

Abstract
Objective
To highlight potential epigenetic risk factors for blood pressure (BP) and ischemic stroke (IS) in loci identified by genome-wide association studies (GWASs).
Methods
We detected DNA methylation for BP (317,756 individuals from UK Biobank) and IS (521,612 individuals from MEGASTROKE) in Europeans by using the summary data–based mendelian randomization (SMR) method. We selected the most relevant gene to validate the association in 1,207 patients with hypertensive IS and 1,269 controls from the Chinese populations.
Results
We first identified 173 CpG sites in 90 genes, 337 CpG sites in 142 genes, and 9 CpG sites in 7 genes that were significantly associated with systolic, diastolic BP, and IS, respectively. The methylation level of cg12760995 in CASZ1 was associated with systolic (PSMR = 1.74 × 10−12), diastolic BP (PSMR = 2.48 × 10−10), and IS (odds ratio [OR] = 0.92 [95% confidence interval [CI]: 0.91–0.94]; PSMR = 2.28 × 10−8) in Europeans. The methylation levels of 17 sites in the promoter of CASZ1 were measured in the Chinese individuals, and 10 of them were significantly associated with IS. The higher methylation level of CASZ1 was associated with a lower risk of IS (adjusted OR = 0.97 [95% CI: 0.96–0.99]). CASZ1 seemed to be hypomethylated in hypertensive cases, and the level was negatively correlated with BP. Systolic and diastolic BP mediated approximately 61.2% (p = 3.49 × 10−6) and 45.0% (p = 0.0029) of the association between CASZ1 methylation and IS, respectively.
Conclusions
This study identified DNA methylations that were associated with BP and IS. CASZ1 was hypomethylated in Chinese patients with hypertensive IS.
Ischemic stroke (IS) is the second leading cause of death worldwide.1,2 Hypertension is identified as one of the most important causal risk factors for cardiovascular diseases.3,4 Genetic and epigenetic factors play important roles in the etiology of hypertension and IS.5 Large-scale genome-wide association studies (GWASs) have successfully identified many loci for blood pressure (BP)6 and IS7,8 and shown that BP and IS shared some of the loci. GWASs have suggested that many disease-associated loci or variants were ethnic specific. However, some genetic loci were shared by Europeans and East Asians, e.g., the CASZ1 (and others) gene variants were found to be associated with BP in both Europeans and East Asians.9
DNA methylation occurs primarily on CpG dinucleotide to control transcription. Studies have shown that blood DNA methylation play important roles in BP regulation.10 It has been known that epigenetic regulation participates in the development of hypertension through a comprehensive mechanism, which targets different levels of complexity including the renin-angiotensin-aldosterone system, the vascular wall, and specific cell types within the vessels.11 Some studies have been performed to identify epigenetic risk factors for stroke.12–14 DNA methylation has also been identified as potential drug targets in the treatment of atherosclerotic diseases, including stroke.15–17 The DNA methylation patterns could be used as potential biomarkers of stroke.18 Even so, the role DNA methylations play in hypertension and stroke remains poorly known.
The functional factors among the GWAS-identified loci were not fully understood. A major challenge remained in the post-GWAS era was uncovering causal factors underlying the GWAS hits. Here, we presented a 2-step study to identified additional epigenetic risk factors for BP and IS. First, we applied the summary data–based mendelian randomization (SMR)19 method to integrate data from large-scale GWASs with methylation quantitative trait loci (mQTL) study to detect BP- and IS-associated methylations in the European populations, among which the public available GWAS summary data are very rich. Second, we examined the associations between the identified methylations and BP and IS among individuals from the Chinese populations.
Methods
Mendelian randomization analysis
Mendelian randomization (MR) is an instrumental variable analysis approach that uses genetic variants as instrumental variables (e.g., mQTLs) to test whether an exposure (e.g., DNA methylation level) has a causal effect on an outcome (e.g., IS). The SMR method can identify potential functionally relevant methylations in the GWAS identified loci by integrating independent GWAS summary statistics with mQTL data.19,20 By using public summary data from large-scale GWAS and mQTL meta-analysis studies, the SMR method evaluated the causal associations in very large samples so that the statistical power was increased. In this study, we first conducted SMR analysis to identified DNA methylations that were causally associated with BP and IS, by which we could prioritize functionally relevant genes in the GWAS loci and find the shared genetic influences.
The BP GWAS data set comprised the summary statistics for the association between more than 10 million single nucleotide polymorphisms (SNPs) and systolic BP (SBP) and diastolic BP (DBP) that were evaluated in 317,756 individuals enrolled in the UK Biobank.21 This data set can be downloaded at ldsc.broadinstitute.org/gwashare/. The IS GWAS, which comprised 521,612 individuals of European ancestry, is the largest GWAS on IS to date.7 Raw data used in the present analysis were the downloaded summary results from this GWAS, which included summary data of almost 8 million SNPs and indels for IS. These data set was available at the MEGASTROKE website (megastroke.org/). The zip file downloaded contained 10 files for the GWAS results of any stroke, any ischemic stroke, large artery stroke, cardioembolic stroke, and small vessel stroke subtypes from Europeans and trans-ethnicity samples. In this study, we restricted the analysis to the IS subtype in Europeans, only the file “MEGASTROKE.2.AIS.EUR.out” inside the zip file was used in the analysis.
The available summary data contained information on the rs number, chromosome, position, effect allele, other allele, sample size, beta, standard error, t statistic, and p value for each SNP. Allele frequencies for the SNPs were obtained from the 1000 Genomes Project (Europeans). The BP and IS GWAS summary data were formatted (the plain file contains 8 columns: rs number, effect allele, other allele, frequency, beta, standard error, p value, and sample size) for the SMR analysis by using the R language.
Another file required for the SMR analysis was the mQTL data file (in binary format). The mQTL summary data were from the study conducted by McRae et al.22 DNA methylation was measured on 614 individuals from the Brisbane Systems Genetics Study and 1,366 individuals from the Lothian Birth Cohorts using Illumina HumanMethylation450 BeadChips. The mQTL summary data in SMR binary format can be downloaded from cnsgenomics.com/software/smr/#DataResource. Only the DNA methylation probes (88,712) with at least a cis-mQTL at p < 5.0 × 10−8 and only SNPs within 2 Mb distance from each probe were available.
SMR jointly analyzed the mQTL and BP and IS GWAS summary statistics to test for association between methylations and BP and IS due to the shared variant at a locus. A heterogeneity in dependent instruments (HEIDI) test for heterogeneity in the resulting association statistics was performed. PHEIDI > 0.05 means that there was no significant heterogeneity underlying the mQTL signals (the basic assumption of MR analysis). We ran SMR (version 0.712) with default parameters in a command line program, which was downloaded from cnsgenomics.com/software/smr/. Genotype data of HapMap r23 CEU were used as a reference panel to calculate the linkage disequilibrium (LD) correlation for SMR analysis.
Study sample from Chinese populations
We conducted a case-control study investigating the association between the DNA methylations of the identified genes and IS among Chinese individuals. The IS cases were randomly selected from the China Antihypertensive Trial in Acute Ischemic Stroke (CATIS) project, a clinical trial registered at clinicaltrials.gov (Identifier: NCT01840072). The CATIS trial was a multicenter, single-blind, blinded end point, randomized clinical trial conducted in 26 hospitals across China from August 2009 to May 2014.23 The study design has been described in a previous study.23 In brief, 4,071 patients aged 22 years or older who had IS confirmed by CT or MRI of the brain within 48 hours of symptom onset and who had an elevated SBP between 140 mm Hg and less than 220 mm Hg were recruited.
The controls were randomly selected from the Suzhou Metabolic Syndrome Study (SMSS).24 The SMSS was an observational cohort study of 18,461 individuals in 6 rural townships in of Suzhou in China's Jiangsu province. Participants who had no evidence of end organ damage, including coronary heart disease, stroke, chronic renal disease, and tumors, and signed informed consent were recruited at baseline. By considering both sequencing cost and statistical power, about 1,200 cases and 1,200 controls were chosen in this study.
Standard protocol approvals, registrations, and patient consents
This study was approved by the institutional review boards or ethical committees at Soochow University. Written consent was obtained from all study participants.
Measurements
Three sitting consecutive BP measurements (30 seconds between each) were taken by trained staff using a standard mercury sphygmomanometer according to a standard protocol, after the participants had been resting for 30 minutes. The first and fifth Korotkoff sounds were recorded as SBP and DBP, respectively. The average of the 3 BP measurements was calculated for each participant and used in the analyses. Hypertension was defined as SBP ≥140 mm Hg and/or DBP ≥90 mm Hg and/or use of antihypertensive medication in the last 2 weeks. For all the participants, overnight fasting blood samples were obtained, plasma (serum) and white blood cell samples were isolated, and DNA was extracted. The specimens were frozen at −80°C until laboratory testing. A modified hexokinase enzymatic method was applied to test fasting blood glucose (FBG) levels. Total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), and triglycerides (TGs) were analyzed enzymatically using a Beckman Synchron CX5 Delta Clinical System (Beckman Coulter, Inc., Fullerton, California) with commercial reagents. Low-density lipoprotein cholesterol (LDL-C) levels were calculated using the Friedewald equation for participants who had less than 400 mg/dL TG.
DNA methylation sequencing
We applied the targeted bisulfite sequencing to measure DNA methylation levels. Briefly, CpG islands adjacent to the promoter region of the gene were analyzed, and based on these CpG islands, the targets were confirmed (figure e-1, links.lww.com/NXG/A313). Genomic DNA passed quality control (concentration ≥20 ng/μL, total DNA ≥ 1 μg, OD260/280 = 1.7–2.0, OD260/230 ≥ 1.8) was bisulfite converted using the EZ DNA Methylation-Gold Kit (ZYMO, CA), and then, the targeted DNA fragments were amplified by PCR. The amplified products were sequenced on an Illumina MiSeq benchtop sequencer (Illumina, CA). The methylation level of each CpG site was calculated as the percentage of the methylated cytosines over the total tested cytosines. The average methylation level for a target was calculated using methylation levels of all measured CpG sites within the target. To test the reliability of methylation levels for each CpG site, 20 samples were randomly selected for duplicate detection.
Statistical analysis
The differences of baseline risk factors between cases and controls were compared, using a Student t test for continuous variables and χ2 tests for categorical variables. The methylation levels of each CpG site in IS cases and controls were assessed by 2-tailed unpaired Student t tests; bar plots depict the means, and the error bars in the figures represent SDs. Linear regression models were used to assess the associations between methylation levels and BP. Logistic regression models were used to calculate odds ratios (ORs) and 95% confidence intervals (CIs) for hypertension and IS for per 1-percent increased with methylation levels. The potential covariates such as age, sex, smoking, drinking, body mass index (BMI) SBP, DBP, TC, and FBG were included in the multivariate models. The analyses were performed in the combined sample and subgroups stratified by sex. Causal mediation analysis was performed to examine the mediation effect of BP on the association between methylation and IS. The mediation analysis was conducted by using the R package “mediation,”25 with 1,000 Monte Carlo draws. Individuals with missing data were deleted in the analysis. All analyses were performed using the R language program (Version 3.5.0).
Data availability
Data are available to researchers on request for purposes of reproducing the results or replicating the procedure by directly contacting the corresponding author.
Results
BP-associated CpG sites
By integrating data from large-scale BP GWAS with mQTL study from European populations, we found 173 CpG sites in 90 genes that were significantly associated with SBP and 337 CpG sites in 142 genes that were significantly associated with DBP (PSMR < 5.67 × 10−7, PHEIDI > 0.05). The total number of CpG sites (or genes) was 470 (199). Among them, 40 (33) were shared by SBP and DBP. The detailed information about these SBP- and DBP-associated CpG sites is presented in tables e-1 and e-2 (links.lww.com/NXG/A313), respectively. Among the identified genes, the top 20 genes that contained the most significant methylations associated with SBP were CNNM2, NT5C2, SLC5A11, FES, TNRC6A, CASZ1, TNXB, FAM109A, ADAMTS8, HOXA13, NEIL2, SLC10A4, SYNPO2L, GATA4, BRAP, SH2B3, RERE, CEP68, MSRA, and GOSR2 (figure 1). The top 20 genes that contained the most significant methylations associated with DBP were FAM109A, NOV, CPEB4, ZSCAN12L1, ZNF389, CNNM2, ZSCAN16, SH2B3, CYP21A2, FES, C6orf10, MSH5, SDCCAG8, RERE, MSRA, HLA-DMA, ULK4, ZNF192, SRRT, and HLA-B (figure 1).
Figure 1. Results for the association between DNA methylations and IS.

The x-axis represents the chromosome positions. The y-axis shows the –log10 p values for the association between levels of DNA methylation sites and BP (UK Biobank) and IS (MEGASTROKE) in European populations. Among the CpG sites analyzed for (A) SBP, (B) DBP, and (C) IS, 430, 581, and 28 passed the suggestive significance threshold of 1.0 × 10-5 (blue line), respectively. Only the top 20 genes were annotated for SBP and DBP. Other CpG sites associated with SBP and DBP are presented in tables e-1 and e-2 (links.lww.com/NXG/A313), respectively. BP = blood pressure; DBP = diastolic BP; IS = ischemic stroke; SBP = systolic BP.
IS-associated CpG sites
By integrating data from large-scale IS GWAS with mQTL study from European populations, we found that more than 6,000 CpG sites were nominally associated with IS (PSMR < 0.05). Among them, p values of 28 CpG sites in 12 loci (14 genes) exceed 1 × 10−5 (figure 1) and 10 of them could be considered significant (PSMR < 5.67 × 10−7, Bonferroni correction). Nine of these significant sites in CASZ1, SLC25A44, FOXQ1, SH3PXD2A, ACAD10, LRCH1, and SLC44A2 passed the HEIDI test (PHEIDI > 0.05) (table 1).
Table 1.
Methylations significantly associated IS identified by mendelian randomization analysis

The selected gene CASZ1
Among the 199 identified BP-related genes, we noticed that CASZ1 SNPs were strongly associated with both SBP (PGWAS = 5.62 × 10−14) and DBP (PGWAS = 2.81 × 10−11) and DNA methylation levels (PmQTL = 1.37 × 10−92), and CASZ1 methylation was significantly associated with both SBP (PSMR = 1.74 × 10−12) and DBP (PSMR = 2.48 × 10−10) in Europeans (tables e-1 and e-2, links.lww.com/NXG/A313, figure 2). Among the 7 identified IS-related genes, CASZ1 SNPs were strongly associated with both IS risk (PGWAS = 5.51 × 10−9) and DNA methylation levels (PmQTL = 1.37 × 10−92), and CASZ1 methylation was significantly associated with IS (beta = −0.08, OR = 0.92 [95% CI: 0.91–0.94]; PSMR = 2.28 × 10−8) in Europeans (table 1, figure 2).
Figure 2. The association between CASZ1 methylation and BP and IS.

The 3 panels present the associations between CASZ1 methylation and SBP (A), DBP (B), and IS (C) in European populations. Each panel consists of 2 parts. The x-axis represents the genomic position (GRCh37.p13). The lower part of each panel shows the results of mQTL. The y-axis represents –log10(P mQTL). In this part, we can see that CASZ1 SNPs were strongly associated with DNA methylation levels (mQTL). The upper part of each panel shows the results of GWAS and SMR analysis. The y-axis represents –log10(P GWAS or SMR). In this part, we can see that CASZ1 SNPs were strongly associated with SBP (A), DBP (B), and IS (C). According to the mendelian randomization analysis, a DNA methylation level of cg12760995 in the CASZ1 gene was significantly associated with SBP (A), DBP (B), and IS (C). DBP = diastolic BP; SBP = systolic BP; GWAS = genome-wide association study; IS = ischemic stroke; SMR = summary data–based mendelian randomization.
CASZ1 gene SNPs have also been reported to be strongly associated with BP, hypertension, and incident hypertension and cardiovascular diseases in the Chinese populations.9,26 Until now, the associations between CASZ1 methylation and BP and IS have not yet been clarified. Therefore, based on these evidence, we conducted a case-control study to investigate the association between the DNA methylations in this gene and IS to identify additional risk factors for IS among Chinese individuals.
Association between CASZ1 methylation and BP and IS in Chinese
A total of 1,207 IS cases and 1,269 controls were included for bisulfite sequencing (table e-3, links.lww.com/NXG/A313). The mean age and the proportion of males, smokers, and drinkers were not different between cases and controls. Compared with the controls, mean SBP, DBP, BMI, TC, LDL-C, TG, and FBG levels were all significantly higher, whereas the HDL-C level was lower in IS cases (p < 0.05). Among the 1,207 patients with IS, 1,005 (83.3%) were thrombotic strokes, 54 (4.5%) were embolic strokes, and 173 (14.3%) were small-vessel lacunar strokes. Three patients were both thrombotic and embolic, 22 were both thrombotic and lacunar. Thrombotic strokes included both intra- and extracranial atherosclerotic strokes. No cryptogenic stroke was included. Among the 1,269 controls, 381 (30.02%) were patients with hypertension (table e-4, links.lww.com/NXG/A313).
We successfully measured the methylation levels of 17 sites inside a CpG island in CASZ1. According to the duplicate detection in 20 individuals, there was no significant difference between the 2 tests for the methylation levels of each of these 17 sites (paired t test p > 0.05). The missing rate of methylation level data was 0.91% (11) for cases and 0.23% (3) for controls. The methylation levels of 10 of these sites were significantly associated with IS in univariate analysis (p < 3.0 × 10−3) (figure e-2, links.lww.com/NXG/A313). The methylation levels of the associated CpG sites were highly correlated (figure e-3, links.lww.com/NXG/A313) and seemed to be hypomethylated in the patients with hypertensive IS (table 2). After adjusted for covariates, the associations between methylation levels of 8 sites and IS were nominally significant, and the associations between methylation levels of CASZ1_70 (adjusted OR = 0.99 [0.98–1.00]; p = 7.26 × 10−4) and CASZ1_67 (adjusted OR = 0.98 [0.98–0.99]; p = 5.54 × 10−5) and IS were significant. These sites were associated with IS in male participants but not females (table 2). The mean methylation level of this tested target was significantly lower in the patients with hypertensive IS than the controls (figure 3A). The higher CASZ1 gene methylation level was significantly associated with a lower risk of IS (adjusted OR = 0.97 [95% CI: 0.96–0.99]; p = 1.59 × 10−4), and the association was significant in males (adjusted OR = 0.96 [95% CI: 0.94–0.98]; p = 4.41 × 10−5) (table 2).
Table 2.
Methylation sites significantly associated with IS in CASZ1

Figure 3. The relationship of mean CASZ1 methylation levels with BP and IS among Chinese.

The mean methylation levels for CASZ1 for each individual were calculated using methylation levels of all measured CpG sites within the genomic fragment sequenced. The bar plots depict the means, and the error bars in the figures represent SDs; the differences were assessed by t tests. (A) This gene was significantly hypomethylated in the 1,207 patients with hypertensive IS compared with the 1,269 controls. (B) This target was hypomethylated in the hypertensive cases (1,207 IS hypertensives plus 381 non-IS hypertensives or only 381 non-IS hypertensives) compared with the 888 controls. The 2 scatter plots present the relationship between the mean methylation level of CASZ1 and SBP (C) and DBP (D). The blue lines represent linear regression lines, and the gray areas represent the corresponding 95% confidence intervals. DBP = diastolic BP; IS = ischemic stroke; SBP = systolic BP.
The methylation levels of 4 and 3 of the 17 sites were significantly associated with SBP and DBP (p < 1.0 × 10−3) in univariate analysis (table e-5, links.lww.com/NXG/A313), respectively. After adjustment for IS status and other covariates, the associations of methylation levels of 3 sites with SBP were nominally significant. The mean methylation level of this target seemed to be hypomethylated in hypertensive cases (figure 3B) and was negatively correlated with SBP and DBP (figure 3, C and D, respectively). The association between the mean methylation level of this target and SBP was still nominally significant after adjustment for IS status and other covariates (beta = −0.3641, p = 0.0341). In the median analysis, the mediation effect estimations were −0.0445 (p = 0.0021) and −0.0315 (p = 0.0062) for SBP and DBP, respectively. SBP and DBP mediated approximately 61.2% (p = 3.49 × 10−6) and 45.0% (p = 0.0029) of the association between CASZ1 methylation and IS, respectively (figure 4).
Figure 4. The indirect effect of CASZ1 methylation on IS.
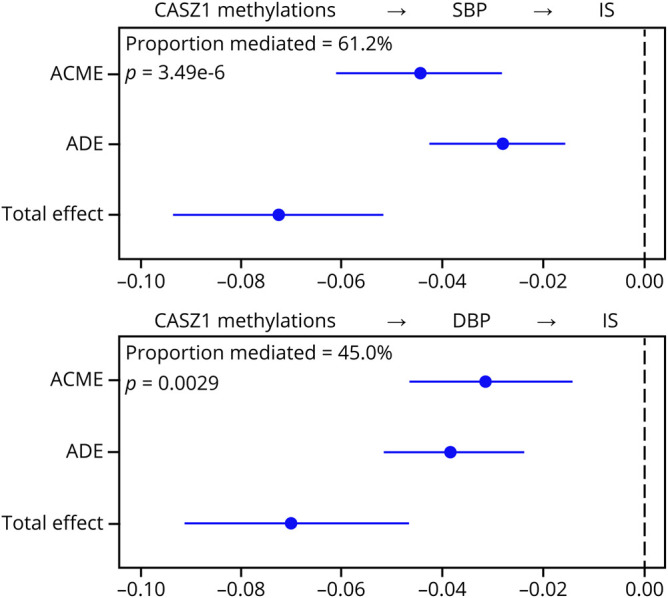
Proportion mediated: the proportion of the association between methylation and ischemic stroke that was explained by the mediator. The analysis was performed on the 1,207 patients with hypertensive IS from the CATIS and 1,269 controls. ACME = average causal mediation effect; ADE = average direct effect; DBP = diastolic blood pressure; SBP = systolic blood pressure.
Discussion
The current study represented an effort to identify potential causal epigenetic factors for BP and IS by integrating data from large-scale GWASs initiative on DNA methylation, BP, and IS risk. Hundreds of CpG sites in important genes (e.g., CASZ1) were found to be causally associated with BP and IS in Europeans by using this strategy. In addition, by applying a target sequencing method, we found that CASZ1 methylations were associated with BP and IS in the Chinese populations.
DNA methylation plays an important role in BP regulation and IS pathology and could be potential therapeutic targets.15–17 Previous studies have identified some DNA methylations for hypertension and IS10,12–14; however, the statistical power of these traditional case-control studies was very limited due to the small sample size, and reverse causation always existed in observational studies. Integration of GWAS data with data from the mQTL study by applying the SMR approach was a possible way to identify relevant methylations in the GWAS loci.19 This methodology not only limited bias due to confounding and was not affected by reverse causation but also increased statistical power. As we showed in the present study, the methylation levels of many genes were detected to be significantly associated with BP and IS. Genes identified in our study were located in the BP and IS GWAS-identified loci, which have implicated the importance of these genes in BP regulation and IS etiology. Our study highlighted the involvement of methylation in these loci. The mQTLs were detected in blood samples, so our study identified blood cell DNA methylations that were associated with BP and IS.
CASZ1 encodes a zinc finger transcription factor. CASZ1 is required to directly regulate an EGFL7/RhoA-mediated pathway to promote vertebrate vascular development27 and plays a key role in cardiac homeostasis and dilated cardiomyopathy.28 The association between CASZ1 methylations and IS has not been reported. The newly identified methylations locate in the CpG Island, which overlaps the promoter in the 5′ region of CASZ1 (figure e-1, links.lww.com/NXG/A313). Moreover, this promoter is connected to several nearby enhancers (GeneHancer regulatory elements29) by long-range interactions. DNA methylations in long-range interactive promoters and enhancers have been shown to affect the long-range interactions and to be associated with disease risk.30–32 Therefore, it seems that the CASZ1 promoter methylation may have the regulatory potentials that affect IS risk.
CASZ1 gene variants have been confirmed to be associated with BP by many GWASs from different populations.9,26,33–35 In SMR analysis, we also detected the association between blood cell DNA methylation of CASZ1 and BP. Therefore, the CASZ1 methylation level should be associated with BP, given the identified relationship of genetic variants and methylation at this locus with BP or hypertension. But although we found that CASZ1 was hypomethylated in patients with hypertensive IS and detected some nominal associations, we did not detect significant association between the CASZ1 methylation level and BP or hypertension in non-IS individuals. The non-IS sample (only 381 non-IS hypertensive individuals) in our study may be too small to detect the association. Therefore, the effect of CASZ1 methylation on BP and whether BP mediates the effect of CASZ1 methylation on IS need to be validated in studies with larger samples.
We observed sex differences in the association between CASZ1 methylation and IS risk in this study. Significant differences between males and females were observed for age, smoking, and drinking in our data. The CASZ1–BP association has suggested to be influenced by age in a sex-specific manner, which documented the modulation of genetic effects on BP by sex and age.34 Besides, sex- and age-specific associations between methylation and diseases for other genes and diseases have been reported.36,37 Blood cell DNA methylation can be influenced by smoking and drinking.38,39 However, in our study, no significant association between sex and methylation was observed in patients with IS or the controls or combined samples. Methylation levels were not associated with age, smoking, or drinking either. No significant interaction effect between methylations and age, smoking, or drinking on IS was found. The associations between methylation levels and IS risk were not significant in subgroups stratified by smoking or drinking. The differences of age, smoking, and drinking between the men and the women in this study might not account for the observation of sex difference. The sex differences in the association between CASZ1 methylation and IS risk needed to clarify in future studies.
In addition to CASZ1, methylations of other genes identified in the MEGASTROKE GWAS were found to be potential causal factors for IS in this study. The original GWAS has linked the genetic variants of these genes to IS risk, whereas the present study associated their methylations with IS. The identified genes have been shown to play certain roles in the cardiovascular system. For example, the SLC25A44 gene, encodes solute carrier family 25 member 44 that is widely expressed in the CNS,40 has been identified to be associated with intracerebral hemorrhage.41 SLC44A2 (solute carrier family 44 member 2), also named CTL2 (choline transporter-like protein 2), codes for a human leukocyte alloantigen. It has been shown that SLC44A2 is expressed on the endothelium, where it expresses an isoform involved in choline transport.42 This gene was a well-known susceptibility gene for thrombosis.43–45 SLC44A2 may be involved in choline uptake in mitochondria, which is the rate-limiting step in DNA methylation.46 Methylation in this gene has been shown to be associated with MS47 but not stroke.
In addition, some of the identified genes have been paid less attention to and not been discussed in the original article. The ACAD10 gene encodes a member of the acyl-CoA dehydrogenase family of enzymes, which participate in the beta-oxidation of fatty acids in mitochondria. ACAD10 gene variants were also associated with platelet count and mean platelet volume,48 coronary artery disease,49 type 2 diabetes, insulin resistance, and lipid oxidation.50 In the MEGASTROKE GWAS, ACAD10 was significantly associated with IS in the gene-based test, and an intronic variant rs4766897 in ACAD10 was a shared genetic influence of large-artery atherosclerotic stroke and small-vessel stroke. But in this genomic region, SH2B3 and TBX3 were the reported associated genes. In our SMR analysis, methylation of CpG site cg08577424 (chr12: 112123256) was significantly associated with IS, without significant heterogeneity. This site locates in a CpG island (chr12:112123247-112124059), which overlaps with the 5′-untranslated region of ACAD10. However, the role of ACAD10 methylation was unknown.
This study showed the advantage of integration of multiomics data in identification of regulatory mechanisms underlying the genetic associations. But there were also several limitations. First, we were unable to validate the results of the SMR analysis in an independent large sample because of lacking in data. For the same reason, we were unable to perform SMR analysis in populations other than Europeans. Second, DNA methylation is known to be tissue specific. We measured methylation levels in peripheral blood cell DNA, so we could have lost signals by not choosing distinct tissues, although peripheral blood cell is commonly used as a surrogate tissue in studies of cardiovascular diseases.14,24 Third, we only tested the CASZ1 gene in our samples due to the high cost of sequencing. Associations between methylations of other identified genes and BP and IS risk are needed to validate in future studies. Finally, the proportions of thrombotic, embolic, and lacunar stroke subtypes in the CATIS participants were 77.9%, 5.0%, and 19.7%, respectively.23 Although the sequenced samples were randomly selected from the CATIS participants, the distribution of subtypes of the selected samples was significantly different from that of the CATIS (χ2 = 18.428, p = 9.96 × 10−5). The proportion of thrombotic stroke was 5.4% higher in our selected patients. Therefore, the interpretation of the association should be cautious.
In summary, the present study found out DNA methylations that were significantly associated with BP and IS risk. CASZ1 methylations were newly identified to be hypomethylated in Chinese patients with hypertensive IS. This study identified new risk factors, and the findings may increase our understanding on the role of DNA methylations in the pathogenesis of IS. Although we have detected significant associations, cell experiments and animal studies are suggested to carry out to confirm the causality and elucidate the mechanism.
Acknowledgment
The authors thank all the participants in this study. The MEGASTROKE project received funding from sources specified at megastroke.org/acknowledgments.html.
Glossary
- BMI
body mass index
- BP
blood pressure
- CATIS
China Antihypertensive Trial in Acute Ischemic Stroke
- CI
confidence interval
- DBP
diastolic BP
- FBG
fasting blood glucose
- GWAS
genome-wide association study
- HDL-C
high-density lipoprotein cholesterol
- HEIDI
heterogeneity in dependent instruments
- IS
ischemic stroke
- LD
linkage disequilibrium
- LDL-C
low-density lipoprotein cholesterol
- MR
mendelian randomization
- OR
odds ratio
- SBP
systolic BP
- SMR
summary data-based mendelian randomization
- SMSS
Suzhou Metabolic Syndrome Study
- SNP
single nucleotide polymorphism
- TC
total cholesterol
- TG
triglyceride
- UTR
untranslated region
Appendix. Authors

Study funding
The study was supported by the Natural Science Foundation of China (81773508 and 81673263), the Key Research Project (Social Development Plan) of Jiangsu Province (BE2016667), the Startup Fund from Soochow University (Q413900313 and Q413900412), and a Project of the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/NG for full disclosures.
References
- 1.GBD 2017 Causes of Death Collaborators. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018;392:1736–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang W, Jiang B, Sun H, et al. Prevalence, incidence, and mortality of stroke in China: results from a nationwide population-based survey of 480 687 adults. Circulation 2017;135:759–771. [DOI] [PubMed] [Google Scholar]
- 3.Zhu Z, Zheng Z, Zhang F, et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat Commun 2018;9:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Westra HJ, Peters MJ, Esko T, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet 2013;45:1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakatochi M, Ichihara S, Yamamoto K, et al. Epigenome-wide association of myocardial infarction with DNA methylation sites at loci related to cardiovascular disease. Clin Epigenetics 2017;9:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duncan CJ, Mohamad SM, Young DF, et al. Human IFNAR2 deficiency: lessons for antiviral immunity. Sci Transl Med 2015;7:307ra154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Battle A, Brown CD, Engelhardt BE, Montgomery SB. Genetic effects on gene expression across human tissues. Nature 2017;550:204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lloyd-Jones LR, Holloway A, McRae A, et al. The genetic architecture of gene expression in peripheral blood. Am J Hum Genet 2017;100:228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu X, Wang L, Lin X, et al. Genome-wide association study in Chinese identifies novel loci for blood pressure and hypertension. Hum Mol Genet 2015;24:865–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richard MA, Huan T, Ligthart S, et al. DNA methylation analysis identifies loci for blood pressure regulation. Am J Hum Genet 2017;101:888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stoll S, Wang C, Qiu H. DNA methylation and histone modification in hypertension. Int J Mol Sci 2018;19:1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilli F, Valentino P, Caldano M, et al. Expression and regulation of IFNalpha/beta receptor in IFNbeta-treated patients with multiple sclerosis. Neurology 2008;71:1940–1947. [DOI] [PubMed] [Google Scholar]
- 13.Frodsham AJ, Zhang L, Dumpis U, et al. Class II cytokine receptor gene cluster is a major locus for hepatitis B persistence. Proc Natl Acad Sci U S A 2006;103:9148–9153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Obora A, Shiratori Y, Okuno M, et al. Synergistic induction of apoptosis by acyclic retinoid and interferon-beta in human hepatocellular carcinoma cells. Hepatology 2002;36:1115–1124. [DOI] [PubMed] [Google Scholar]
- 15.Byrne MM, Murphy RT, Ryan AW. Epigenetic modulation in the treatment of atherosclerotic disease. Front Genet 2014;5:364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu Z, Zhong B, Tan J, Chen C, Lei Q, Zeng L. The emerging role of epigenetics in cerebral ischemia. Mol Neurobiol 2017;54:1887–1905. [DOI] [PubMed] [Google Scholar]
- 17.Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet 2018;50:693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gomez-Uriz AM, Milagro FI, Mansego ML, et al. Obesity and ischemic stroke modulate the methylation levels of KCNQ1 in white blood cells. Hum Mol Genet 2015;24:1432–1440. [DOI] [PubMed] [Google Scholar]
- 19.Zhu Z, Zhang F, Hu H, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 2016;48:481–487. [DOI] [PubMed] [Google Scholar]
- 20.Pavlides JM, Zhu Z, Gratten J, McRae AF, Wray NR, Yang J. Predicting gene targets from integrative analyses of summary data from GWAS and eQTL studies for 28 human complex traits. Genome Med 2016;8:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamai T, Yanai Y, Arai K, et al. Increased interferon alpha receptor 2 mRNA levels is associated with renal cell carcinoma metastasis. BMC Cancer 2007;7:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McRae AF, Marioni RE, Shah S, et al. Identification of 55,000 replicated DNA methylation QTL. Sci Rep 2018;8:17605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He J, Zhang Y, Xu T, et al. Effects of immediate blood pressure reduction on death and major disability in patients with acute ischemic stroke: the CATIS randomized clinical trial. JAMA 2014;311:479–489. [DOI] [PubMed] [Google Scholar]
- 24.Kato N, Loh M, Takeuchi F, et al. Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nat Genet 2015;47:1282–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tingley D, Yamamoto T, Hirose K, Keele L, Imai K. Mediation: R package for causal mediation analysis. J Stat Softw 2014;59:1–38.26917999 [Google Scholar]
- 26.Lu X, Huang J, Wang L, et al. Genetic predisposition to higher blood pressure increases risk of incident hypertension and cardiovascular diseases in Chinese. Hypertension 2015;66:786–792. [DOI] [PubMed] [Google Scholar]
- 27.Charpentier MS, Christine KS, Amin NM, et al. CASZ1 promotes vascular assembly and morphogenesis through the direct regulation of an EGFL7/RhoA-mediated pathway. Dev Cell 2013;25:132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kennedy L, Kaltenbrun E, Greco TM, et al. Formation of a TBX20-CASZ1 protein complex is protective against dilated cardiomyopathy and critical for cardiac homeostasis. PLoS Genet 2017;13:e1007011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fishilevich S, Nudel R, Rappaport N, et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford) 2017;2017:bax028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park SM, Choi EY, Bae M, Choi JK, Kim YJ. A long-range interactive DNA methylation marker panel for the promoters of HOXA9 and HOXA10 predicts survival in breast cancer patients. Clin Epigenetics 2017;9:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flavahan WA, Drier Y, Liau BB, et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016;529:110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schoenfelder S, Fraser P. Long-range enhancer-promoter contacts in gene expression control. Nat Rev Genet 2019;20:437–455. [DOI] [PubMed] [Google Scholar]
- 33.Kato N, Takeuchi F, Tabara Y, et al. Meta-analysis of genome-wide association studies identifies common variants associated with blood pressure variation in east Asians. Nat Genet 2011;43:531–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takeuchi F, Isono M, Katsuya T, et al. Blood pressure and hypertension are associated with 7 loci in the Japanese population. Circulation 2010;121:2302–2309. [DOI] [PubMed] [Google Scholar]
- 35.Levy D, Ehret GB, Rice K, et al. Genome-wide association study of blood pressure and hypertension. Nat Genet 2009;41:677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bozdag S, Li A, Riddick G, et al. Age-specific signatures of glioblastoma at the genomic, genetic, and epigenetic levels. PLoS One 2013;8:e62982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qin X, Li J, Wu T, et al. Overall and sex-specific associations between methylation of the ABCG1 and APOE genes and ischemic stroke or other atherosclerosis-related traits in a sibling study of Chinese population. Clin Epigenetics 2019;11:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park SL, Patel YM, Loo LWM, et al. Association of internal smoking dose with blood DNA methylation in three racial/ethnic populations. Clin Epigenetics 2018;10:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilson LE, Xu Z, Harlid S, et al. Alcohol and DNA methylation: an epigenome-wide association study in blood and normal breast tissue. Am J Epidemiol 2019;188:1055–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haitina T, Lindblom J, Renstrom T, Fredriksson R. Fourteen novel human members of mitochondrial solute carrier family 25 (SLC25) widely expressed in the central nervous system. Genomics 2006;88:779–790. [DOI] [PubMed] [Google Scholar]
- 41.Woo D, Falcone GJ, Devan WJ, et al. Meta-analysis of genome-wide association studies identifies 1q22 as a susceptibility locus for intracerebral hemorrhage. Am J Hum Genet 2014;94:511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kommareddi PK, Nair TS, Thang LV, et al. Isoforms, expression, glycosylation, and tissue distribution of CTL2/SLC44A2. Protein J 2010;29:417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Germain M, Chasman DI, de Haan H, et al. Meta-analysis of 65,734 individuals identifies TSPAN15 and SLC44A2 as two susceptibility loci for venous thromboembolism. Am J Hum Genet 2015;96:532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hinds DA, Buil A, Ziemek D, et al. Genome-wide association analysis of self-reported events in 6135 individuals and 252 827 controls identifies 8 loci associated with thrombosis. Hum Mol Genet 2016;25:1867–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heestermans M, Cunha ML, Reitsma PH, Zeerleder SS, Middeldorp S, van Vlijmen BJ. Circulating nucleosomes and elastase alpha1-antitrypsin complexes and the novel thrombosis susceptibility locus SLC44A2. Thromb Res 2016;142:8–10. [DOI] [PubMed] [Google Scholar]
- 46.Nagashima F, Nishiyama R, Iwao B, et al. Molecular and functional characterization of choline transporter-like proteins in esophageal cancer cells and potential therapeutic targets. Biomol Ther (Seoul) 2018;26:399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maltby VE, Lea RA, Graves MC, et al. Genome-wide DNA methylation changes in CD19(+) B cells from relapsing-remitting multiple sclerosis patients. Sci Rep 2018;8:17418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qayyum R, Snively BM, Ziv E, et al. A meta-analysis and genome-wide association study of platelet count and mean platelet volume in african americans. PLoS Genet 2012;8:e1002491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee JY, Lee BS, Shin DJ, et al. A genome-wide association study of a coronary artery disease risk variant. J Hum Genet 2013;58:120–126. [DOI] [PubMed] [Google Scholar]
- 50.Bian L, Hanson RL, Muller YL, et al. Variants in ACAD10 are associated with type 2 diabetes, insulin resistance and lipid oxidation in Pima Indians. Diabetologia 2010;53:1349–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available to researchers on request for purposes of reproducing the results or replicating the procedure by directly contacting the corresponding author.