

Abstract
Purpose of review
Mammals have two complete sets of chromosomes, one from each parent with equal autosomal gene expression. Less than one percentage of human genes are imprinted or show expression from only one parent without changing gene structure, usually by DNA methylation, but reversible in gametogenesis. Many imprinted genes affect fetal growth and development accounting for several human disorders reviewed in this report.
Recent findings
Disorders include Prader–Willi and Angelman syndromes, the first examples of imprinting errors in humans, chromosome 15q11.2-q13.3 duplication, Silver–Russell syndrome, Beckwith–Weidemann syndrome, GNAS gene-related inactivation disorders (e.g. Albright hereditary osteodystrophy), uniparental chromosome 14 disomy, chromosome 6q24-related transient neonatal diabetes mellitus, parent of origin effects in 15q11.2 BP1–BP2 deletion (Burnside–Butler) syndrome and 15q11-q13 single gene imprinted disorders.
Summary
Periconceptional and intrauterine life can be influenced by environmental factors and nutrition impacting DNA methylation. This process not only alters development of the fetus, but pregnancy complications may result from large fetal size. Epigenetic processes control imprinted gene functions and regulation with susceptibility to diseases as described. A better understanding of these processes will impact on care and treatment of affected individuals.
Keywords: 15q11.2 BP1–BP2 deletion, Angelman, Beckwith–Weidemann, Prader–Willi, Silver–Russell syndromes, chromosome 6q24-related transient neonatal diabetes mellitus, genomic imprinting and epigenetics, GNAS-related gene inactivation, uniparental chromosome 14 disomy
INTRODUCTION
Genomic imprinting and X-inactivation are classical epigenetic phenomena that involve transcriptional silencing of one parental gene allele with major consequences on both fetal growth and placental development [1■■,2–4,5■]. Genes contributed by the mother generally replicate or express at different rates than genes from the father with the ability to exert counteracting effects on growth of the embryo during development [6,7]. The expression pattern of imprinted genes results from methylation changes at cytosine DNA bases at CpG dinucleotides which are key regulatory functional gene elements and can be tissue and stage specific [1■■,2,8].
Evidence suggests that genomic imprinting may have evolved over 150 million years ago after divergence form egg-laying animals in a common live-born mammalian ancestor and errors now contribute to over 100 separate disorders with the first recognized imprinted gene (H19) reported in 1992 [9]. Tucci et al. [1■■] summarized the number of imprinted genes in humans at 165 and 197 genes in mice with about one-third overlapping between humans and mice. In addition, the number of maternally imprinted genes in mice (44%) is double of maternally imprinted genes in humans (21%) while two-thirds of human genes are paternally imprinted compared with 53% in mice [1■■]. Mouse embryos containing only a paternal genome with a diploid set of paternal chromosomes have reduced fetal growth and proliferation of extra-embryonic tissue such as the placenta while embryos containing only a diploid set of maternal chromosomes produce relatively normal fetal growth but exhibit poor extra-embryonic tissue or placental growth. The process of turning on and off genes, is ongoing throughout mammalian life and influenced by timing with tissue specificity [1■■,7,10,11]. Many additional imprinted genes have been studied and reportedly involved in human development and/or disease including cancer, obesity, overgrowth disorders and diabetes.
Imprinting disturbances due to epigenetics have been reported in classical disorders including Prader–Willi, Angelman and Beckwith–Weidemann syndromes [12–17]. The incidence of these syndromes is reportedly increased with the help of assisted reproductive technology (ART) which may impact the imprinting process and DNA methylation status suppressing gene expression by changing the regulation of expression of imprinted genes early in development impacted by pre and periconceptional environmental factors [18]. In the USA, ART-conceived infants contribute to about 5% of all infants with low birth weight and in some states more than 10% of all infants had low birth weight when conceived with ART [19].
Examples of genomic imprinting human disorders will be reviewed, besides Prader–Willi and Angleman syndromes and chromosome 15q11.2-q13.3 duplications, but also Silver–Russell and Beckwith–Wiedemann syndromes (BWSs), Albright hereditary osteodystrophy (AHO) and other related GNAS gene inactivation disorders, uniparental chromosome 14 disomy and chromosome 6q24-related transient neonatal diabetes mellitus (TNDM). Parent of origin effects have been reported in Burnside–Butler syndrome (15q11.2 BP1–BP2 deletion) involving four genes and single imprinted gene conditions, Schaaf–Yang syndrome (MAGEL2) and central precocious puberty 2 (MKRN3); both genes paternally expressed and located in the chromosome 15q11-q13 region [13,20–24].
EXAMPLES OF GENOMIC IMPRINTING DISORDERS IN HUMANS
Chromosome 15 disorders, genomic imprinting and parent of origin effects
Prader–Willi syndrome
Prader–Willi syndrome (PWS) is characterized initially by infantile hypotonia, a poor suck with feeding difficulties, short stature, small hands and feet and hypogonadism due to growth and other hormone deficiencies [40]. Mild intellectual disability is common, food seeking and hyperphagia onset occurs between 6 and 8 years resulting in marked obesity, if not controlled. Craniofacial features include a narrow bifrontal diameter, dolichocephaly, strabismus, a small upturned nose, a thin upper lip and down-turned corners of mouth with sticky saliva and enamel hypoplasia [13,25,26] (Fig. 1). Behavioral problems begin in childhood with self-injury and skin picking, along with outbursts, stubbornness and temper tantrums. Psychiatric problems can occur during childhood but generally later in adolescence or young adulthood requiring medical attention and treatment [13]. Additional behavioral problems are anxiety, mood disorders, psychosis and autism that may correlate with specific PWS genetic subtypes or molecular classes [27]. Most cases of PWS are sporadic and noted in all ethnic groups and both sexes. PWS is estimated to affect one in 10 000–30 000 livebirths [13,26] and worldwide accounts for about 400 000 individuals with an estimated 20 000 living in the USA [28].
FIGURE 1.
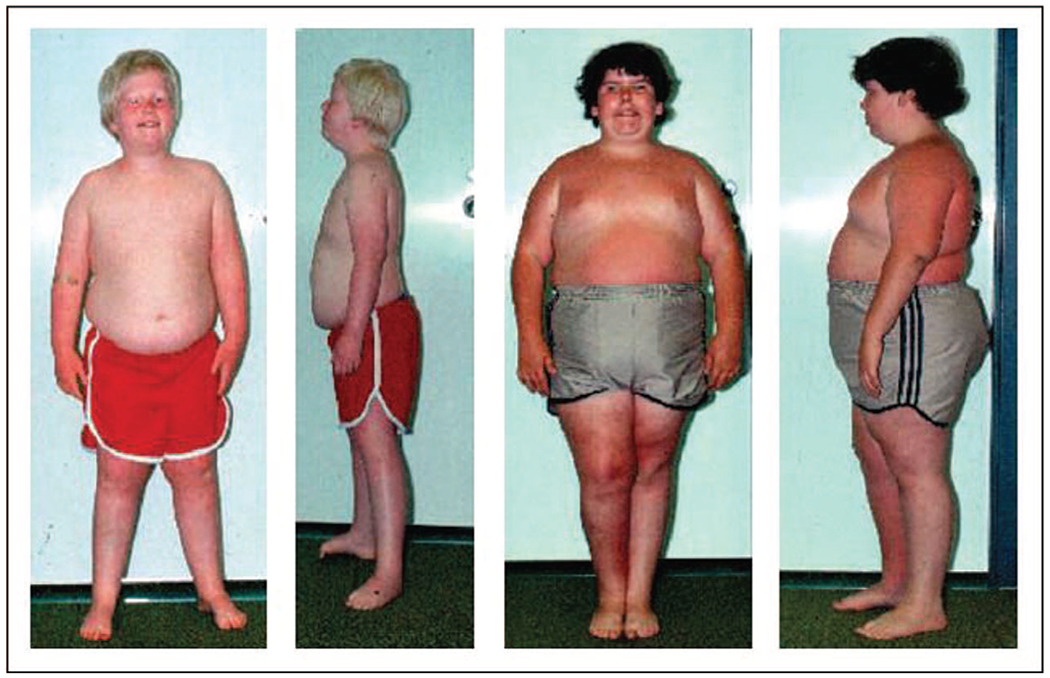
Frontal and profile views of two males with Prader–Willi syndrome patient A on the left with the chromosome 15q11–q13 deletion patient B is 11 years of age with maternal disomy 15). Note the typical facial appearance (e.g. narrow bifrontal diameter, almond-shaped eyes, triangular mouth), small hands and feet with characteristic obesity seen in both patients while hypopigmentation is seen in patient A with the 15q11–q13 deletion. Modified from [52]. Reprinted with permission from Butler M.G., Hanchett J.M., Thompson T. (2006). Clinical Findings and Natural History of Prader–Willi Syndrome. In: Butler M.G., Lee P.D.K., Whitman B.Y. (eds) Management of Prader–Willi Syndrome. Springer, New York, NY.
Although first reported in 1956 [29], Ledbetter et al. [30] reported the cause in 1981 as a 15q11-q13 deletion in the majority of PWS patients using high-resolution chromosome studies (Fig. 2). Butler and Palmer in 1983 [31] reported the 15q11-q13 deletion was de novo with the chromosome 15 having the deletion donated by the father in all cases. Later, Nicholls et al. in 1989 [12] did not find the anticipated submicroscopic deletion on chromosome 15 using polymorphic chromosome 15 DNA markers in those with normal appearing chromosomes. But, both chromosome 15s came from the mother and no 15 inherited from the father. This unusual finding was coined uniparental maternal disomy 15 and now recognized as the second most common cause of PWS. Pregnancy studies have shown the origin of maternal disomy 15 from an embryo with trisomy 15 and a trisomic 15 rescue event in early pregnancy led to the missing chromosome 15 from the father. Trisomic 15 pregnancies do correlate with advanced maternal age and an extra maternal chromosome 15 [27] that generally spontaneously abort. However, if a trisomic 15 rescue event occurs, then the pregnancy continues with the baby having PWS and maternal disomy 15 [32].
FIGURE 2.

A prometaphase or high-resolution chromosome analysis at greater than 550 band level first developed in the early 1980s and used to identify small cytogenetic deletions not detectable with routine chromosome banding methods and represented by a chromosome 15 ideogram (left). The chromosome 15 arms (p for short arm and q for long arm) are designated as bands and shown with two representative chromosome 15 pairs from two patients (right). The arrows on the ideogram indicate the deletion breakpoints at bands 15q11 and 15q13. The 15q12 band is indicated by the arrow on each member of the chromosome 15 pair. The left member of the chromosome 15 pair shows the deletion from an 8.5-year-old male with Prader–Willi syndrome. Modified from Butler, Meaney, and Palmer, American Journal of Medical Genetics 1986;23:793–809. Reprinted with permission from Butler M.G., Hanchett J.M., Thompson T. (2006). Clinical Findings and Natural History of Prader–Willi Syndrome. In: Butler M.G., Lee P.D.K., Whitman B.Y. (eds) Management of Prader–Willi Syndrome. Springer, New York, NY.
There are over one dozen genes and transcripts in the 15q11-q13 region which appear to play a role in the causation of PWS and/or Angelman syndrome (Fig. 3). Genes and transcripts between the proximal 15q11.2 breakpoints BP1 and BP2 are TUBGCP5, CYFIP1, NIPA1 and NIPA2 and those between BP2 and the distal 15q13 breakpoint BP3 are MRKN3, MAGEL2, NDN, NIPAP1, SNURF-SNRPN, noncoding RNAs (SNORDs), UBE3A, ATP10A, GABRB3, GABRA5, GABRG3, OCA2 and HERC2. Imprinted MRKN3, MAGEL2, NDN, NIPAP1 and SNURF-SNRPN genes are paternally expressed and when disturbed may cause features of PWS (e.g. [33]). Patients have also been reported with a PWS phenotype with small microdeletions of the noncoding SNORD116 transcript [34] and other similar deletions in the region. Patients with these small microdeletions affect the SNORD transcripts and/or the imprinting controlling center with findings summarized by Hassan and Butler [35] and Tan et al. [36].
FIGURE 3.

Chromosome 15 ideogram showing the location of genes and transcripts. Those causing Prader–Willi syndrome are imprinted and paternally expressed (blue) and Angelman syndrome are imprinted and maternally expressed (red). The location of the 15q11.2 BP1–BP2 deletion (Burnside–Butler) syndrome, the typical larger 15q11–q13 Type I deletion involving breakpoints BP1 and BP3, and the typical smaller 15q1-q13 Type II deletion involving breakpoints BP2 and BP3 as well as the location of other breakpoints are shown. Breakpoints BP4 and BP5 are distal to BP3 in the proximal long arm of chromosome 15. IC: Imprinting center location controlling the activity of imprinted genes in the 15q11-q13 region involving Prader–Willi syndrome and Angelman syndrome.
Butler et al. [27] characterized three PWS molecular classes in 510 subjects including the typical paternal 15q11-q13 deletion of about 5–6 MB in size in 303 (60%) of cases with 118 (38.9%) having the larger typical Type I deletion involving breakpoints BP1 and BP3 and 165 individuals (54.5%) with the smaller typical Type II deletion involving BP2 and BP3. Twenty individuals had an atypical (larger or smaller) deletion (6.6%). In those without a deletion, 185 individuals (36%) had maternal disomy 15 with 13 individuals (12.5%) with the total isodisomy 15 subclass where the entire chromosome 15 showed loss of heterozygosity (LOH) due to errors in maternal meiosis II detected by high-resolution single nucleotide polymorphism (SNP) chromosomal microarrays; 60 (57.7%) showed the segmental isodisomy subclass from crossover events in maternal meiosis I where only segments of LOHs are present on chromosome 15 and 31 showed heterodisomy 15 (29.8%) with no LOHs due to lack of crossover events in meiosis I. No SNP microarray analysis was performed on 81 individuals. PWS imprinting defects were found in 22 individuals (4%) without deletions or evidence of maternal disomy 15 and 13 (76.5%) had a nondeletion epimutation status, four individuals (23.5%) had a microdeletion of the imprinting center while the remaining five individuals did not have the type of imprinting defect analyzed. Rarely, other chromosome 15q abnormalities are found including inversions and translocations [27].
The presence of maternal disomy 15 and specific subclasses may impact diagnosis and medical care surveillance. For exmaple, a second genetic condition may be present if the mother is a carrier of a recessive gene located in the LOH region (segmental or total isodisomy) found in her PWS child and at risk of having two identical gene copies. Hundreds of potentially disease-causing genes are found on chromosome 15.
Angelman syndrome
Angelman syndrome is characterized by developmental delay, mild hypotonia and with difficult to control seizures occurring at about 6 months of age with a characteristic EEG pattern. Later, tremor, lack of speech, a wide-based gait and ataxia can occur along with a characteristic happy demeanor and fascination with water [37]. A sleep disorder is reported in more than 80% of individuals with a decreased need for sleep [38].
Four recognized Angelman syndrome molecular classes are identified and categorized by the impact on methylation of chromosome 15. The most common defect is the typical 15q11-q13 deletion as seen in PWS but of maternal origin and found in approximately 70% of individuals with Angelman syndrome [39]. The larger typical 15q11-q13 deletion encompassing breakpoints BP1 and BP3 is seen in about 40% of those with deletions. Atypical 15q deletions are also seen in Angelman syndrome as seen in PWS.
Uniparental paternal disomy 15 accounts for 5–7% of individuals with Angelman syndrome while imprinting defects account for 3–5% of cases and caused by errors in the imprinting center [40] that fails to change the epigenetic marking in germline by properly switching from a paternal pattern with silenced UBE3A expression to a maternal pattern with UBE3A expression. Up to 50% of these cases may have a mutation in the imprinting center. Mosaic cases of Angelman syndrome have been reported involving imprinting center defects. The final genetic defect does not impact DNA methylation, but involves a maternally inherited UBE3A gene mutation and account for 11% of Angelman syndrome cases [41].
Chromosome 15q11.2-q13.3 duplication, 15q11.2 BP1–BP2 deletion, Schaaf–Wang syndrome and central precocious puberty 2
Chromosome 15 imprinting disorders depend on the parental source of the typical 15q11-q13 deletions seen in both Prader–Willi and Angelman syndromes, but separate clinical disorders are found in those with chromosome 15q duplications in the same or larger chromosome 15q regions, parent of origin effects in the 15q11.2 BP1–BP2 deletion and 15q11-q13 single imprinted gene disorders (Schaaf–Wang syndrome and central precocious puberty 2) [13,20,22–24,42–46]. Patients with PWS or Angelman syndrome may present with variable phenotypes depending on the molecular class with potentially different treatment and surveillance approaches needed. The clinician evaluating these patients should be aware of available genetic testing options beginning with DNA methylation assays for both PWS and Angelman syndrome. Identifying the PWS or Angelman syndrome molecular class or other chromosome 15 anomalies and parent of origin often requires advanced genetic testing including high-resolution SNP microarrays for better prognostic indicators and more detailed genetic counseling. For example, a microdeletion imprinting center defect can lead to a 50% risk for additionally affected children if the father (in PWS families) or the mother (in Angelman syndrome) carry the same imprinting defect. Autistic features and psychoses are more common in those with PWS and maternal disomy 15 possibly related to specific disomic subclasses. Angelman syndrome and PWS patients with the larger typical 15q11-q13 deletion may be more severely affected (e.g. seizures in Angelman syndrome and compulsions in PWS) [39,47–50] and those with the deletion may have hypopigmentation due to lose of the OCA2 gene for pigment production found in the 15q11-q13 region [51,52] (see Fig. 1). A laboratory testing flowchart for use in the clinical setting was recently reported by Butler and Duis [33].
A maternally derived chromosome 15q11.2-q13.3 duplication involving imprinted genes in the region can lead to a weak cry, hypotonia, hypermobility, developmental delay including speech and language, intellectual disability, seizures, schizophrenia and autism spectrum disorder. Subtle facial features may be present but they do not have a PWS or Angelman syndrome phenotype. The pathogenicity of paternal duplications is unclear. Duplications are de novo in 50% of cases including an isodicentric chromosome 15 (idic15) having more severe problems such as seizures than the typical 15q11.2-q13.3 duplication [45,46].
The chromosome 15q11.2 BP1–BP2 deletion (Burnside–Butler; OMIM #615656) syndrome is an emerging neurodevelopmental disorder [24,44]. This condition includes the deletion of four protein-coding genes (TUBGCP5, CYFIP1, NIPA1, NIPA2) with evidence of parent of origin effect. These genes are deleted in both Angelman syndrome and PWS with the larger 15q11-q13 Type I deletion but intact in the smaller Type II deletion. All four genes in the BP1–BP2 region are highly conserved and, when disturbed, are associated with neurological, motor, intellectual and behavioral problems (e.g. [24]). This deletion was the most common cytogenetic finding in a recent study of consecutive patients presenting with neurodevelopmental disorders using chromosomal microarrays [53].
Clinical features reported in over 200 cases reviewed by Butler [44] include developmental and speech delays (70% of cases); dysmorphic features (congenital anomalies) including ear and palatal problems (46%); reading, writing, memory problems and verbal intelligence quotient (IQ) less than 75 (50%); behavioral problems (55%) and abnormal brain imaging (43%). Dyslexia, dyscalculia and brain changes were seen in both structural gray and white matter [5■].
To identify if parent-of-origin effects in 15q11.2 BP1–BP2 deletions and associated with clinical differences in individuals inheriting the deletion, Davis et al. [54] collected phenotypic data from 71 individuals with the deletion and known inheritance patterns. Among all probands, maternal deletions were associated with epilepsy, macrocephaly and autism spectrum disorder, while paternal deletions were associated with congenital heart disease and abnormal muscular phenotypes.
Other disorders with parent of origin effects include mutations in the MAGEL2 gene causing neonatal hypotonia, developmental delay, arthrogryposis, autistic features, a poor suck and obesity known as Schaaf–Yang syndrome [2]. Mutations in the MRKN3 gene causes central precocious puberty 2 [23]. The 15q11.2 BP1–BP2 deletion causes Burnside–Butler syndrome, emerging disorder with parent of origin effects leading to neurodevelopmental – autism phenotypes [24,44].
Silver–Russell syndrome
Silver–Russell syndrome (SRS) was first reported by Silver et al. in 1953 [55] and Russell in 1954 [56] affecting about one in 75 000 livebirths. SRS patients present with pre and postnatal growth retardation, a small, triangular face with narrow chin, frontal bossing and normal size but large appearing head resembling hydrocephaly [57]. Growth asymmetry and fifth finger clinodactyly are common with late closure of the anterior fontanel, immature bone development and excessive sweating particularly of the head and upper trunk. Low glucose levels may be present in infancy and early childhood with feeding difficulties requiring aggressive medical management. Café au lait spots, cardiac defects, precocious puberty, hypospadias or cryptorchidism maybe present. Although these patients are generally underweight with feeding problems, they due gain weight, but growth hormone (GH) deficiency is reported requiring endocrine and dietary consulation and treatment. The average adult height if not treated with GH is 3.1 ± 1.4 SD below the mean [57–59].
Abnormalities of chromosomes 7, 8, 15, 17 and 18 indicating genetic heterogeneity are historically rings, deletions, translocations and disomy, but the majority have a normal karyotype. No single gene appears to be responsible for SRS, but evidence suggests involvement of two separate regions on chromosome 7 (7p11.2-p13 and 7q31-qter) having imprinted genes and only paternal expression involving growth stimulation within the 7p13 band including MEST (mesoderm-specific transcript), PEG1 (paternally expressed gene 1), carboxypeptidase A4 (CPA4), coatomer protein complex subunit gamma 2 (COPG2) and two imprinted noncoding RNAs (MESTIT, C1T2/COPG2IT1). GRB10 (growth factor receptor-bound protein 10) gene is maternally expressed and also located in chromosome 7p with other genes involved in human growth and development. GRB10 acts as a growth suppressor through interaction with either IGF1 receptor or the GH receptor. Possibly, maternal disomy 7 causes features of SRS and growth anomalies in the presence of two functional maternal copies instead of the normal one copy and/or lack of paternally expressed growth promoter genes (e.g. MEST/PEG1) [59].
A second form of SRS includes mutations in imprinting centers on chromosome 11p15 seen in about 60% of patients [57]. The 11p15 band contains a cluster of imprinted genes crucial for fetal growth control with expression regulated by two imprinting regions (ICR1 and ICR2). The telomeric ICR1 domain appears to control expression of H19, possibly to function as a microRNA precursor involved in posttranscriptional regulation of specific mRNAs during vertebrate development. A second gene in this region (IGF2) is paternally expressed and involved with stimulating fetal growth and development. Chromosome 11p15 epimutations are typically due to hypomethylation of the ICR1 domain; thus, suppressing IGF2 growth factor activity reduces growth seen in individuals with SRS. Hypomethylation of ICR1 is found in 35–50% of SRS individuals, while maternal disomy 7 is found in 7–10% of cases. Those with maternal disomy 7 may also have fewer features and a milder phenotype [22,33]. SRS patients with duplications, deletions or translocations involve imprinting centers at 11p15.5 or on chromosome 7 including variants or disturbances of CDKN1C, IGF2, PLAG1 and HMGA2 genes, but approximately 40% of patients have negative genetic results [57,60].
Beckwith–Wiedemann syndrome
BWS was first reported by Wiedemann in 1964 [61] and Beckwith in 1969 [62] is generally sporadic with autosomal dominant transmission reported in about 10% of cases. BWS affects one in about 10 000 livebirths [63]. Major features include macrosomia, with a large muscle mass at birth, macroglossia with prominent eyes and periorbital fullness with characteristic ear creases and/or pits uniquely seen in this disorder. Hemihyperplasia may be present affecting segmental body regions or selected organs/tissues. Other features include capillary nevus flammeus over the central forehead and eyelids; a large fontanel; accelerated bone age with growth asymmetry; organomegaly involving kidneys, liver, pancreas, and spleen and an omphalocele with increased intra-abdominal tumors such as Wilms or hepatoblastoma estimated at 10–20%. Neonatal hypoglycemia may be present in about one-third of cases along with cardiovascular defects and cryptorchidism. Early death may occur from complications of prematurity, hypoglycemia, cardiomyopathy, macroglossia which leads to feeding and breathing problems and tumors [58,59,63,64].
Majority of patients with BWS have normal chromosome findings but abnormal methylation patterns are seen for genes in the 11p15.5 region, specifically H19 and IGF2. The 11p15.5 chromosome region contains more than a dozen imprinted genes [e.g. IGF2 – paternally expressed encoding a fetal mitogen stimulating growth; H19 – maternally expressed acting as a growth suppressor; CDKN1C – maternally expressed; KVLQT1 – maternally expressed and KCNQ10T1 (LIT1) – paternally expressed] and organized into two separately controlled imprinted domains [telomeric (ICR1 or IC1) and centromeric (ICR2 or IC2)]. The telomeric ICR1 domain controls transcription and regulation of both IGF2 and H19 genes [57,59]. A reciprocal coordinated relationship does exist between the IGF2 and H19 genes which impact both cellular growth and development.
Hemihypertrophy and hypoglycemia found in BWS with altered methylation of both KCNQ10T1 (LIT1) and H19 genes are associated with specific phenotypes. Those with tumors and BWS are described with H19 gene methylation alterations, while macrosomia and midline abdominal wall defects can be found in those with KCNQ10T1 (LIT1) transcript altered methylation. Hence, imprinting interaction of contiguous genes clustered in the 11p15.5 region is involved in BWS, an overgrowth disorder and genetically opposite effects are seen in SRS, a disorder with growth retardation [65,66■].
Chromosome abnormalities can alter gene expression leading to BWS in about 10% of subjects when using SNP microarray technology. Increased IGF2 expression is noted in maternally derived translocations and inversions of 11p15, duplications of paternal 11p15, paternal disomy 11 (about 20% of cases) and imprinting anomalies such as hypermethylation of the maternal ICR1 domain (about 5% of cases). The centromeric-located ICR2 domain regulates the expression of CDKN1C, KCNQ1 and other genes on the maternal allele, while noncoding RNA in the 11p15 region, KCNQ10T1 (LIT1), localized in intron 9 of the KCNQ1 gene is expressed on the paternal allele. This action probably represses the function of the CDKN1C gene. Loss of methylation of the maternal ICR1 domain is correlated with expression of KCNQ10T1 (LIT1). Mutations of the CDKN1C gene account for about 40% of familial BWS cases and 5–10% of sporadic cases [63]. Hypomethylation of ICR2 and point mutations of CDKN1C can lead to reduced expression and cause overgrowth in humans. Loss of methylation at ICR2 (maternal) also accounts for about 50% of BWS cases.
GNAS inactivation-related disorders
Albright [67] first reported an osteodystrophy condition in 1942 with end-organ resistance to actions of parathyroid hormone (PTH) and other hormones such as gonadotropins [lutenizing hormone (LH) and follicle stimulating hormone (FSH)], thyroid stimulating hormone, GH releasing hormone (GHRH). Central nervous system (CNS) neurotransmitters can also be involved leading to obesity, short stature and an adult height of 54–60 inches with variable mental deficiency and an average IQ of 60. There are two major variants of the alpha subunit of the membrane bound trimeric G subunit-regulatory protein (GNAS) gene with inactivation described for pseudohypoparathyroidism (PHP; PHP-Ia, PHP-Ib) and pseudo-PHP (PHPP). Individuals having PHP-Ia are reported with features of AHO and hypocalcemia with hyperphosphatemia despite elevated serum PTH levels caused by mutations in exons of the imprinted GNAS gene leading to gene inactivity. Autosomal dominant PHP-Ib is caused by heterozygous mutations that disrupt the imprinting center of the GNAS gene and not within the exons [59,68–70].
Patients with PPHP have AHO features, but without resistance to parathyroid or other hormones. AHO consists of short stature, moderate obesity, mental deficiency, a round face with a short nose and neck, delayed dental eruption with enamel hypoplasia, short metacarpals and metatarsals (brachydactyly), especially of the fourth and fifth digits and a short distal phalanx of the thumb. Osteoporosis, subcutaneous ossifications including the basal ganglia and variable hypocalcemia can be seen with occasional cataracts and hypothyroidism [58,59,71].
PHP-Ia and PPHP have been reported in the same families dependent on the parent of origin with both variants resulting from decreased GNAS protein and function. The function of this GNAS protein is to couple membrane receptors for adenyl cyclase activity for stimulating secondary messenger, cyclic adenosine monophosphate (cAMP) [57,70] required for several cellular functions. Different forms and genetic defects can affect the GNAS gene located at chromosome 20q13.11. The imprinted GNAS gene is complex and produces four different transcripts using alternative promoters including G protein subunit alpha impacting AHO production, paternally expressed XLAS, maternally expressed NESP55 encoding a chromogranin-like neuroendocrine secretory protein, and the paternally expressed A/B transcript.
Individuals with PHP can be grouped according to their clinical finding into PHP-Ia and PHP-Ib depending on the presence of hormone resistance along with the AHO phenotype. Nearly all patients with PHP-Ia have mild hypothyroidism, hypogonadism and abnormal treatment response to GHRH [72–74]. Those patients with PHP-Ia and features of AHO have mutations of the GNAS gene or deletions of chromosome 20q including the GNAS gene. Patients with PHPP or those AHO patients without evidence of hormone resistance also carry heterozygous mutations that inactivate GNAS. Maternal inheritance of such mutations can lead to a diagnosis of PHP-Ia or AHO with hormone resistance. They can have high urine levels of cAMP and a normal increase in urinary cAMP after PTH infusion [75]. Paternal inheritance of the same mutation can lead to PHPP or AHO alone. Patients with PHP presenting with PTH-resistance, but lacking AHO features are diagnosed as having the PHP-Ib subtype [73]. Most PHP-Ib cases are sporadic in origin but in some families the inheritance pattern is autosomal dominant with incomplete penetrance. These cases typically lack GNAS gene mutations but inherit alterations from a female involving the imprinting process of the GNAS locus. The most consistent or common defect is loss of methylation in the controlling elements regulating the imprint of the GNAS gene. PHP-Ib can also result from paternal chromosome 20 disomy.
The imprinted pattern of inheritance for hormone resistance in this disorder could best be explained by predominantly maternal expression of GNAS in specific tissues. Patients with PHP-Ia lacking GNAS mutations display gene disturbances due to imprinting defects with loss of imprint at the exon A/B differentially methylated region (DMR). A unique 3-KB microdeletion has been reported that disrupts the nearby STX16 gene which is close to the differentially methylated domain causing PHP-I and when disturbed prevents proper maternal imprint of GNAS [57,76,77]. Chromosomal microarrays can identify defects in about 10% of affected patients [78].
Progressive osseous heteroplasia (POH) is another GNAS gene related disorder consisting of dermal ossification starting in infancy and later increased and extensive bone formation found in deep muscle tissue and fascia layers [57]. Osteoma cutis is also characterized by extra-skeletal ossifications found in the dermis and subcutaneous tissues, as well. POH and osteoma cutis are considered more restrictive variants of PPHP and lack of expression/function of the protein encoded by the paternal GNAS gene allele due to an inactivating pathogenic variant.
Uniparental chromosome 14 disomy
Wang et al. [79] and Temple et al. [80] in 1991 first described different clinical phenotypes in individuals with either paternal or maternal chromosome 14 disomy. Characteristics of maternal disomy 14 now referred to as Tempe syndrome include prenatal and postnatal growth retardation with a reduced final adult height, congenital hypotonia, a short nose and down-turned mouth, joint laxity, gross motor delay with mild-to-moderate intellectual disabilities, early onset of puberty, truncal obesity and minor dysmorphic features of the face with small hands and feet; reminiscent of PWS [81–84]. Maternal disomy 14 often involves a chromosome 14 translocation (e.g. Robertsonian translocation 13, 14). Congenital hypotonia, motor delay, small hands and feet and early puberty are seen in greater than 80% of affected subjects. Mild intellectual disability and obesity is seen in about 50% of cases but at risk for type 2 diabetes [83]. Rapid postnatal head growth can be present due to hydrocephalus which is arrested spontaneously in about 30% of patients. Paternal disomy 14 has a more severe phenotype including polyhydramnios, thoracic and abdominal wall defects, growth retardation, a characteristic face and severe developmental delay now known as Kagami–Ogata syndrome [85].
Errors in imprinting on chromosome 14 are likely causes of the phenotypes while segmental uniparental disomy 14 with imprinted genes in the 14q32 region spans about 1 Mb and include the paternally expressed DLK1 (delta, Drosophila homologue-like 1) encoding a transmembrane signaling protein as a growth regulator homologous to proteins in the Notch/delta pathway [86] under control of a paternally methylated region along with MEG3 (maternally expressed 3) which is an imprinted long noncoding RNA with several isoforms generated by alternative splicing [87]. Regulation of an imprint is via the intergenic DMR (IGD-MR). This IG-DMR lies between DLK1 and MEG3 with the MEG3-DMR at the MEG3 promoter area. Both DMRs are methylated after paternal transmission and unmethylated after maternal transmission in the body but a different pattern is seen in the placenta [85]. Loss of methylation at the IG-DMR on chromosome 14 is reported in about 10% of cases (e.g. [88]). The clinical phenotypes are due to dysregulation of imprinted genes utilizing several mechanisms including uniparental chromosome 14 disomy, copy-number changes in imprinted genes in the chromosome, disruption of regulatory sequences or mutations of a single active allele on chromosome 14.
Chromosome 6q24-related transient neonatal diabetes mellitus
Features of the chromosome 6q24-related TNDM is found early in life and caused by genetic errors in the 6q24 complex imprinted locus and accounting for about two-thirds of all TNDM [89]. Clinical findings of this rare imprinting disorder include severe intrauterine growth retardation and hyperglycemia beginning in the neonatal period and generally resolved by 18 months of age along with dehydration and absence of ketoacidosis. Umbilical hernias and an increased tongue size may also be present with marked hypotonia, congenital heart disease, deafness, epilepsy and renal malformations, if more than one 6q24 imprinting error is involved. Intermittent levels of hyperglycemia can also occur during childhood and females with 6q24-related TNDM with multiloci errors are at risk of reoccurrence of findings during pregnancy [57,89,90]. The diagnosis is established when hypomethylation is detected within the 6q24 DMR leading to overexpression of two imprinted genes (PLAGL1 and HYMAI). The methylated region is present within the shared promoter of these two genes and impacts both genes when disturbed. Generally, expression of maternal alleles of the PLAGL1 and HYMAI genes is silenced by DMR methylation and only the paternal alleles of PLAGL1 and HYMAI genes are expressed at twice the normal dosage causing this unique form of diabetes mellitus.
There are three recognized mechanisms leading to genomic imprinting errors at the 6q24 locus including paternal disomy 6, chromosome 6q24 paternal duplication and hypomethylation of the maternal alleles for either or both PLAGL1 and HYMAI genes resulting in abnormal expression patterns. These disturbances lead to a range of presentations beyond diabetes in TNDM. Involvement of another gene (ZFP57), a transcription factor required for adequate methylation during early embryonic development is maternally hypomethylated in about one-half of affected individuals with multiple imprinting disturbances and other associated diseases [57,89,90].
CONCLUSION
Two complete sets of chromosomes are inherited, one from the father and one from the mother, with vast majority of autosomal genes expressed from both maternal and paternal gene alleles. Imprinted genes show an expression from only one member of the gene pair with expression determined by the parent during the time of gamete production through an epigenetics process involving DNA methylation but represent only a small subset of mammalian genes. Imprinted genes account for multiple disorders affecting growth with Prader–Willi and Angelman syndromes as the first examples in human. Genomic imprints are reversible and erased in both male and female germlines and reset accordingly. Periconceptional and intrauterine life can be influenced by environmental factors and nutrition particularly DNA methylation impacting fetal development and growth, pregnancy complications due to fetal overgrowth, classical disorders, metabolism and even malignancies. Hence, epigenetic processes are important in controlling imprinting and gene regulation with susceptibility to diseases. A better understanding of these processes will shed light on better care and treatment for those affected.
KEY POINTS.
Basic information, principles and mechanisms of genomic imprinting in humans will be described.
Clinical features are characterized in several classical genomic imprinting disorders including Prader–Willi and Angelman syndromes, first examples of errors in imprinting in humans.
Chromosome and molecular defects in imprinted genes observed in humans involving several imprinting disorders are described and illustrated.
Acknowledgements
I thank Grace Graham for expert preparation of the article.
Financial support and sponsorship
Support is recognized from the National Institute of Child Health and Human Development (NICHD) grant number HD02528.
Footnotes
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
■ of special interest
■■ of outstanding interest
- 1.Tucci V, Isles AR, Kelsey G, et al. Genomic imprinting and physiological processes in mammals. Cell 2019; 176:952–965. [DOI] [PubMed] [Google Scholar]; ■■ The reference discusses the background of genomic imprinting and current understanding of mechanisms as well as comparison of imprinted genes in humans and mice.
- 2.Bartolomei MS, Tilghman SM. Genomic imprinting in mammals. Annu Rev Genet 1997; 31:493–525. [DOI] [PubMed] [Google Scholar]
- 3.Walter J, Paulsen M. Imprinting and disease. Semin Cell Dev Biol 2003; 14:101–110. [DOI] [PubMed] [Google Scholar]
- 4.Chen Z, Zhang Y. Maternal H3K27me3-dependent autosomal and X chromosome imprinting. Nat Rev Genet 2020; 21:555–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bina M Discovering candidate imprinted genes and imprinting control regions in the human genome. BMC Genomics 2020; 21:378. [DOI] [PMC free article] [PubMed] [Google Scholar]; ■ The reference discusses the research and mechanisms leading to the discovery of candidate imprinted genes and their controlling elements.
- 6.Haig D, Graham C. Genomic imprinting and the strange case of the insulin-like growth factor. Cell 1991; 64:1045–1046. [DOI] [PubMed] [Google Scholar]
- 7.Monk D, Mackay DJG, Eggermann T, et al. Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat Rev Genet 2019; 20:235–248. [DOI] [PubMed] [Google Scholar]
- 8.Delaval K, Wagschal A, Feil R. Epigenetic deregulation of imprinting in congenital disease of aberrant growth. Bioessays 2006; 28:453–459. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Tycko B. Monoallelic expression of the human H19 gene. Nat Genet 1992; 1:40–44. [DOI] [PubMed] [Google Scholar]
- 10.Barton SC, Surani MA, Norris ML. Role of paternal and maternal genomes in mouse development. Nature 1984; 311:374–376. [DOI] [PubMed] [Google Scholar]
- 11.Cattanach BM, Kirk M. Differential activity of maternally and paternally derived chromosome regions in mice. Nature 1985; 315:496–798. [DOI] [PubMed] [Google Scholar]
- 12.Nicholls RD, Knoll JH, Butler MG, et al. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader–Willi syndrome. Nature 1989; 342:281–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butler MG, Lee PDK, Whitman BY. Management of Prader–Willi syndrome. New York, NY: Springer; 2006. [Google Scholar]
- 14.DeBaun MR, Neimitz EL, Feinberg AP. Association of in vitro fertilization with Beckwith–Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am J Hum Genet 2003; 72:156–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manialviratn S, DeCherny S, Segars J. Imprinting disorders and assisted reproductive technology. Fertil Steril 2009; 91:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cox GF, Burger JL, Mau UA. Intracytoplasmic sperm injection may increase the risk of imprinting defects. Am J Hum Genet 2002; 71:62–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gold JA, Ruth C, Osann K, et al. Frequency of Prader–Willi syndrome in births conceived via assisted reproductive technology. Genet Med 2014; 16:164–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niemitz EL, Feinberg AP. Epigenetics and assisted reproductive technology: a call for investigation. Am J Hum Genet 2004; 74:599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marjonen H, Auvinen P, Kahila H, et al. rs10732516 Polymorphism at the IGF2/H19 locus associates with genotype-specific effects on placental DNA methylation and birth weight of newborns conceived by assisted reproductive technology. Clin Epigenetics 2018; 10:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fountain MD, Schaaf CP. Prader–Willi syndrome and Schaaf–Yang syndrome: neurodevelopmental diseases intersecting at the MAGEL2 gene. Diseases 2016; 4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Temple K, Shrubb V, Lever M, et al. Isolated imprinting mutation of the DLK1/GTL2 locus associated with a clinical presentation of maternal uniparental disomy of chromosome 14. J Med Genet 2007; 44:637–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Butler MG. Genomic imprinting disorders in humans: a mini-review. J Assist Reprod Genet 2009; 26:477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abreu AP, Dauber A, Macedo DB, et al. Central precocious puberty caused by mutations in the imprinted gene MKRN3. N Engl J Med 2013; 368:2467–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rafi SK, Butler MG. The 15q11.2 BP1–BP2 microdeletion (Burnside–Butler) syndrome: in silico analyses of the four coding genes reveal functional associations with neurodevelopmental phenotypes. Int J Mol Sci 2020; 21:3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cassidy SB, Schwartz S, Miller JL, et al. Prader–Willi syndrome. Genet Med 2012; 14:10–26. [DOI] [PubMed] [Google Scholar]
- 26.Angulo MA, Butler MG, Cataletto ME. Prader–Willi syndrome: a review of clinical, genetic and endocrine findings. J Endocrinol Invest 2015; 38:1249–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Butler MG, Hartin SN, Hossain WA, et al. Molecular genetic classification in Prader–Willi syndrome: a multisite cohort study. J Med Genet 2019; 56:149–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Butler M, Thompson T. Prader–Willi syndrome: clinical and genetic findings. Endocrinologist 2000; 10:3s–16s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prader A, Labhart A, Willi H. A syndrome characterized by obesity, small stature, cryptorchidism and oligophrenia following a mytotonia-like status in infacy. Schweiz Med Wochenschr 1956; 86:1260–1261. [Google Scholar]
- 30.Ledbetter DH, Riccardi VM, Airhart SD, et al. Deletions of chromosome 15 as a cause of the Prader–Willi syndrome. N Engl J Med 1981; 304:325–329. [DOI] [PubMed] [Google Scholar]
- 31.Butler MG, Palmer CG. Parental origin of chromosome 15 deletion in Prader–Willi syndrome. Lancet 1983; 1:1285–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cassidy SB, Lai LW, Erickson RP, et al. Trisomy 15 with loss of the paternal 15 as a cause of Prader–Willi syndrome due to maternal disomy. Am J Hum Genet 1992; 51:701–708. [PMC free article] [PubMed] [Google Scholar]
- 33.Butler MG, Duis J. Chromosome 15 imprinting disorders: genetic laboratory methodology and approaches. Front Pediatr 2020; 8:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sahoo T, Bacino CA, German JR, et al. Identification of novel deletions of 15q11q13 in Angelman syndrome by array – CGH: molecular characterization and genotype-phenotype correlations. Eur J Hum Genet 2007; 15:943–949. [DOI] [PubMed] [Google Scholar]
- 35.Hassan M, Butler MG. Prader–Willi syndrome and atypical submicroscopic 15q11-q13 deletions with or without imprinting defects. Eur J Med Genet 2016; 59:584–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tan Q, Potter KJ, Burnett LC, et al. Prader–Willi-like phenotype caused by an atypical 15q11.2 microdeletion. Genes 2020; 25:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams CA, Driscoll DJ, Dagli AI. Clinical and genetic aspects of Angelman syndrome. Genet Med 2010; 12:385–395. [DOI] [PubMed] [Google Scholar]
- 38.Trickett J, Oliver C, Heald M, et al. Multimethod assessment of sleep in children with Angelman syndrome: a case–controlled study. Front Psychiatry 2019; 10:874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lossie AC, Whitney MM, Amidon D, et al. Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J Med Genet 2001; 38:834–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buiting K, Williams C, Horsthemke B. Angelman syndrome – insights into a rare neurogenetic disorder. Nat Rev Neurol 2016; 12:584–593. [DOI] [PubMed] [Google Scholar]
- 41.Margolis SS, Sell GL, Zbinden MA, Bird LM. Angelman syndrome. Neurotherapeutics 2015; 12:641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burnside RD, Pasion R, Mikhail FM, et al. Microdeletion, microduplication of proximal 15q11.2 between BP1 and BP2: a susceptibility region for neurological dysfunction including developmental and language delay. Hum Genet 2011; 130:517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cox DM, Butler MG. The 15q11.2 BP1–BP2 syndrome: a review. Int J Mol Sci 2015; 16:4068–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Butler MG. Clinical and genetic aspects of the 15q11.2 BP1–BP2 microdeletion disorder. J Intellect Disabil Res 2017; 61:568–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kalsner L, Chamberlain SJ. Prader–Willi, Angelman, and 15q11-q13 duplication syndromes. Pediatr Clin North Am 2015; 62:587–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Isles AR, Ingason A, Lowther C, et al. Parental origin of interstitial duplications at 15q11.2-q13.3 in schizophrenia and neurodevelopmental disorders. PLoS Genet 2016; 12:e1005993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moncla A, Malzac P, Voelckel MA, et al. Phenotype–genotype correlation in 20 deletion and to nondeletion Angelman syndrome patients. Eur J Hum Genet 1999; 7:131–139. [DOI] [PubMed] [Google Scholar]
- 48.Bonello D, Camilleri F, Callega-Agius J. Angelman syndrome: identification and management. Neonatal Netw 2017; 36:142–151. [DOI] [PubMed] [Google Scholar]
- 49.Carson RP, Bird L, Childers AK, et al. Preserved expressive language as a phenotypic determinant of mosaic Angelman syndrome. Mol Genet Genomic Med 2019; 7:e837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bindels-de Heus KGCB, Mouse SE, Hooven-Rastaake MT, et al. An overview of health issues and development in a large clinical cohort of children with Angelman syndrome. Am J Med Genet 2020; 182:53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Butler MG. Hypopigmentation: a common feature of Prader–Labhart–Willi syndrome. Am J Hum Genet 1989; 45:140–146. [PMC free article] [PubMed] [Google Scholar]
- 52.Butler MG. Prader–Willi syndrome: current understanding of cause and diagnosis. Am J Med Genet 1990; 35:319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ho KS, Wassman ER, Butler MG. Chromosomal microarray analysis of consecutive individuals with autism spectrum disorders using an ultra-high resolution chromosomal microarray optimized for neurodevelopmental disorders. Int J Mol Sci 2016; 17:2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davis KW, Serrano M, Butler MG, et al. Parent-of-origin effects in 15q11.2 BP1–BP2 microdeletion (Burnside–Butler) syndrome. Int J Mol Sci 2019; 20:1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Silver HK, Kiyasu W, George J, et al. Syndrome of congenital hemihypertrophy, shortness of stature, and elevated urinary gonadotropins. Pediatrics 1953; 12:268–276 [PubMed] [Google Scholar]; Butler MG. Hypopigmentation: a common feature of Prader–Labhart–Willi syndrome. Am J Hum Genet 1989;45:140–146. [PubMed] [Google Scholar]
- 56.Russell A. A syndrome of intra-uterine dwarfism recognizable at birth with cranio-facial dysostosis, disproportionately short arms, and other anomalies (5 examples). Proc R Soc Med 1954; 47:1040–1044. [PubMed] [Google Scholar]
- 57.Adam MP, Ardinger HH, Pagon RA, et al. Genereviews.org [Internet]. Seattle, WA. National Center for Biotechonology Information; [updated 2020; cited July 1 2020]. Available from: https://www.genereviews.org. [Google Scholar]
- 58.Jones KL. Smith’s recognizable patterns of human malformation. 7th ed Philadelphia: W.B. Saunders Company; 2013; 1–954. [Google Scholar]
- 59.National Library of Medicine, William H. Welch Medical Library at Johns Hopkins. Omim.org [Internet]. Baltimore, MD: Online Mendelian Inheritance in Man; [updated 2020; cited July 1 2020]. Available from: https://omim.org. [Google Scholar]
- 60.Bullman H, Lever M, Robinson DO, et al. Mosaic maternal uniparental disomy of chromosome 11 in a patient with Silver–Russell syndrome. J Med Genet 2008; 45:396–399. [DOI] [PubMed] [Google Scholar]
- 61.Wiedemann HR. Familial malformation complex with umbilical hernia and macroglossia - a “new syndrome”? J Genet Hum 1964; 13:223. [PubMed] [Google Scholar]
- 62.Beckwith JB. Macroglossia, omphalocele, adrenal cytomegaly, gigantism, and hyperplasic visceromegaly. Birth Defects 1969; 5:188. [Google Scholar]
- 63.Wang KH, Kupa J, Duffy KA, Kalish JM. Diagnosis and management of Beckwith–Wiedemann syndrome. Front Pediatr 2020; 7:562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Edmonson A, Kalish JM. Overgrowth syndromes. J Pediatr Genet 2015; 4:136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eggermann T, Eggermann K, Schoenherr N. Growth retardation versus overgrowth: Silver–Russell syndrome is genetically opposite to Beckwith–Wiedemann syndrome. Trends Genet 2008; 24:195–204. [DOI] [PubMed] [Google Scholar]
- 66.Neuheuser L, Meyer R, Begemann M, et al. Next generation sequencing and imprinting disorders: current applications and future perspectives: lessons from Silver–Russell syndrome. Mol Cell Probes 2019; 44:1–7. [DOI] [PubMed] [Google Scholar]; ■ The reference discusses the use of advanced genomic technology including next-generation sequencing and application to understanding the epigenomics mechanisms and defects in Silver–Russell syndrome, a classical genomic imprinting disorder in humans.
- 67.Albright F, Burnett CH, Smith PH, Parson W. Pseudo-hypoparathyroidism – an example of ‘Seabright-bantam syndrome’: report of three cases. Endocrinology 1942; 30:922–932. [Google Scholar]
- 68.Juppner H Genetic and epigenetic defects at the GNAS locus cause different forms of pseudohypoparathyroidism. Ann Endocrinol (Paris) 2015; 76:92–97. [DOI] [PubMed] [Google Scholar]
- 69.Mantovani G, Spada A, Elli FM. Pseudohypoparathyroidism and Gsa-cAMP-linked disorder: current view and open issues. Nat Rev Endocrinol 2016; 12:347–356. [DOI] [PubMed] [Google Scholar]
- 70.Schernthaner-Retier MH, Trivellin G, Stratakis C. Chaperones, somatotroph tumors and the cyclic AMP (cAMP)-dependent protein kinase (PKA) pathway. Mol Cell Endocrinol 2020; 499:110607. [DOI] [PubMed] [Google Scholar]
- 71.Levine MA. Albright’s hereditary osteodystrophy: historical credit. Endocr Pract 2000; 6:115. [PubMed] [Google Scholar]
- 72.Joseph AW, Shoemaker AH, Germain-Lee EL. Increased prevalence of Carpal Tunnel syndrome in Albright hereditary osteodystrophy. J Clin Endocrinol Metab 2011; 96:2065–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sbrocchi AM, Rauch F, Lawson ML, et al. Osteosclerosis in two brothers with autosomal dominant pseudohypoparathyroidism type 1b: bone histomorphometric analysis. Eur J Endocrinol 2011; 164:295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Levine MA. An update on the clinical and molecular characteristics of pseudohypoparathyroidism. Curr Opin Endocrinol Diabetes Obes 2012; 19:443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chase LR, Melson GL, Aurbach GD. Pseudohypoparathyroidism: defective excretion of 3′,5′-AMP in response to parathyroid hormone. J Clin Invest 1969; 48:1832–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bastepe M, Juppner H. GNAS locus and pdeusohypoparathyroidism. Adv Exp Med Biol 2005; 63:65–74. [Google Scholar]
- 77.Bastepe M The GNAS locus and pseudohypoparathyroidism. Adv Exp Med Biol 2008; 626:27–40. [DOI] [PubMed] [Google Scholar]
- 78.Takatani R, Minagawa M, Molinaro A, et al. Similar frequency of paternal uniparental disomy involving chromosome 20q (patUPD20q) in Japanese and Caucasian patients affect by sporadic pseudohypoparathyroidism Type Ib (sporPHP1B). Bone 2015; 79:15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang JC, Passage MB, Yen PH. The uniparental heterodisomy for chromosome 14 in a phenotypically abnormal familial balanced 13/14 Robertsonian translocation carried. Am J Hum Genet 1991; 48:1069–1074. [PMC free article] [PubMed] [Google Scholar]
- 80.Temple IK, Cockwell A, Hassold T, et al. Maternal uniparental disomy for chromosome 14. J Med Genet 1991; 28:511–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Butler MG. Imprinting disorders: non-Mendelian mechanisms affecting growth. J Pediatr Endocrinol Metab 2002; 15:1279–1288. [PMC free article] [PubMed] [Google Scholar]
- 82.Falk MJ, Curtis CA, Bass NE, et al. Maternal uniparental disomy chromosome 14: case report and literature review. Pediatr Neurol 2005; 32:116–120. [DOI] [PubMed] [Google Scholar]
- 83.Ioannides Y, Lokulo-Sodipe K, Mackay DJG, et al. Temple syndrome: improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: an analysis of 51 published cases. J Med Genet 2014; 51:495–501. [DOI] [PubMed] [Google Scholar]
- 84.Briggs TA, Lokulo-Sodipe K, Chandler KE, et al. Temple syndrome as a result of isolated hypomethylation of the 14q32 imprinted DLK1/MEG3 region. Am J Med Genet A 2016; 170A:170–175. [DOI] [PubMed] [Google Scholar]
- 85.Ogata T, Kagami M. Kagami–Ogata syndrome: a clinically recognized upd(14)pat and related disorder affecting the chromosome 14q32.3 imprinted region. J Hum Genet 2016; 61:87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Howard M, Charalambous M. Molecular basis of imprinting disorders affecting chromosome 14: lesson from murine models. Reproduction 2015; 149:R237–R249. [DOI] [PubMed] [Google Scholar]
- 87.Zhang X, Rice K, Wang Y, et al. Maternally expressed gene 3 (MEG3) noncoding ribonucleic acid: isoform structure, expression, and functions. Endocrinology 2010; 151:939–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Temple IK, Shrubb V, Lever M, et al. Isolated imprinting mutation of the DLK1/GTL2 locus associated with a clinical presentation of maternal uniparental disomy of chromosome 14. J Med Genet 2007; 44:637–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yorifuji T, Higuchi S, Hosokawa Y, Kwakita R. Chromosome 6q24-related diabetes mellitus. Clin Pediatr Endocrinol 2018; 27:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Touati A, Errea-Dorronsoro J, Nouri S, et al. Transient neonatal diabetes mellitus and hypomethylation at additional imprinted loci: novel ZFP57 mutation and review on the literature. Acta Biabetol 2019; 56:301–307. [DOI] [PubMed] [Google Scholar]