

Abstract
Sarcoidosis is a multisystem disease with heterogeneity in manifestations and outcomes. System-level studies leveraging “omics” technologies are expected to define mechanisms contributing to sarcoidosis heterogeneous manifestations and course. With improvements in mass spectrometry (MS) and bioinformatics, it is possible to study protein abundance for a large number of proteins simultaneously. Contemporary fast-scanning MS enables the acquisition of spectral data for deep coverage of the proteins with data-dependent or data-independent acquisition MS modes. Studies leveraging MS-based proteomics in sarcoidosis have characterized BAL fluid (BALF), alveolar macrophages, plasma, and exosomes. These studies identified several differentially expressed proteins, including protocadherin-2 precursor, annexin A2, pulmonary surfactant A2, complement factors C3, vitamin-D–binding protein, cystatin B, and amyloid P, comparing subjects with sarcoidosis with control subjects. Other studies identified ceruloplasmin, complement factors B, C3, and 1, and others with differential abundance in sarcoidosis compared with other interstitial lung diseases. Using quantitative proteomics, most recent studies found differences in PI3K/Akt/mTOR, MAP kinase, pluripotency-associated transcriptional factor, and hypoxia response pathways. Other studies identified increased clathrin-mediated endocytosis and Fcγ receptor–mediated phagocytosis pathways in sarcoidosis alveolar macrophages. Although studies in mixed BAL and blood cells or plasma are limited, some of the changes in lung compartment are detected in the blood cells and plasma. We review proteomics for sarcoidosis with a focus on the existing MS data acquisition strategies, bioinformatics for spectral data analysis to infer protein identity and quantity, unique aspects about biospecimen collection and processing for lung-related proteomics, and proteomics studies conducted to date in sarcoidosis.
Keywords: proteomics, sarcoidosis, biomarkers, pulmonary sarcoidosis
Sarcoidosis is a rare multisystem disease of unknown cause characterized by granulomatous inflammation with variable severity and outcomes. The pathologic hallmark of sarcoidosis is epithelioid granuloma associated with the infiltration of CD4+ T cells, scattered macrophages, giant cells, CD8+ T cells, and B cells around the granuloma. Despite a greater understanding of sarcoidosis pathogenesis, the mechanisms contributing to the heterogeneity of disease manifestations and predictors of disease outcomes are poorly defined (1). A crucial step to narrow the gaps in the understanding of sarcoidosis includes the use of “omics” and systems biology research for the molecular characterization of sarcoidosis phenotypes and to uncover novel disease mechanisms that could represent new disease biomarkers or therapeutic targets.
Powerful high-throughput technologies make it possible to investigate large numbers of biological functions at multiple levels, which can be particularly useful in a polygenic condition such as sarcoidosis (2). Transcriptional studies in sarcoidosis have found genes that regulate the IFN-α, β, and γ signaling (STAT1), consistent with the Th1-type inflammatory response (3). In a genome-wide analysis of lung tissue, networks corresponding with cell movement and immune function, including STAT1, IL-7, IL-15, and specific chemokine genes, were overexpressed in subjects with sarcoidosis (4). However, proteins are the main effectors of cellular function, and their alterations result in the disruption of biologic systems. To gain a comprehensive understanding in sarcoidosis, systematic testing of the protein changes is essential.
Several platforms identify proteins by detecting antigenic epitopes. Meso scale discovery (5), proximity extension assays (6) and aptamer-based tools (7) are available to quantify candidate proteins. A limitation of methodologies that detect antigens is that they are limited to an a priori set of proteins. With the improvement in the mass spectrometers and bioinformatics, it is now possible to quantify thousands of proteins in complex protein mixtures. For this review, we will focus on workflows for mass spectrometry (MS)-based proteomics, which beneficially provides comprehensive identification and quantification of unknown proteins, and their application to sarcoidosis.
Biospecimens for Lung Proteomics
In proteomics research, sample handling and processing are crucial for robust results. The lung has inflammatory cells as well as epithelial lining fluid (ELF) consisting of mucins and surfactants. These “matrix” biomolecules in ELF make the MS analysis challenging, as they interfere with spectral data acquisition. The wide dynamic range also limits the detection of less abundant proteins (8), and high abundant protein depletion increases protein detection (9). The recent American Thoracic Soceity document provides guidance for the preparation of samples for proteomic studies (10). A critical issue is to prevent ex vivo changes. For this reason, standard operating procedures are necessary. These standard operating procedures should include sample transportation on ice, aliquot in usable sample volumes to avoid future freeze and thaw cycles, immediate processing for freezing (−80°C) until the time of assay, and long-term storage in liquid nitrogen (greater than 6 mo) (10).
For sarcoidosis, it would be ideal to have affected tissue for analysis; however, the patchy nature of involvement and limited tissue availability is a barrier to examining changes in the areas of granulomatous inflammation. Using blood or urine offers the benefit of repeated sampling, but the lung-specific signal is diluted. Consequently, sputum (11), ELF (12), lung edema fluid (13), exhaled breath condensate (EBC) (14), BAL fluid (BALF), and BALF-derived exosomes (15) have been investigated (Table 1). BAL cells provide insights into the immune mechanisms of inflammatory lung diseases. In sarcoidosis, although gene expression studies have identified the activation of alveolar resident immune processes such as mechanisms involved in T-cell, IL-12, IL-23, and IL-17 signaling (16) and lower concentrations of steroid receptor coactivator-3 in cases needing treatment (17), the investigation of BAL cells is underused, with only a few reports implementing comprehensive protein evaluation (18, 19) (described later). Examining EBC is challenging because water vapor constitutes more than 99% of its content (20). Higher EBC TNF-α concentrations, as a marker of alveolar macrophage activity, were observed by ELISA in sarcoidosis patients with sarcoidosis (21). In chronic obstructive pulmonary disease (COPD), differences in EBC cytokines, cytokeratin (22), and surfactant proteins indicated distinct COPD variants (23). Such studies could identify subtypes in sarcoidosis. Sputum offers ease of collection but is a challenging biospecimen because of variable viscosity; components such as inflammatory cells, microbes and their peptides, cellular debris, and most importantly, mucins; and contamination by salivary proteins. The mucins and highly charged proteins form crosslinks with other sputum proteins (24) and make detection by MS challenging. Specific sputum sample preparation identified the protein changes characteristic of current smoking status and thirteen differentially expressed proteins between asymptomatic smokers and COPD cases. Blood-based proteomics is attractive, and several studies in sarcoidosis have used multiplexed platforms that detected a priori identified antigens (25, 26), and only a few studies have used MS (27). The high dynamic range of the plasma/serum has made it challenging to examine the global protein changes.
Table 1.
Biospecimens for Proteomic Investigations in Sarcoidosis
| Advantages | Limitations | Sample-Specific Recommendation for Sarcoidosis | Role in Sarcoidosis | |
|---|---|---|---|---|
| Lung/other tissue | Samples the site of
injury Highest likelihood to capture biological changes |
Granulomatous changes are
patchy and prone to missed sampling Limited amount of tissue available Need invasive procedures for biopsy Contamination of tissue samples by blood and plasma proteins |
Snap-freeze lung tissue
promptly Washing before sample preparation for proteomics |
Determine biological
mechanism of granulomatous inflammation Promising for integration with other omics, including genomics |
| BALF | Easy to obtain via
bronchoscopy Samples epithelial lining fluid (most proximate to the site of injury) Samples proteins from diverse cellular sources Used with standardized protocols by several investigators |
High salt content needs
removal (10) Variable dilution because of unpredictable volume of return High dynamic range because of albumin and other proteins Surfactant protein may impact spectral data acquisition |
Enrichment of specific
proteins of interest or removal of higher abundance
proteins Standardized protocols for sample collection and storage Salt removal before analysis Avoid freeze-thaw |
Better suited for
developing biomarkers for diagnosis and
prognosis Promising for integrative omics, such as metabolomics, but not genomics because of limited availability of RNA |
| BAL cells | Easy to obtain via
bronchoscopy Identifies immune mechanisms by sampling inflammatory cells |
Large volume lavage to
ensure adequate cellular yield Contamination by red blood cells Variable proportion of cells depending on disease states and environmental exposures |
Account for cell proportion
during assay and analysis Cryopreservation may alter cellular proteins Avoid freeze-thaw |
Determine biological
mechanism of granulomatous inflammation Promising for integration with other omics, including genomics |
| EBC | Can be obtaining without
any procedures Could be repeated without difficulty |
Because EBC is >99%
condensed water vapor (20), most analytes are close to the detection limit
(10, 91) Variable collection methods alter EBC analytes and concentrations |
Standard procedures to minimize the variability in collection (20) | Limited role unless standardized sample preparation and improved resolution of MS instruments |
| Sputum | Easy to obtain | Variable viscosity from
sampling methods and sputum components Large amounts of mucins Contamination by oral microbes and microbial peptides |
Adding reducing agents such
as DTT (20),
size-exclusion with or without dialysis (24), or gel-cleanup
(92) to remove
mucins Standardized approach to sample collection and processing Acetone precipitation for protein isolation |
Unexplored but challenging for sarcoidosis pathogenesis and biomarker discovery |
| Blood-based proteomics | Easy to obtain without
special procedures Better to capture systemic changes beyond organ-specific changes |
High dynamic range of
plasma Low-abundance proteins or those with a small change may be difficult to quantify reliably High-abundance protein removal techniques may remove other proteins Semple preparation could decrease throughput for discovery studies |
Plasma is preferred over
serum with EDTA (over heparin) (93) Removal of high-abundance proteins to improve depth of coverage (94) Fractionation before MS analysis (95) Removal of disease-relevant proteins bound to high-abundance proteins remains a challenge |
Biomarker in blood would be
idea for diagnosis and prognosis Targeted proteomics approaches offer promise to develop diagnostic tools and clinical assay |
Definition of abbreviations: BALF = BAL fluid; EBC = exhaled breath condensate; MS = mass spectrometry.
The official report of the American Thoracic Society workgroup on metabolomics and proteomics offers guidance on techniques and challenges and recommendations for sample handling and processing (10).
BALF is performed with normal saline, and therefore, removal of high salt content is necessary before MS analysis. During BAL, ELF is diluted up to 100-fold in an unpredictable manner that is difficult to account for. Although some recommend using BALF urea (10, 28), others conclude that no method can sufficiently account for the variability in the dilution (29). Standardizing the instilled volume (29), the number of input aliquots (29), and the site of lavage (29) and using a single-cycle lavage (30) could decrease the variability in ELF dilution. ELF is rich in plasma-derived proteins (albumin, transferrin, etc.), especially in conditions with increased epithelial permeability. The enrichment of specific proteins of interest or removal of higher abundance proteins are commonly used strategies when examining BALF (31–33). Despite challenges of sample processing, global or targeted protein studies have provided valuable insights into the biology of lung diseases such as idiopathic pulmonary fibrosis (IPF) (34, 35), acute respiratory distress syndrome (13, 36), asthma (37, 38), COPD (39), and lung cancer (40, 41). The studies in sarcoidosis are described later.
Disease models provide excitement for the identification of biological systems that participate in the formation of sarcoid granuloma. Recently, in an innovative human granuloma model, the incubation of peripheral blood mononuclear cells from sarcoidosis cases with bacillus Calmette-Guérin–coated beads resulted in the formation of cell aggregates with centralized macrophage aggregation and peripheral T-cell clusters, as is observed in sarcoidosis human tissue (42). The genomic profile of the in vitro sarcoidosis granulomas conforms to human sarcoidosis tissue granulomas, including the mTORc1 signaling pathway, IL-13, and STAT6 as important upstream regulators of genes and pathways active in human lung and lymph node sarcoidosis tissues (43). Because of limited access to tissue with granulomatous inflammation, examining the landscape of protein changes in the in vitro granuloma models offers promise for the identification of both biomarkers for diagnosis and novel mechanisms that dictate sarcoidosis development and course.
Mass Spectrometry–based Proteomics
With advances in MS, MS instruments are ever increasing in sensitivity, speed, and selectivity. MS instrumentation, coupled with upfront ion funneling technologies, such as gas-phase ion mobility fractionation, substantially increases the ability to comprehensively detect and quantify peptides across the dynamic range of the proteome (44–46). In combination with evolving sample preparation workflows for organelle enrichment and/or membrane structure enrichment (47, 48), these techniques now provide opportunities to specifically investigate membrane or mitochondrial proteins. There are also powerful in vivo labeling methods emerging to target specific membrane structures and enrich these for proteomics analysis (49, 50). In sarcoidosis, MS proteomics could be performed for discovery studies that analyze a large number of proteins or for the targeted measurement of specific proteins. Discovery proteomics analyze either intact proteins (top-down) (51) or peptides from proteolytic digests (bottom-up). Bottom-up proteomics offers protein inference through tandem MS (MS/MS) peptide spectral matching using traditional data-dependent acquisition (DDA), which has failed to identify low-abundance proteins such as transcription factors, cell receptors, and lipid-bound or mitochondrial proteins. Novel data-independent acquisition (DIA) MS provides an alternative method that comprehensively detects and robustly quantifies medium- and low-abundance proteins (52, 53).
Three top-down MS applications exist: matrix-assisted laser desorption Ionization-time of flight (MALDI-TOF), surface-enhanced laser desorption ionization (SELDI)-TOF, and electrospray (ESI)-MS. Both MALDI-TOF and SELDI-TOF can screen and compare protein profiles, but neither method can afford protein identification. Protein identification from MS/MS is feasible with ESI-MS/MS or liquid chromatography (LC)-ESI-MS/MS, but this technique is available in limited laboratories because specialized MS instrumentation is needed. However, high-throughput abilities of top-down MS make it appealing for discovery applications despite protein interference that occurs because of dynamic ranges in the proteome. Therefore, additional sample preparation, such as solid-phase extraction and chip-based techniques, is used to decrease sample complexity (54, 55). In contrast, bottom-up proteomics exploits two sample preparation methods (in-gel digestion and in-solution digestion) and permits relative and absolute protein quantitation. Such experiments are executed in the following steps: 1) proteolytic digestion, 2) chromatographic separation, 3) peptide MS/MS, 4) database search for peptide identification, and 5) protein assembly and quantification. The peptide MS/MS data acquisition can be acquired via DDA-MS or DIA-MS techniques. During DDA-MS, MS1 scans, known as “survey scans,” are acquired for all real-time precursor ions eluting during a time-course LC gradient. Subsequently, MS/MS scans (or MS2 scans) are triggered on the most intense precursor ions from the survey scan, using either top-speed or top-N mode (Figure 1A). This cycle is repeated over the duration of the LC gradient. Upstream sample separation methods, such as two-dimensional LC orthogonal separation is employed to reduce sample complexity and improve protein sequence coverage. Two-dimensional LC separation techniques often include offline high pH reverse-phase (RP) fractionation followed by low pH RP separation in line to ESI-MS (56). Discovery-based proteomics employs both label-free (57) and labeled-based (58, 59) approaches for relative quantification. Label-free quantitation (Figure 2A) can be determined in one of the following two ways: 1) integration of peak area of MS1 ions or 2) peptide spectral counting (60). Both techniques require replicate measurements of each sample to validate the results statistically. Label-free methods experience drawbacks because of chromatographic reproducibility, spectral variability, sample complexity, and throughput.
Figure 1.
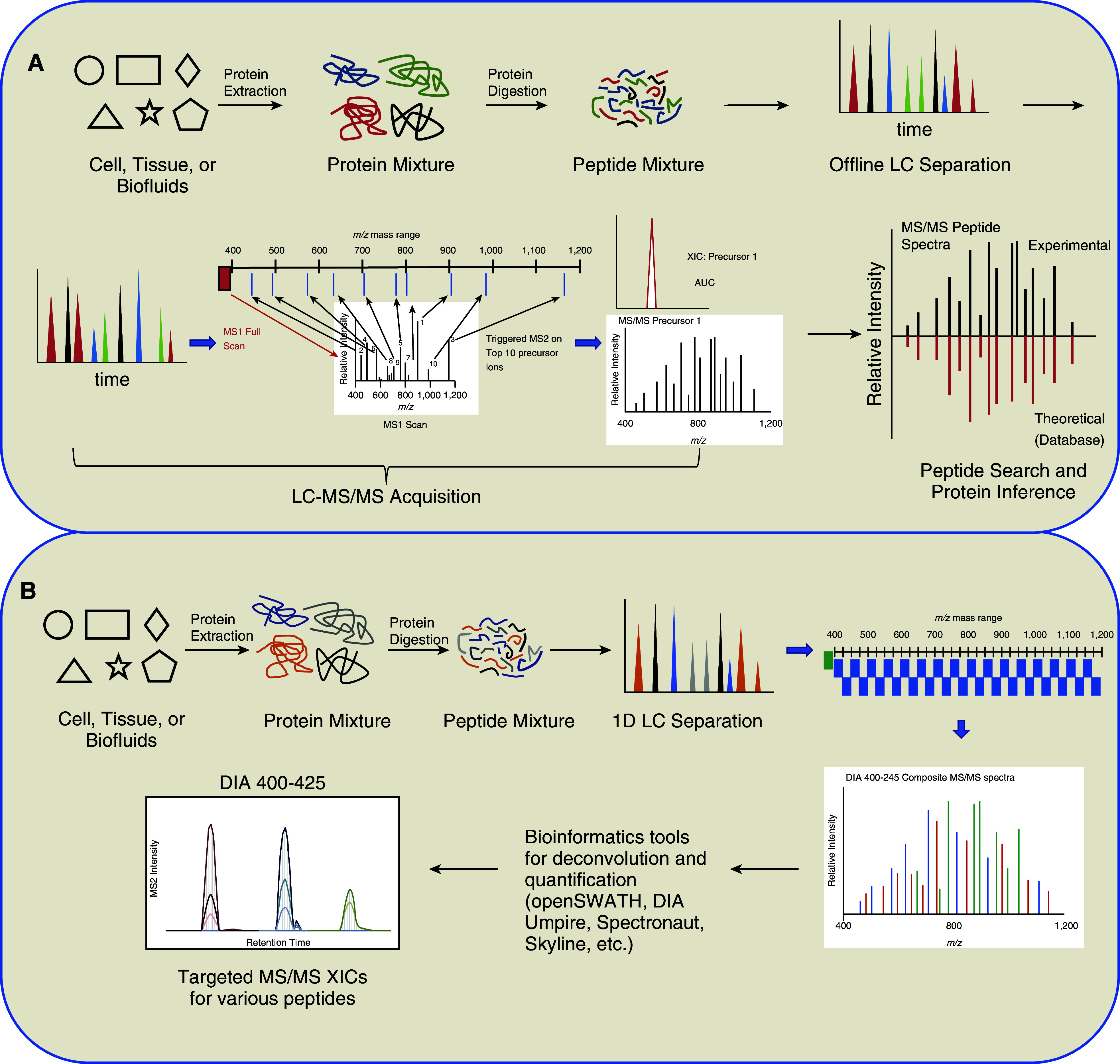
Data-dependent and data-independent mass spectrometry (MS). (A) Data-dependent acquisition (DDA) MS. During DDA-MS, protein samples are proteolytically digested into peptides. The peptides are then subjected to complementary offline liquid chromatography separation and subsequently concatenated into fractions. Fractions are then each separated using low pH liquid chromatography coupled to MS. Each duty cycle over a time-course gradient will include one precursor scan over the entire mass range. The precursor spectral intensity will then dictate which MS1 ions are selected for tandem MS (MS/MS). A dynamic exclusion time parameter can be used to prohibit duplication of MS/MS for a predefined time, allowing new MS/MS to be acquired. Effectively, DDA will acquire MS/MS spectra on the most abundant peptides in a mixture. Raw data are then processed using traditional peptide assignment software using the theoretical peptide values expected from genomic databases. Lastly, protein inference is assigned based principle of parsimony, in which the lowest number of proteins accounting for the peptides identified. (B) Data-independent acquisition (DIA) MS. In contrast to DDA, DIA omits real-time ion selection but obtains MS/MS spectra on all precursor ions irrespective of MS1 spectral intensity. DIA-MS uses small precursor windows ranging from 10 to 25 m/z to produce MS/MS spectra. The duty cycle will encompass successive windows of precursor isolation and fragmentation until the complete m/z range is completed. This process will repeat for the entire time course. The MS/MS spectra of wider isolations windows will result in a convolution of MS/MS spectra, which results in multiple peptides being fragmented. For bioinformatics analysis of DIA-MS spectral dataset, an upstream spectral library generation via DDA methods and specialized computational algorithms will deconvolute spectra and extract peptide extracted ion chromatogram (XIC) at MS1 and MS/MS levels for quantification. DIA-MS workflows have a high computational complexity. AUC = area under the curve; LC = liquid chromatography.
Figure 2.
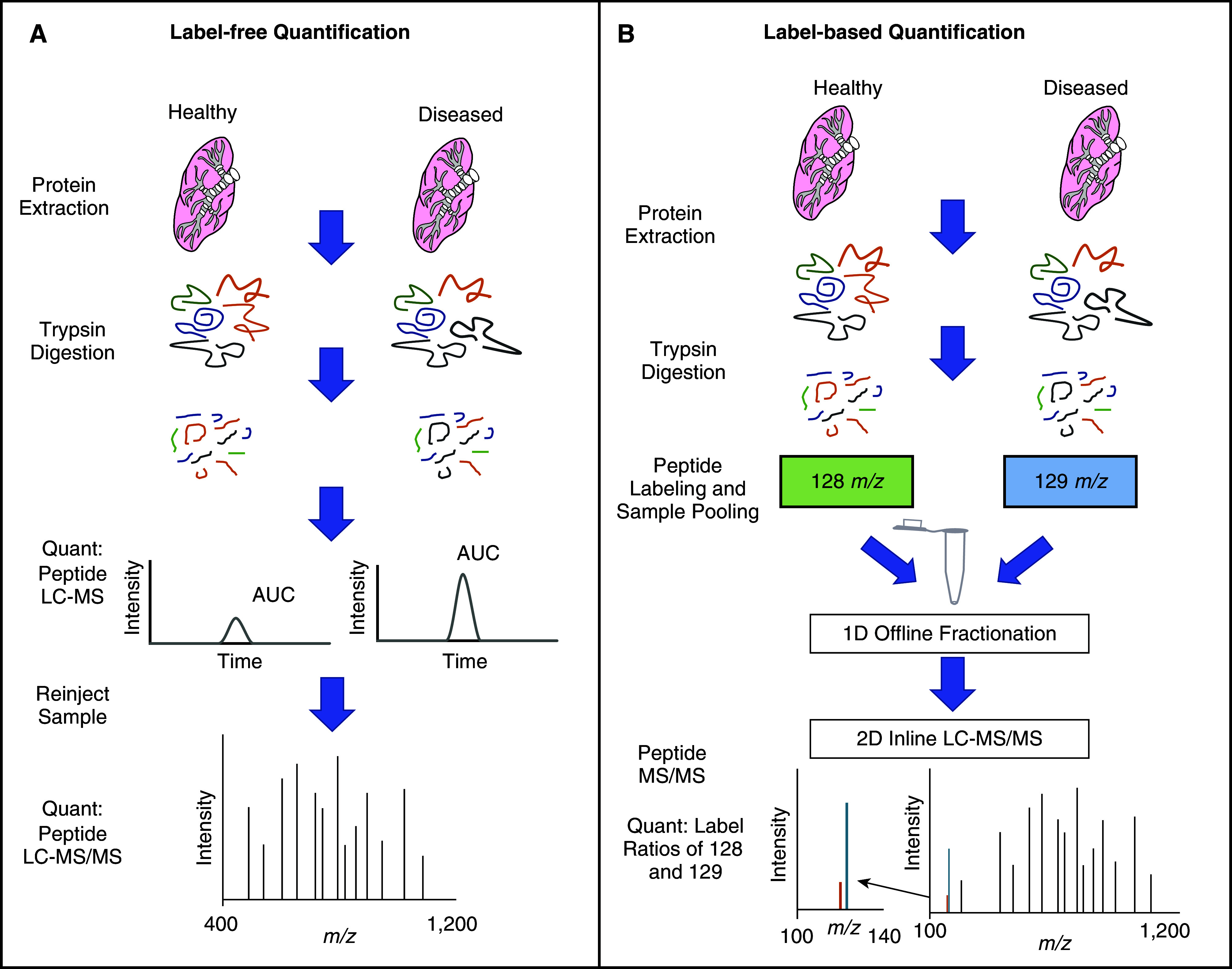
Quantitative proteomics approaches. Techniques for label-free quantitation (A) Peptide peak AUC or peptide spectral counting. The AUC for peptides can be calculated using the analyte intensity in combination with retention time profiles. The liquid chromatography elution profiles for peptide features within defined retention time window and mass accuracy settings are integrated. Peptide features are aligned and matched to proteins by their corresponding MS/MS and finally, peptide signals intensities between sample types are compared for protein abundance differences. Relative peptide amounts present with healthy and disease state are proportional to the peak AUC. Peptide spectral counting can use spectral counting of precursor (MS1) ions. The frequency of MS/MS spectra is proportional to the relative abundance of the peptide being surveyed. Targeted peptide identification is confirmed with MS/MS on a subsequent sample injection. (B) Label-based quantification involves the incorporation of metabolic or chemical tags for estimating protein abundance. Here, we demonstrate the use of chemical tags with two tandem mass tag labels. Healthy and diseased states are lysed and digested in similar ways as label-free approaches. The digested peptide from each sample is labeled with a tandem mass tag and fractionated offline. The fractionated samples are then individually subjected to liquid chromatography MS/MS. Under MS/MS conditions, each tandem mass tag label will cleave and generate a reporter signal. The ratio of these reported signals is indicative of peptide abundance within a labeled sample.
The principal of label-based quantification is the incorporation of metabolic or chemical tags for estimating protein abundance. Stable isotope labeling by amino acids in cell culture (SILAC) is metabolic labeling that incorporates stable heavy isotopes into proteins via labeled amino acids to compare two or three conditions of interest. During culture, cells are exposed to growth media containing heavy- or light-labeled amino acids that are globally incorporated into the proteome (61). In the case of a dual comparison, one sample condition is prepared with “light” media, which does not contain heavy-labeled amino acids. In the second condition, cells are cultured from “heavy” media. After protein extraction, equal amounts of light and heavy proteins are combined, proteolytically digested, and analyzed by ESI-MS. The resulting MS peak intensities for light and heavy are used for relative protein quantification. Isobaric labeling with tandem mass tags (TMT) or isobaric tags for relative and absolute quantification (iTRAQ) (Figure 2B) are used when biological applications may warrant multiple conditions be analyzed in an experiment. Isobaric tag labeling technologies use a differential chemical tag (reporter ion) to label the free amine group of peptides. Each tag comprises a heavy isotope–labeled functional group that contains a variation in the composition of heavy isotopes, resulting in the labeling of up to 16 samples simultaneously. Because the chemical tag is isobaric, peptides common to each sample type have identical precursor m/z values. Relative intensities of the reporter ions coincide with peptide abundance in each condition. Isobaric labeling offers major advantages in reducing MS variability, improving sample throughput, and decreasing laborious data processing.
In contrast to DDA, DIA omits using real-time ion selection dependent on precursor scans to obtain MS/MS spectra (Figure 1B) (62–64). Thus, the fragmentation of peptide ions is independent of survey scan intensity. Instead, all ionized species of a sample that fall within a specified mass range (e.g., m/z windows; i.e., 25 m/z) are fragmented systematically and impartially. Subsequently, MS/MS acquisition will represent all fragment ions generated from ionizable analytes in the defined window. The acquisition of the next MS/MS is a result of a new precursor isolation window. A spectral library is used to match the MS/MSall spectral dataset and infer protein identification.
Bioinformatics for DDA-MS and DIA-MS
The MS/MS spectra are assigned to peptide spectral match via peptide spectral matching. Publicly available species-specific repositories, which contain in silico translated genomic sequences, are used as databases in computational algorithms for determining peptide spectral matches. These theoretical in silico fragments ions are compared with experimental data, which is scored based on mass accuracy, consecutive ion matches, relative intensity, and number of unmatched fragment ions. The scored PSMs are then ranked, and the highest scoring PSMs are used to determine inferred proteins. Typically, one or two high-scoring peptide matches are sufficient evidence for the detection of a protein (65). Statistical validation of peptide and protein identification is determined by calculating false discovery rates when protein sequence databases are concatenated with reversed or scrambled databases (66). In comparison with DDA-MS bioinformatic algorithms, DIA-MS workflows have a high computational complexity because of wide isolation windows that generate fragment ions from multiple precursor ions (62, 67). Computational tools are available in which analyses can be performed in a peptide-centric or data-centric approach. Peptide-centric approaches (OpenSWATH, SWATH2.0, Skyline, and Spectronaut) require MS/MS spectral libraries to extract groups of information that reliably represent a particular peptide and use statistical methods to determine true matches (68). A data-centric software package, DIA-Umpire, constructs pseudo-MS/MS spectra that are subsequently identified and quantified with traditional database-searching and protein inference tools without an assay library (69). DIA offers promise as a reliable quantification method that can be scaled to clinical diagnostics. Although no published studies employing DIA-MS are available, it is a promising MS platform for improveing the understanding of sarcoidosis pathogenesis.
Discovery-based proteomics approaches are limited by their sensitivity, specificity, throughput, and cost-effectiveness for clinical applications. When specific signature features are known, targeted MS can be employed to provide absolute and/or relative quantification. Targeted assays improve the limits of detection and quantification and expand the working dynamic range when compared with DDA and DIA methods. Targeted assays require considerable method development but if used for absolute quantification, can be highly reproducible (70), and they are ideal for clinical assay development. Targeted MS employs multiple reaction monitoring (MRM) and selective reaction monitoring (SRM) or parallel reaction monitoring (PRM)–MS. For MRM or SRM, both precursor and fragment ions must be determined during the experimental design to ensure the appropriate precursor charge states and fragment ions are chosen. During analysis, precursor ions are selected and isolated in the first quadrupole. These ions are fragmented in a collision cell to generate fragment ions, which are selectively transmitted to the mass analyzer. MRM/SRM can monitor thousands of peptide transitions with appropriate chromatographic window scheduling for signature peptides SRM/MRM can monitor thousands of peptide transitions with appropriate chromatographic window scheduling for signature peptides. This targeted application excels in analyte sensitivity, sample throughput, and short acquisition times. MRM/SRM is well suited for validating selected protein and peptides form discovery studies in biomarker development and for clinical assay development. The MRM-based clinical classifier was developed to differentiate malignant lung nodules from benign lung nodules (71), but such studies have not been undertaken in sarcoidosis. The advantage of PRM over SRM/MRM is that it eliminates the need to preselect an optimum precursor to fragment PRM-MS requires the high-resolution MS/MS acquisition of all fragment ions after precursor ion isolation and fragmentation, typically on an orbitrap MS instrument. With full MS/MS spectra, PRM data can be searched with traditional DDA software algorithms, unlike SRM/MRM data. Ultimately, PRM assays have many uses, including biomarker discovery and validation in sarcoidosis.
Inferences from Proteomic Studies
Studies employing global proteomics provide high-dimensional data that could be leveraged to identify subtypes, to develop models (classifiers), and to gain insights into mechanisms contributing to sarcoidosis development and progression. Coupling contemporary proteomic platforms that offer deep coverage with appropriately powered studies and machine-learning algorithms will provide an opportunity to develop in silico disease models. The machine-learning principles that could be used for such studies include classification methods (Bayesian classifier, decision trees, random forest, rule-based learners, support vector machines, and artificial neural networks) and feature selection methods. The stability of the inferences could be tested by using partitioning tools, including cross-validation. These approaches are reviewed in prior publications (72, 73).
To gain novel insights into the mechanisms implicated in sarcoidosis development and progression, computational biology algorithms can be applied on proteomics datasets to provide insights into protein pathways and networks. Pathway analysis can organize proteins into existing canonical modules (signaling, gene-regulatory, or metabolic) to infer function. Networks use principles of graph theory to visualize and infer protein interactions using prior knowledge or de novo construction based on shared behaviors (such as regulation by transcription factors). Several currently available tools provide varying degrees of information about pathways and networks. These include tools that offer basic pathway functional information and basic network topological features. Such tools focus on the functional annotation of an unordered list of proteins by identifying statistically significant proteins first and functionally annotating these proteins, thus organizing interesting proteins into biological functions (most commonly, gene ontology terms). Several existing tools have this capability such as the Database for Annotation, Visualization and Integrated Discovery (DAVID), BioCarta, Reactome, GoMiner and others. Other tools provide rich pathway information by integrating statistical significance into functional integration by analyzing a ranked list of proteins in functional context, as done by our group previously (74). These tools (e.g., Gene Set Enrichment Analysis) provide information regarding the enrichment of the dataset proteins among eight curated gene sets, such as those predicted by targets for microRNA (regulatory target gene sets) or immunological gene sets. In comparison with basic pathway information tools, rich pathway information tools add information regarding shared behaviors, e.g., regulation or function. Bioinformatics algorithms can also provide basic or rich network information, including topological connections of proteins and events (protein interaction/regulation), that are carefully assembled into a graph. A number of tools including Cystoscope and Ingenuity Pathway Analysis are capable of identifying network topology and high-connectivity “hub” features that may be critical for network function. Previous publications summarize tools that may provide principles and available tools for pathway and network analysis of proteomics datasets that could be used for future studies in sarcoidosis (75, 76). To keep up with the ever evolving high-throughput platforms, novel bioinformatic algorithms can integrate multiomics datasets, which have positioned the scientific community to gain deeper insights into disease mechanisms that are not possible with reductionist approaches.
Proteomic Studies in Sarcoidosis
Several studies have examined system-level changes using genome-wide analysis, RNA sequencing, and protein changes and are summarized in prior reviews (77, 78). In lung tissue, the genes involved in Th1 (4, 79) and Th17 (80) immune responses were implicated in sarcoidosis and were drivers of fibrosis (81). These studies identified the crucial role of STAT1- and STAT3-related chemokines (CXCL9, CXCL10, and CXCL11) regulated by INF-γ and other genes, such as CXR5, GBP5, AIM2, ICAM1, JAK2, IL-5, IL-7, IL-12, and IL-15. Although significant overlap was observed in whole blood and lung tissue (3), some changes of pulmonary sarcoidosis are compartmentalized to the lung. In blood, the genes participating in IFN signaling (STAT1, STAT2, and GBPI) and pattern recognition (TLR2, TLR4, and TLR8), TCRs (CD28, ITK, and LEF1), and CXCL9 were linked to sarcoidosis, and CXCL9 and TCR genes were associated with progressive sarcoidosis (82). Some gene expression changes have also been detected at the proteome level. Table 2 lists proteomic studies in sarcoidosis that characterized BALF (26, 55, 83–88) and a few studies that examined serum/plasma (25–27), alveolar macrophages (18, 19), and BALF exosomes (15).
Table 2.
Summary of Studies Investigating Protein Changes in Sarcoidosis
| Study | Tissue Type Studied | Sarcoidosis | Control Subjects | Proteomics Method | Findings |
|---|---|---|---|---|---|
| Magi 2002 (83) | BALF | Active sarcoidosis (n = 6) | IPF (n = 6) | 2DE | Thirty-two protein spots with differential expression, of which 15 were more abundant in sarcoidosis |
| Sabounchi-Schütt 2003 (84) | BALF | Acute onset LS (n = 6) | Healthy (n = 4) | 2DE-MALDI-TOF | Seventeen protein spots different in sarcoidosis; seven proteins not previously reported in BALF were also identified |
| Sabounchi-Schütt 2004 (27) | Serum | Sarcoidosis (n = 6) | Healthy (n = 4) | 2DE and MALDI-TOF | Twenty-two protein spots were different between subjects with sarcoidosis and control subjects, 14 were more abundant and eight were less abundant in sarcoidosis. Nineteen spots had a statistically significant difference. Eleven proteins were identified from thee protein spots |
| Kriegova 2006 (55) | BALF | Stage I (n = 32 [LS = 14; NLS = 18]), stage II (n = 22), and stage III (n = 11) | Healthy (n = 23) | SELDI-TOF MS | Forty differentially expressed protein peaks, of which three were identified to be protococadherein-2 precursor, HSA, and alpha-1 antitrypsin |
| Rottoli 2005 (88) | BALF | Active sarcoidosis (n = 11) | IPF (n = 10) and SSc pulmonary fibrosis (n = 10) | 2DE and cellular profile by flow cytometry. | An increase in plasma protein in BALF from sarcoidosis and SSc and more locally produced lower molecular weight proteins in IPF |
| Rottoli 2005 (87) | BALF | Active sarcoidosis | IPF (n = 10) and SSc pulmonary fibrosis (n = 10) | Carbonylated proteins assessed by ELISA | Increase in carbonylated proteins in the subjects with diffuse lung diseases compared with control subjects. Patients with IPF had more carbonylation protein targets, including plasmatic proteins and transferrin immunoglobulin light chain, and IgA. Albumin and immunoglobulin heavy chain target carbonylated in all cases and control subjects and may act as a buffer and prevent other proteins from oxidant damage |
| Silva 2007 (86) | BALF | Sarcoidosis (n = 4) | Chronic beryllium disease (n = 4) and control subjects (n = 5) | DIGE coupled with MALDI-TOF | Sixteen protein spots and five proteins were different in subjects with sarcoidosis versus control subjects. Seventeen protein spots and eight proteins were different in subjects with CBD and control subjects, and one protein was downregulated in sarcoidosis compared with CBD. One protein was more abundant in sarcoidosis (compared with CBD) and could not be identified |
| Landi 2013 (89) | BALF | Sarcoidosis (n = 9) | IPF (n = 7), PLCH (n = 9), and SSc pulmonary fibrosis, n = 7 | 2DE with MALDI-TOF MS/MS | An average of 1,000 spots were identified in each gel, and 154 spots were differentially expressed in various comparisons corresponding with 77 proteins. The proteins mapped to biological pathways involved in defense, inflammation, and response to stress |
| Silva 2013 (19) | Soluble protein fraction from alveolar macrophages | Sarcoidosis (n = 7) | Healthy (n = 7) | DIGE followed by MALDI-TOF | Sixty-nine protein spots were significantly different, corresponding with 25 unique proteins (17 upregulated and eight downregulated) in sarcoidosis. Clathrin mediated endocytosis and Fcχ receptor mediated phagocytosis |
| Du 2015 (25) | Serum | Sarcoidosis (n = 20) | Tuberculosis (n = 20) and healthy control subjects (n = 20) | Human cytokine antibody array | ICAM-1 and leptin discriminate cases with sarcoidosis from those with tuberculosis. |
| Landi 2015 (85) | BALF | Stage II/III active pulmonary sarcoidosis (n = 9) | Nonsmokers (n = 10) and smokers (n = 8) | 2DE and MALDI-TOF MS or LC MS/MS | Differentially expressed proteins map to PI3K/Akt/mTOR, MAP kinase, pluripotency-associated transcriptional factor, and hypoxia response pathway |
| Häggmark 2015 (26) | BALF and plasma | LS (n = 139) and NLS (n = 140) | Asthma (n = 17) and healthy subjects (n = 49) | Planar antigen microarray | Sarcoidosis cases have higher reactivity to zinc finger protein 688 and mitochondrial protein L43 |
| Kjellin 2016 (18) | Alveolar macrophages | Sarcoidosis (n = 8) | Control subjects (n = 6) | iTRAQ LC MS/MS | Eighty differentially expressed proteins were identified. There was upregulation of two phagocytotic pathways (Fcγ receptor–mediated phagocytosis and clathrin-mediated endocytosis signaling). Pyruvate metabolism was upregulated in sarcoidosis cases. Oxidative phosphorylation pathway was upregulated in patients with LS and downregulated in patients without LS |
| Martinez-Bravo 2017 (15) | BALF exosomes | Sarcoidosis (n = 15) | Healthy (n = 5) | iTRAQ LC MS/MS on BALF exosomes | Six-hundred and ninety proteins were quantified consistently in exosomes. Proteins more abundant in patients with sarcoidosis are involved in innate and adaptive immune response, protein maturation, and homeostasis. There were indications of downregulation of endocytosis, catabolism (such as peptidase and endopeptidase activity and ubiquitination), and metabolic processes |
Definition of abbreviations: 2DE = two-dimensional electrophoresis; CBD = chronic beryllium disease; DIGE = difference gel electrophoresis; ICAM-1 = intercellular adhesion molecule-1; IPF = idiopathic pulmonary fibrosis; iTRAQ = isobaric tags for relative and absolute quantification; LC = liquid chromatography; LS = Lofgren’s syndrome; MALDI = matrix-assisted laser desorption ionization; MAP = mitogen-actived protein; MS = mass spectrometry; MS/MS = tandem MS; NLS = non–Lofgren’s syndrome; PLCH = pulmonary Langerhans cell histiocytosis; SELDI = surface-enhanced laser desorption ionization; SSc = systemic sclerosis; TOF = time of flight.
Early studies implemented either two-dimensional electrophoresis or top-down proteomics in sarcoidosis. Magi and colleagues (83) identified 32 differentially abundant (DA) proteins between sarcoidosis and IPF. Sabounchi-Schut and colleagues (27) observed 21 DA spots comparing subjects with Lofgren’s syndrome (LS) with healthy control subjects. Seventeen proteins were identified; many were nonplasmatic and participate in the inflammatory and redox processes. Twelve had lower intensity in patients with sarcoidosis, including some antioxidant proteins. In a larger study, Kriegova and colleagues (55) compared subjects with pulmonary sarcoidosis with control subjects using SELDI-TOF and detected 40 DA protein peaks, including 26 that were upregulated in sarcoidosis. The protein pattern among patients with chest X-ray (CXR) stage I disease was dependent on a diagnosis of LS, suggesting that the CXR stage influences protein abundance. Rottoli and colleagues assessed the differences in oxidant stress in diffuse lung diseases (DLDs [IPF, sarcoidosis, and systemic sclerosis (SSc)]) by quantifying BALF carbonylation (87). A higher oxidant burden was noted in subjects with DLDs compared with control subjects. A limited number of carbonylated proteins were present in SSc and sarcoidosis compared with IPF, suggesting differences in oxidant stress in DLDs. Landi and colleagues (89) compared BALF from patients with sarcoidosis with IPF and SSc as well as pulmonary langerhans cell histiocytosis (PLCH). Seventy-seven unique DA protein spots were selected for identification. These proteins included those involved in immune response, inflammation, coagulation, oxidative stress, and antiprotease activity. Silva and colleagues (86) compared BALF from patients with sarcoidosis and chronic beryllium disease (CBD) and control subjects. Five unique proteins were DA between subjects with sarcoidosis and control subjects, three were DA between subjects with sarcoidosis and those with CBD. These studies demonstrate the ability of MS proteomics to detect clinically meaningful differences in sarcoidosis.
With the improvement in the MS instrumentation, several studies have obtained deeper proteome coverage to provide insights into disease biology. Leveraging MALDI-TOF MS, Landi and colleagues (85) identified 34 DA proteins between nine patients without LS and 18 healthy control subjects in BALF. DA proteins map to PI3K/Akt/mTOR, MAP kinase, pluripotency-associated transcriptional factor, and hypoxia response pathways. Kjellin and colleagues (18) examined alveolar macrophage membrane-associated proteins employing iTRAQ LC MS/MS. In contrast to the limited coverage observed in prior studies, they detected 1,656 proteins, 423 of which were identified with at least two high-confidence peptides. Eighty proteins were DA between eight patients with sarcoidosis (four with LS) and six control subjects. There was upregulation of two phagocytotic pathways and pyruvate metabolism in sarcoidosis. Oxidative phosphorylation was downregulated in patients with sarcoidosis (mostly in the group without LS). Martinez-Bravo and colleagues (15) compared BALF exosomes from 15 patients with sarcoidosis with those from five healthy control subjects using three iTRAQ LC MS/MS and detected an average of 1,400 proteins in each run. Of the 690 consistent proteins, 144 proteins were DA. The proteins more abundant in sarcoidosis included those mapping to cell-to-cell interaction, LPS binding, and vitamin D binding. One specific protein, vitamin D–binding protein, was more abundant in sarcoidosis and was validated in plasma. These studies demonstrate the value of contemporary tools to gain novel insights into sarcoidosis biology as well as to develop biomarkers that could have utility in diagnosis, prognosis, and monitoring disease activity.
Conclusions and Future Directions
The field of MS proteomics has improved substantially because of advances in the instrumentation and sophistication of bioinformatics algorithms. Substantial knowledge gaps remain in the understanding of mechanisms that explain the heterogeneity of sarcoidosis manifestation and course (1). The implementation of system-level studies using “omics” tools is expected to narrow the knowledge gaps by identifying biomarkers and mechanisms that contribute to high-risk manifestations in sarcoidosis (90). MS proteomic studies conducted in sarcoidosis have provided insights into disease mechanisms (15, 85) and clues to biomarkers, despite being limited by small sample sizes. Systematic studies characterizing the proteins in well-phenotyped patients are needed. Ongoing efforts by the Foundation of Sarcoidosis Research and other organizations to develop sarcoidosis cohorts offer promise for undertaking proteomic studies. However, close attention will be needed for the collection and storage of biospecimens from these cohorts. Examining the post-translational modification will be crucial to gaining insights into the mechanisms contributing to sarcoidosis progression and identifying precise therapeutic targets. The integration of gene–protein data will identify genetic contributions to the heterogenicity. Applying network analysis to proteomic data can elucidate connections between pathways not previously recognized. The current state of proteomic technology is well positioned to address knowledge gaps that exist in the pathobiology of sarcoidosis.
Acknowledgments
Acknowledgment
The authors thank Craig Solid, Solid Research Group, LLC for facilitating the writing of this manuscript.
Footnotes
Author Contributions: C.R.G., T.J.G., L.H., and C.P.N.: literature review, editing, and writing proteomics aspects of this manuscript. L.A.M., D.M.P., and M.B.: literature review, writing, and editing sarcoidosis and lung proteome–related section of this manuscript.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0070PS on August 17, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Maier LA, Crouser ED, Martin WJ, II, Eu J. Executive summary of the NHLBI workshop report: leveraging current scientific advancements to understand sarcoidosis variability and improve outcomes. Ann Am Thorac Soc . 2017;14:S415–S420. doi: 10.1513/AnnalsATS.201707-563OT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Crouser ED, Fingerlin TE, Yang IV, Maier LA, Nana-Sinkam P, Collman RG, et al. Application of “omics” and systems biology to sarcoidosis research. Ann Am Thorac Soc . 2017;14:S445–S451. doi: 10.1513/AnnalsATS.201707-567OT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Koth LL, Solberg OD, Peng JC, Bhakta NR, Nguyen CP, Woodruff PG. Sarcoidosis blood transcriptome reflects lung inflammation and overlaps with tuberculosis. Am J Respir Crit Care Med . 2011;184:1153–1163. doi: 10.1164/rccm.201106-1143OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Crouser ED, Culver DA, Knox KS, Julian MW, Shao G, Abraham S, et al. Gene expression profiling identifies MMP-12 and ADAMDEC1 as potential pathogenic mediators of pulmonary sarcoidosis. Am J Respir Crit Care Med . 2009;179:929–938. doi: 10.1164/rccm.200803-490OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Russello SV. Assessing cellular protein phosphorylation: high throughput drug discovery technologies. Assay Drug Dev Technol . 2004;2:225–235. doi: 10.1089/154065804323056567. [DOI] [PubMed] [Google Scholar]
- 6. Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature . 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 7. Gold L. Unbiased affinity-based proteomics: Somascan applications for healthcare. FEBS J . 2013;280:632. [Google Scholar]
- 8. Vasarmidi E, Sarantoulaki S, Trachalaki A, Margaritopoulos G, Bibaki E, Spandidos DA, et al. Investigation of key autophagy-and mitophagy-related proteins and gene expression in BALF cells from patients with IPF and RA-ILD. Mol Med Rep . 2018;18:3891–3897. doi: 10.3892/mmr.2018.9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Prieto DA, Johann DJ, Jr, Wei BR, Ye X, Chan KC, Nissley DV, et al. Mass spectrometry in cancer biomarker research: a case for immunodepletion of abundant blood-derived proteins from clinical tissue specimens. Biomarkers Med . 2014;8:269–286. doi: 10.2217/bmm.13.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bowler RP, Wendt CH, Fessler MB, Foster MW, Kelly RS, Lasky-Su J, et al. American Thoracic Society Workgroup on Metabolomics and Proteomics. New strategies and challenges in lung proteomics and metabolomics: an official American Thoracic Society workshop report. Ann Am Thorac Soc . 2017;14:1721–1743. doi: 10.1513/AnnalsATS.201710-770WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gray RD, MacGregor G, Noble D, Imrie M, Dewar M, Boyd AC, et al. Sputum proteomics in inflammatory and suppurative respiratory diseases. Am J Respir Crit Care Med . 2008;178:444–452. doi: 10.1164/rccm.200703-409OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kipnis E, Hansen K, Sawa T, Moriyama K, Zurawel A, Ishizaka A, et al. Proteomic analysis of undiluted lung epithelial lining fluid. Chest . 2008;134:338–345. doi: 10.1378/chest.07-1643. [DOI] [PubMed] [Google Scholar]
- 13. Bowler RP, Duda B, Chan ED, Enghild JJ, Ware LB, Matthay MA, et al. Proteomic analysis of pulmonary edema fluid and plasma in patients with acute lung injury. Am J Physiol Lung Cell Mol Physiol . 2004;286:L1095–L1104. doi: 10.1152/ajplung.00304.2003. [DOI] [PubMed] [Google Scholar]
- 14. Griese M, Noss J, von Bredow C. Protein pattern of exhaled breath condensate and saliva. Proteomics . 2002;2:690–696. doi: 10.1002/1615-9861(200206)2:6<690::AID-PROT690>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 15. Martinez-Bravo MJ, Wahlund CJ, Qazi KR, Moulder R, Lukic A, Rådmark O, et al. Pulmonary sarcoidosis is associated with exosomal vitamin D-binding protein and inflammatory molecules. J Allergy Clin Immunol . 2017;139:1186–1194. doi: 10.1016/j.jaci.2016.05.051. [DOI] [PubMed] [Google Scholar]
- 16. Gharib SA, Malur A, Huizar I, Barna BP, Kavuru MS, Schnapp LM, et al. Sarcoidosis activates diverse transcriptional programs in bronchoalveolar lavage cells. Respir Res . 2016;17:93. doi: 10.1186/s12931-016-0411-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barna BP, Culver DA, Kanchwala A, Singh RJ, Huizar I, Abraham S, et al. Alveolar macrophage cathelicidin deficiency in severe sarcoidosis. J Innate Immun . 2012;4:569–578. doi: 10.1159/000339149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kjellin H, Silva E, Branca RM, Eklund A, Jakobsson PJ, Grunewald J, et al. Alterations in the membrane-associated proteome fraction of alveolar macrophages in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis . 2016;33:17–28. [PubMed] [Google Scholar]
- 19. Silva E, Souchelnytskyi S, Kasuga K, Eklund A, Grunewald J, Wheelock AM. Quantitative intact proteomics investigations of alveolar macrophages in sarcoidosis. Eur Respir J . 2013;41:1331–1339. doi: 10.1183/09031936.00178111. [DOI] [PubMed] [Google Scholar]
- 20. Horváth I, Hunt J, Barnes PJ, Alving K, Antczak A, Baraldi E, et al. ATS/ERS Task Force on Exhaled Breath Condensate. Exhaled breath condensate: methodological recommendations and unresolved questions. Eur Respir J . 2005;26:523–548. doi: 10.1183/09031936.05.00029705. [DOI] [PubMed] [Google Scholar]
- 21. Mohan N, Akter R, Bryant K, Herbert C, Chow S, Thomas PS. Exhaled breath markers of alveolar macrophage activity in sarcoidosis. Inflamm Res . 2016;65:471–478. doi: 10.1007/s00011-016-0929-y. [DOI] [PubMed] [Google Scholar]
- 22. Fumagalli M, Dolcini L, Sala A, Stolk J, Fregonese L, Ferrari F, et al. Proteomic analysis of exhaled breath condensate from single patients with pulmonary emphysema associated to alpha1-antitrypsin deficiency. J Proteomics . 2008;71:211–221. doi: 10.1016/j.jprot.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 23. Fumagalli M, Ferrari F, Luisetti M, Stolk J, Hiemstra PS, Capuano D, et al. Profiling the proteome of exhaled breath condensate in healthy smokers and COPD patients by LC-MS/MS. Int J Mol Sci . 2012;13:13894–13910. doi: 10.3390/ijms131113894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nicholas B, Djukanović R. Induced sputum: a window to lung pathology. Biochem Soc Trans . 2009;37:868–872. doi: 10.1042/BST0370868. [DOI] [PubMed] [Google Scholar]
- 25. Du SS, Zhao MM, Zhang Y, Zhang P, Hu Y, Wang LS, et al. Screening for differentially expressed proteins relevant to the differential diagnosis of sarcoidosis and tuberculosis. PLoS One . 2015;10:e0132466. doi: 10.1371/journal.pone.0132466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Häggmark A, Hamsten C, Wiklundh E, Lindskog C, Mattsson C, Andersson E, et al. Proteomic profiling reveals autoimmune targets in sarcoidosis. Am J Respir Crit Care Med . 2015;191:574–583. doi: 10.1164/rccm.201407-1341OC. [DOI] [PubMed] [Google Scholar]
- 27. Sabounchi-Schütt F, Mikko M, Eklund A, Grunewald J, AStröm J. Serum protein pattern in sarcoidosis analysed by a proteomics approach. Sarcoidosis Vasc Diffuse Lung Dis . 2004;21:182–190. [PubMed] [Google Scholar]
- 28. Rennard SI, Basset G, Lecossier D, O’Donnell KM, Pinkston P, Martin PG, et al. Estimation of volume of epithelial lining fluid recovered by lavage using urea as marker of dilution. J Appl Physiol (1985) . 1986;60:532–538. doi: 10.1152/jappl.1986.60.2.532. [DOI] [PubMed] [Google Scholar]
- 29. Haslam PL, Baughman RP. Report of ERS Task Force: guidelines for measurement of acellular components and standardization of BAL. Eur Respir J . 1999;14:245–248. doi: 10.1034/j.1399-3003.1999.14b01.x. [DOI] [PubMed] [Google Scholar]
- 30. van der Vliet A, O’Neill CA, Cross CE, Koostra JM, Volz WG, Halliwell B, et al. Determination of low-molecular-mass antioxidant concentrations in human respiratory tract lining fluids. Am J Physiol . 1999;276:L289–L296. doi: 10.1152/ajplung.1999.276.2.L289. [DOI] [PubMed] [Google Scholar]
- 31. Bhargava M, Becker TL, Viken KJ, Jagtap PD, Dey S, Steinbach MS, et al. Proteomic profiles in acute respiratory distress syndrome differentiates survivors from non-survivors. PLoS One . 2014;9:e109713. doi: 10.1371/journal.pone.0109713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bhargava M, Viken K, Wang Q, Jagtap P, Bitterman P, Ingbar D, et al. Bronchoalveolar lavage fluid protein expression in acute respiratory distress syndrome provides insights into pathways activated in subjects with different outcomes. Sci Rep . 2017;7:7464. doi: 10.1038/s41598-017-07791-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bhargava M, Viken KJ, Dey S, Steinbach MS, Wu B, Jagtap PD, et al. Proteome profiling in lung injury after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant . 2016;22:1383–1390. doi: 10.1016/j.bbmt.2016.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Landi C, Bargagli E, Carleo A, Bianchi L, Gagliardi A, Prasse A, et al. A system biology study of BALF from patients affected by idiopathic pulmonary fibrosis (IPF) and healthy controls. Proteomics Clin Appl . 2014;8:932–950. doi: 10.1002/prca.201400001. [DOI] [PubMed] [Google Scholar]
- 35. Foster MW, Morrison LD, Todd JL, Snyder LD, Thompson JW, Soderblom EJ, et al. Quantitative proteomics of bronchoalveolar lavage fluid in idiopathic pulmonary fibrosis. J Proteome Res . 2015;14:1238–1249. doi: 10.1021/pr501149m. [DOI] [PubMed] [Google Scholar]
- 36. Chang DW, Hayashi S, Gharib SA, Vaisar T, King ST, Tsuchiya M, et al. Proteomic and computational analysis of bronchoalveolar proteins during the course of the acute respiratory distress syndrome. Am J Respir Crit Care Med . 2008;178:701–709. doi: 10.1164/rccm.200712-1895OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Foster MW, Thompson JW, Que LG, Yang IV, Schwartz DA, Moseley MA, et al. Proteomic analysis of human bronchoalveolar lavage fluid after subsgemental exposure. J Proteome Res . 2013;12:2194–2205. doi: 10.1021/pr400066g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cederfur C, Malmström J, Nihlberg K, Block M, Breimer ME, Bjermer L, et al. Glycoproteomic identification of galectin-3 and -8 ligands in bronchoalveolar lavage of mild asthmatics and healthy subjects. Biochim Biophys Acta . 2012;1820:1429–1436. doi: 10.1016/j.bbagen.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 39. Tu C, Mammen MJ, Li J, Shen X, Jiang X, Hu Q, et al. Large-scale, ion-current-based proteomics investigation of bronchoalveolar lavage fluid in chronic obstructive pulmonary disease patients. J Proteome Res . 2014;13:627–639. doi: 10.1021/pr4007602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Almatroodi SA, McDonald CF, Collins AL, Darby IA, Pouniotis DS. Quantitative proteomics of bronchoalveolar lavage fluid in lung adenocarcinoma. Cancer Genomics Proteomics . 2015;12:39–48. [PubMed] [Google Scholar]
- 41. Ortea I, Rodríguez-Ariza A, Chicano-Gálvez E, Arenas Vacas MS, Jurado Gámez B. Discovery of potential protein biomarkers of lung adenocarcinoma in bronchoalveolar lavage fluid by SWATH MS data-independent acquisition and targeted data extraction. J Proteomics . 2016;138:106–114. doi: 10.1016/j.jprot.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 42. Crouser ED, White P, Caceres EG, Julian MW, Papp AC, Locke LW, et al. A novel in vitro human granuloma model of sarcoidosis and latent tuberculosis infection. Am J Respir Cell Mol Biol . 2017;57:487–498. doi: 10.1165/rcmb.2016-0321OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Locke LW, Crouser ED, White P, Julian MW, Caceres EG, Papp AC, et al. Il-13-regulated macrophage polarization during granuloma formation in an in vitro human sarcoidosis model. Am J Respir Cell Mol Biol . 2019;60:84–95. doi: 10.1165/rcmb.2018-0053OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pfammatter S, Bonneil E, McManus FP, Prasad S, Bailey DJ, Belford M, et al. A novel differential ion mobility device expands the depth of proteome coverage and the sensitivity of multiplex proteomic measurements. Mol Cell Proteomics . 2018;17:2051–2067. doi: 10.1074/mcp.TIR118.000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hebert AS, Prasad S, Belford MW, Bailey DJ, McAlister GC, Abbatiello SE, et al. Comprehensive single-shot proteomics with faims on a hybrid orbitrap mass spectrometer. Anal Chem . 2018;90:9529–9537. doi: 10.1021/acs.analchem.8b02233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Meier F, Brunner AD, Koch S, Koch H, Lubeck M, Krause M, et al. Online parallel accumulation-serial fragmentation (pasef) with a novel trapped ion mobility mass spectrometer. Mol Cell Proteomics . 2018;17:2534–2545. doi: 10.1074/mcp.TIR118.000900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hou R, Li Y, Sui Z, Yuan H, Yang K, Liang Z, et al. Advances in exosome isolation methods and their applications in proteomic analysis of biological samples. Anal Bioanal Chem . 2019;411:5351–5361. doi: 10.1007/s00216-019-01982-0. [DOI] [PubMed] [Google Scholar]
- 48. Li Y, Qin H, Ye M. An overview on enrichment methods for cell surface proteome profiling. J Sep Sci . 2020;43:292–312. doi: 10.1002/jssc.201900700. [DOI] [PubMed] [Google Scholar]
- 49. Kwak C, Shin S, Park JS, Jung M, Nhung TTM, Kang MG, et al. Contact-ID, a tool for profiling organelle contact sites, reveals regulatory proteins of mitochondrial-associated membrane formation. Proc Natl Acad Sci USA . 2020;117:12109–12120. doi: 10.1073/pnas.1916584117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gingras AC, Abe KT, Raught B. Getting to know the neighborhood: using proximity-dependent biotinylation to characterize protein complexes and map organelles. Curr Opin Chem Biol . 2019;48:44–54. doi: 10.1016/j.cbpa.2018.10.017. [DOI] [PubMed] [Google Scholar]
- 51. Han X, Jin M, Breuker K, McLafferty FW. Extending top-down mass spectrometry to proteins with masses greater than 200 kilodaltons. Science . 2006;314:109–112. doi: 10.1126/science.1128868. [DOI] [PubMed] [Google Scholar]
- 52. Searle BC, Pino LK, Egertson JD, Ting YS, Lawrence RT, MacLean BX, et al. Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nat Commun . 2018;9:5128. doi: 10.1038/s41467-018-07454-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cho KC, Clark DJ, Schnaubelt M, Teo GC, Leprevost FDV, Bocik W, et al. Deep proteomics using two dimensional data independent acquisition mass spectrometry. Anal Chem . 2020;92:4217–4225. doi: 10.1021/acs.analchem.9b04418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang Y, Wroblewski M, Hertz MI, Wendt CH, Cervenka TM, Nelsestuen GL. Analysis of chronic lung transplant rejection by MALDI-TOF profiles of bronchoalveolar lavage fluid. Proteomics . 2006;6:1001–1010. doi: 10.1002/pmic.200500105. [DOI] [PubMed] [Google Scholar]
- 55. Kriegova E, Melle C, Kolek V, Hutyrova B, Mrazek F, Bleul A, et al. Protein profiles of bronchoalveolar lavage fluid from patients with pulmonary sarcoidosis. Am J Respir Crit Care Med . 2006;173:1145–1154. doi: 10.1164/rccm.200507-1126OC. [DOI] [PubMed] [Google Scholar]
- 56. Yang F, Shen Y, Camp DG, II, Smith RD. High-pH reversed-phase chromatography with fraction concatenation for 2D proteomic analysis. Expert Rev Proteomics . 2012;9:129–134. doi: 10.1586/epr.12.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chelius D, Bondarenko PV. Quantitative profiling of proteins in complex mixtures using liquid chromatography and mass spectrometry. J Proteome Res . 2002;1:317–323. doi: 10.1021/pr025517j. [DOI] [PubMed] [Google Scholar]
- 58. Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol . 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 59. Barr JR, Maggio VL, Patterson DG, Jr, Cooper GR, Henderson LO, Turner WE, et al. Isotope dilution--mass spectrometric quantification of specific proteins: model application with apolipoprotein A-I. Clin Chem . 1996;42:1676–1682. [PubMed] [Google Scholar]
- 60. Tabb DL, MacCoss MJ, Wu CC, Anderson SD, Yates JR., III Similarity among tandem mass spectra from proteomic experiments: detection, significance, and utility. Anal Chem . 2003;75:2470–2477. doi: 10.1021/ac026424o. [DOI] [PubMed] [Google Scholar]
- 61. Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics . 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 62. Weisbrod CR, Eng JK, Hoopmann MR, Baker T, Bruce JE. Accurate peptide fragment mass analysis: multiplexed peptide identification and quantification. J Proteome Res . 2012;11:1621–1632. doi: 10.1021/pr2008175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Venable JD, Dong MQ, Wohlschlegel J, Dillin A, Yates JR. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat Methods . 2004;1:39–45. doi: 10.1038/nmeth705. [DOI] [PubMed] [Google Scholar]
- 64. Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, et al. Targeted data extraction of the ms/ms spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics . 2012;11:O111.016717. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gupta N, Pevzner PA. False discovery rates of protein identifications: a strike against the two-peptide rule. J Proteome Res . 2009;8:4173–4181. doi: 10.1021/pr9004794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Käll L, Storey JD, MacCoss MJ, Noble WS. Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J Proteome Res . 2008;7:29–34. doi: 10.1021/pr700600n. [DOI] [PubMed] [Google Scholar]
- 67. Plumb RS, Johnson KA, Rainville P, Smith BW, Wilson ID, Castro-Perez JM, et al. UPLC/MS(E); a new approach for generating molecular fragment information for biomarker structure elucidation. Rapid Commun Mass Spectrom . 2006;20:1989–1994. doi: 10.1002/rcm.2550. [DOI] [PubMed] [Google Scholar]
- 68. Reiter L, Rinner O, Picotti P, Hüttenhain R, Beck M, Brusniak MY, et al. mProphet: automated data processing and statistical validation for large-scale SRM experiments. Nat Methods . 2011;8:430–435. doi: 10.1038/nmeth.1584. [DOI] [PubMed] [Google Scholar]
- 69. Tsou CC, Avtonomov D, Larsen B, Tucholska M, Choi H, Gingras AC, et al. Dia-umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat Methods . 2015;12:258–264. doi: 10.1038/nmeth.3255. 257 p following 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bereman MS, MacLean B, Tomazela DM, Liebler DC, MacCoss MJ. The development of selected reaction monitoring methods for targeted proteomics via empirical refinement. Proteomics . 2012;12:1134–1141. doi: 10.1002/pmic.201200042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Vachani A, Pass HI, Rom WN, Midthun DE, Edell ES, Laviolette M, et al. Validation of a multiprotein plasma classifier to identify benign lung nodules. J Thorac Oncol . 2015;10:629–637. doi: 10.1097/JTO.0000000000000447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Swan AL, Mobasheri A, Allaway D, Liddell S, Bacardit J. Application of machine learning to proteomics data: classification and biomarker identification in postgenomics biology. OMICS . 2013;17:595–610. doi: 10.1089/omi.2013.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bi Q, Goodman KE, Kaminsky J, Lessler J. What is machine learning? A primer for the epidemiologist. Am J Epidemiol . 2019;188:2222–2239. doi: 10.1093/aje/kwz189. [DOI] [PubMed] [Google Scholar]
- 74. Bhargava M, Dey S, Becker T, Steinbach M, Wu B, Lee SM, et al. Protein expression profile of rat type two alveolar epithelial cells during hyperoxic stress and recovery. Am J Physiol Lung Cell Mol Physiol . 2013;305:L604–L614. doi: 10.1152/ajplung.00079.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wu X, Hasan MA, Chen JY. Pathway and network analysis in proteomics. J Theor Biol . 2014;362:44–52. doi: 10.1016/j.jtbi.2014.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Khatri P, Sirota M, Butte AJ. Ten years of pathway analysis: current approaches and outstanding challenges. PLOS Comput Biol . 2012;8:e1002375. doi: 10.1371/journal.pcbi.1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Schupp JC, Vukmirovic M, Kaminski N, Prasse A. Transcriptome profiles in sarcoidosis and their potential role in disease prediction. Curr Opin Pulm Med . 2017;23:487–492. doi: 10.1097/MCP.0000000000000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Landi C, Carleo A, Cillis G, Rottoli P. Sarcoidosis: proteomics and new perspectives for improving personalized medicine. Expert Rev Proteomics . 2018;15:829–835. doi: 10.1080/14789450.2018.1528148. [DOI] [PubMed] [Google Scholar]
- 79. Rosenbaum JT, Pasadhika S, Crouser ED, Choi D, Harrington CA, Lewis JA, et al. Hypothesis: sarcoidosis is a STAT1-mediated disease. Clin Immunol . 2009;132:174–183. doi: 10.1016/j.clim.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Judson MA, Marchell RM, Mascelli M, Piantone A, Barnathan ES, Petty KJ, et al. Molecular profiling and gene expression analysis in cutaneous sarcoidosis: the role of interleukin-12, interleukin-23, and the t-helper 17 pathway. J Am Acad Dermatol . 2012;66:901–910, 910.e1–2. doi: 10.1016/j.jaad.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 81. Lockstone HE, Sanderson S, Kulakova N, Baban D, Leonard A, Kok WL, et al. Gene set analysis of lung samples provides insight into pathogenesis of progressive, fibrotic pulmonary sarcoidosis. Am J Respir Crit Care Med . 2010;181:1367–1375. doi: 10.1164/rccm.200912-1855OC. [DOI] [PubMed] [Google Scholar]
- 82. Su R, Li MM, Bhakta NR, Solberg OD, Darnell EP, Ramstein J, et al. Longitudinal analysis of sarcoidosis blood transcriptomic signatures and disease outcomes. Eur Respir J . 2014;44:985–993. doi: 10.1183/09031936.00039714. [DOI] [PubMed] [Google Scholar]
- 83. Magi B, Bini L, Perari MG, Fossi A, Sanchez JC, Hochstrasser D, et al. Bronchoalveolar lavage fluid protein composition in patients with sarcoidosis and idiopathic pulmonary fibrosis: a two-dimensional electrophoretic study. Electrophoresis . 2002;23:3434–3444. doi: 10.1002/1522-2683(200210)23:19<3434::AID-ELPS3434>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 84. Sabounchi-Schütt F, Aström J, Hellman U, Eklund A, Grunewald J. Changes in bronchoalveolar lavage fluid proteins in sarcoidosis: a proteomics approach. Eur Respir J . 2003;21:414–420. doi: 10.1183/09031936.03.00060902. [DOI] [PubMed] [Google Scholar]
- 85. Landi C, Bargagli E, Carleo A, Bianchi L, Gagliardi A, Cillis G, et al. A functional proteomics approach to the comprehension of sarcoidosis. J Proteomics . 2015;128:375–387. doi: 10.1016/j.jprot.2015.08.012. [DOI] [PubMed] [Google Scholar]
- 86. Silva E, Bourin S, Sabounchi-Schütt F, Laurin Y, Barker E, Newman L, et al. A quantitative proteomic analysis of soluble bronchoalveolar fluid proteins from patients with sarcoidosis and chronic beryllium disease. Sarcoidosis Vasc Diffuse Lung Dis . 2007;24:24–32. [PubMed] [Google Scholar]
- 87. Rottoli P, Magi B, Cianti R, Bargagli E, Vagaggini C, Nikiforakis N, et al. Carbonylated proteins in bronchoalveolar lavage of patients with sarcoidosis, pulmonary fibrosis associated with systemic sclerosis and idiopathic pulmonary fibrosis. Proteomics . 2005;5:2612–2618. doi: 10.1002/pmic.200401206. [DOI] [PubMed] [Google Scholar]
- 88. Rottoli P, Magi B, Perari MG, Liberatori S, Nikiforakis N, Bargagli E, et al. Cytokine profile and proteome analysis in bronchoalveolar lavage of patients with sarcoidosis, pulmonary fibrosis associated with systemic sclerosis and idiopathic pulmonary fibrosis. Proteomics . 2005;5:1423–1430. doi: 10.1002/pmic.200301007. [DOI] [PubMed] [Google Scholar]
- 89. Landi C, Bargagli E, Bianchi L, Gagliardi A, Carleo A, Bennett D, et al. Towards a functional proteomics approach to the comprehension of idiopathic pulmonary fibrosis, sarcoidosis, systemic sclerosis and pulmonary Langerhans cell histiocytosis. J Proteomics . 2013;83:60–75. doi: 10.1016/j.jprot.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 90. Sauer WH, Stern BJ, Baughman RP, Culver DA, Royal W. High risk sarcoidosis: current concepts and research imperatives. Ann Am Thorac Soc . 2017;14:S437–S444. doi: 10.1513/AnnalsATS.201707-566OT. [DOI] [PubMed] [Google Scholar]
- 91. Davis MD, Montpetit AJ. Exhaled breath condensate: an update. Immunol Allergy Clin North Am . 2018;38:667–678. doi: 10.1016/j.iac.2018.06.002. [DOI] [PubMed] [Google Scholar]
- 92. Pelaia G, Terracciano R, Vatrella A, Gallelli L, Busceti MT, Calabrese C, et al. Application of proteomics and peptidomics to COPD. BioMed Res Int . 2014;2014:764581. doi: 10.1155/2014/764581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Omenn GS, States DJ, Adamski M, Blackwell TW, Menon R, Hermjakob H, et al. Overview of the HUPO Plasma Proteome Project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database. Proteomics . 2005;5:3226–3245. doi: 10.1002/pmic.200500358. [DOI] [PubMed] [Google Scholar]
- 94. Beer LA, Ky B, Barnhart KT, Speicher DW. In-depth, reproducible analysis of human plasma using igy 14 and supermix immunodepletion. Methods Mol Biol . 2017;1619:81–101. doi: 10.1007/978-1-4939-7057-5_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Cao Z, Tang HY, Wang H, Liu Q, Speicher DW. Systematic comparison of fractionation methods for in-depth analysis of plasma proteomes. J Proteome Res . 2012;11:3090–3100. doi: 10.1021/pr201068b. [DOI] [PMC free article] [PubMed] [Google Scholar]