

Abstract
Interindividual variability in drug efficacy and toxicity is a major challenge in clinical practice. Variations in drug pharmacokinetics (PKs) and pharmacodynamics (PDs) can be, in part, explained by polymorphic variants in genes encoding drug metabolizing enzymes and transporters (absorption, distribution, metabolism, and excretion) or in genes encoding drug receptors. Pharmacogenomics (PGx) has allowed the identification of predictive biomarkers of drug PKs and PDs and the current knowledge of genome‐disease and genome‐drug interactions offers the opportunity to optimize tailored drug therapy. High‐throughput PGx genotyping, from targeted to more comprehensive strategies, allows the identification of PK/PD genotypes to be developed as clinical predictive biomarkers. However, a biomarker needs a robust process of validation followed by clinical‐grade assay development and must comply to stringent regulatory guidelines. We here discuss the methodological challenges and the emerging technological tools in PGx biomarker discovery and validation, at the crossroad among molecular genetics, bioinformatics, and clinical medicine.
The interindividual heterogeneity in drug response is difficult to predict and manage 1 due to its multifactorial nature, partly attributed to patients’ genetics. Polymorphic variants in genes involved in drug absorption, distribution, metabolism, and excretion (ADME) may impact either pharmacokinetics (PKs) and pharmacodynamics (PDs) of a drug interfering at different dynamic biological levels of the human body (metabolome, epigenome, transcriptome, and proteome) 2 and affect, therefore, clinical outcome. The variability in drug response has a strong impact on dosing, therapeutic efficacy or toxicity, risk for hypersensitivity reactions, and drug resistance. From the first observations in PK studies of different phenotypes of metabolizers, candidate gene studies led to the hypothesis that polymorphic variants in ADME genes could have a strong effect on drug action. In recent years, hundreds of genomewide association studies (GWASs) have explored the associations between common genetic variations and drug response in large cohorts of individuals and in different populations. All published GWAS and association results since 2005 are included in the public catalog GWAS Catalog (www.ebi.ac.uk/gwas) produced and developed by the National Human Genome Research Institute (NHGRI) and the European Bioinformatics Institute (EMBL‐EBI). 3 Allelic variants in HLA genes have been also correlated with susceptibility and resistance to different diseases, as well as adverse drug reactions. 4 In oncology, in addition to germline variations (inherited), pharmacogenomic (PGx) biomarkers from tumor genome (acquired) are used to tailor drug therapy and predict disease outcome. According to strong genotype‐phenotype evidence, the US Food and Drug Administration (FDA) has recognized more than 250 biomarkers of known PGx value providing recommendations for therapeutic management (https://www.fda.gov). Information on PGx variants and related guidelines are also available in databases, such as the Clinical Pharmacogenetics Implementation Consortium (CPIC; https://cpicpgx.org/), the Pharmacogenomics Knowledge Base (PharmGKB; https://www.pharmgkb.org/), the Dutch Pharmacogenetics Working Group (DPWG; https://www.pharmgkb.org/page/dpwg), and the Canadian Pharmacogenomics Network for Drug Safety (CPNDS; http://cpnds.ubc.ca/). The knowledge of human genome sequence and the availability of novel technologies have allowed high‐throughput screening of large number of individuals, generating large amount of data with short turn‐around and competitive costs. Hence, PGx marker genotyping represents a crucial tool for different applications, such as drug discovery and development, drug prescription based on genomic information, and design of customized companion diagnostics. Genome sequencing and methylation analyses have led to the discovery of rare genetic and epigenetic biomarkers potentially correlated to the pathogenesis or risk assessment of several diseases, including cancer, or to drug resistance. 5 However, many PGx associations, such as CYP1A2*F and colon cancer risk, 6 could not be validated when tested in larger and independent cohorts and the introduction in daily practice of PGx biomarkers is still an unmet need. The myriad of PGx variants without unclear functional roles need to be tested in studies designed to support the added value of PGx testing for a tailored prescription. Identified biomarkers need a robust process of validation and regulatory qualification until the phase of biomarker assay development. Here, we provide a state‐of‐the‐art review of the current technologic tools for biomarker discovery and the complex biomarker validation process for widespread clinic translation.
GENETIC VARIATIONS IN PGX
Germline variations, such as single nucleotide polymorphisms (SNPs), insertions/deletions (INDEL), copy number variations (CNVs) and small tandem repeats, may have a functional role when occurring in coding sequences or regulatory regions of a pharmacogene. These variants can cause (i) higher or lower drug exposure, (ii) high level of toxic metabolites, (iii) modification of effect on drug target, or (iv) idiosyncratic drug toxicity due to immune system activation. Drug efficacy and toxicity is often due to germline variations in genes encoding phase I and II enzymes, regulatory and modifier genes, drug transporters, and HLA molecules. Classical examples of genetic variants in PK genes include CYP2C19*17, which has been associated with bleeding during clopidogrel therapy, DPYD variants associated with increase plasma concentrations and toxicity risk of 5‐fluorouracil, and other fluoropyrimidines, such as capecitabine, TPMT variants linked to thiopurine toxicity, and UGT1A*28 associated to irinotecan toxicity. Among drug transporters, an example of genetic variants impacting plasma levels is the common SLCO1B1*5 variant that has been associated with elevated simvastatin plasma concentrations and an increased risk for simvastatin toxicity. HLA‐B*5701 associated to the risk of hypersensitivity reaction to the antiretroviral abacavir, and VKORC1 variants associated to warfarin resistance are two examples of PD mechanisms. In PGx, the impact of a genetic variant is not only dependent on the strength of the association with a drug phenotype but also on its frequency. Using GWAS, hundreds of thousands of common variants (minor allele frequency (MAF) ≥ 5%) can be simultaneously investigated with the aim to elucidate disease risk or trait variability. However, variability in drug response cannot be linked to common variants only. It was estimated that low‐frequency variants (0.1% ≤ MAF < 5%) and rare variants (MAF < 0.1%) account for 30–40% of highly gene and drug‐specific functional alterations. 7 For DPYD, hundreds of rare variants have been described, such as DPYD*2A (rs3918290) and DPYD*9A (rs1801265; https://www.pharmvar.org/gene/DPYD). DPYD*9A varies significantly among different populations (expressed by 40.3%, 21.8%, and 5.7% individuals of African, European, and Asian ancestry, respectively). 8 In addition, most genes with relevance to drug PK and PD are highly polymorphic (i.e., genes of the CYP superfamily). In oncology, PGx biomarkers guide the choice of drug therapy targeting specific genetic variations in the somatic tumor genome. However, also germline variants can influence dosing strategy for various chemotherapeutics, including fluoropyrimidines or thiopurines. An example of integration of somatic and germline variations in the therapeutic decision is the use of cetuximab, in elderly patients with metastatic colorectal cancer, in combination with FOLFIRI (irinotecan plus 5‐fuorouracil/leucovorin) or FOLFOX (oxaliplatin plus 5‐fuorouracil/leucovorin). This schedule requires the assessment of RAS mutations as well as DPYD and UGT polymorphisms before treatment. 9
PGX GENOTYPING STRATEGIES
Traditionally, GWAS was performed by genomewide SNP‐arrays, which simultaneously interrogate hundreds of thousands of genetic variants. In array‐based design, genomic variants are representative SNPs (also called tag SNPs) in linkage disequilibrium (LD) with other undetected SNPs, that can provide the same information. In this way, by selecting a small number of tag SNPs, it is possible to identify others among the thousand SNPs in LD. 10 The design of contemporary platforms is guided by advanced algorithms providing a greater imputation power and coverage compared with a tag SNP design approach. An alternative strategy to “genomewide” is the use of targeted SNP panels that are specifically designed to analyze a predefined list of common genetic variants in genes involved in drug response. An important question in “targeted” and “genomewide” is the limited detection of rare variants. Rare variants have low levels of pairwise LD with common variants on SNP platforms, with consequently low power to detect association signals. 5 Presently, the introduction, although slow in clinical practice, of next‐generation sequencing (NGS), leads to the opportunity to generate information on novel common and rare variants, which might represent targetable variants for new or already used drugs, overcoming several biases of SNP approaches. In Table 1 are summarized advantages and disadvantages of the three methods.
Table 1.
Advantages and disadvantages of PGx genotyping methods
| Platforms | Advantages | Disadvantages |
|---|---|---|
|
Targeted genotyping: Array SNP panel (i.e., DMET Plus, PharmacoScan solution) |
|
|
|
Sequencing SNP panel (i.e., PGR‐seq Panel, Ion AmpliSeq PGx Panel) |
|
|
|
Genomewide genotyping: (i.e., Axiom Precision Diversity Research Array, Axiom UK Biobank Array, Infinium Global Screening Array, Infinium Omni 5M) |
|
|
| NGS (WES/WGS) |
|
|
CNV, copy number variation; DMET, Drug Metabolizing Enzymes and Transporters; GC, guanine‐cytosine; NGS, next‐generation sequencing; PCR, polymerase chain reaction; PGx, pharmacogenomic; SNP, single nucleotide polymorphism; VUS, variant of unknown clinical significance; WES/WGS, whole exome sequencing/whole genome sequencing.
Genomewide genotyping
Genotyping arrays have been widely used in large‐scale population studies with the aim to interrogate several hundreds of thousands of variations across the human genome. Array design is performed to maximize genomic coverage by indirect genotyping of unobserved variants in high LD. The International HapMap Project has been used as reference dataset for the design of the first‐generation arrays for GWAS. Another strategy to increase genome coverage is imputation of unmeasured variants or missing genotypes using haplotypes from densely reference panels (i.e., 1000 Genomes Project phase III, International HapMap Project phase III, Haplotype Reference Consortium, and Trans‐Omics for Precision Medicine). In this way, haplotypes from the sample are matched to haplotypes in the reference panel. Genotype imputation allows increase of statistical power inferring causal variants so imputed SNPs that exhibit large associations may become candidates for replication studies. There is also the possibility to perform a meta‐analysis of different data that use different genotyping platforms. For common and low‐frequency variants, imputation can be performed with sufficient accuracy using the above‐mentioned ethnically heterogeneous reference panels. However, imputation accuracy for rare variants is lower than common variants because of their poor representation in the reference panel and this may be overcome when an ethnically matched reference panel is used. 11 Most of the above‐mentioned reference panels focused primarily on white populations and data for African populations and admixed populations containing African ancestry is limited. The Consortium on Asthma among African ancestry populations in the Americas reference panel, the African Genome variation project, and the African Genome Resource are three available resources for more accurate imputation of African populations. There are many software products able to perform imputation in admixed and specific reference panels, such as Beagle4.1, IMPUTE2, MACH + Minimac3, and SHAPEIT2 + IMPUTE2. Additionally, some genotyping platforms have been customized to represent African populations (i.e., H3Africa Array, Axiom Genome‐Wide PanAFR Genotyping Bundle, and The African Diaspora Power SNP Chip). Density of genotyping array is another limiting factor in imputation accuracy so the design of contemporary arrays (second‐generation) is done using dedicated algorithms to optimize imputation power. The majority of second‐generation SNP‐arrays are commercialized by two companies, Thermo Fisher Scientific (i.e., Axiom Precision Diversity Research Array and Axiom UK Biobank Array) and Illumina (i.e., Infinium Global Screening Array and Infinium Omni 5M). Many of these platforms show comparable performance in terms of call rate, concordance, and reproducibility. By imputation, it is possible to cover common and rare variants in PGx genes and disease‐associated genes in the major ancestral populations, whereas the Axiom UK Biobank Array is only optimized for European ancestry using the EUR panel defined as the GBR, CEU, FIN, IBS, and TSI samples from the 1000 Genomes Project.
Targeted genotyping
Targeted genotyping tests are designed to cover the most common allelic variants and/or variants known to have a functional impact in a predefined or custom panel of selected pharmaco‐genes. Targeted approaches may also include genes with low/no evidence for gene drug associations included in the current PGx databases (i.e., PharmGKB and CPIC). The main advantage of a targeted approach is its focus on ADME genes and HLA genes known to be important in immune responses, becoming “actionable” for known high‐risk drug‐gene interactions and suitable for small sample sized studies. A potential constrain is the need to be constantly updated. Various technologies for genotyping include real‐time multiplexed polymerase chain reaction (PCR)‐based methods using TaqMan probe chemistry, microarrays, mass spectroscopy arrays, and targeted sequencing. One of the first high‐throughput genotyping assays was the Drug Metabolizing Enzymes and Transporters (DMET) Plus microarray launched by Affymetrix (now Thermo Fisher Scientific, Waltham, MA) to detect 1,936 genetic variants (SNPs, INDELs, and CNVs) in 231 genes (no longer available). 12 , 13 , 14 Another assay from the same company is the PharmacoScan solution, considered the successor of the DMET array. This panel is able to genotype 4,627 markers in 1,191 PGx genes, including phase I and phase II enzymes, regulatory and modifier genes, drug target genes, phase III transporter genes, and HLA genes. An example of matrix‐assisted laser desorption ionization‐time of flight mass spectroscopy platform is the Sequenom MassARRAY system combined with the iPLEX ADME PGx Pro Panel (Agena Bioscience, San Diego, CA). This panel analyzes 192 relevant ADME markers in 36 genes to detect SNPs, INDELs, and CNVs. An alternative high‐throughput approach is the use of capture libraries for sequencing of PGx genes through NGS technology with reduction in cost and increased coverage. 15 The Ion AmpliSeq PGx Panel is an example of multiplex PCR assay performed for NGS library construction focused on selected genomic regions, including 136 markers in 40 relevant PGx genes and sequenced on Ion Torrent platform (Thermo Fisher Scientific). The PGRN‐seq Panel is a targeted sequencing platform, developed by the PGRN network and used for several projects, including the electronic Medical Records and Genomics (eMERGE) initiative. This panel includes 84 actionable genes based on information available by the CPIC and/or PharmGKB. 16
Genomewide next generation sequencing
In recent years, NGS has emerged as a comprehensive approach to profile pharmacogenes with relevance to drug‐treatment outcome. In most cases, the interindividual difference in PKs or PDs cannot be explained by known genetic variants. Thus, broader approaches, such as whole exome sequencing (WES) or whole genome sequencing (WGS), may represent a more reliable and efficient tool to discover both unknown common and rare genetic variations as compared with classical genotyping approaches. 17 In addition, it is known that about 2% of CYP‐genes harbor functional variations outside protein‐coding exons (i.e., CYP1A2*1C, CYP1A2*1F, CYP2C19*17, CYP3A4*22, CYP3A5*3, and UGT1A1*28) and to identify these intronic variants it is useful to plain WGS respect to WES or targeted sequencing. 18 However, not all variants identified will have functional and/or clinical evidence, and, therefore, may be classified as variant of unknown clinical significance. 18 Lack of evidence in clinical associations or familial segregation studies, in functional characterization, and in silico predictions lead to clinical “nonactionable” outcomes of variant of unknown clinical significance. Moreover, NGS analysis using short reads can generate miscalled sequencing variants in complex loci with high guanine‐cytosine‐content or highly homolog genes and pseudogenes. In particular, CYP2D6 is a critical highly polymorphic PGx gene located on chromosome 22 and flanked by pseudogenes (i.e., CYP2D7 and CYP2D8). Sequencing by NGS of this region is not yet technically feasible due to partial or complete gene duplication, translocation, or recombination between CYP2D6 and nearby pseudogenes. Therefore, identifying CNVs in addition to detecting single nucleotide variations is critical to predict CYP2D6 metabolizer phenotype. In addition, NGS‐based HLA sequencing has been technically difficult due to the high level of sequence homology and high variability. 18
PGX BIOMARKERS: FROM RESEARCH TO CLINICAL PRACTICE
Technological advancements in genomics have broadened the identification of variations in DNA sequence to be considered as potential PGx biomarkers for drug discovery and development, risk assessment and outcome prediction of human diseases, and drug efficacy or toxicity. Examples of biomarkers known to be predictive of drug response or adverse drug reactions are HLA‐B*1502 for carbamazepine severe skin toxicity, maculopapular exanthema, and drug reaction with eosinophilia and systemic symptoms, CYP2C9, and VKORC1 for warfarin dosing, maintenance, and response recommendations. Biomarker development provides multiple processes, involving discovery and external validation in basic studies, analytical validation, clinical utility, and clinical implementation.
Biomarker discovery and external validation
Through the previously described high‐throughput approaches, the identification starts with agnostic perspective in a target population selected in specific clinical context (training set) based on research design and objectives. A case/control design might (for instance) compare patients experiencing toxicity vs. no‐toxicity treated matched controls. Subsequently, selected genetic variants must undergo internal validation (by cross‐validation‐based methods) and finally, after technical confirmation using orthogonal approaches, must undergo external validation in an independent patient series (validation set). Study design, end points, data analysis, and reproducibility of a biomarker study can be key source of bias. In fact, small sample size or the use of clinically invalid surrogate end points from retrospective studies might preclude biomarker development. Prospective larger‐scale trials in an independent population or prospective‐retrospective study could allow biomarker clinical validation. Moreover, enriched expansion cohorts based on biomarker‐adaptive threshold design, also within phase I multi‐institutional platforms, could be a potential alternative, especially in a proof‐of‐concept perspective. 19 In a subset of PGx biomarker‐selected patients, the comparison of conventional dosing of a drug with a dose‐adjustment on gene variant‐based functional prediction, might validate the in silico assumption. Examples are reported by Antoniou et al. 20 All these studies have important methodological and statistical issues 21 and must follow regulatory guidelines (guideline on good pharmacogenomic practice European Medicines Agency (EMA)/Committee for Medicinal Products (CHMP)/718998/2016).
Biomarker assay development and analytical validation
After discovery and external validation, potential biomarkers are adapted to clinical grade assay platforms to move toward analytical and clinical validation. The analytical validation is focused on development of assay technology, which has to prove its accuracy and reliability to marker measurement. In this phase, a well‐performed assay requires the best reproducibility, technical accuracy, precision, sensitivity, specificity, and stability of the method used for biomarker detection (for instance a real‐time PCR assay for single mutation analysis or DNA sequencing by pyrosequencing). The biomarker assay should have a quick turnaround and affordable costs.
The clinical validation is on robustness and reliability of the biomarker assay on the actual performance within the specific clinical context and on the expected outcome.
Biomarker clinical utility
Despite analytical and clinical validation, a biomarker should demonstrate clinical utility, that means a clear effect in improving patient management and should demonstrate an added value to the available instruments for decision making for patient care.
Commercial and clinical implementation
After analytical and clinical validation, biomarker assay moves toward implementation in clinical care through regulatory approval, different between countries, commercialization, reimbursement, and the adoption of harmonized guidelines for clinical interpretation. The “assay intended use” may be designed as a customized test or selected panel of validated biomarkers related to a specific drug or disease. Many molecular diagnostics providers use analytical platforms or individual companion diagnostic tests, performed in certified laboratories according to Clinical Laboratory Improvement Amendment (CLIA) (http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM262327.pdf; Figure 1 ).
Figure 1.
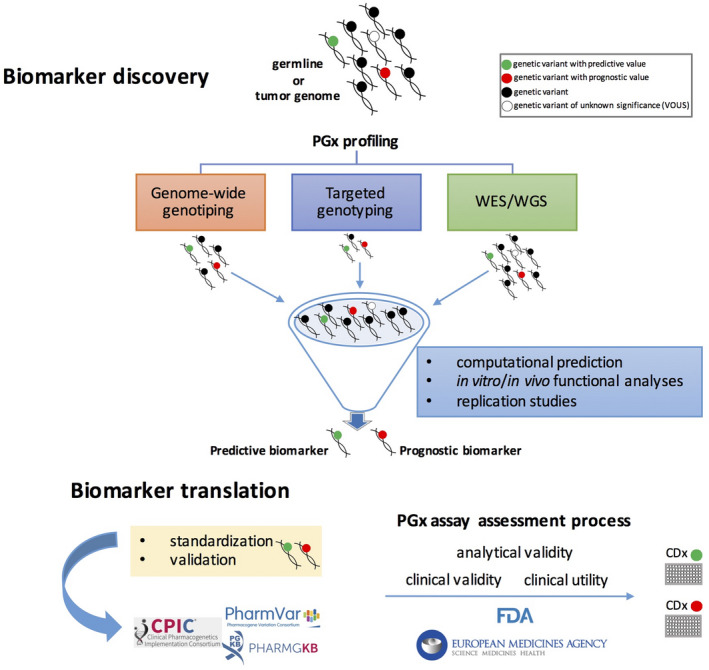
Pharmacogenomic (PGx) biomarker discovery and validation process. The workflow from biomarker discovery to assay for clinical use (companion diagnostic, CDx) starts from different individual DNA sources for identification of genomic variations, high‐throughput PGx genotyping strategies (targeted, Genome‐Wide or WES/WGS). Selected annotated or unknown genomic variants, after a complex process of validation and standardization, could became a predictive or prognostic biomarkers to be translated in clinical practice as validated CDx assay for tailored drug prescriptions.
However, among proposed predictive biomarkers, only a limited number have overcome the validation and qualification process and have changed the standard of practice and patient’s outcome. One example is DPYD testing prior to starting fluoropyrimidine‐based therapy, today strictly required by the FDA and EMA. However, although strong evidence for UGT1A1*28 irinotecan‐related toxicity, which is described in the labeling, no recommendation for pre‐emptive UGT1A1*28 test has been issued. 22 At the end of 2019, the FDA published an updated list of nearly 400 gene‐drug interactions for which PGx biomarker information is available in each drug labeling (https://www.fda.gov/drugs/science‐and‐research‐drugs/table‐pharmacogenomic‐biomarkers‐drug‐labeling). Among these, 110 genes/drugs pairs are FDA‐approved drug labels of “actionable pgx,” 7 of “genetic testing recommended,” and 15 of “genetic testing required.” The drug therapeutic area spans from oncology, hematology, psychiatry, and infectious disease to toxicology (Figure 2a,b ).
Figure 2.
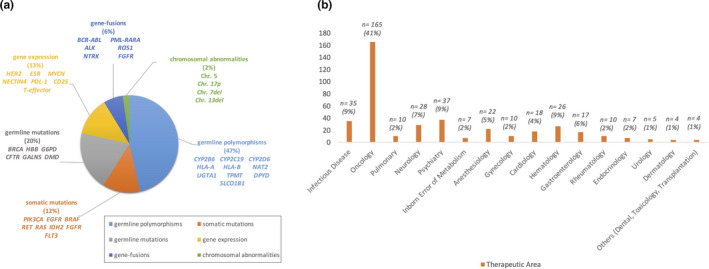
Germline and somatic PGx biomarkers (a); Therapeutic areas of drugs with PGx information in U.S. FDA (b).
DISCUSSION
Current clinical practice is moving toward a tailored medicine designed according to patients’ characteristics, lifestyle, comorbidity, and PGx profile. Rapid technologic advances in biomarker research have enhanced the discovery of actionable genetic variants as predictive and prognostic factors related to drug efficacy/toxicity and clinical outcome. Genotyping strategies described here are performed with agnostic perspective and generate large amounts of biological data whose interpretation and translation in clinical practice is still challenging. They are not equivalent or interchangeable and the choice of one or the other is conditioned by different aspects, such as the end point of the PGx study, the sample size, the familiarity with technology, and the number of markers tested besides higher costs of analysis. Targeted sequencing, with its limitations and cost considerations, generates a manageable dataset compared with broader approaches, simplifying data analysis. On the other hand, the opportunity to adopt genomewide genotyping combined with imputation represents a more comprehensive approach for biomarker discovery and a challenge for a prospective use in PGx clinical implementation (Table 2 ). However, with integrative genomic studies through data sharing from public and private databases, genome privacy needs to be carefully addressed, due to the risk of re‐identification and access by third parties for unintended purposes. WGS or WES allow the better coverage of genome or coding sequences and the study of the effects of unknown genetic variants on drugs response at a large scale and with higher precision. 17 The major limit of NGS is the high costs, complex statistics, and the need of robust computational analysis due to the large amount of generated data. The use of SNP arrays combined with imputation to large WGS reference panels will be complementary to the study of rare variants and will provide major advances in the field of complex disease genetics. Artificial intelligence and computational prediction by bioinformatics tools and deep learning algorithms will allow the integrative association of apparently unrelated variants derived from different platforms, offering an opportunity for precision medicine in cancer or rare diseases. 23 , 24 , 25
Table 2.
Challenges and Opportunities of PGx biomarker development
| Challenges |
|
| Opportunities |
|
ADR, adverse drug reaction; PGx, pharmacogenomic.
CONCLUSIONS
Although only a limited number of predictive biomarkers have high priority for dose adjustment, PGx discovery and validation of predictive biomarkers is one of the greatest challenges for diseases management, drug development, prediction of patient’s outcome, and reduction of health care costs in the vision of precision medicine (Table 2 ). In this complex scenario, additional efforts are needed for overcoming barriers in PGx implementation and the demonstration of usefulness and cost‐effectiveness of PGx test for clinicians will allow translation in clinical practice.
Conflict of Interest
All authors declared no competing interests for this work.
Funding
No funding was received for this work.
Contributor Information
Mariamena Arbitrio, Email: mariamena.arbitrio@irib.cnr.it.
Pierosandro Tagliaferri, Email: tagliaferri@unicz.it.
References
- 1. Giardina, C. et al Adverse drug reactions in hospitalized patients: results of the FORWARD (Facilitation of Reporting in Hospital Ward) Study. Front. Pharmacol. 9, 350 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arbitrio, M. et al Pharmacogenomic profiling of ADME genetic variants: current challenges and validation perspective. High Throughput 7, 40 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Buniello, A. et al The NHGRI‐EBI GWAS Catalog of published genome‐wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Illing, P.T. , Purcell, A.W. & Mccluskey, J. The role of HLA genes in pharmacogenomics: unravelling HLA associated adverse drug reactions. Immunogenetics 69, 617–630 (2017). [DOI] [PubMed] [Google Scholar]
- 5. Ingelman‐Sundberg, M. , Mkrtchian, S. , Zhou, Y. & Lauschke, V.M. Integrating rare genetic variants into pharmacogenetic drug response predictions. Hum. Genomics 12, 26 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. He, X.F. , Wei, J. , Liu, Z.Z. et al Association between CYP1A2 and CYP1B1 polymorphisms and colorectal cancer risk: a meta‐analysis. PLoS One 9, e100487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lauschke, V.M. , Milani, L. & Ingelman‐Sundberg, M. Pharmacogenomic biomarkers for improved drug therapy—recent progress and future developments. AAPS J. 20, 4 (2018). [DOI] [PubMed] [Google Scholar]
- 8. Thavaneswaran, S. , Rath, E. , Tucker, K. et al Therapeutic implications of germline genetic findings in cancer. Nat. Rev. Clin. Oncol. 16, 386–396 (2019). [DOI] [PubMed] [Google Scholar]
- 9. Battaglin, F. et al Pharmacogenomics in colorectal cancer: current role in clinical practice and future perspectives. J. Cancer Metastasis Treat. 4, 12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johnson, G.C. et al Haplotype tagging for the identification of common disease genes. Nat. Genet. 29, 233–237 (2001). [DOI] [PubMed] [Google Scholar]
- 11. Das, S. , Abecasis, G.R. & Browning, B.L. Genotype imputation from large reference panels. Annu. Rev. Genomics Hum. Genet. 19, 73–96 (2018). [DOI] [PubMed] [Google Scholar]
- 12. Di Martino, M.T. et al Genetic variants associated with gastrointestinal symptoms in Fabry disease. Oncotarget 7, 85895–85904 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Scionti, F. et al Genetic variants associated with Fabry disease progression despite enzyme replacement therapy. Oncotarget 8, 107558–107564 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arbitrio, M. et al Polymorphic variants in NR1I3 and UGT2B7 predict taxane neurotoxicity and have prognostic relevance in patients with breast cancer: a case‐control study. Clin. Pharmacol. Ther. 106, 422–431 (2019). [DOI] [PubMed] [Google Scholar]
- 15. Jensen, L. , Borsting, C. , Dalhoff, K. & Morling, N. Evaluation of the iPLEX(R) ADME PGx Pro Panel and allele frequencies of pharmacogenetic markers in Danes. Clin. Biochem. 49, 1299–1301 (2016). [DOI] [PubMed] [Google Scholar]
- 16. Gordon, A.S. et al PGRNseq: a targeted capture sequencing panel for pharmacogenetic research and implementation. Pharmacogenet. Genomics 26, 161–168 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schwarze, K. , Buchanan, J. , Taylor, J.C. & Wordsworth, S. Are whole‐exome and whole‐genome sequencing approaches cost‐effective? A systematic review of the literature. Genet. Med. 20, 1122–1130 (2018). [DOI] [PubMed] [Google Scholar]
- 18. Ji, Y. , Si, Y. , McMillin, G.A. & Lyon, E. Clinical pharmacogenomics testing in the era of next generation sequencing: challenges and opportunities for precision medicine. Expert Rev. Mol. Diagn. 18, 411–421 (2018). [DOI] [PubMed] [Google Scholar]
- 19. Jiang, W. , Freidlin, B. & Simon, R. Biomarker‐adaptive threshold design: a procedure for evaluating treatment with possible biomarker‐defined subset effect. J. Natl. Cancer Inst. 99, 1036–1043 (2007). [DOI] [PubMed] [Google Scholar]
- 20. Antoniou, M. et al Biomarker‐guided trials: challenges in practice. Contemp. Clin. Trials Commun. 16, 100493 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ross, S. , Anand, S.S. , Joseph, P. & Pare, G. Promises and challenges of pharmacogenetics: an overview of study design, methodological and statistical issues. JRSM Cardiovasc. Dis. 1, cvd.2012.012001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. CAMPTOSAR® (Irinotecan). <https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/020571s048lbl.pdf>.
- 23. Tagliaferri, P. et al BRCA1/2 genetic background‐based therapeutic tailoring of human ovarian cancer: hope or reality? J. Ovarian Res. 2, 14 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Agapito, G. et al DMETTM genotyping: tools for biomarkers discovery in the era of precision medicine. High Throughput 9, 8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gallo Cantafio, M.E. et al From single level analysis to multi‐omics integrative approaches: a powerful strategy towards the precision oncology. High Throughput 7, 33 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]