP1 KO mice. These observations are consistent with our data from PARP1\G972R. Remarkably, PARP1/Y986H, which produces the strongest alterations in PAR structure (16-fold higher branching ratio combined with reduced chain length), showed a similar or even slightly beneficial phenotype compared to PARP1\WT. Accordingly, significantly higher amounts of branching can be easily tolerated - at least by HeLa cells - while reduced branching seems to have severe consequences on cellular physiology. A positive role of branching is further supported by previous studies, revealing that histones preferentially bind to branched PAR (49). APLF, which has previously been shown to exhibit histone chaperone activities and function as an important scaffold protein within the non-homologous end joining pathway of DNA repair (77,78), has recently also been reported as a reader of branched PAR chains (51). Earlier studies already suggested that its tandem PBZ domains are able to bind multiple ADP-ribose units along a PAR polymer via specific conserved residues that reduce or abolish PAR binding upon mutation (79,80). Together, this suggests that branching facilitates chromatin remodeling, e.g. during DNA damage repair (49,51).
It is generally assumed that automodification of PARP1 results in its dissociation from DNA, which can be inhibited by PARP inhibitor treatment, i.e. a process known as PARP trapping (81). However, the exact mechanism of PARP1 dissociation and trapping is still unknown. Laser microirradiation experiments revealed that PARP1\Y986H displays similar recruitment and dissociation kinetics to and from sites of laser-induced DNA damage compared to PARP1\WT, while dissociation kinetics of PARP1\G972R and PARP1\Y986S mutants were delayed. Therefore, our data demonstrate that PARP1 dissociation is not only dependent on PAR formation, but also on the PAR quality with regards to chain length and branching. As dissociation is probably mainly driven by electrostatic repulsion, short and linear PAR chains might not reach the required threshold. Since high levels of branching seem to compensate a reduced PAR chain length, this suggests that not only electrostatic but probably also steric repulsion is responsible for PARP1 dissociation. The slight differences in PARP1 recruitment we observed between different experiments could be explained by the different systems we applied to study PARP1 recruitment alone or in combination with the downstream factor XRCC1. While our first experiments comprised PARP1 KO cells reconstituted with eGFP-tagged PARP1 variants, recruitment in combination with the downstream factor XRCC1 was analyzed in double-reconstituted PARP1/XRCC1 DKO cells, using eGPF-tagged PARP1 and mRFP-tagged XRCC1, in order to avoid possible confounding effects of endogenous PARP1 and XRCC1. Nonetheless, recruitment studies in both systems consistently showed delayed accumulation of PARP1\G972R at sites of laser-induced DNA damage compared to the other variants, while dissociation was decelerated especially for PARP1\G972R and PARP1\Y986S. The observation that PARP1 recruitment was delayed for PARP1\G972R, while the velocity of downstream XRCC1 recruitment on the other hand was not, suggests that initial PAR formation at the site of DNA damage is not only necessary for later second-wave recruitment of PARP1 (14), but might also be sufficient for initial recruitment of the downstream DNA repair factor. PAR structure and especially chain length however seem essential for XRCC1 recruitment, as especially PARP1\Y986S was not able to recruit XRCC1 as efficiently.
Viewed together, our data point towards a regulatory function of PAR structure in cells. Yet, it remains elusive how cells can specifically regulate PAR chain length and branching with spatial as well as temporal resolution. In this regard, we identified the degradation process as a mechanism for the temporal control of PAR branching frequency. By using our previously developed mass spectrometric approach (60,61), we detected an enrichment of branching points during the catabolic phase of the genotoxic stress-induced PARylation response in cells, which is in accordance with previous data using low-resolution techniques (55,56). A plausible reason for an increase of branching points might be a preferential degradation of linear chains by PARG. Consistently, results from our in vitro PARG degradation assay revealed faster degradation for PAR synthesized by PARP1\G972R and PARP1\Y986S in comparison to PAR from PARP1\WT or PARP1\Y986H. This suggests that degradation efficiency decreases with longer chains or increasing branching frequencies. This seems contradictory to previous results suggesting an early rapid degradation of large polymers and a late slow degradation of small polymers (54). Yet, it was also suggested that PARG mainly performs endoglycosidic cleavage in the first phase of cleavage (55), which is not detectable with our in vitro PARG degradation assay. Still, it is surprising that highly branched PAR from PARP1\Y986H was degraded with kinetics similar to PAR from PARP1\WT. This not only became apparent in the in vitro PARG degradation assay, but also in the time-course immunofluorescence analyses of PARP1-reconstituted cells demonstrating similar formation and degradation kinetics for PAR from PARP1\WT and PARP1\Y986H. Braun et al. suggested that endoglycosidic cleavage of PARG produces linear polymers from branched polymers, while leaving the branching sites themselves intact (55). Accordingly, branching sites might function as recognition sites for PARG, which trigger endoglycosidic cleavage but are not targeted themselves. Thus, an enrichment of branching points might occur even though linear and branched chains are degraded with similar efficiencies. Therefore, it is tempting to speculate that branching points themselves may function as downstream signaling factors. First evidence in this direction is given by Chen et al., who reported APLF as a factor that binds specifically to diribosyladenosine and thereby regulates chromatin remodeling and DNA damage response (51).
Notably, by analyzing PAR of different mouse organs, we provide evidence for a biological significance of PAR heterogeneity on an organismic level. The finding that PAR levels did not correlate with branching frequencies suggests distinct regulatory mechanisms of PAR branching in different tissues and cell types. Highest PAR levels were observed in skin. In accordance with these results, a previous study identified PAR in human skin with especially high PAR levels in hair follicle cells (82). It was suggested that high PAR levels in hair follicle cells occur due to their rapid cycling and the well-known role of PARP1 in cell proliferation (83). Alternatively, high PAR levels might help to maintain genomic integrity in epidermal cells, which are constantly exposed to exogenous stress. Lowest PAR levels were observed in spleen and lung, which is consistent with previous data on mouse organs (61). Highest branching frequencies were detected in stomach and colon, suggesting that PAR branching fulfills distinct functions in cells involved in food digestion and absorption. Following up on this observation, the analysis of PAR levels and branching in cell types corresponding to the already tested organs would be an important step to further understand this connection. Similar to skin, stomach and colon are constantly exposed to exogenous stress stimuli. Therefore, not only high levels of PAR but also high levels of branching might have a protective function, which would be fully consistent with our data from cellular studies. Yet, branching ratios were only moderately increased in cells from the small intestine, which is also part of the gastrointestinal tract. Considering the central role of PARPs in genome maintenance, these findings are consistent with the fact that malignancies of stomach and colon occur frequently, while malignancies of the small intestine are very rare. As PARP inhibitors are FDA approved for the treatment of breast, ovarian and pancreatic cancers, the future evaluation of PAR levels and branching in corresponding tissues would be of considerable interest to elucidate the potential link between PARP activity, PAR quality, tumor formation propensity, and possible treatment options. Although we can be reasonably sure about an organismic significance of PAR quality, as of this moment, we can only speculate about the reasons for varying PAR levels and PAR branching across different tissues in vivo. Besides different degradation kinetics of PAR chains – as mentioned before – the regulation of PARP1 via post-translational modifications could be one contributing factor. Various modification sites have so far been identified within PARP1, including phosphorylation sites within its catalytic domain (84,85) and several of these modifications have already been shown to alter PARP1 activity in general (86,87). Additionally, a level of regulation via accessory factors has recently been discovered, showing that HPF1 extends the catalytic core of PARP enzymes by forming a composite active site and thereby switching the amino acid specificity for PARylation from aspartate and glutamate to serine residues in case of DNA damage (12). A striking example for the importance of allosteric interactions is given by Zandarashvili et al., demonstrating how the affinity of PARP1 to DNA strand breaks can be modulated by allosteric signals conveyed via its helical domain (88). Taken together, it appears rather likely that some modifications and/or interactions might also influence PARP1 activity with regards to branching and chain length. Relating to the point of regulation, another possible explanation for the observed PAR heterogeneity in mammalian tissues could be differential expression and/or activity of members of the PARP family. First evidence in this direction is provided by Chen et al. (51), connecting PARP2 to the formation of branched PAR.
CONCLUSION
This study provides insights into a potential biological function of PAR heterogeneity. By applying PARP1 variants as a tool to analyze differently structured PAR chains, we demonstrate that distinct modulations of PARP1 activity can have severe effects on cellular physiology, such as cell adhesion, proliferation, cell cycle regulation and cell death, as well as intracellular molecular processes, such as protein localization dynamics and gene expression profiles. Since complete inhibition of PARP1 catalytic activity was better tolerated by cells than expression of the PARP1\G972R variant, the significance of a correct PAR structure becomes strikingly apparent. We hypothesize that PAR branching may be at a certain level of saturation in a cellular system, with no additional beneficial effect of more strongly branched PAR, but detrimental effects of lesser branched PAR. Accordingly, the development of compounds, which modulate PARP1 activity instead of inhibiting it, and - taking into account possible differences in expression and activity of PARP family members in various tissues - further development of homolog-specific inhibitors and organ-specific inhibition of PARPs, might offer strategies for new therapeutic approaches.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge the Bioimaging Center and Flow Cytometry Center (FlowKon) of the University of Konstanz for the use of scientific instruments and the expert technical support, and Xianyue Ai for assisting in microirradiation experiments. Furthermore, we thank Alfred Leitenstorfer for his continuous support regarding fiber laser technology and Beatrice Hämmerle for support during her Master's thesis project.
Author contributions: Conceptualization and data interpretation, L.A., A.K., J.M.R., E.F.M., A.M.; experimental research and data analysis, L.A., A.K., J.M.R., M.L., M.H., M.S., I.R.E.A.T., S.S., T.Z., K.S.; intellectual input, scientific know-how, reagents, Andreas Marx, A.H., A.B., E.F.M.; writing - original draft, L.A., A.K., J.M.R., A.M.; writing - review and editing, all authors; overall project supervision, A.M.
Contributor Information
Lisa Aberle, Department of Biology, University of Konstanz, 78457 Konstanz, Germany.
Annika Krüger, Department of Biology, University of Konstanz, 78457 Konstanz, Germany.
Julia M Reber, Department of Biology, University of Konstanz, 78457 Konstanz, Germany.
Michelle Lippmann, Department of Biology, University of Konstanz, 78457 Konstanz, Germany.
Matthias Hufnagel, Department of Food Chemistry and Toxicology, Institute for Applied Biosciences, Karlsruhe Institute of Technology (KIT), 76131 Karlsruhe, Germany.
Michael Schmalz, Department of Physics, University of Konstanz, 78457 Konstanz, Germany.
Irmela R E A Trussina, Department of Biology, University of Konstanz, 78457 Konstanz, Germany.
Sarah Schlesiger, Department of Chemistry, University of Konstanz, 78457 Konstanz, Germany.
Tabea Zubel, Department of Biology, University of Konstanz, 78457 Konstanz, Germany.
Karina Schütz, Department of Biology, University of Konstanz, 78457 Konstanz, Germany.
Andreas Marx, Department of Chemistry, University of Konstanz, 78457 Konstanz, Germany.
Andrea Hartwig, Department of Food Chemistry and Toxicology, Institute for Applied Biosciences, Karlsruhe Institute of Technology (KIT), 76131 Karlsruhe, Germany.
Elisa Ferrando-May, Department of Biology, University of Konstanz, 78457 Konstanz, Germany.
Alexander Bürkle, Department of Biology, University of Konstanz, 78457 Konstanz, Germany.
Aswin Mangerich, Department of Biology, University of Konstanz, 78457 Konstanz, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
German Research Foundation (DFG) [MA4905/4-1, INST 38/537-1]; A.K., J.M.R., T.Z. were supported by the DFG-funded Konstanz Research School ‘Chemical Biology’ [GSC218]; A.K. was further supported by the Zukunftskolleg Konstanz. Funding for open access charge: University of Konstanz and DFG.
Conflict of interest statement. None declared.
REFERENCES
- 1. Leung A.K.L. PARPs. Curr. Biol. 2017; 27:R1256–R1258. [DOI] [PubMed] [Google Scholar]
- 2. Gupte R., Liu Z., Kraus W.L.. PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev. 2017; 31:101–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Slade D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020; 34:360–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lord C.J., Ashworth A.. PARP inhibitors: synthetic lethality in the clinic. Science. 2017; 355:1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Luscher B., Butepage M., Eckei L., Krieg S., Verheugd P., Shilton B.H.. ADP-Ribosylation, a multifaceted posttranslational modification involved in the control of cell physiology in health and disease. Chem. Rev. 2018; 118:1092–1136. [DOI] [PubMed] [Google Scholar]
- 6. Alemasova E.E., Lavrik O.I.. Poly(ADP-ribosyl)ation by PARP1: reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019; 47:3811–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang Y., Wang J., Ding M., Yu Y.. Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nat. Methods. 2013; 10:981. [DOI] [PubMed] [Google Scholar]
- 8. Bonfiglio J.J., Fontana P., Zhang Q., Colby T., Gibbs-Seymour I., Atanassov I., Bartlett E., Zaja R., Ahel I., Matic I.. Serine ADP-Ribosylation depends on HPF1. Mol. Cell. 2017; 65:932–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martello R., Leutert M., Jungmichel S., Bilan V., Larsen S.C., Young C., Hottiger M.O., Nielsen M.L.. Proteome-wide identification of the endogenous ADP-ribosylome of mammalian cells and tissue. Nat. Commun. 2016; 7:12917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Palazzo L., Leidecker O., Prokhorova E., Dauben H., Matic I., Ahel I.. Serine is the major residue for ADP-ribosylation upon DNA damage. eLife. 2018; 7:e34334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leslie Pedrioli D.M., Leutert M., Bilan V., Nowak K., Gunasekera K., Ferrari E., Imhof R., Malmstrom L., Hottiger M.O.. Comprehensive ADP-ribosylome analysis identifies tyrosine as an ADP-ribose acceptor site. EMBO Rep. 2018; 19:e45310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Suskiewicz M.J., Zobel F., Ogden T.E.H., Fontana P., Ariza A., Yang J.-C., Zhu K., Bracken L., Hawthorne W.J., Ahel D. et al.. HPF1 completes the PARP active site for DNA damage-induced ADP-ribosylation. Nature. 2020; 579:598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gagné J.-P., Ethier C., Defoy D., Bourassa S., Langelier M.-F., Riccio A.A., Pascal J.M., Moon K.-M., Foster L.J., Ning Z. et al.. Quantitative site-specific ADP-ribosylation profiling of DNA-dependent PARPs. DNA Repair (Amst.). 2015; 30:68–79. [DOI] [PubMed] [Google Scholar]
- 14. Mortusewicz O., Ame J.C., Schreiber V., Leonhardt H.. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res. 2007; 35:7665–7675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krüger A., Bürkle A., Hauser K., Mangerich A.. Real-time monitoring of PARP1-dependent PARylation by ATR-FTIR spectroscopy. Nat. Commun. 2020; 11:2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eustermann S., Wu W.-F., Langelier M.-F., Yang J.-C., Easton Laura E., Riccio Amanda A., Pascal John M., Neuhaus D.. Structural basis of detection and signaling of DNA single-strand breaks by human PARP-1. Mol. Cell. 2015; 60:742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Langelier M.-F., Planck J.L., Roy S., Pascal J.M.. Structural basis for DNA damage–dependent poly(ADP-ribosyl)ation by human PARP-1. Science (New York, N.Y.). 2012; 336:728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu L., Kong M., Gassman N.R., Freudenthal B.D., Prasad R., Zhen S., Watkins S.C., Wilson S.H., Van Houten B.. PARP1 changes from three-dimensional DNA damage searching to one-dimensional diffusion after auto-PARylation or in the presence of APE1. Nucleic Acids Res. 2017; 45:12834–12847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Steffen J.D., McCauley M.M., Pascal J.M.. Fluorescent sensors of PARP-1 structural dynamics and allosteric regulation in response to DNA damage. Nucleic Acids Res. 2016; 44:9771–9783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mendoza-Alvarez H., Alvarez-Gonzalez R.. Poly(ADP-ribose) polymerase is a catalytic dimer and the automodification reaction is intermolecular. J. Biol. Chem. 1993; 268:22575–22580. [PubMed] [Google Scholar]
- 21. Pion E., Ullmann G.M., Ame J.C., Gerard D., de Murcia G., Bombarda E.. DNA-induced dimerization of poly(ADP-ribose) polymerase-1 triggers its activation. Biochemistry. 2005; 44:14670–14681. [DOI] [PubMed] [Google Scholar]
- 22. Ali A.A.E., Timinszky G., Arribas-Bosacoma R., Kozlowski M., Hassa P.O., Hassler M., Ladurner A.G., Pearl L.H., Oliver A.W.. The zinc-finger domains of PARP1 cooperate to recognize DNA strand breaks. Nat. Struct. Mol. Biol. 2012; 19:685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fischbach A., Krüger A., Hampp S., Assmann G., Rank L., Hufnagel M., Stöckl M.T., Fischer J.M.F., Veith S., Rossatti P et al.. The C-terminal domain of p53 orchestrates the interplay between non-covalent and covalent poly(ADP-ribosyl)ation of p53 by PARP1. Nucleic Acids Res. 2018; 46:804–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kamaletdinova T., Fanaei-Kahrani Z., Wang Z.Q.. The enigmatic function of PARP1: From PARylation activity to PAR readers. Cells. 2019; 8:1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Teloni F., Altmeyer M.. Readers of poly(ADP-ribose): designed to be fit for purpose. Nucleic Acids Res. 2016; 44:993–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pleschke J.M., Kleczkowska H.E., Strohm M., Althaus F.R.. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J. Biol. Chem. 2000; 275:40974–40980. [DOI] [PubMed] [Google Scholar]
- 27. Haince J.F., McDonald D., Rodrigue A., Dery U., Masson J.Y., Hendzel M.J., Poirier G.G.. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008; 283:1197–1208. [DOI] [PubMed] [Google Scholar]
- 28. Ahel I., Ahel D., Matsusaka T., Clark A.J., Pines J., Boulton S.J., West S.C.. Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature. 2008; 451:81–85. [DOI] [PubMed] [Google Scholar]
- 29. Timinszky G., Till S., Hassa P.O., Hothorn M., Kustatscher G., Nijmeijer B., Colombelli J., Altmeyer M., Stelzer E.H., Scheffzek K. et al.. A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 2009; 16:923–929. [DOI] [PubMed] [Google Scholar]
- 30. Wang Z., Michaud G.A., Cheng Z., Zhang Y., Hinds T.R., Fan E., Cong F., Xu W.. Recognition of the iso-ADP-ribose moiety in poly(ADP-ribose) by WWE domains suggests a general mechanism for poly(ADP-ribosyl)ation-dependent ubiquitination. Genes Dev. 2012; 26:235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O'Sullivan J., Tedim Ferreira M., Gagne J.P., Sharma A.K., Hendzel M.J., Masson J.Y., Poirier G.G.. Emerging roles of eraser enzymes in the dynamic control of protein ADP-ribosylation. Nat. Commun. 2019; 10:1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eisemann T., Pascal J.M.. Poly(ADP-ribose) polymerase enzymes and the maintenance of genome integrity. Cell. Mol. Life Sci.: CMLS. 2020; 77:19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ray Chaudhuri A., Nussenzweig A.. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017; 18:610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rank L., Veith S., Gwosch E.C., Demgenski J., Ganz M., Jongmans M.C., Vogel C., Fischbach A., Buerger S., Fischer J.M. et al.. Analyzing structure-function relationships of artificial and cancer-associated PARP1 variants by reconstituting TALEN-generated HeLa PARP1 knock-out cells. Nucleic Acids Res. 2016; 44:10386–10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aleksandrov R., Dotchev A., Poser I., Krastev D., Georgiev G., Panova G., Babukov Y., Danovski G., Dyankova T., Hubatsch L. et al.. Protein dynamics in complex DNA lesions. Mol. Cell. 2018; 69:1046–1061. [DOI] [PubMed] [Google Scholar]
- 36. Strickfaden H., McDonald D., Kruhlak M.J., Haince J.F., Th’ng J.P., Rouleau M., Ishibashi T., Corry G.N., Ausio J., Underhill D.A. et al.. Poly(ADP-ribosyl)ation-dependent transient chromatin decondensation and histone displacement following laser microirradiation. J. Biol. Chem. 2016; 291:1789–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schmutz I., Timashev L., Xie W., Patel D.J., de Lange T.. TRF2 binds branched DNA to safeguard telomere integrity. Nat. Struct. Mol. Biol. 2017; 24:734. [DOI] [PubMed] [Google Scholar]
- 38. Slade D. Mitotic functions of poly(ADP-ribose) polymerases. Biochem. Pharmacol. 2019; 167:33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hanzlikova H., Kalasova I., Demin A.A., Pennicott L.E., Cihlarova Z., Caldecott K.W.. The importance of poly(ADP-ribose) polymerase as a sensor of unligated okazaki fragments during DNA replication. Mol. Cell. 2018; 71:319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hanzlikova H., Caldecott K.W.. Perspectives on PARPs in S phase. Trends Genet. 2019; 35:412–422. [DOI] [PubMed] [Google Scholar]
- 41. Ke Y., Wang C., Zhang J., Zhong X., Wang R., Zeng X., Ba X.. The role of PARPs in inflammation-and metabolic-related diseases: molecular mechanisms and beyond. Cells. 2019; 8:1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kunze F.A., Hottiger M.O.. Regulating immunity via ADP-ribosylation: therapeutic implications and beyond. Trends Immunol. 2019; 40:159–173. [DOI] [PubMed] [Google Scholar]
- 43. Ying Y., Padanilam B.J.. Regulation of necrotic cell death: p53, PARP1 and cyclophilin D-overlapping pathways of regulated necrosis. Cell. Mol. Life Sci.: CMLS. 2016; 73:2309–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fahrer J., Kranaster R., Altmeyer M., Marx A., Bürkle A.. Quantitative analysis of the binding affinity of poly(ADP-ribose) to specific binding proteins as a function of chain length. Nucleic Acids Res. 2007; 35:e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Popp O., Veith S., Fahrer J., Bohr V.A., Bürkle A., Mangerich A.. Site-specific noncovalent interaction of the biopolymer poly(ADP-ribose) with the Werner syndrome protein regulates protein functions. ACS Chem. Biol. 2013; 8:179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fischer J.M.F., Popp O., Gebhard D., Veith S., Fischbach A., Beneke S., Leitenstorfer A., Bergemann J., Scheffner M., Ferrando‐May E. et al.. Poly(ADP‐ribose)‐mediated interplay of XPA and PARP1 leads to reciprocal regulation of protein function. FEBS J. 2014; 281:3625–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Krietsch J., Rouleau M., Pic E., Ethier C., Dawson T.M., Dawson V.L., Masson J.Y., Poirier G.G., Gagne J.P.. Reprogramming cellular events by poly(ADP-ribose)-binding proteins. Mol. Aspects Med. 2013; 34:1066–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Leung A.K.L. Poly(ADP-ribose): a dynamic trigger for biomolecular condensate formation. Trends Cell Biol. 2020; 30:370–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Panzeter P.L., Realini C.A., Althaus F.R.. Noncovalent interactions of poly(adenosine diphosphate ribose) with histones. Biochemistry. 1992; 31:1379–1385. [DOI] [PubMed] [Google Scholar]
- 50. Krüger A., Stier A., Fischbach A., Bürkle A., Hauser K., Mangerich A.. Interactions of p53 with poly(ADP-ribose) and DNA induce distinct changes in protein structure as revealed by ATR-FTIR spectroscopy. Nucleic Acids Res. 2019; 47:4843–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen Q., Kassab M.A., Dantzer F., Yu X.. PARP2 mediates branched poly ADP-ribosylation in response to DNA damage. Nat. Commun. 2018; 9:3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rippmann J.F., Damm K., Schnapp A.. Functional characterization of the poly(ADP-ribose) polymerase activity of tankyrase 1, a potential regulator of telomere length. J. Mol. Biol. 2002; 323:217–224. [DOI] [PubMed] [Google Scholar]
- 53. Alvarez-Gonzalez R., Mendoza-Alvarez H.. Dissection of ADP-ribose polymer synthesis into individual steps of initiation, elongation, and branching. Biochimie. 1995; 77:403–407. [DOI] [PubMed] [Google Scholar]
- 54. Hatakeyama K., Nemoto Y., Ueda K., Hayaishi O.. Purification and characterization of poly(ADP-ribose) glycohydrolase. Different modes of action on large and small poly(ADP-ribose). J. Biol. Chem. 1986; 261:14902–14911. [PubMed] [Google Scholar]
- 55. Braun S.A., Panzeter P.L., Collinge M.A., Althaus F.R.. Endoglycosidic cleavage of branched polymers by poly(ADP-ribose) glycohydrolase. Eur. J. Biochem. 1994; 220:369–375. [DOI] [PubMed] [Google Scholar]
- 56. Malanga M., Althaus F.R.. Poly(ADP-ribose) molecules formed during DNA repair in vivo. J. Biol. Chem. 1994; 269:17691–17696. [PubMed] [Google Scholar]
- 57. Rolli V., O’Farrell M., Menissier-de Murcia J., de Murcia G.. Random mutagenesis of the poly(ADP-ribose) polymerase catalytic domain reveals amino acids involved in polymer branching. Biochemistry. 1997; 36:12147–12154. [DOI] [PubMed] [Google Scholar]
- 58. Langelier M.F., Planck J.L., Servent K.M., Pascal J.M.. Purification of human PARP-1 and PARP-1 domains from Escherichia coli for structural and biochemical analysis. Methods Mol. Biol. 2011; 780:209–226. [DOI] [PubMed] [Google Scholar]
- 59. Niesen F.H., Berglund H., Vedadi M.. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007; 2:2212–2221. [DOI] [PubMed] [Google Scholar]
- 60. Zubel T., Martello R., Bürkle A., Mangerich A.. Quantitation of poly(ADP-ribose) by isotope dilution mass spectrometry. Methods Mol. Biol. 2017; 1608:3–18. [DOI] [PubMed] [Google Scholar]
- 61. Martello R., Mangerich A., Sass S., Dedon P.C., Bürkle A.. Quantification of cellular poly(ADP-ribosyl)ation by stable isotope dilution mass spectrometry reveals tissue- and drug-dependent stress response dynamics. ACS Chem. Biol. 2013; 8:1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wallrodt S., Simpson E.L., Marx A.. Investigation of the action of poly(ADP-ribose)-synthesising enzymes on NAD+ analogues. Beilstein J. Org. Chem. 2017; 13:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Putt K.S., Hergenrother P.J.. An enzymatic assay for poly(ADP-ribose) polymerase-1 (PARP-1) via the chemical quantitation of NAD+: application to the high-throughput screening of small molecules as potential inhibitors. Anal. Biochem. 2004; 326:78–86. [DOI] [PubMed] [Google Scholar]
- 64. Kawamitsu H., Hoshino H., Okada H., Miwa M., Momoi H., Sugimura T.. Monoclonal antibodies to poly(adenosine diphosphate ribose) recognize different structures. Biochemistry. 1984; 23:3771–3777. [DOI] [PubMed] [Google Scholar]
- 65. Veith S., Schink A., Engbrecht M., Mack M., Rank L., Rossatti P., Hakobyan M., Goly D., Hefele T., Frensch M. et al.. PARP1 regulates DNA damage-induced nucleolar-nucleoplasmic shuttling of WRN and XRCC1 in a toxicant and protein-specific manner. Sci. Rep. 2019; 9:10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schmalz M.F., Wieser I., Schindler F., Czada C., Leitenstorfer A., Ferrando-May E.. Highly standardized multicolor femtosecond fiber system for selective microphotomanipulation of deoxyribonucleic acid and chromatin. Opt. Lett. 2018; 43:2877–2880. [DOI] [PubMed] [Google Scholar]
- 67. Fischer B.M., Neumann D., Piberger A.L., Risnes S.F., Koberle B., Hartwig A.. Use of high-throughput RT-qPCR to assess modulations of gene expression profiles related to genomic stability and interactions by cadmium. Arch. Toxicol. 2016; 90:2745–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. García-Saura A.G., Zapata-Pérez R., Hidalgo J.F., Sánchez-Ferrer Á. Comparative inhibitory profile and distribution of bacterial PARPs, using Clostridioides difficile CD160 PARP as a model. Sci. Rep. 2018; 8:8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Langelier M.F., Zandarashvili L., Aguiar P.M., Black B.E., Pascal J.M.. NAD(+) analog reveals PARP-1 substrate-blocking mechanism and allosteric communication from catalytic center to DNA-binding domains. Nat. Commun. 2018; 9:844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Dawicki-McKenna J.M., Langelier M.-F., DeNizio J.E., Riccio A.A., Cao C.D., Karch K.R., McCauley M., Steffen J.D., Black B.E., Pascal J.M.. PARP-1 activation requires local unfolding of an autoinhibitory domain. Mol. Cell. 2015; 60:755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Buntz A., Wallrodt S., Gwosch E., Schmalz M., Beneke S., Ferrando-May E., Marx A., Zumbusch A.. Real-Time cellular imaging of protein Poly(ADP-ribos)ylation. Angew. Chem. Int. Ed. Engl. 2016; 55:11256–11260. [DOI] [PubMed] [Google Scholar]
- 72. Rafehi H., Orlowski C., Georgiadis G.T., Ververis K., El-Osta A., Karagiannis T.C.. Clonogenic assay: adherent cells. J. Vis. Exp. 2011; 2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kwon Y.T., Ciechanover A.. The ubiquitin code in the Ubiquitin-Proteasome system and autophagy. Trends Biochem. Sci. 2017; 42:873–886. [DOI] [PubMed] [Google Scholar]
- 74. Zhao X., Lutz J., Höllmüller E., Scheffner M., Marx A., Stengel F.. Identification of proteins interacting with ubiquitin chains. Angew. Chem. Int. Ed. 2017; 56:15764–15768. [DOI] [PubMed] [Google Scholar]
- 75. Ruf A., Rolli V., de Murcia G., Schulz G.E.. The mechanism of the elongation and branching reaction of Poly(ADP-ribose) polymerase as derived from crystal structures and mutagenesis. J. Mol. Biol. 1998; 278:57–65. [DOI] [PubMed] [Google Scholar]
- 76. Schuhwerk H., Bruhn C., Siniuk K., Min W., Erener S., Grigaravicius P., Krüger A., Ferrari E., Zubel T., Lazaro D. et al.. Kinetics of poly(ADP-ribosyl)ation, but not PARP1 itself, determines the cell fate in response to DNA damage in vitro and in vivo. Nucleic Acids Res. 2017; 45:11174–11192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mehrotra P.V., Ahel D., Ryan D.P., Weston R., Wiechens N., Kraehenbuehl R., Owen-Hughes T., Ahel I.. DNA repair factor APLF is a histone chaperone. Mol. Cell. 2011; 41:46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Grundy G.J., Rulten S.L., Zeng Z., Arribas-Bosacoma R., Iles N., Manley K., Oliver A., Caldecott K.W.. APLF promotes the assembly and activity of non-homologous end joining protein complexes. EMBO J. 2013; 32:112–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Li G.Y., McCulloch R.D., Fenton A.L., Cheung M., Meng L., Ikura M., Koch C.A.. Structure and identification of ADP-ribose recognition motifs of APLF and role in the DNA damage response. PNAS. 2010; 107:9129–9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Eustermann S., Brockmann C., Mehrotra P.V., Yang J.C., Loakes D., West S.C., Ahel I., Neuhaus D.. Solution structures of the two PBZ domains from human APLF and their interaction with poly(ADP-ribose). Nat. Struct. Mol. Biol. 2010; 17:241–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shen Y., Aoyagi-Scharber M., Wang B.. Trapping poly(ADP-ribose) polymerase. J. Pharmacol. Exp. Ther. 2015; 353:446–457. [DOI] [PubMed] [Google Scholar]
- 82. Gehl Z., Bai P., Bodnar E., Emri G., Remenyik E., Nemeth J., Gergely P., Virag L., Szabo E.. Poly(ADP-ribose) in the skin and in melanomas. Histol. Histopathol. 2012; 27:651–659. [DOI] [PubMed] [Google Scholar]
- 83. Jubin T., Kadam A., Jariwala M., Bhatt S., Sutariya S., Gani A.R., Gautam S., Begum R.. The PARP family: insights into functional aspects of poly (ADP-ribose) polymerase-1 in cell growth and survival. Cell Prolif. 2016; 49:421–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Du Y., Yamaguchi H., Wei Y., Hsu J.L., Wang H.L., Hsu Y.H., Lin W.C., Yu W.H., Leonard P.G., Lee G.R.T. et al.. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat. Med. 2016; 22:194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Piao L., Fujioka K., Nakakido M., Hamamoto R.. Regulation of poly(ADP-Ribose) polymerase 1 functions by post-translational modifications. Front. Biosci. (Landmark Ed.). 2018; 23:13–26. [DOI] [PubMed] [Google Scholar]
- 86. Kauppinen T.M., Chan W.Y., Suh S.W., Wiggins A.K., Huang E.J., Swanson R.A.. Direct phosphorylation and regulation of poly(ADP-ribose) polymerase-1 by extracellular signal-regulated kinases 1/2. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:7136–7141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wright R.H., Castellano G., Bonet J., Le Dily F., Font-Mateu J., Ballare C., Nacht A.S., Soronellas D., Oliva B., Beato M.. CDK2-dependent activation of PARP-1 is required for hormonal gene regulation in breast cancer cells. Genes Dev. 2012; 26:1972–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zandarashvili L., Langelier M.F., Velagapudi U.K., Hancock M.A., Steffen J.D., Billur R., Hannan Z.M., Wicks A.J., Krastev D.B., Pettitt S.J. et al.. Structural basis for allosteric PARP-1 retention on DNA breaks. Science. 2020; 368:eaax6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Putt K.S., Hergenrother P.J.. A nonradiometric, high-throughput assay for poly(ADP-ribose) glycohydrolase (PARG): application to inhibitor identification and evaluation. Anal. Biochem. 2004; 333:256–264. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.