

Abstract
Aims
During atherosclerosis, smooth muscle cells (SMCs) accumulate in the intima where they switch from a contractile to a synthetic phenotype. From porcine coronary artery, we isolated spindle-shaped (S) SMCs exhibiting features of the contractile phenotype and rhomboid (R) SMCs typical of the synthetic phenotype. S100A4 was identified as a marker of R-SMCs in vitro and intimal SMCs, in pig and man. S100A4 exhibits intra- and extracellular functions. In this study, we investigated the role of extracellular S100A4 in SMC phenotypic transition.
Methods and results
S-SMCs were treated with oligomeric recombinant S100A4 (oS100A4), which induced nuclear factor (NF)-κB activation. Treatment of S-SMCs with oS100A4 in combination with platelet-derived growth factor (PDGF)-BB induced a complete SMC transition towards a pro-inflammatory R-phenotype associated with NF-κB activation, through toll-like receptor-4. RNA sequencing of cells treated with oS100A4/PDGF-BB revealed a strong up-regulation of pro-inflammatory genes and enrichment of transcription factor binding sites essential for SMC phenotypic transition. In a mouse model of established atherosclerosis, neutralization of extracellular S100A4 decreased area of atherosclerotic lesions, necrotic core, and CD68 expression and increased α-smooth muscle actin and smooth muscle myosin heavy chain expression.
Conclusion
We suggest that the neutralization of extracellular S100A4 promotes the stabilization of atherosclerotic plaques. Extracellular S100A4 could be a new target to influence the evolution of atherosclerotic plaques.
Keywords: Extracellular S100A4, ApoE−/−, TLR4, RAGE, NF-κB, α-Smooth muscle actin, Smooth muscle myosin heavy chains, CD68
Graphical Abstract

1. Introduction
In atherosclerosis, smooth muscle cells (SMCs) accumulate in the intima where they undergo a transition from a contractile to a synthetic phenotype. This phenomenon includes a process of cell dedifferentiation characterized by altered expression of contractile proteins, as well as increased production of extracellular matrix components. During the last two decades, synthetic SMCs have been considered as beneficial players in atherosclerotic plaque development essentially by contributing to fibrous cap formation that protects plaques from rupture. However, SMCs exhibit remarkable plasticity. Dependent on environmental cues/signals, SMCs can acquire inflammatory cell properties1–4 and give rise to macrophage-like cells in the atherosclerotic plaque microenvironment. In human coronary artery atherosclerotic plaques, at least 50% of foam cells derive from SMCs.5 Moreover, about 40% of macrophages, identified by CD68 specific marker, express α-smooth muscle actin (α-SMA) and not CD45, a leukocyte specific lineage marker; hence these cells are suggested to derive from SMCs.5,6 In human coronary arteries, ∼30% of macrophage marker-positive cells derive from SMCs,7 as shown by using the specific epigenetic SMC marker, H3K4me2. By generating SMC lineage tracing apolipoprotein E-knockout (ApoE−/−) mouse, Owens and collaborators7 have shown that, in advanced atherosclerotic lesions, 80% of SMCs were undetectable with the classical SMC markers and, among these 80%, 30% express macrophage-like cell markers. Moreover, Francis and collaborators have recently demonstrated in ApoE−/− mice that a large part of foam cells were derived from SMCs.6
We isolated morphologically distinct SMC populations from porcine coronary artery, spindle-shaped (S) and rhomboid (R), typical of the contractile and synthetic phenotype, respectively.8 We identified S100A4 as a marker of the SMC synthetic phenotype in vitro (i.e. R-phenotype) and of intimal SMCs in vivo in pigs and humans,9–11 later confirmed by other groups.12–16 Furthermore, S100A4, also known as fibroblast-specific protein-1, is expressed by cancer cells, fibroblasts, and various inflammatory cells, including macrophages.17,18 For instance, in infarcted and pressure-overloaded myocardium induced in the GFP-expressing transgenic mice under the S100A4 promoter, S100A4 is expressed by various cell types, including fibroblasts, vascular cells, and inflammatory leucocytes.19 S100A4 belongs to a large family of low-molecular weight calcium-binding S100 proteins characterized by a pair of EF-hands and exhibits both intra- and extracellular functions. Seminal studies demonstrated that intracellular S100A4 is correlated with poor cancer prognosis in humans and is a mediator of cancer metastasis through cytoskeleton rearrangement, p53 regulation, Wnt/β-catenin, and AKT pathways.17,18 Extracellular S100A4 is present in tumour interstitial fluid of human breast cancer biopsies.20,21 as well as in fluid and plasma from patients with rheumatoid arthritis,22 idiopathic inflammatory myopathies,23 and systemic lupus erythematosus.24 Extracellular S100A4 triggers not only the metastatic potential of cancer cells but also pro-inflammatory processes. It has recently been shown that it is a potent activator of inflammatory molecules, such as serum amyloid A (SAA), granulocyte-colony stimulating growth factor (G-CSF), interleukine (IL)-1, IL-6, tumour necrosis factor (TNF)-α, and matrix metalloproteinases (MMPs).18 In vitro, the functional extracellular S100A4 exhibits an oligomeric conformation.25 A number of receptors associated with pro-inflammatory pathways as the receptor for advanced glycation end products (RAGE), a member of the immunoglobulin superfamily, toll-like receptor-4 (TLR4), and epidermal growth factor receptor (EGFR) have been described as receptors for S100A4.26–29 We have recently shown that extracellular S100A4-rich conditioned medium (CM, collected from SMCs transfected with S100A4-containing plasmid) induced an S- to R-phenotype transition associated with RAGE up-regulation, NF-κB activation, and changes in the expression of MMPs and their inhibitors.30
In this study, we aimed at deciphering the role of extracellular S100A4 in the phenotypic transition of SMCs and explored the effects of S100A4 neutralization in vivo on established atherosclerotic plaques.31 We show that oligomeric S100A4 (oS100A4) in synergy with platelet-derived growth factor (PDGF)-BB (a well-known factor responsible for SMC dedifferentiation)1–4 induce a complete SMC transition towards an R-phenotype. This is associated with NF-κB activation and substantial pro-inflammatory response through TLR4. In vivo, neutralization of extracellular S100A4 decreases plaque area and promotes plaque stabilization, characterized by increased expression of α-SMA and smooth muscle myosin heavy chains (SMMHCs) as well as decreased expression of CD68 in the intima.
2. Methods
2.1 Animals
All animal studies were performed after approval by the Swiss Federal Veterinary Office and were in accordance with the established Swiss guidelines and regulations. These animal studies conform to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. ApoE−/− mice on a C57BL/6J background, known to develop atherosclerotic lesions under high cholesterol diet (HCD), were purchased at Charles River Laboratories (France) and were further bred in our animal facility. All mice were kept in conventional housing. Fourteen-week-old ApoE−/− mice were fed with HCD (1.25% cholesterol, 0% cholate; Research Diets Inc.) for 9 weeks. During the last 3 weeks of HCD, mice were injected intraperitoneally three times per week with 7.5 mg/kg of a neutralizing mouse monoclonal IgG1 antibody specific for S100A4 (clone 6B12, n = 11)32 or with the corresponding mouse monoclonal IgG1 isotype control (clone MOPC-21, BioXcell, n = 12) at the same concentration. Usual anaesthetic agent ketarom (10 mg/kg xylazine mixed with 100 mg/kg ketamine, 0.1 mL/20 g) was injected intraperitoneally at 2.5 times higher concentration (0.25 mL/20 g) to euthanize mice. Blood was collected by cardiac puncture and serum samples were prepared for lipid level and SAA analysis. Mice were bled by right atrium transection followed by perfusion with 0.9% NaCl solution through the left ventricle. Aorta and heart were collected. Thoracic-abdominal aortas were fixed in 4% paraformaldehyde (PFA, Fluka) overnight at 4°C and then rinsed with phosphate-buffered saline (PBS). Aortic roots were embedded in OCT compound (Tissue-Tek), snap-frozen, and prepared as 4 µm thick sections for further analysis.
2.2 Cell culture and treatment
Coronary arteries of 8-month-old pigs were obtained from a nearby slaughterhouse. S-SMCs were isolated from the porcine coronary artery media by using enzymatic digestion.8 SMCs between the 6th and 11th passages were plated at a density of 60 cells/mm2 in 35 mm culture dishes containing Dulbecco’s-modified eagle medium (DMEM Glutamax, Gibco-Invitrogen) supplemented with 10% foetal calf serum (FCS, Amimed, Bioconcept). Each experiment was performed with biological triplicates (individual cell populations were isolated from different animals).
For the preparation of control CM and S100A4 rich-CM,30 transfection with pcDNA3-empty vector or pcDNA3-S100A4 vector (1 μg/mL), respectively, was performed on adherent SMCs by using Lipofectamine 2000 (2 μL/mL, Gibco-Invitrogen) in OptiMEM (Gibco-Invitrogen). After 6 h, the medium was replaced with DMEM containing 10% FCS for 48 h. The medium was not changed during the time of the experiments. Medium of S-SMC transfected with pcDNA3-empty or pcDNA3-S100A4 vector for 48 h was used as source of CM. S-SMCs used as target cells were plated at a density of 60 cells/mm2 in 60 mm culture dishes for 24 h. The culture medium was replaced by the fresh CM collected from S-SMCs transfected with pcDNA3-empty or pcDNA3-S100A4 vector for 96 h. CM-treated S-SMCs were fixed for immunostaining or processed for protein and RNA extraction.
Human recombinant N-terminal His-tagged S100A4 was produced in a bacterial expression system and purified by affinity chromatography. Different fractions of S100A4 conformations were isolated by size exclusion chromatography. The fraction corresponding to molecular masse of 22 kDa is considered as dS100A4, while pooled fractions from 44 to 200 kDa as oligomeric S100A4 (oS100A4).25 Twenty-four hours after plating, S-SMCs were treated with (i) 30 ng/mL of human recombinant PDGF-BB (Sigma), (ii) 1 µg/mL of human dS100A4 or oS100A4,25 (iii) PDGF-BB plus dS100A4 (PDGF-BB/dS100A4), or PDGF-BB plus oS100A4 (PDGF-BB/oS100A4) for 1 h for NF-κB activation experiments, 4 h for RNA sequencing, and 96 h to follow phenotypic changes of SMCs. To neutralize the activity of oS100A4, S-SMCs were treated with a mix of oS100A4 (1 µg/mL) and neutralizing S100A4 antibody (clone 6B12, 5 µg/mL). All materials (recombinant PDGF-BB, S100A4 and neutralizing S100A4 antibody) were tested for the presence of endotoxins by using the Pierce LAL Chromogenic Endotoxin Quantitation Kit (ThermoFischer Scientific) according to the manufacturer’s protocol. We confirmed the absence of endotoxins in all samples.33
2.3 Protein content per cell
After counting the cells by using a haemocytometer, the protein content was determined by using BCA protein assay (Pierce) according to the manufacturer’s protocol. The protein content per cell was calculated by dividing the total protein content by the total number of cells
2.4 Blood analysis
Total cholesterol and triglyceride levels were measured in mouse serum samples after HCD using a Cobas C111 (Mouse Metabolic Facility of Center for Integrative Genomic, University of Lausanne, Switzerland). SAA levels in serum samples were assessed by using the Mouse SAA ELISA kit (Thermo Fisher Scientific) according to manufacturer’s instructions.
2.5 Sudan-IV staining
Thoracic-abdominal aortas were incubated overnight in Sudan-IV solution for lipid deposition analysis.34 The vessels were opened longitudinally and images were taken with a Nikon SMZ1000 stereomicroscope equipped with a Nikon 1 J5 camera. The extent of atherosclerotic lesions was determined by using the image processing program ImageJ v1.49 software (NIH, http://rsb.info.nih.gov/ij/). The atherosclerotic positive area was defined by dividing the Sudan-IV-positive area with the total area of the thoracic-abdominal aorta.
2.6 Histology
Haematoxylin and eosin, as well as Sirius red staining for collagen deposition were performed on aortic root cryosections.35 Sirius red-stained slides were scanned by a fully automated Axio Scan.Z1 equipped with a Plan-Apochromat 20×/0.8 objective M27 (Carl Zeiss) and analysed by the ImageJ v1.49 software. The cross-sectional area of the intima was manually drawn, whereas the Sirius red staining was automatically detected. Results were calculated as area of immunostaining/total intima area.
2.7 Immunofluorescence staining
Double-immunofluorescence staining was performed on adherent SMCs and mouse cryosections of aortic roots. Primary antibodies, including homemade anti-α-SMA36 and anti-S100A4,9 are listed in Supplementary material online, Table S1. Cells and cryosections were fixed for 15 min in 1% PFA, then rinsed in PBS, and further incubated for 5 min in ice-cold methanol for most of the antibodies except for anti-CD68 antibody, for which cryosections were fixed in ice-cold acetone for 5 min. Staining with anti-cleaved caspase-3 required the fixation in 1% PFA for 15 min, followed by permeabilization with 0,1% Triton X-100 for 2 min. After washing in PBS, cells and cryosections were stained with the primary antibody followed by FITC-conjugated goat anti-mouse IgG2a, rhodamine-conjugated goat anti-mouse IgG1, rhodamine-conjugated goat anti-mouse IgM, FITC- or rhodamine-conjugated goat anti-rabbit IgG (Southern Lab), and fluorescein rabbit anti-rat IgG antibody (Vector Laboratories). Nuclei were stained with DAPI (Sigma). Slides were mounted in buffered polyvinyl alcohol.
Images of cells were taken on Axioskop 2 microscope (Carl Zeiss) equipped with an oil plan-neofluar ×40/1.4 objective and a high sensitivity, high-resolution digital colour camera (Axiocam, Carl Zeiss) by using the ZEN program (Carl Zeiss). Immunostainings of aortic root cryosections were scanned with a fully automated Axio Scan.Z1 equipped with a Plan-Apochromat 10×/0.8 objective M27 (Carl Zeiss) and processed by using Adobe Photoshop. Quantification was performed by using ImageJ v1.49 software. Cross-sectional areas of the intima were manually drawn, whereas the positive cells within the cross-sectional area were automatically discriminated from the unstained portions of the specimens according to red, green, and blue components. Results were calculated as the area of immunostaining/total intima area. In addition, the necrotic core was identified as the region lacking DAPI-positive nuclei and was manually drawn. Results were calculated as area of necrotic core/total intima area.
2.8 Small-interfering RNA
Specific siRNA targeting the coding sequence of porcine TLR4 (TLR4 siRNA) from nucleotide position 1947 to 196737 and RAGE (RAGE siRNA) from nucleotide position 469 to 48730 were selected; Silencer Negative Control No. 1 scramble siRNA (Thermo Fisher Scientific) was used as a negative control. Transfection of siRNA (120 nM for TLR4 and 160 nM for RAGE) was performed on adherent SMCs by using Lipofectamine 2000 (2 μL/mL, Gibco-Invitrogen) in OptiMEM (Gibco-Invitrogen). After 6 h, the medium was replaced with DMEM containing 10% FCS. Forty-eight hours post-transfection, treatments with PDGF-BB, oS100A4, or PDGF-B/oS100A4 were performed for 1 h (to assess NF-κB activation). Cells were fixed and processed for immunofluorescence staining or harvested for Western blotting and real-time PCR.
2.9 Protein extraction, electrophoresis, and Western blotting
SMCs were trypsinized and proteins were extracted as previously described.9 Proteins were separated by SDS–PAGE on 12% mini gels (Bio-Rad) and stained with Coomassie brilliant blue (R250, Fluka). For Western blotting (see Supplementary material online, Table S1, for antibodies), 1 μg of proteins for α-SMA and 12 μg of proteins for total and phospho-NF-κB were electrophoresed and transferred to a nitrocellulose membrane (Protran® 0.2 μm; Schleicher and Schuell). About 12 μg of proteins for S100A4 were electrophoresed and transferred on PVDF membrane (0.45 μm, Immobilon™-P, Millipore Corporation). α-Tubulin was used as a housekeeping protein. Horseradish peroxidase-conjugated goat anti-mouse IgG or IgM and goat anti-rabbit IgG were used as secondary antibodies. Enhanced chemiluminescence was used for detection (Amersham). Signals were digitized with Epson perfection 4990 photo scanner and analysed by using ImageJ v1.49 software. Results were normalized to tubulin expression.
2.10 RNA extraction, reverse-transcription, and real-time quantitative PCR
Total RNA was extracted from treated SMCs by using the NucleoSpin kit (Macherey-Nagel) and processed for reverse transcription and real-time SYBR Green fluorescent PCR. The cDNA was synthesized from total RNA with random hexamers and TAKARA Reverse Transcription (Gibco-Invitrogen). Reverse transcription was performed at 37°C for 15 min and then at 85°C for 5 s. The forward and reverse primers (Sigma) used in experiments are listed in Supplementary material online, Table S2. Real-time SYBR Green fluorescent PCR was performed in an iCycler iQ® Real-TimePCR Detection System (Bio-Rad) in a final volume of 10 μL comprising 5 μL of cDNA. Each couple of primers was used at a final concentration of 0.86 μM. Denaturation was performed for 10 min at 95°C and then DNA was amplified for 40 cycles of 15 s at 95°C, 45 s at 60°C, and 5 min at 72°C followed by 70 cycles of 10 s from 60°C to 95°C (+0.5°C/cycle) for the dissociation curve. Amplifications were repeated in triplicates. Results were normalized to amplified GAPDH transcripts in the same samples and were expressed as fold-change vs. the corresponding control.
2.11 Microarray analysis
To select the appropriate time point to perform microarray analysis, S-SMCs were treated with CM containing or not extracellular S100A4 for 0.5, 1, 2, 4, and 24 h. Then, RNA was isolated as described above to assess mRNA expression of several genes that are well known to be up-regulated (PDGF-BB and c-Myc) or down-regulated SM22-α during SMC phenotypic modulation.1–4 The time point of 4 h was selected to perform a porcine microarray analysis by using MessageAmp™ II-Biotin Enhanced kit (Ambion). Experiments were performed with cells isolated from three different animals. Quality of the samples was verified by using the Agilent 2100 Bioanalyzer with the Agilent RNA 6000 Nano Kit (Agilent Technologies). Complementary RNAs from S-SMCs were generated and labelled by using the GeneChip Porcine genome array (Affymetrix). Samples were hybridized to a porcine array (Affymetrix). The resulting expression data were normalized with the Partek Genomics Suite (Partek) and analysed by ANOVA test. Bioinformatics analysis was performed by using Kyoto Encyclopedia of Genes and Genomes (KEGG) database.
2.12 Luciferase assay
S-SMCs were transfected with pNFkB Tluc16-DD Vector for Luciferase Assays (0.5 μg/mL), by using Lipofectamine 2000 (2 μL/mL, Gibco-Invitrogen) in OptiMEM (Gibco-Invitrogen). After 6 h, cells were trypsinized, resuspended in complete media, and 20 000 cells/well were replated in a 96-well plate. After 36 h, cells were treated with (i) 30 ng/mL of PDGF-BB, (ii) 1 µg/mL of oS100A4, or (iii) PDGF-BB/oS100A4 for 1 h to activate NF-κB. TurboLuc One-Step Glow Assay (Thermo Fischer Scientific) was performed according to the manufacturer’s protocol. The luciferase signal was detected by using the POLARstar Omega plate reader (BMG Labtech).
2.13 RNA sequencing
Cells were treated with PDGF-BB, oS100A4, and PDGF-BB/oS100A4 for 4 h, the same time point selected for microarray data, and total RNA was extracted by using the NucleoSpin kit (Macherey-Nagel). Experiments were performed with cells isolated from three different animals. Quality of the samples was verified by using the Agilent 2100 Bioanalyzer with the Agilent RNA 6000 Nano Kit (Agilent Technologies). cDNA libraries were constructed by the Genomic platform of the University of Geneva by using the Illumina TruSeq RNA sample Preparation Kit according to the manufacturer’s protocol. Libraries were sequenced by using single-end (100 nt-long) on Illumina HiSeq2000. Mapping was performed by using STAR version 2.4.0j. Only reads that were mapped once to the genome were considered for the read allocation to genomic features. Library size normalizations and differential gene expression calculations were performed by using the package edgeR.38 The fold-change and the Benjamini–Hochberg corrected P-value thresholds were set to 2 and 0.05, respectively. Gene set enrichment analysis was used to predict up- or down-regulated biological pathways.39
2.14 Transcription factor binding site enrichment
Transcription factor binding site (TFBS) enrichment Pscan was used to identify potential TFBSs (JASPAR database) that are overrepresented between nucleotides −1000 and +500 relative to the transcription start site (http://159.149.160.51/pscan/). TFBSs with P-values <0.05 were considered to be significantly overrepresented.40,41
2.15 Statistical analysis
Statistics were performed in Graph Pad Prism 8, by using the two-tailed unpaired Student’s t-tests with a confidence level of 95%. Multiple group comparisons were performed by using one-way ANOVA, followed by Tukey’s multiple comparison test. Results are shown as mean ± SEM. Differences were considered statistically significant at values of P ≤ 0.05.
3. Results
3.1 S-SMCs treated with S100A4-rich CM acquire pro-inflammatory properties
Using a proteomic approach, we previously identified S100A4 as a marker of R-SMCs (the synthetic phenotype) in vitro in porcine coronary artery and of intimal SMCs in vivo in both pigs and humans.9,10 More recently, we have shown that S-SMCs treated with S100A4-rich CM (collected from SMCs transfected with S100A4-containing vector) acquired an R-phenotype associated with NF-κB activation and changes in expression of proteolytic enzymes and inhibitors.30 To further study the role of extracellular S100A4 on SMC phenotypic transition in porcine SMCs, we performed a microarray analysis of S-SMCs treated with S100A4-rich or control CM. To select the most appropriate duration of treatment, we treated S-SMCs (n = 3) with S100A4-rich or control CM for 0.5, 1, 2, 4, and 24 h and assessed mRNA expression of several genes that are well known to be up-regulated (PDGF-BB and c-Myc) or down-regulated (SM22-α, i.e. transgelin) during SMC phenotypic transition.1–4 Real-time PCR demonstrated that the most important changes were detected in S-SMCs treated with extracellular S100A4-rich CM for 4 h. This time point was selected to carry out the microarray analysis (Supplementary material online, Figure S1). The microarray analysis showed marked differences in gene expression between the two conditions, which were reproducible and statistically accurate: 82 genes showed 2- to 6-fold reduction and 195 genes exhibited 2- to 30-fold increase in expression upon S100A4-rich CM treatment as compared with the control CM (n = 3). Among those genes, 23 showed more than two-fold increase and were involved in cell motility or inflammatory responses. The granulocyte macrophage colony-stimulating factor (GM-CSF) was the most up-regulated gene (30.38-fold increase) in this analysis. The most differentially expressed genes are shown in Supplementary material online, Table S3. Bioinformatic analysis using the KEGG database demonstrated that the majority of activated signalling pathways was related to inflammation, including the NF-κB and TLR pathways. These results show that SMCs treated with S100A4-rich CM acquire pro-inflammatory properties.
3.2 Oligomeric S100A4 in combination with PDGF-BB promotes SMC phenotypic transition
To decipher the specific role of extracellular S100A4 in the phenotypic changes observed in SMCs after treatment with S100A4-rich CM, we treated porcine S-SMCs (n = 3), which exhibit the classical ‘hill and valley’ pattern,8 with dimeric S100A4 (dS100A4) and oS100A425 alone or in combination with PDGF-BB for 4 days. By phase-contrast microscopy, PDGF-BB-treated S-SMCs appeared thinner in shape (Figure 1A). As expected, PDGF-BB significantly increased the proliferation of SMCs (Figure 1B). By immunofluorescence staining (Figure 1A) and Western blotting (Figure 1D and F), we observed an increase of intracellular S100A4 expression after PDGF-BB treatment. Treatment with oS100A4 did not affect S-SMC proliferation (Figure 1B), phenotypic transition, α-SMA, or S100A4 expression as compared with control conditions (Figure 1A and C–F). When we treated S-SMCs with oS100A4 in combination with PDGF-BB, we observed by phase-contrast microscopy a full phenotypic transition (Figure 1A) associated with an increase in cell proliferation (Figure 1B). By immunofluorescence staining, we observed disorganization of α-SMA fibres associated with a decrease of α-SMA expression and an increase of intracellular S100A4 expression (Figure 1A). Using Western-blot analysis, we confirmed a significant decrease of α-SMA expression (Figure 1C and E) and a significant increase of intracellular S100A4 expression (Figure 1D and F). Noteworthy, PDGF-BB/oS100A4 increased the expression of intracellular S100A4 to a greater extent compared with PDGF-BB alone. In addition, PDGF-BB and PDGF-BB/oS100A4 induced SMC dedifferentiation as reflected by a decrease in SMMHC (typical marker of well-differentiated SMCs)1,2 expression and an increase in connexin 43 (Cx43, marker of dedifferentiated SMCs)42 expression (Supplementary material online, Figure S2). The protein content per cell was decreased after PDGF-BB and PDGF-BB/oS100A4 treatment (Supplementary material online, Figure S3). When S-SMCs were treated with dS100A4, no change in morphology, proliferation, α-SMA, or S100A4 expression was observed (Supplementary material online, Figure S4Aand B). When dS100A4 and PDGF-BB were used in combination, we observed changes that were similar to that of PDGF-BB alone, confirming that dS100A4 did not affect SMC phenotype (Supplementary material online, Figure S4A and B). These results indicate that oS100A4 and PDGF-BB act in synergy to induce S-SMC phenotypic changes towards an R-phenotype and recapitulate the findings observed after treatment of S-SMCs with S100A4-rich CM.
Figure 1.

Oligomeric S100A4 in combination with PDGF-BB promotes SMC phenotypic transition. Phase-contrast photomicrographs and double-immunofluorescence staining for α-SMA and S100A4 (A), quantification of proliferating cells normalized to control condition (B), representative Western blots for α-SMA (C) and corresponding quantification of α-SMA (E), representative Western blots for S100A4 (D) and corresponding quantification of S100A4 (F) in S-SMCs treated with PDGF-BB, oS100A4, or PDGF-BB/oS100A4 for 4 days. Note that the full phenotypic transition is acquired only in PDGF-BB/oS100A4 condition (A). In (D), the band detected above S100A4 corresponds to the exogenous His-tagged S100A4. In (A), nuclei are stained in blue by DAPI. In (C and D), α-tubulin is used as a housekeeping protein. Bar = 75 μm for phase contrast, bar = 25 μm for immunofluorescence, n = 3 biological replicates for each experiment. Comparisons were performed by using one-way ANOVA [B, F = 6.951, F(DFn, DFd) = 0.9611 (3,8); E, F = 10.68, F(DFn, DFd) = 2 (3, 8); F, F = 29.86 F(DFn, DFd) = 0.8042 (3, 8)].
3.3 Oligomeric S100A4 activates SMCs through TLR4-dependent NF-κB pathway
Previous studies showed that S100A4 activates the NF-κB pathway both in a RAGE-dependent or independent manner.17,28–30 To define the pathways involved in PDGF-BB/oS100A4 induced SMC phenotypic transition, S-SMCs were treated with PDGF-BB, dS100A4, oS100A4, PDGF-BB/dS100A4, or PDGF-BB/oS100A4 for 1 h and NF-κB activation was investigated by immunofluorescence and Western blotting. When treated with oS100A4 or PDGF-BB/oS100A4, NF-κB was translocated into the nuclei (Figure 2A). Moreover, Western blotting showed that phosphorylated NF-κB was significantly increased after treatment with oS100A4 alone or in combination with PDGF-BB (Figure 2B and E), while total NF-κB expression did not change (Figure 2B and H). By using luciferase promoter assay, we showed that oS100A4 and PDGF-BB/oS100A4 activated the NFkB promoter (Figure 2K). Silencing of RAGE did not prevent oS100A4- or PDGF-BB/oS100A4-induced NF-κB translocation (Figure 2A), or phosphorylation of NF-κB (Figure 2C and F), while total NF-κB levels were unchanged (Figure 2C and I). After treatment with PDGF-BB, dS100A4, or both in combination, we did not observe NF-κB translocation into the nuclei (Supplementary material online, Figure S5). To test whether S100A4 signals through the TLR4,33,43 cells were treated with PDGF-BB alone, oS100A4 alone, or both in combination after TLR4 silencing. TLR4 mRNA expression was decreased in SMCs transfected with siTLR4 compared with scrambled small-interfering RNA (siRNA, 0.14 ± 0.03 vs. 0.87 ± 0.12, arbitrary units, respectively). Results showed that NF-κB translocation, as well as increased NF-κB phosphorylation, were abolished after TLR4 silencing (Figure 2A, D, G, and J). The pre-treatment of oS100A4 with a neutralizing S100A4 antibody prevented NF-κB translocation, confirming that oS100A4 is exclusively responsible for NF-κB activation, ruling out the presence of endotoxins (Supplementary material online, Figure S6A and B).
Figure 2.

Oligomeric S100A4 activates SMCs through TLR4-dependent NF-κB pathway. Immunofluorescence staining (A) and representative Western blots for NF-κB and pNF-κB (B–D) followed by quantification (E–J) after transfection of S-SMCs with scramble siRNA, siRAGE, or siTLR4 followed by treatments with PDGF-BB, oS100A4, or PDGF-BB/oS100A4 for 1 h. Note that only by silencing of TLR4, oS100A4 and PDGF-BB/oS100A4-induced NF-κB activation is prevented (A, D, G, and J). In (B–D), α-tubulin is used as a housekeeping protein. In (A), nuclei are stained in blue by DAPI. Luciferase promoter reporter assay (K) after transfection of S-SMCs with pNFκB Tluc16-DD vector followed by treatments with PDGF-BB, oS100A4, or PDGF-BB/oS100A4 for 1 h. Note that promoter is activated after the treatment with oS100A4 and PDGF-BB/oS100A4. Bar = 25 µm, n = 3 biological replicates for each experiment. Comparisons were performed by using one-way ANOVA [E, F = 18.36, F(DFn, DFd) = 0.4488 (3,8); F, F = 16.63, F(DFn, DFd) = 1.201 (3,8); G, F = 1.021, F(DFn, DFd) = 0.6769 (3,8); H, F = 1.504, F(DFn, DFd) = 1.012 (3,8); I, F = 0.1510, F(DFn, DFd) = 0.7588 (3,8); J, F = 0.5399, F(DFn, DFd) = 0.5658 (3,8); K, F = 11.66, F(DFn, DFd) = 1.994 (3,8)].
3.4 PDGF-BB amplifies the pro-inflammatory properties induced by oligomeric S100A4
To characterize the factors/pathways that regulate the pro-inflammatory-like SMC phenotypic changes, we performed RNA sequencing in S-SMCs (n = 3) treated with PDGF-BB, oS100A4, or PDGF-BB/oS100A4 for 4 h. The top 15 up- and down-regulated genes after each treatment are summarized in Supplementary material online, Table S4. The total number and relations of differentially expressed genes are depicted in the Venn diagrams in Figure 3A and B. The most prominent differences were present among the up-regulated genes. Many of these genes were previously shown to be relevant in the field of atherosclerosis and vascular SMC plasticity. After PDGF-BB treatment, most up-regulated genes were related to growth response or to extracellular matrix component proteases (Supplementary material online, Table S4). Early growth response protein 3 (EGR3), known to be implicated in PDGF–PDGFR pathway,44 was among the top three up-regulated genes (Figure 3C). After treatment with oS100A4, most up-regulated genes were related to inflammation (Supplementary material online, Table S4). In particular, chemokine C-C motif ligand 20 (CCL20), known to be important in the recruitment of monocytes and macrophages in atherosclerosis,45 and GM-CSF (CSF2, Figure 3D) known to be involved in the differentiation of monocytes into M1-polarized macrophages as well as of dendritic cells,46 were the top up-regulated genes. Interestingly, treatment with PDGF-BB/oS100A4 amplified the level of up-regulation of certain genes that were up-regulated by oS100A4 alone and not PDGF-BB alone (e.g. GM-CSF, Figure 3D). This strongly indicates a synergistic and not cumulative effect of PDGF-BB and oS100A4. Remarkably, when analysing differentially expressed genes after PDGF-BB/oS100A4 treatment, we found 129 down-regulated and 134 up-regulated genes unique for this condition and absent in PDGF-BB or oS100A4 alone (Figure 3A and B). A high majority of up-regulated genes in this condition was related to inflammation (e.g. CCL1; Figure 3E), cancer, and cell–cell interaction processes. Altogether, our results indicate that PDGF-BB and oS100A4 work in synergy and that this treatment recapitulates the effects of S100A4-rich CM, that is phenotypic transition associated with pro-inflammatory properties.
Figure 3.

PDGF-BB amplifies the pro-inflammatory properties induced by oligomeric S100A4. EdgeR analysis of RNA sequencing data represented with Venn diagrams showing the total number and relations of down-regulated (A) or up-regulated (B) genes in S-SMCs after treatments with PDGF-BB, oS100A4, or PDGF-BB/oS100A4 for 4 h and histograms showing the fold-changes vs. the control conditions for EGR3 (C), GM-CSF (D), and CCL1 (E). Note that PDGF-BB amplifies oS100A4-induced up-regulation of GM-CSF (D) and CCL1 (E) is exclusively expressed after PDGF-BB/oS100A4 treatment. The fold-change and the Benjamini–Hochberg corrected P-value thresholds were set to 2 and 0.05, respectively. Experiment was performed with biological triplicates.
Insight into the synergistic mechanisms of PDGF-BB/oS100A4 was addressed by TFBS analysis. TFBS enrichment was examined in the promoters of the genes, which were either up- or down-regulated after PDGF-BB, oS100A4, or PDGF-BB/oS100A4 treatment. TFBS enrichment analysis clearly demonstrated that the most noticeable and significant differences in enrichment of TFBSs was induced by up-regulated genes after PDGF-BB/oS100A4 treatment (Figure 4). Among them, we identified TFs known to be key regulators in SMC phenotypic transition such as Krüppel-like factor 4 (KLF4), specificity protein 1 (SP1), EGR1, and NF-kB. In addition, immunofluorescence staining for KLF4 expression in SMCs after PDGF-BB, oS100A4, or PDGF-BB/oS100A4 treatment for 4 days demonstrated the highest nuclear expression of KLF4 after PDGF-BB/oS100A4 treatment (Supplementary material online, Figure S7). Taken together, these data show that PDGF-BB/oS100A4 treatment lead to enrichment of promoter sites that are essential for inducing phenotypic transition of SMCs and are involved in the pathogenesis of atherosclerosis.
Figure 4.
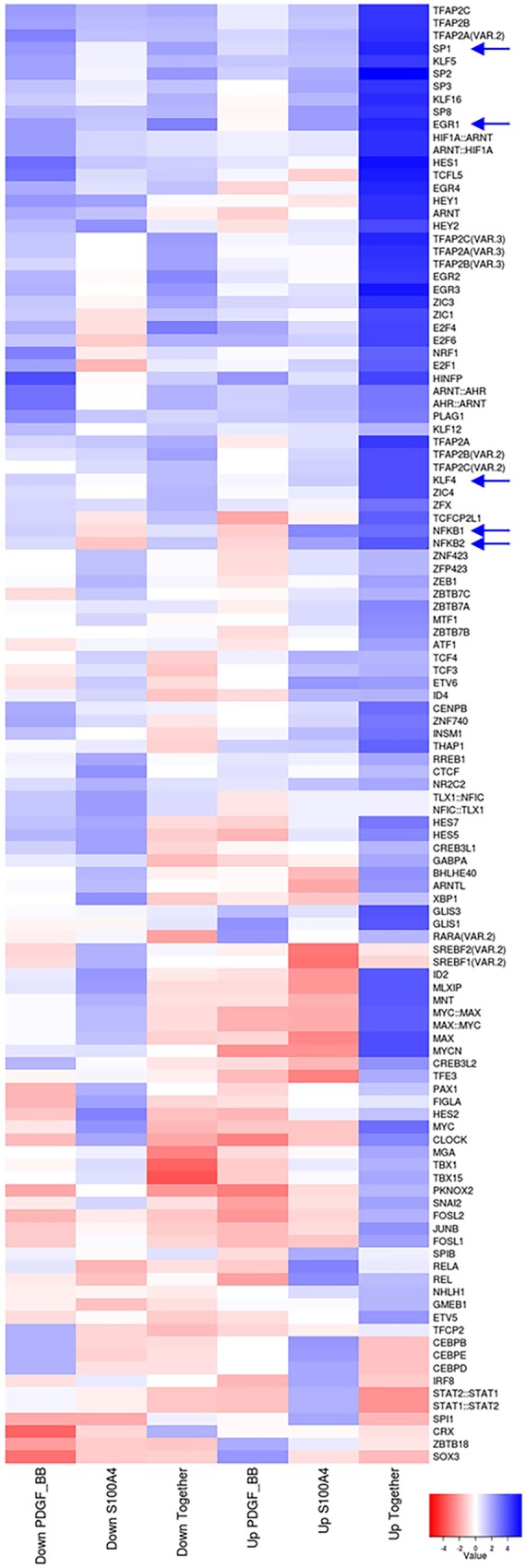
Oligomeric S100A4 in synergy with PDGF-BB induces enrichment in TFBSs essential for SMC phenotypic transition. Heat map representing TFBS enrichment in promoter regions of genes that were down- or up-regulated after treatments of SMCs with PDGF-BB, oS100A4, or PDGF-BB/oS100A4 for 4 h. TFBSs were defined according to JASPAR. The heat map shows the relative enrichment (z-score) of TFBSs that are significantly overrepresented (P < 0.05). Blue colour represents up-regulated and red colour down-regulated TFBSs. Arrows indicate important TFBSs in SMC phenotypic transition. Note that KLF4, SP1, EGR1, and NF-kB are significantly enriched only after PDGF-BB/oS100A4 treatment. Experiment was performed with biological triplicates.
3.5 Neutralization of S100A4 decreases systemic inflammation and atherosclerotic lesion area
It has been previously shown that the S100A4 antibody 6B12 specifically neutralizes extracellular S100A4 and suppresses tumour progression in a mouse model of spontaneous breast cancer32 and inflammatory responses in an allergy model.47 Mice were fed with HCD for 9 weeks (from 14 to 23 weeks of age) to induce atherosclerosis. The S100A4-neutralizing antibody (n = 11 mice) or IgG1 isotype control (n = 12 mice) were injected during the last 3 weeks of HCD (Figure 5A). Of note, the treatment was applied after 6 weeks of the HCD when atherosclerotic lesions were already formed in ApoE−/− mice.48 Neutralization of S100A4 strongly reduced systemic inflammation by decreasing the serum concentration of SAA compared with control conditions (Figure 5B). By using a rhodamine-conjugated anti-IgG1, high positive staining was observed in aortic roots when mice were treated with S100A4-neutralizing antibody compared to control mice, strongly suggesting that S100A4 antibody localizes in the lesions (Supplementary material online, Figure S8). Notably, neutralization of S100A4 did not affect body weight or cholesterol and triglyceride levels in treated and control mice (Figure 5C–E). To define the burden of atherosclerosis, we performed Sudan IV staining on thoracic-abdominal aortas. We observed that neutralization of S100A4 significantly decreased Sudan IV positive area (Figure 5F and G). These data demonstrate that neutralization of S100A4 results in decreased systemic inflammation associated with decreased atherosclerotic lesion area without affecting the lipid metabolism.
Figure 5.

Neutralization of S100A4 decreases systemic inflammation and atherosclerotic lesion area without affecting lipid metabolism. Scheme representing the experimental setup, in which mice were fed with HCD for 9 weeks to induce the development of atherosclerosis; the IgG1 control or anti-S100A4 were injected during the last 3 weeks of HCD (A). Quantification of SAA levels (B; n = 11 for IgG1 control and n = 10 for anti-S100A4), body weight (C; n = 12 for IgG1 control and n = 11 for anti-S100A4), cholesterol (D; n = 11 for IgG1 control and n = 10 for anti-S100A4), and triglyceride (E; n = 12 for IgG1 control and n = 10 for anti-S100A4) levels in mouse serum samples. En face Sudan IV staining (F) and quantification of Sudan IV positive area of thoracic-abdominal aorta (G) in ApoE−/− mice treated with IgG1 control (n = 10) and anti-S100A4 (n = 11). Bar = 0.2 cm. Comparisons were performed by using two-tailed unpaired Student’s t-test (B, t = 2.476, df = 19, C: t = 0.2139, df = 21; D, t = 1.267, df = 19; E, t = 0.1440, df = 20; G, t = 2.636, df = 19).
3.6 Neutralization of S100A4 induces beneficial changes in atherosclerotic lesion composition
We first confirmed that intracellular S100A4 was expressed in atherosclerotic plaques, whereas it was hardly detectable in the underlying media (Figure 6E and J). To characterize the atherosclerotic plaque composition, we analysed aortic root sections by haematoxylin and eosin staining (Figure 6A), Sirius red staining (Figure 6C) as well as antibodies specific for SMC differentiation markers (α-SMA and SMMHCs; Figure 6E–I). The necrotic core area was decreased after neutralization of S100A4 (Figure 6B).
Figure 6.

Neutralization of S100A4 increases the amount and differentiation of SMCs and decreases the size of the necrotic core and the burden of inflammatory cells within the lesions. Haematoxylin and eosin staining (A), quantification of necrotic core (B; n = 9 for IgG1 control and anti-S100A4), Sirius red staining (C; n = 9 for IgG1 control and n = 8 for anti-S100A4), and quantification of S100A4+ area (D; n = 9 for IgG1 control and anti-S100A4). Representative double-immunofluorescence staining for α-SMA and S100A4 (E) followed by the quantification of α-SMA+ area (F; n = 8 for IgG1 control and anti-S100A4). Representative double-immunofluorescence staining for α-SMA and SMMHCs (G) followed by the quantification of SMMHCs+ (H; n = 9 for IgG1 control and anti-S100A4) or α-SMA+SMMHCs+ area (I; n = 9 for IgG1 control and anti-S100A4). Representative double-immunofluorescence staining for CD68 and S100A4 (J) followed by the quantification of CD68+ area (K; n = 9 for IgG1 control and anti-S100A4). All analyses were performed on aortic root sections. Dashed lines highlight the media. The box indicates the region magnified in the right panels. Bar = 250 μm for low magnification, and bar = 100 μm for high magnification. Comparisons were performed by using two-tailed unpaired Student’s t-tests (B, t = 3.660, df = 16, C: t = 0.4565, df = 15; D, t = 0.1552, df = 16; F, t = 4.731, df = 14; H, t = 2.141, df = 16; I, t = 2.321, df = 16; K, t = 3.409, df = 16).
The treatment did not affect the collagen content in the lesion (Figure 6C). While the S100A4+ area did not change (Figure 6D), we observed a robust increase in α-SMA+ as well as SMMHC+ area (Figure 6E–H) within the intima when S100A4 was neutralized compared with control mice. This increase was noticeable in the fibrous cap area. α-SMA+ cells within the intima were always S100A4+ (Figure 6E). α-SMA+ cells coexpressed SMMHCs and α-SMA+SMMHC+ area increased when S100A4 was neutralized compared with control mice (Figure 6G and I). Staining for CD68 showed a significant decrease in macrophage content in intima after S100A4 neutralization (Figure 6J and K). Notably, we could not detect α-SMA+CD68+ cells in the lesions (data not shown). Our results indicate that neutralization of S100A4 has beneficial effects on atherosclerotic lesions by increasing the area and differentiation level of SMCs and by decreasing burden of inflammatory cells within the lesions.
To determine whether the increase in intimal α-SMA+ SMCs was a result of increased proliferation or reduced apoptosis, we performed staining for Ki67 and cleaved caspase-3, respectively. Unexpectedly, proliferation of α-SMA+ SMCs was strongly decreased after S100A4 neutralization compared to control mice (Figure 7A and B), whereas apoptotic cells were hardly detectable within atherosclerotic lesions in treated or control mice (data not shown).
Figure 7.

Neutralization of S100A4 decreases intimal SMC proliferation. Representative double-immunofluorescence staining for α-SMA and Ki67 within intima (A) followed by the quantification of Ki67 positive SMCs (B; n = 9 for IgG1 control and n = 8 for anti-S100A4). Bar = 50 µm. Comparisons were performed by using two-tailed unpaired Student’s t-tests (t = 2.774, df = 15).
4. Discussion
Our results show that a combination of PDGF-BB and extracellular oS100A4 triggered the full pro-inflammatory phenotypic transition of SMCs, which was characterized by an R-phenotype associated with NF-κB activation. Moreover, oS100A4-induced NF-κB activation was mediated by TLR4 and not RAGE. It has been proposed that S100 proteins act as damage-associated molecular pattern (DAMP) molecules, through diverse scavenger receptors, including TLR4.49 Therefore, in our model, S100A4 triggers similar DAMP-related pathways by executing inflammation-stimulatory functions.18 The importance of TLR4 in S100A4-mediated inflammation is well documented.33,50–52 Extracellular S100A4 induces enhanced production of IL-1β, IL-6, and TNF-α in rheumatoid arthritis patient-derived peripheral blood mononuclear cells. It acts, at least partly, through TLR4 and NF-κB activation, whereas RAGE is not involved.33 Moreover, in tumour cell lines, extracellular S100A4 stimulates inflammatory acute-phase protein SAA.50,51 again through TLR4 and NF-κB activation.50 In contrast, in the context of pulmonary hypertension, S100A4 released by human pulmonary artery SMCs induces cell proliferation and migration through NF-κB activation in a RAGE-dependent manner.53
Extracellular oS100A4 is not only necessary for the SMC phenotypic transition associated with pro-inflammatory properties but is also sufficient, on its own, to induce the pro-inflammatory profile. The latter was demonstrated by means of TLR4-dependent NF-κB activation and RNA sequencing. Unexpectedly, GM-CSF was the top up-regulated gene after treatment of S-SMCs with either S100A4-rich CM or extracellular oS100A4 but not with PDGF-BB alone. GM-CSF was increased to a greater extent after treatment with PDGF-BB/oS100A4 in combination, showing a synergistic rather than a cumulative effect of these two proteins. Interestingly, GM-CSF has only been sporadically shown to be secreted by SMCs. Thus, it is weakly expressed in cultured human arterial SMCs under basal conditions and its production can be stimulated by TNF-α, IL-1, or oxidized low-density lipoproteins.54 GM-CSF is known to be involved in the differentiation of monocytes into M1-polarized macrophages as well as of dendritic cells, which accumulate in rupture-prone areas of atherosclerotic plaques.46,55–57 Moreover, oS100A4 induced marked up-regulation of other pro-inflammatory genes like CCL20. Remarkably, treatment of SMCs with PDGF-BB/oS100A4 in combination resulted in a high and exclusive up-regulation of a variety of genes related to inflammation and immune responses, such as CCL1, an inducer of the inflammatory phenotype and chemotaxis in vascular SMCs.58 Our observations indicate that pro-inflammatory SMCs activate monocytes, thereby shedding new light on the crucial role of SMCs in plaque vulnerability.
Besides, RNA sequencing showed that genes known to be up-regulated by PDGF-BB-(e.g. growth response factors, extracellular matrix components, and MMPs) were not modified by PDGF-BB/oS100A4 treatment. This suggests that the PDGF-BB pathway is not influenced by oS100A4. In sharp contrast, the synergistic effect of PDGF-BB/oS100A4 on GM-CSF described above suggests that the oS100A4 pathway is influenced by PDGF-BB, further underlining a complex crosstalk between these two proteins.
TFBS analysis predicted that EGR1, SP1, and KLF4, known to be key players in SMC phenotypic transition,1–4 were enriched after the treatment with PDGF-BB/oS100A4. EGR1 is elevated in mouse and human atherosclerosis and is a mediator of IL-1β expression in SMCs, as well as a promoter of pulmonary artery SMC proliferation. SP1 mediates phenotypic transition by decreasing SM22-α. KLF4 is a pluripotency transcription factor promoting phenotypic modulation of SMCs associated with pro-inflammatory properties.7 By immunofluorescence staining, we confirmed the expected KLF4 activation after treatment with PDGF-BB and to a greater extent with PDGF-BB/oS100A4.
Our in vivo results proved for the first time that neutralization of extracellular S100A4 in ApoE−/− mice with established atherosclerosis promoted stabilization of atherosclerotic plaques. This was supported by several observations: (i) the atherosclerotic plaque area decreased, (ii) α-SMA+/SMMHC+ area increased, and (iii) inflammatory burden both systemically (SAA concentration) and locally within the atherosclerotic lesions (CD68+ area) decreased. The increase of α-SMA+/SMMHC+ area was particularly noticeable in the fibrous cap. Strikingly, this was correlated with decreased intimal α-SMA+ SMC proliferation. One of the mechanisms underlying this phenomenon could be related to EGFR, which is involved in SMC proliferation59,60 and known to be a receptor of S100A4.50,61–63 One can assume that neutralization of S100A4 prevents activation of EGFR signalling cascade hereby decreasing intimal α-SMA+ SMC proliferation. This result also raises the possibility that a subpopulation of intimal cells, which are allegedly SMCs having lost their typical markers,7,64 undergo a transition towards a highly differentiated phenotype related to plaque stabilization, implying a process of SMC redifferentiation. Alternatively, neutralization of extracellular S100A4 could prevent SMC dedifferentiation. This well-differentiated phenotype could be related to a myofibroblast-like phenotype,65,66 functionally dedicated to tissue repair.67 It should be noticed that all α-SMA+ cells we observed in the intimal thickening were positive for intracellular S100A4, whereas SMCs of the media were nearly devoid of S100A4, confirming S100A4 as a marker of intimal SMCs.9–11
Noteworthy, the above described beneficial effects of S100A4 neutralization resulting in plaque stabilization together with decreased systemic inflammation were not causally related to serum lipid levels. Hence, this rules out the possibility that S100A4 neutralization acts through lipid lowering, and such treatment would therefore not compete with the well-established statin drugs but could be complementary instead. Besides, we observed reduced systemic inflammation by detecting a decreased serum concentration of SAA. This is consistent with previous reports showing that SAA is a transcriptional target of S100A4 again via TLR4/NF-κB signalling.50
Our in vivo studies indicate that extracellular S100A4 is causally related to atherosclerotic plaque progression putting it forward as a prospective therapeutic target for plaque stabilization and/or regression. The unexpected observation that SMCs could undergo a process of redifferentiation has drawn our attention to future experiments deciphering the mechanisms responsible for this process. Our in vitro studies identified extracellular S100A4 as a key regulator of SMC phenotypic transition associated with pro-inflammatory properties. Further exploration of the synergistic effect of oS100A4 and PDGF-BB should be instrumental in shedding light on the mechanisms involved in SMC phenotypic transition.
Data availability
The data underlying this article are available in the article and in its Supplementary material online. RNA sequencing data have been submitted to GEO. The GEO accession number for these data is GSE143296.
Supplementary Material
Acknowledgements
We thank Anita Hiltbrunner, Bernard Foglia, and Graziano Pelli for excellent technical assistance and Luis Miguel Santos for helpful discussions.
Conflict of interest: none declared.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This study was supported by the Swiss National Science Foundation (Grant No. 310030_185370/1) and by the Foundation Centre de Recherches Médicales Carlos and Elsie de Reuter.
Translational Perspective
Our studies indicate that extracellular S100A4 is causally related to atherosclerotic plaque progression putting it forward as a prospective therapeutic target for plaque stabilization and/or regression.
References
- 1. Tabas I, García-Cardeña G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol 2015;209:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Allahverdian S, Chaabane C, Boukais K, Francis GA, Bochaton-Piallat ML. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc Res 2018;114:540–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res 2016;118:692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Basatemur GL, Jorgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol 2019;16:727–744. [DOI] [PubMed] [Google Scholar]
- 5. Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 2014;129:1551–1559. [DOI] [PubMed] [Google Scholar]
- 6. Wang Y, Dubland JA, Allahverdian S, Asonye E, Sahin B, Jaw JE, Sin DD, Seidman MA, Leeper NJ, Francis GA. Smooth muscle cells contribute the majority of foam cells in ApoE (Apolipoprotein E)-deficient mouse atherosclerosis. Arterioscler Thromb Vasc Biol 2019;39:876–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med 2015;21:628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hao H, Ropraz P, Verin V, Camenzind E, Geinoz A, Pepper MS, Gabbiani G, Bochaton-Piallat ML. Heterogeneity of smooth muscle cell populations cultured from pig coronary artery. Arterioscler Thromb Vasc Biol 2002;22:1093–1099. [DOI] [PubMed] [Google Scholar]
- 9. Brisset AC, Hao H, Camenzind E, Bacchetta M, Geinoz A, Sanchez JC, Chaponnier C, Gabbiani G, Bochaton-Piallat ML. Intimal smooth muscle cells of porcine and human coronary artery express S100A4, a marker of the rhomboid phenotype in vitro. Circ Res 2007;100:1055–1062. [DOI] [PubMed] [Google Scholar]
- 10. Coen M, Marchetti G, Palagi PM, Zerbinati C, Guastella G, Gagliano T, Bernardi F, Mascoli F, Bochaton-Piallat ML. Calmodulin expression distinguishes the smooth muscle cell population of human carotid plaque. Am J Pathol 2013;183:996–1009. [DOI] [PubMed] [Google Scholar]
- 11. Coen M, Burkhardt K, Bijlenga P, Gabbiani G, Schaller K, Kovari E, Rufenacht DA, Ruiz DS, Pizzolato G, Bochaton-Piallat ML. Smooth muscle cells of human intracranial aneurysms assume phenotypic features similar to those of the atherosclerotic plaque. Cardiovasc Pathol 2013;22:339–344. [DOI] [PubMed] [Google Scholar]
- 12. Bockmeyer CL, Kern DS, Forstmeier V, Lovric S, Modde F, Agustian PA, Steffens S, Birschmann I, Traeder J, Dammrich ME, Schwarz A, Kreipe HH, Brocker V, Becker JU. Arteriolar vascular smooth muscle cell differentiation in benign nephrosclerosis. Nephrol Dial Transplant 2012;27:3493–3501. [DOI] [PubMed] [Google Scholar]
- 13. Cao J, Geng L, Wu Q, Wang W, Chen Q, Lu L, Shen W, Chen Y. Spatiotemporal expression of matrix metalloproteinases (MMPs) is regulated by the Ca2+-signal transducer S100A4 in the pathogenesis of thoracic aortic aneurysm. PLoS One 2013;8:e70057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liang M, Wang Y, Liang A, Mitch WE, Roy-Chaudhury P, Han G, Cheng J. Migration of smooth muscle cells from the arterial anastomosis of arteriovenous fistulas requires Notch activation to form neointima. Kidney Int 2015;88:490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rocchiccioli S, Cecchettini A, Ucciferri N, Terreni M, Viglione F, Trivella MG, Citti L, Parodi O, Pelosi G. Site-specific secretome map evidences VSMC-related markers of coronary atherosclerosis grade and extent in the hypercholesterolemic swine. Dis Markers 2015;2015:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Choe N, Kwon DH, Shin S, Kim YS, Kim YK, Kim J, Ahn Y, Eom GH, Kook H. The microRNA miR-124 inhibits vascular smooth muscle cell proliferation by targeting S100 calcium-binding protein A4 (S100A4). FEBS Lett 2017;591:1041–1052. [DOI] [PubMed] [Google Scholar]
- 17. Bresnick AR, Weber DJ, Zimmer DB. S100 proteins in cancer. Nat Rev Cancer 2015;15:96–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ambartsumian N, Klingelhofer J, Grigorian M. The multifaceted S100A4 protein in cancer and inflammation. Methods Mol Biol 2019;1929:339–365. [DOI] [PubMed] [Google Scholar]
- 19. Kong P, Christia P, Saxena A, Su Y, Frangogiannis NG. Lack of specificity of fibroblast-specific protein 1 in cardiac remodeling and fibrosis. Am J Physiol Heart Circ Physiol 2013;305:H1363–H1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schmidt-Hansen B, Klingelhofer J, Grum-Schwensen B, Christensen A, Andresen S, Kruse C, Hansen T, Ambartsumian N, Lukanidin E, Grigorian M. Functional significance of metastasis-inducing S100A4(Mts1) in tumor-stroma interplay. J Biol Chem 2004;279:24498–24504. [DOI] [PubMed] [Google Scholar]
- 21. Ambartsumian N, Klingelhofer J, Grigorian M, Christensen C, Kriajevska M, Tulchinsky E, Georgiev G, Berezin V, Bock E, Rygaard J, Cao R, Cao Y, Lukanidin E. The metastasis-associated Mts1(S100A4) protein could act as an angiogenic factor. Oncogene 2001;20:4685–4695. [DOI] [PubMed] [Google Scholar]
- 22. Klingelhofer J, Senolt L, Baslund B, Nielsen GH, Skibshoj I, Pavelka K, Neidhart M, Gay S, Ambartsumian N, Hansen BS, Petersen J, Lukanidin E, Grigorian M. Up-regulation of metastasis-promoting S100A4 (Mts-1) in rheumatoid arthritis: putative involvement in the pathogenesis of rheumatoid arthritis. Arthritis Rheum 2007;56:779–789. [DOI] [PubMed] [Google Scholar]
- 23. Pleštilová L, Mann H, Andrés Cerezo L, Pecha O, Vencovský J, Šenolt L. The metastasis promoting protein S100A4 levels associate with disease activity rather than cancer development in patients with idiopathic inflammatory myopathies. Arthritis Res Ther 2014;16:468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sumova B, Cerezo LA, Szczukova L, Nekvindova L, Uher M, Hulejova H, Moravcova R, Grigorian M, Pavelka K, Vencovsky J, Senolt L, Zavada J. Circulating S100 proteins effectively discriminate SLE patients from healthy controls: a cross-sectional study. Rheumatol Int 2019;39:469–478. [DOI] [PubMed] [Google Scholar]
- 25. Novitskaya V, Grigorian M, Kriajevska M, Tarabykina S, Bronstein I, Berezin V, Bock E, Lukanidin E. Oligomeric forms of the metastasis-related Mts1 (S100A4) protein stimulate neuronal differentiation in cultures of rat hippocampal neurons. J Biol Chem 2000;275:41278–41286. [DOI] [PubMed] [Google Scholar]
- 26. Leclerc E. Measuring binding of S100 proteins to RAGE by surface plasmon resonance. Methods Mol Biol 2013;963:201–213. [DOI] [PubMed] [Google Scholar]
- 27. Leclerc E, Heizmann CW. The importance of Ca2+/Zn2+ signaling S100 proteins and RAGE in translational medicine. Front Biosci 2011;3:1232–1262. [DOI] [PubMed] [Google Scholar]
- 28. Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, Geczy CL. Functions of S100 proteins. Curr Mol Med 2013;13:24–57. [PMC free article] [PubMed] [Google Scholar]
- 29. Gross SR, Sin CG, Barraclough R, Rudland PS. Joining S100 proteins and migration: for better or for worse, in sickness and in health. Cell Mol Life Sci 2014;71:1551–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chaabane C, Heizmann CW, Bochaton-Piallat ML. Extracellular S100A4 induces smooth muscle cell phenotypic transition mediated by RAGE. Biochim Biophys Acta 2015;1853:2144–2157. [DOI] [PubMed] [Google Scholar]
- 31. Baylis RA, Gomez D, Owens GK. Shifting the focus of preclinical, murine atherosclerosis studies from prevention to late-stage intervention. Circ Res 2017;120:775–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Klingelhofer J, Grum-Schwensen B, Beck MK, Knudsen RS, Grigorian M, Lukanidin E, Ambartsumian N. Anti-S100A4 antibody suppresses metastasis formation by blocking stroma cell invasion. Neoplasia 2012;14:1260–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cerezo LA, Remakova M, Tom Ik M, Gay S, Neidhart M, Lukanidin E, Pavelka K, Grigorian M, Vencovsky J, Enolt L. The metastasis-associated protein S100A4 promotes the inflammatory response of mononuclear cells via the TLR4 signalling pathway in rheumatoid arthritis. Rheumatology (Oxford) 2014;53:1520–1526. [DOI] [PubMed] [Google Scholar]
- 34. Wong CW, Christen T, Roth I, Chadjichristos CE, Derouette JP, Foglia BF, Chanson M, Goodenough DA, Kwak BR. Connexin37 protects against atherosclerosis by regulating monocyte adhesion. Nat Med 2006;12:950–954. [DOI] [PubMed] [Google Scholar]
- 35. Kwak BR, Veillard N, Pelli G, Mulhaupt F, James RW, Chanson M, Mach F. Reduced connexin43 expression inhibits atherosclerotic lesion formation in low-density lipoprotein receptor-deficient mice. Circulation 2003;107:1033–1039. [DOI] [PubMed] [Google Scholar]
- 36. Skalli O, Ropraz P, Trzeciak A, Benzonana G, Gillessen D, Gabbiani G. A monoclonal antibody against alpha-smooth muscle actin: a new probe for smooth muscle differentiation. J Cell Biol 1986;103:2787–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bi J, Song S, Fang L, Wang D, Jing H, Gao L, Cai Y, Luo R, Chen H, Xiao S. Porcine reproductive and respiratory syndrome virus induces IL-1beta production depending on TLR4/MyD88 pathway and NLRP3 inflammasome in primary porcine alveolar macrophages. Mediators Inflamm 2014;2014:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zambelli F, Pesole G, Pavesi G. Pscan: finding over-represented transcription factor binding site motifs in sequences from co-regulated or co-expressed genes. Nucleic Acids Res 2009;37:W247–W252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sandelin A, Alkema W, Engstrom P, Wasserman WW, Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res 2004;32:D91–D94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chadjichristos CE, Morel S, Derouette JP, Sutter E, Roth I, Brisset AC, Bochaton-Piallat ML, Kwak BR. Targeting connexin 43 prevents platelet-derived growth factor-BB-induced phenotypic change in porcine coronary artery smooth muscle cells. Circ Res 2008;102:653–660. [DOI] [PubMed] [Google Scholar]
- 43. Bjork P, Kallberg E, Wellmar U, Riva M, Olsson A, He Z, Torngren M, Liberg D, Ivars F, Leanderson T. Common interactions between S100A4 and S100A9 defined by a novel chemical probe. PLoS One 2013;8:e63012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu E, Palmer N, Tian Z, Moseman AP, Galdzicki M, Wang X, Berger B, Zhang H, Kohane IS. Comprehensive dissection of PDGF-PDGFR signaling pathways in PDGFR genetically defined cells. PLoS One 2008;3:e3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zernecke A, Weber C. Chemokines in atherosclerosis: proceedings resumed. Arterioscler Thromb Vasc Biol 2014;34:742–750. [DOI] [PubMed] [Google Scholar]
- 46. Wolf D, Zirlik A, Ley K. Beyond vascular inflammation-recent advances in understanding atherosclerosis. Cell Mol Life Sci 2015;72:3853–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bruhn S, Fang Y, Barrenas F, Gustafsson M, Zhang H, Konstantinell A, Kronke A, Sonnichsen B, Bresnick A, Dulyaninova N, Wang H, Zhao Y, Klingelhofer J, Ambartsumian N, Beck MK, Nestor C, Bona E, Xiang Z, Benson M. A generally applicable translational strategy identifies S100A4 as a candidate gene in allergy. Sci Transl Med 2014;6:218ra214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Veillard NR, Steffens S, Burger F, Pelli G, Mach F. Differential expression patterns of proinflammatory and antiinflammatory mediators during atherogenesis in mice. Arterioscler Thromb Vasc Biol 2004;24:2339–2344. [DOI] [PubMed] [Google Scholar]
- 49. Xia C, Braunstein Z, Toomey AC, Zhong J, Rao X. S100 proteins as an important regulator of macrophage inflammation. Front Immunol 2017;8:1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hansen MT, Forst B, Cremers N, Quagliata L, Ambartsumian N, Grum-Schwensen B, Klingelhofer J, Abdul-Al A, Herrmann P, Osterland M, Stein U, Nielsen GH, Scherer PE, Lukanidin E, Sleeman JP, Grigorian M. A link between inflammation and metastasis: serum amyloid A1 and A3 induce metastasis, and are targets of metastasis-inducing S100A4. Oncogene 2015;34:424–435. [DOI] [PubMed] [Google Scholar]
- 51. Bettum IJ, Vasiliauskaite K, Nygaard V, Clancy T, Pettersen SJ, Tenstad E, Mælandsmo GM, Prasmickaite L. Metastasis-associated protein S100A4 induces a network of inflammatory cytokines that activate stromal cells to acquire pro-tumorigenic properties. Cancer Lett 2014;344:28–39. [DOI] [PubMed] [Google Scholar]
- 52. Li Q, Dai C, Xue R, Wang P, Chen L, Han Y, Erben U, Qin Z. S100A4 protects myeloid-derived suppressor cells from intrinsic apoptosis via TLR4-ERK1/2 signaling. Front Immunol 2018;9:388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Spiekerkoetter E, Guignabert C, de Jesus Perez V, Alastalo TP, Powers JM, Wang L, Lawrie A, Ambartsumian N, Schmidt AM, Berryman M, Ashley RH, Rabinovitch M. S100A4 and bone morphogenetic protein-2 codependently induce vascular smooth muscle cell migration via phospho-extracellular signal-regulated kinase and chloride intracellular channel 4. Circ Res 2009;105:639–647, 613p following 647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Filonzi EL, Zoellner H, Stanton H, Hamilton JA. Cytokine regulation of granulocyte-macrophage colony stimulating factor and macrophage colony-stimulating factor production in human arterial smooth muscle cells. Atherosclerosis 1993;99:241–252. [DOI] [PubMed] [Google Scholar]
- 55. Di Gregoli K, Johnson JL. Role of colony-stimulating factors in atherosclerosis. Curr Opin Lipidol 2012;23:412–421. [DOI] [PubMed] [Google Scholar]
- 56. Ait-Oufella H, Sage AP, Mallat Z, Tedgui A. Adaptive (T and B cells) immunity and control by dendritic cells in atherosclerosis. Circ Res 2014;114:1640–1660. [DOI] [PubMed] [Google Scholar]
- 57. Legein B, Temmerman L, Biessen EA, Lutgens E. Inflammation and immune system interactions in atherosclerosis. Cell Mol Life Sci 2013;70:3847–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Haque NS, Fallon JT, Pan JJ, Taubman MB, Harpel PC. Chemokine receptor-8 (CCR8) mediates human vascular smooth muscle cell chemotaxis and metalloproteinase-2 secretion. Blood 2004;103:1296–1304. [DOI] [PubMed] [Google Scholar]
- 59. Schreier B, Hunerberg M, Rabe S, Mildenberger S, Bethmann D, Heise C, Sibilia M, Offermanns S, Gekle M. Consequences of postnatal vascular smooth muscle EGFR deletion on acute angiotensin II action. Clin Sci (Lond) 2016;130:19–33. [DOI] [PubMed] [Google Scholar]
- 60. Yamanaka Y, Hayashi K, Komurasaki T, Morimoto S, Ogihara T, Sobue K. EGF family ligand-dependent phenotypic modulation of smooth muscle cells through EGF receptor. Biochem Biophys Res Commun 2001;281:373–377. [DOI] [PubMed] [Google Scholar]
- 61. Klingelhofer J, Moller HD, Sumer EU, Berg CH, Poulsen M, Kiryushko D, Soroka V, Ambartsumian N, Grigorian M, Lukanidin EM. Epidermal growth factor receptor ligands as new extracellular targets for the metastasis-promoting S100A4 protein. FEBS J 2009;276:5936–5948. [DOI] [PubMed] [Google Scholar]
- 62. Cho CC, Chou RH, Yu C. Amlexanox blocks the interaction between S100A4 and epidermal growth factor and inhibits cell proliferation. PLoS One 2016;11:e0161663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pankratova S, Klingelhofer J, Dmytriyeva O, Owczarek S, Renziehausen A, Syed N, Porter AE, Dexter DT, Kiryushko D. The S100A4 protein signals through the ErbB4 receptor to promote neuronal survival. Theranostics 2018;8:3977–3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chappell J, Harman JL, Narasimhan VM, Yu H, Foote K, Simons BD, Bennett MR, Jorgensen HF. Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circ Res 2016;119:1313–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hao H, Gabbiani G, Camenzind E, Bacchetta M, Virmani R, Bochaton-Piallat ML. Phenotypic modulation of intima and media smooth muscle cells in fatal cases of coronary artery lesion. Arterioscler Thromb Vasc Biol 2006;26:326–332. [DOI] [PubMed] [Google Scholar]
- 66. Rizzo S, Coen M, Sakic A, De Gaspari M, Thiene G, Gabbiani G, Basso C, Bochaton-Piallat ML. Sudden coronary death in the young: evidence of contractile phenotype of smooth muscle cells in the culprit atherosclerotic plaque. Int J Cardiol 2018;264:1–6. [DOI] [PubMed] [Google Scholar]
- 67. Bochaton-Piallat ML, Gabbiani G, Hinz B. The myofibroblast in wound healing and fibrosis: answered and unanswered questions. F1000Res 2016;5:752. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its Supplementary material online. RNA sequencing data have been submitted to GEO. The GEO accession number for these data is GSE143296.