

Abstract
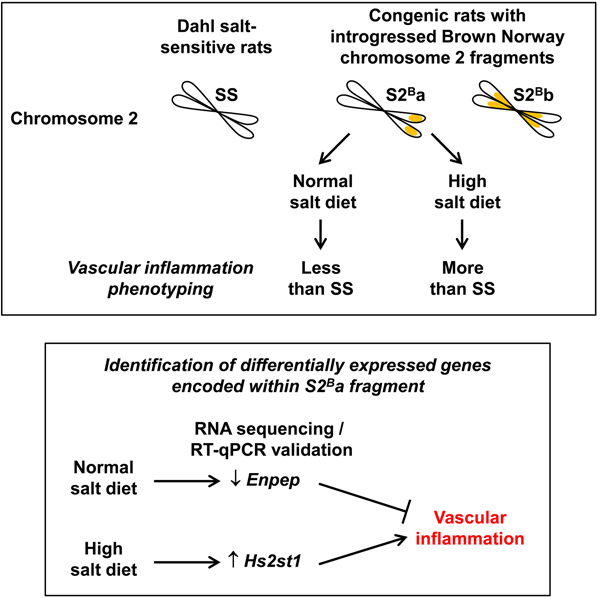
Chromosome 2 introgression from normotensive Brown Norway (BN) rats into hypertensive Dahl salt-sensitive (SS) background (SS-Chr 2BN/Mcwi; consomic S2B) reduced blood pressure (BP) and vascular inflammation under a normal salt diet (NSD). We hypothesized that BN chromosome 2 contains anti-inflammatory genes that could reduce BP and vascular inflammation in rats fed NSD or high salt diet (HSD). Four- to 6-week old male SS and congenic rats containing the BN Chromosome 2 distal portion (SS.BN-(rs13453786-rs66377062)/Aek; S2Ba) and middle segment (SS.BN-(rs106982173-rs65057186)/Aek; S2Bb) were fed NSD or HSD (4% NaCl) up to age 12–13 weeks. Systolic BP determined by telemetry was higher in SS rats fed HSD versus NSD. Systolic BP was lower in both congenic rats than in SS under NSD, but similar under HSD versus SS. Reactive oxygen species generation using dihydroethidium staining, expression of vascular cell adhesion molecule 1 and monocyte chemoattractant protein-1, and immune cell infiltration by immunofluorescence demonstrated that S2Ba rats present less inflammation under NSD and more under HSD versus SS rats. RNA sequencing and reverse transcription-quantitative PCR identified two differentially expressed genes encoded within BN chromosome 2 distal portion that could act as regulators of vascular inflammation. These were down-regulated glutamyl aminopeptidase (Enpep) that was anti-inflammatory under NSD and up-regulated heparan sulfate 2-O-sulfotransferase 1 (Hs2st1) that was pro-inflammatory under HSD. In conclusion, two differentially expressed genes encoded within introgressed BN chromosome 2 distal fragment were identified: Enpep associated with reduced vascular inflammation under NSD, and Hs2st1, associated with increased vascular inflammation under HSD.
Keywords: congenic rats, RNA sequencing, blood pressure, vascular inflammation
Graphical Abstract
INTRODUCTION
Hypertension is a major risk factor for cardiovascular and renal disease. Although the direct causes leading to development of primary human hypertension remain complex and elusive, research has demonstrated that mechanisms of elevation of blood pressure (BP) are multifactorial, including genetic susceptibility and environmental factors such as high salt intake, which leads to an inflammatory process.1 Multiple loci and genes are associated with hypertension.2 However, the efficiency of genome-wide association studies (GWAS) to identify risk loci for hypertension is limited, and investigation of specific genetic variations contributing to different heritable hypertensive phenotypes remains challenging.
Chromosomal substitution strains allow discovering regions of the rat genome that contain genes related to cardiovascular quantitative trait loci (QTLs) with multiple and complex connections between genes that influence the phenotype.3 For example, this approach has demonstrated that rat chromosome (Chr) 2 contains QTLs for BP.4–7 We have previously shown using Chr substitutions that the rat Chr 2 may contain genes regulating vascular inflammation.8 Chr 2 introgression from normotensive Brown Norway (BN) rats into hypertensive Dahl salt-sensitive (SS) background (SS-Chr 2BN/Mcw renamed here as S2B) reduced BP and vascular inflammation under a normal salt diet (NSD). However, the BN Chr 2 anti-inflammatory genes have not been identified as yet.
We hypothesized that BN Chr 2 contains anti-inflammatory genes that reduce BP elevation and vascular inflammation in rats fed a NSD and high salt diet (HSD). To test this hypothesis, we first generated consomic S2Ba and S2Bb rats containing respectively the BN Chr 2 distal and middle segments to map the region of BN Chr 2 responsible for vascular inflammatory responses. Systolic BP, aortic remodeling, oxidative stress and inflammation were determined in SS, S2Ba and S2Bb rats under NSD and HSD. Small (microRNA) and total RNA sequencing (RNA-seq) of aortic RNA was performed to demonstrate the gene expression patterns associated with the introgression of the different fragments of BN Chr 2 into SS rats fed NSD and HSD. Finally, differentially expressed (DE) genes associated with vascular inflammatory responses, encoded within the introgressed BN Chr 2 fragment, were validated by reverse transcription-quantitative PCR (RT-qPCR) and candidate genes regulating vascular inflammation identified by comparing their expression in SS, S2Ba and S2Bb rats under NSD and HSD.
METHODS
Expanded Methods are provided in the online Data Supplement. The data and analytic methods but not the study materials, due to limited amount of tissues and RNA available, will be/have been made available to other researchers for the purpose of reproducing the results or replicating the procedure. The sequencing data have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE149516). The congenic strains have been donated to the Rat Research and Resource Center where they are cryopreserved (RRRC #s 684 and 685). The authors declare that all supporting data are available within the article and the online Data Supplement.
Experimental design
All experimental procedures were approved by the Animal Care Committee of the Lady Davis Institute for Medical Research (LDI), McGill University, and followed the recommendations of the Canadian Council of Animal Care, and were as well in agreement with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
In order to map the region of BN Chr 2 responsible for the vascular inflammatory response, congenic S2Ba and S2Bb rats were generated by Dr. Anne E. Kwitek at the University of Iowa (IA, USA) through an F2 intercross between consomic SS-Chr 2BN/1McwiAek and parental Dahl SS/Mcwi salt-sensitive rats (Supplemental Figure S1A).9–11
Two sets of rats were used for BN Chr 2 mapping of vascular inflammatory response and for microRNA and total RNA profiling using the HiSeq 2500 sequencing system (Illumina, San Diego, CA) at the Sainte-Justine University Hospital Integrated Centre for Pediatric Clinical Genomics (Supplemental Figure S1B). A first set of SS, S2Ba and S2Bb male rats were maintained on NSD (Teklad Global 18% protein rodent diet with 0.2% of NaCl, Envigo) and used at ages 12–13 weeks. Thereafter, a second set of rats were fed NSD until 4–6 weeks old and then switched to HSD (TD.92034 4% NaCl, Envigo) up to age 12–13 weeks or until they developed a stroke as manifested by seizures. BP was determined by telemetry for 2 days before the end of the study in NSD-fed rats and after 6 weeks in HSD-fed rats. Note that the two sets of rats were studied at different times. However, the RNA profiling of both sets of rats was performed at the same time using frozen tissues.
Comparison and validation of DE genes encoded within BN Chr 2 distal portion were used to identify the down-regulated glutamyl aminopeptidase (Enpep) under NSD and up-regulated heparan sulfate 2-O-sulfotransferase 1 (Hs2st1) under HSD as regulators of vascular inflammation (Supplemental Figure S1C). A similar approach was used to tentatively identify DE genes encoded within BN Chr 2 middle portion associated with greater BP in S2Bb rats under HSD.
Statistical analyses
Acquisition of data was done by experimenters blinded to group/treatment assignments. Results are presented as means ± SEM. Incidence of seizures was determined by Kaplan-Meier survival analysis using a log-rank test followed by the Holm-Sidak test for multiple comparisons. Comparisons of BP data were carried out using two-way analysis of variance (ANOVA) for repeated measures or by comparing the sum of squares using one-way ANOVA. Other comparisons between more than 2 groups were done using one-way or two-way ANOVA as appropriate. All ANOVA tests were followed by a Student-Newman-Keuls post-hoc test. In absence of normal distribution, a Kruskal-Wallis one-way ANOVA on Ranks followed by a Dunn’s multiple comparison post hoc test was performed. Statistical tests were done in SigmaPlot version 13 (Systat Software, San Jose, CA). P<0.05 was considered statistically significant. Differential expression analysis was performed using an ANOVA-like test in EdgeR based on generalized linear models. Differential expression was defined with a threshold of fold change > 1.3 and false discovery rate (FDR) <0.05.
RESULTS
Generation of congenic S2Ba and S2Bb rat strains
In order to map the region of the S2B Chr 2 containing anti-inflammatory genes, five congenic rat strains were generated from S2B consomic rats. Of these, 3 were initially selected to map Chr 2 with a minimum of overlap, but one could not be maintained and was lost. The study was performed with the remaining 2 congenic rat strains (S2Ba and S2Bb) that allowed nevertheless a good coverage of Chr 2 (Figure 1). S2Ba and S2Bb rats possess a BN Chr 2 distal segment of ~24 Mb and a middle segment of ~270 Mb, respectively, with some overlap. The simple sequence length polymorphism markers used to genotype S2B x SS F2 offspring in the generation of congenic S2B strains, and the single nucleotide polymorphism markers used to define the boundaries of BN Chr 2 distal and middle fragments introgressed in Dahl salt-sensitive rats are shown in Supplemental Table S1 and 2.
Figure 1. Map of rat chromosome 2 of S2B congenic strains.

Chromosome 2 of Dahl salt-sensitive (SS) and congenic S2Ba and S2Bb rats with reference single nucleotide polymorphism (RefSNP) makers and their position in megabases (Mb) in the rat (Rattus norvegicus) genome assembly version rn6.0 are presented. SS and introgressed Brown Norway intervals are indicated in white and black, respectively. Hatched regions are in putative recombination intervals. All other chromosomes (1, 3–20, X and Y) are fixed for the SS genotype.
BP was lower in S2Ba and S2Bb rats under NSD, but higher in S2Bb rats under HSD compared to SS rats
No introgressed BN Chr 2 segment was mapped for lower BP observed previously in S2B rats under NSD.8 Systolic BP (SBP) was ~20 mm Hg lower in S2Ba and S2Bb compared to SS rats under NSD (Figure 2A). Pulse pressure was also lower in S2Ba (by ~12 mm Hg) and S2Bb (by ~20 mm Hg) compared to SS rats (Supplemental Figure S2). However, the 3 groups of rats presented similar mean BP, diastolic BP (DBP) and heart rate. Some rats had seizures during the 8 weeks of HSD feeding (Figure 2B). Seizures started to be observed after 4 weeks of HSD feeding, with numbers of rats 4-fold higher in S2Bb compared to SS after 8 weeks of HSD. Because of the high incidence of seizures in the last weeks of the HSD study, BP was determined after 6 weeks of HSD. SS rats presented >20 mm Hg systolic BP after 6 weeks of HSD compared to NSD-fed rats (Figure 2A). SBP was similar in S2Ba rats and tended to be higher by ~20 mm Hg in S2Bb compared to SS and S2Ba rats. Furthermore, S2Bb rats presented higher mean BP (by ~20 mm Hg) and DBP (by ~30 mm Hg) compared to SS and S2Ba rats (Supplemental Figure S2). Pulse pressure and heart rate were similar in the 3 groups of rats. Accordingly, it could be concluded that higher BP under HSD was mapped to the introgressed middle fragment of the BN Chr 2.
Figure 2. Systolic blood pressure and Kaplan-Meier seizure-free survival curves.

A. Systolic blood pressure (BP) was measured by telemetry in Dahl salt-sensitive (SS) and congenic S2Ba and S2Bb rats fed a normal salt diet (NSD) or after 6 weeks high salt diet (HSD). Daytime and nighttime are highlighted by open and close horizontal bars, respectively. B. The incidence of stroke manifesting as seizures was determined in congenic and SS rats fed HSD. Data are presented as means ± SEM, n = 5–8 in A and n=30–36 in B. Data were analyzed by two-way ANOVA for repeated measures followed by a Student-Newman-Keuls post hoc test in A and by Log-Rank test followed by the Holm-Sidak test for multiple comparisons in B. *P<0.05 and **P<0.01 vs. SS.
Media cross-sectional area, collagen content and fibronectin levels
The media cross-sectional area (MCSA) of thoracic aorta was unaffected in S2Ba and S2Bb compared to SS under NSD and HSD (Supplemental Figure S3A). Aortic media collagen content was similar in S2Ba compared to SS rats under NSD, whereas it was decreased by 63% in S2Bb (Supplemental Figures S3B and S4). Under HSD, aortic media collagen content was similar in the 3 groups of rats. Aortic fibronectin levels were unchanged in congenic rats compared to SS under NSD and HSD (Supplemental Figures S3C and S5).
Vascular inflammation in S2Ba rats was lower under NSD, but greater under HSD, compared to SS rats.
Under NSD, S2Ba rats presented less pro-inflammatory features than SS and S2Bb rats. Reactive oxygen species generation was 60% lower in S2Ba, and 30% lower in S2Bb, compared to SS rats (Figure 3A and Supplemental Figure S6). Vascular cell adhesion molecule-1 (VCAM-1) and monocyte chemotactic protein-1 (MCP-1) expression levels were decreased by >35% in S2Ba and similar in S2Bb compared to SS rats (Figures 3B and C and Supplemental Figure S7). Monocyte/macrophage (MoMΦ) and T cell infiltration in aortic perivascular adipose tissue were similar in the 3 groups of rats fed a NSD (Figures 3D and E and Supplemental Figure S8).
Figure 3. Introgression of Brown Norway chromosome 2 distal fragment was associated with lower vascular inflammation under normal salt diet and greater vascular inflammation under high salt diet.

Reactive oxygen species generation by dihydroethidium (DHE) staining (A), vascular cell adhesion molecule (VCAM)-1 (B) and monocyte chemoattractant protein (MCP)-1 expression (C) in aortic media and monocyte/macrophage (MoMΦ, D) and T cell infiltration using immunofluorescence (E) were determined in Dahl salt-sensitive (SS) and congenic S2Ba and S2Bb rats fed normal salt diet (NSD) and high salt diet (HSD). Data are presented as means ± SEM, n = 5–9 in A, 5–8 in B and E, 5–6 in C and D. Data were analyzed using one-way ANOVA followed by a Student-Newman-Keuls post hoc test in A-D, and Kruskal-Wallis test with Dunn’s multiple comparison post hoc test in E. *P<0.05 and **P<0.001 vs. SS, †P<0.05 vs. S2Ba.
Under HSD the results were reversed, and S2Ba rats demonstrated more pro-inflammatory features than S2Bb and SS rats. Reactive oxygen species generation was twice greater in S2Ba rats and 25% lower in S2Bb rats compared to SS rats (Figure 3A and Supplemental Figure S6). Aortic VCAM-1 expression levels were 2.3 times greater in S2Ba rats compared to SS rats, whereas MCP-1 was unchanged (Figures 3B and C, and Supplemental Figure S7). In S2Bb, aortic VCAM-1 tended to increase and MCP-1 decrease compared to SS rats. S2Ba rats presented 1.6 times more infiltrating MoMΦ, whereas S2Bb rats had less infiltrating T cells compared to SS rats (Figures 3D and E and Supplemental Figure S8).
Altogether, these results suggest that the distal but not the middle segment of BN chr 2 contains genes regulating vascular inflammation under NSD or HSD.
BN Chr 2 distal segment integration into SS background induced differential gene expression under NSD and HSD
In order to identify differentially expressed (DE) genes associated with BN Chr 2 introgressed fragments leading to changes in vascular inflammatory status under NSD or HSD, the expression profiling of microRNAs, messenger RNAs (mRNAs) and other non-coding RNAs (ncRNAs, such as microRNA precursors [pre- and pri-microRNAs], long ncRNA [lncRNA], small nuclear RNA [snRNA], small nucleolar RNA [snoRNA], processed transcripts and pseudogenes) was determined using small and total RNA-seq in thoracic descending aorta of SS, S2Ba and S2Bb rats under NSD and HSD. Small and total RNA libraries were constructed using total RNA with RNA integrity numbers >8 (Supplemental Table S3). An average of 20.8±1.2 million single-end reads (51 bp) were obtained in small RNA-seq, 83.3 ± 0.9% of which mapped to a single locus, and 8.9 ± 0.3% to multiple loci. An average of 24.5 ± 0.3 million paired-end reads (75 bp) were obtained from total RNA-seq, of which 81.8 ± 0.3% mapped to a single locus, and 13.2± 0.3% to multiple loci. A total of 32,754 genes and 1,043 microRNAs with >10 reads per million mapped reads per sample in 5 samples in at least one group were selected for analysis.
Differential expression analysis of small and total RNA-seq data identified DE genes in S2Ba vs. SS uniquely under NSD (27) and HSD (608) and under both diets (7). In S2Bb vs. SS DE genes were identified uniquely under NSD (133) and HSD (9), and under both diets (12) (Figure 4A and Supplemental Table S4 and S5). Three novel microRNAs predicted with miRDeep2 were DE (Supplemental Table S5 and S6).
Figure 4. Congenic rats present different gene expression profiles under both diets.

Venn diagrams showing the number of differentially expressed (DE) genes determined by small (microRNAs) and total RNA sequencing (messenger RNAs [mRNAs] and non-coding RNAs [ncRNAs]) in the aorta of congenic S2Ba (A and B) and S2Bb (A and C) vs. Dahl salt-sensitive (SS) rats under normal-salt diet (NSD) and high-salt diet (HSD). DE genes encoded in the whole genome and the introgressed BN chromosome 2 distal and middle fragments are shown in A, B and C, respectively. n = 5–6. The number of DE genes and microRNAs were identified with fold change >1.3 and false discovery rate <0.05.
Hierarchical clustering analysis of DE genes (mRNAs and ncRNAs) clustered S2Bb rats and partially SS and S2Ba rats under NSD, whereas it clustered SS rats and partially S2Ba and S2Bb rats under HSD (Supplemental Figure S9). However, hierarchical clustering analysis of DE microRNAs showed only partial clustering in the 3 groups of rats.
The results of the gene enrichment analysis of DE genes are shown in Supplemental Table S7. DE genes encoding transcription factors are listed in Supplemental Table S8. Molecular networks integrating DE microRNAs and genes, the predicted interactions between DE microRNAs and inversely DE messenger RNA and non-coding RNAs as well as interactions between DE transcription factors and DE genes, gene ontology term enrichment groups and DE genes encoded in BN Chr 2 fragments (distal or middle) are presented in Supplemental Figures S10–S13.
Identification of DE genes encoded within BN Chr 2 distal fragment associated with less and more vascular inflammation in S2Ba rats under NSD and HSD, respectively
Since associations were demonstrated between the S2Ba but not S2Bb rats and vascular inflammatory status under NSD or HSD, DE genes encoded within BN Chr 2 distal fragment were investigated to identify vascular inflammatory modulators. S2Ba rats presented DE mRNAs uniquely associated to NSD (2), uniquely under HSD (8), and under both diets (3), all encoded within the BN introgressed Chr 2 distal fragment (Figure 4B, Supplemental Table S5 and S9).
DE genes encoded within the BN Chr 2 distal portion were further studied by RT-qPCR since they could contain genes affecting vascular inflammation under NSD and HSD. The oligonucleotide primers used for qPCR are depicted in Supplemental Table S10. Using RNA-seq, down-regulation of glutamyl aminopeptidase (Enpep) mRNA and up-regulation of G protein subunit gamma 5 (Gng5) mRNA were shown in S2Ba compared to SS rats uniquely under NSD (Figure 5a and Supplemental Figure S14). RT-qPCR confirmed the down-regulation of Enpep but not the up-regulation of Gng5 in S2Ba compared to SS rats under NSD. In addition, it confirmed that Enpep was higher in S2Bb compared to S2Ba rats and not different from SS under NSD. RT-qPCR also confirmed that Enpep was up-regulated by HSD in both S2Ba and S2Bb rats. Enpep was up-regulated in S2Bb compared to SS and S2Ba rats under HSD. Altogether these results suggest that Enpep down-regulation in S2Ba rats under NSD may play a role in the reduction of vascular inflammation.
Figure 5. Validation of differentially expressed genes encoded in chromosome 2 distal fragment by reverse transcription-quantitative PCR.

Differentially expressed genes encoded within introgressed Brown Norway chromosome 2 distal portion revealed by RNA sequencing (RNA-seq) in thoracic aorta of congenic S2Ba rats under normal salt diet (NSD) or high salt diet (HSD) were reanalyzed by reverse transcription-quantitative PCR (RT-qPCR) in Dahl salt-sensitive (SS) and congenic S2Ba and S2Bb fed a NSD or NSD. The mRNA expression was normalized by ribosomal protein S16 (Rps16) mRNA levels and expressed as fold change over control. Data are presented as means ± SEM, n = 6–9 for centrosome-associated protein E (Cenpe) and 5–9 for Unc-5 netrin receptor C (Unc5c) RT-qPCR and 5–6 for all the other data. RNA-seq data were analyzed using an ANOVA one-way-like test in EdgeR based on generalized linear models with threshold of fold change >1.3 and false discovery rate <0.05. All RT-qPCR data except those of Cenpe were analyzed using two-way ANOVA followed by a Student-Newman-Keuls post hoc test. Cenpe RT-qPCR data were analyzed using a Kruskal-Wallis one-way ANOVA on Ranks followed by a Dunn’s multiple comparison post hoc test. *P, q<0.05 and **P, q<0.001 vs. respective SS, †P, q<0.05 and ††P, q<0.001 vs. respective NSD and ‡P, q<0.05 and ‡‡P, q<0.001 vs. respective S2Ba group. Aimp1, aminoacyl tRNA synthetase complex-interacting multifunctional protein 1; Bcl10, B-cell CLL/lymphoma 10; Ctbs, chitobiase; Ddah1, dimethylarginine dimethylaminohydrolase 1; Enpep, glutamyl aminopeptidase; H2afz, H2A histone family member Z; Hs2st1, heparan sulfate 2-O-sulfotransferase 1; and Pla2g12a, phospholipase A2, group XIIA.
RNA-seq showed that dimethylarginine dimethylaminohydrolase 1 (Ddah1) and mannosidase beta (Manba) mRNAs were decreased and phospholipase A2 group XIIA (Pla2g12a) mRNA was increased in S2Ba compared to SS rats under both diets (Figure 5B and Supplementals Figure S14). RT-qPCR validated the down-regulation of Ddah1 in S2Ba rats under HSD and up-regulation of Pla2g12a in S2Ba rats under both diets (Figure 5B). It also confirmed that Ddah1 was higher in S2Bb compared to S2Ba rats and revealed that it was higher than SS rats under NSD. In addition, it validated that HSD reduced Ddah1 in S2Bb but not S2Ba rats, and demonstrated that Ddah1 was down-regulated in S2Bb compared to SS rats under HSD. RT-qPCR also demonstrated that Pla2g12a was up-regulated in S2Bb compared to SS rats under NSD. Furthermore, it confirmed that Pla2g12a was lower in S2Bb compared to S2Ba rats but not different from SS rats under HSD. However, RT-qPCR did not validate the down-regulation of Manba in S2Ba compared to SS rats under both diets (Supplemental Figure S14). Accordingly, whether Ddah1 and Pla2g12a play a role in regulation of vascular inflammation in S2Ba rats under NSD and HSD remains uncertain.
RNA-seq showed that the mRNAs encoding aminoacyl tRNA synthetase complex-interacting multifunctional protein 1 (Aimp1), B-cell CLL/lymphoma 10 (Bcl10), centrosome-associated protein E (Cenpe), chitobiase (Ctbs), H2A histone family member Z (H2afz), and heparan sulfate 2-O-sulfotransferase 1 (Hs2st1) were up-regulated and that the mRNA encoding Unc-5 Netrin Receptor C (Unc5c) and synapse defective Rho GTPase homolog 2 (Syde2) were down-regulated in S2Ba compared to SS rats uniquely under HSD (Figure 5C and Supplemental Figure S14). RT-qPCR confirmed that Hs2st1 was up-regulated in S2Ba compared to SS rats uniquely under HSD and that it was up-regulated by HSD in S2Ba rats, which makes it a good pro-inflammatory candidate (Figure 5C). It also demonstrated that Hs2st1 was down-regulated in S2Bb compared to S2Ba rats under HSD. RT-qPCR indicated that Aimp1 was up-regulated and Unc5c down-regulated in S2Ba compared to SS rats under both diets. It also confirmed that Unc5c was upregulated in S2Bb compared to S2Ba rats under both diets. Therefore, whether Aimp1 and Unc5c are involved in regulation of vascular inflammation in S2Ba rats under NSD or HSD is unclear. RT-PCR did not validate that Syde2 was down-regulated by HSD in S2Bb rats Supplemental Figure S14. For the other genes, RT-qPCR demonstrated their up-regulation in S2Ba compared to SS rats under HSD and showed or validated that Bcl10, Cenpe, Ctbs, and H2afz were up-regulated by HSD in S2Bb rats compared to HSD-treated SS rats (Figure 5C). Although RT-qPCR showed almost similar results as RNA-seq for Cenpe, the changes did not reach significance due to dispersion of the data from the congenic rats under HSD. Since Bcl10, Cenpe, Ctbs, and H2afz were up-regulated by HSD in both S2Ba and S2Bb rats, they were not retained as vascular pro-inflammatory candidates.
Identification of DE genes encoded within BN Chr 2 middle fragment associated with higher blood pressure in S2Bb rats under HSD
Since we observed an association between higher BP under HSD and the introgressed middle fragment of the BN Chr 2, DE genes under HSD encoded within BN Chr 2 middle fragment were investigated to identify BP modulators. RNA-seq revealed that 3 mRNAs including arylsulfatase E (Arse), gap junction alpha-5 protein (Gjar, also known as connexin 40 [Cx40]) and high affinity cAMP-specific 3’,5’-cyclic phosphodiesterase 7A (Pde7a), were DE in S2Bb compared to SS rats only under HSD (Figure 4C). However, DE Pde7a was also DE in S2Ba rats under HSD (Supplemental Table S9). RNA-seq showed that Arse was decreased in S2Bb rats fed a HSD compared to SS rats fed a HSD and S2Bb rats fed a NSD, whereas Gjar was increased in S2Bb rats fed HSD compared to SS rats fed a HSD, S2Bb rats fed a NSD and S2Ba rats fed a HSD (Supplemental Figure S15). Arse and Gjar were further studied by RT-qPCR. The oligonucleotide primers used for qPCR are presented in Supplemental Table S10. RT-qPCR did not validate the lower expression of Arse in S2Bb rats compared to SS under HSD. However, it confirmed that Arse was decreased in S2Bb rats fed HSD compared to S2Bb rats fed a NSD, and revealed that Arse was down-regulated in S2Ba compared to both SS and S2Bb rats under NSD. Therefore, Arse could not be associated with the higher BP in S2Bb compared to the other groups of rats under HSD. RT-qPCR failed to validate the higher expression of Gja5 in S2Bb rats compared to S2Ba and SS rats under HSD. The increase in expression of Gja5 in S2Bb rats compared to S2Ba and SS rats under HSD did not reach significance due to dispersion of the data from the S2Bb rats. However, RT-qPCR confirmed that Gja5 was increased in S2Bb rats fed a HSD compared to NSD. It also demonstrated similar changes in the other groups: the Gja5 expression was increased in S2Ba rats and tended to be elevated in SS rats by HSD compared to NSD. Accordingly, it is uncertain whether Gja5 could be associated with the higher BP in S2Bb compared to the other groups of rats under HSD.
Discussion
The precise mechanisms regulating the molecular processes of aortic inflammation in congenic rats are complex. Genomic approaches represent powerful methods to discover the molecular basis of these mechanisms. We have previously shown that consomic S2B rats had reduced BP and vascular inflammation compared with SS rats under NSD.8 In the present study, we generated two congenic rat stains containing the BN Chr 2 distal (S2Ba) and middle segments (S2Bb), and mapped the reduced vascular inflammatory features of S2B rats under NSD to the BN Chr 2 distal portion. Unexpectedly, we discovered that introgression of the BN Chr 2 distal portion in S2Ba rats conferred increased vascular inflammation compared to SS rats under HSD. Finally, using RNA-seq and RT-qPCR, we identified two DE genes encoded within BN Chr 2 distal portion that could act as regulators of vascular inflammation: down-regulated Enpep as anti-inflammatory under NSD and up-regulated Hs2st1 as pro-inflammatory under HSD. Higher mean and diastolic BP under HSD were associated with the introgression of the BN Chr 2 middle segment, but no DE gene encoded within this BN Chr 2 fragment that was associated with this phenotype could be identified.
Two congenic rat strains with different introgressed BN Chr 2 segments were first generated to map the BN Chr 2 portion containing gene(s) regulating vascular inflammation. No BN Chr 2 portion was mapped for BP under NSD. Both congenic rat stains displayed lower BP than SS rats under NSD as previously observed for S2B rats.8 However, the middle fragment of BN Chr 2 mapped for higher BP under HSD. S2Ba rats presented similar BP as SS rats under HSD as previously observed for S2B rats,12 but S2Bb rats displayed higher mean and DBP. The higher BP in S2Bb rats could partly explain the greater incidence of seizures in this congenic strain compared to SS rats. The lower vascular inflammation observed in S2B rats under NSD8 was mapped to the BN Chr 2 distal portion. This was characterized by less oxidative stress, VCAM-1 and MCP-1 in aorta of S2Ba compared with SS fed NSD. Although there was no difference in BP between S2Ba and SS groups under HSD, increased vascular inflammation revealed by increased aortic oxidative stress, VCAM-1 and aortic perivascular adipose tissue MoMΦ infiltration compared to SS rats was also mapped to the BN Chr 2 distal portion. Unexpectedly, the higher BP in S2Bb rats was not associated with greater vascular inflammation, but a slight decrease in aorta ROS generation and a reduction in perivascular fat T cell infiltration. Vascular inflammation had never been reported for S2B rats fed a HSD. However, renal inflammation was demonstrated in S2B rats. Oxalate-induced tubule injury, oxalate crystal deposition and renal immune cell infiltration in BN rats were previously mapped to BN Chr 2.13
The comparison of congenic rats with SS rats demonstrated that the introgressed BN Chr 2 distal segment contains DE genes that are anti-inflammatory under NSD and pro-inflammatory under HSD. An unbiased approach, RNA-seq, was used to identify the genes regulating vascular inflammation under NSD and HSD. The whole genome expression profiling revealed distinct expression profiles for S2Ba and S2Bb rats compared to SS rats under NSD and HSD. Introgression of BN Chr 2 distal portion in S2Ba rats was associated with few DE genes under NSD whereas the largest number of DE genes were observed in S2Ba rats under HSD. The comparison and validation of DE genes encoded within BN Chr 2 distal portion allowed the identification of down-regulated Enpep under NSD and up-regulated Hs2st1 under HSD as regulators of vascular inflammation.
The down-regulation of Enpep was associated with lower vascular inflammation in S2Ba rats under NSD. In humans, genome-wide association studies revealed an association between ENPEP and hypertension, SBP or DBP.14–16 In mice, Enpep knockout was associated with elevated SBP.17 In this study, although down-regulated Enpep was not associated with BP, it was contained within the gene ontology term enrichment group “regulation of BP” (Supplemental Figure S10). Enpep encodes the membrane-bound metalloprotease glutamyl aminopeptidase (also know as aminopeptidase A [APA], EC 3.4.11.7). APA cleaves the N-terminal glutamatic and aspartatic amino acid residues from peptides. The Enpep murine homolog B-lymphocyte differentiation antigen BP-1/6C3 was originally cloned from mouse pre-B cells.18 BP-1/6C3 northern blot and APA immunohistochemistry demonstrated that APA is expressed in many tissues including spleen, lung, brain, kidney and gut, which suggests diverse physiological roles.18,19 APA plays a role in the renin-angiotensin system by converting angiotensin II to angiotensin III through cleavage of the N-terminal aspartic acid residue, and could play a role in BP regulation.20,21 APA could counteract angiotensin II-induced BP elevation by degrading angiotensin II.21 Exaggerated BP elevation in response to infusion of a low angiotensin II dose has been demonstrated in Enpep null mice deficient in APA.17 Bolus injection of recombinant APA decreased SBP in spontaneously hypertensive rats (SHR) but not in normotensive Wistar-Kyoto rats.22 HSD-induced BP elevation in SS rats was associated with a transient increase in renal APA activity at 10-weeks old that returns to basal levels by age 18 weeks, whereas HSD did not affect SBP in Dahl salt-resistant rats, but caused a sustained SBP elevation from 10 to 18 weeks of age.23 Development of specific inhibitors of APA (EC33) and aminopeptidase N (EC27 and PC18), the latter that degrades angiotensin III into angiotensin IV, has demonstrated that angiotensin III is the main brain renin-angiotensin system effector that causes BP elevation by inducing sympathetic activation, synaptic inhibition of the baroreflex in the nucleus of the tractus solitarius, and vasopressin release.20,24 Intracerebroventricular injection of EC33 decreased BP in SHR and deoxycorticosterone acetate (DOCA)-salt treated rats.25 However, intravenous injection of EC33 did not affect BP in SHR, which is in contradiction with the above observations. Central ENPEP/APA may be a main regulator of BP. Furthermore, treatment of SHR and DOCA-salt rats with firibastat (RB150), an EC33 prodrug that crosses the gastrointestinal and blood-brain barriers, enters the brain where it generates 2 active molecules of EC33 and blocks brain APA activity, reducing BP.26,27 It should be noted that firibastat had no BP effects on normotensive rats. This may explain the lack of BP effect in S2Ba rats fed a normal salt diet compared to S2Bb rats if Enpep expression is also reduced in the brain. S2Bb rats presented similar normal BP, but higher Enpep expression compared to S2Bb rats. Furthermore, it is possible that brain Enpep down-regulation reduced vascular inflammation in S2Ba rats fed a normal salt diet by decreasing sympathetic activation. It has been demonstrated in mice that increased sympathetic activation participates in immune cells activation and migration to target organs.28,29 It is noteworthy that the high salt diet increased Enpep expression in all the congenic rat groups, but to a much higher level in S2Bb rats. This may have contributed to exaggerating the high salt diet-induced BP elevation and seizures in these rats. However, it is unclear why this did not result in increased vascular inflammation compared to SS rats. The activation of another gene, such as Hs2st1, may be necessary (see below). Blocking brain APA is a novel anti-hypertensive therapeutic approach that is currently under investigation. Phase I clinical trials have demonstrated that firibastat is clinically and biologically well tolerated in healthy volunteers, and phase II clinical trials have demonstrated efficacy of firibastat in reducing BP in hypertensive patients.24 Aortic APA/angiotensin III may have other effects than BP regulation and Enpep down-regulation could reduce vascular inflammation by decreasing local action of angiotensin III. SBP was similarly lower in both congenic rats, but Enpep was decreased only in S2Ba rats. Recently, angiotensin III was shown to induce vascular smooth muscle cell proliferation through activation of p38 mitogen-activated kinase, c-Jun N-terminal kinase and interleukin 6 via activation of Janus kinase-2/signal transducer and activators of transcription-3.30–32 Furthermore, APA may participate in the generation of other peptides that are pro-inflammatory, and thus Enpep down-regulation could contribute to decrease vascular inflammation, protection that may be lost under HSD due in part to Enpep up-regulation under the latter conditions of salt intake. Interestingly, a genome-wide association study has identified fibrinogen A-α as an ENPEP target.33
The up-regulated gene Hs2st1 that was contained within the gene ontology term “enrichment group developmental process” (Supplemental Figure S11) was associated with enhanced vascular inflammation in S2Ba rats under HSD. Hs2st1 encodes heparan sulfate (HS) 2-O-sulfotransferase 1 that participates in HS biosynthesis.34–36 HS occurs in proteoglycans (HSPGs) that reside on the plasma membrane and are a major component of the extracellular matrix. HSPGs bind to a plethora of proteins such as members of the fibroblast growth factor (FGF) family and their receptors, transforming growth factors, bone morphogenetic proteins, Wnt proteins, chemokines and cytokines. HS is a member of the glycosaminoglycan family of carbohydrates and is constituted of a variety of sulfated repeated disaccharide unit that are important for the binding of proteins. HS2ST1 catalyzes the transfer of a sulfate group to the C2-position of the iduronic acid residue of HS. HSPGs are involved in a variety of biological activities such as developmental processes, cell signaling and morphogenesis, cellular crosstalk, and tissue injury and repair. Hs2st1 knockout in mice caused failure of kidney development.36,37 Endothelium-restricted Hs2st1 knockout increased neutrophil infiltration in acute inflammation.38 There is evidence that HS2ST1 participates in vascular injury. Deficiency in N-deacetylase N-sulfotransferase1, which is upstream of HS2ST1,35 caused reduction in N- and 2-O sulfation, decreased femoral artery smooth muscle number and vessel size, and reduced wire-induced vascular injury.39 Indeed, N-sulfate groups and iduronate-2-sulfate are essential for strong binding of FGF-2 to HS.40 In endothelial cells hypoxia-inducible factor-1a increased the expression of the key regulatory enzyme responsible for HS chain synthesis, 1,4 N-acetylglucosamine transferase and HS2ST. This was associated with increases in the content of HS, the number of low-affinity HS-FGF2 binding sites and enhanced cell sensitivity to FGF2-induced cell proliferation.41 Removal of HS by heparinase treatment inhibits FGF2-induced smooth muscle proliferation in balloon-injured rat carotid artery.42 Therefore, it can be speculated that HSD-induced increase in HS2ST could disturb HS protein binding function and signaling, causing changes in gene expression and increased vascular inflammation. In this study, apart from increased vascular inflammation, increased Hs2st1 expression in S2Ba rats under HSD was not associated with increased aortic MCSA, collagen or fibronectin content. However, S2Ba rats displayed similar high BP as SS rats, which could explain, at least in part, the lack of vascular remodeling and changes in collagen deposition and fibronectin expression. However, elevated Hs2st1 expression in S2Ba rats under HSD may contribute to development of further vascular injury over time.
In humans, HS2ST1 has been associated with SBP and DBP by analysis of the Genetic Analysis Workshop 18 data using a penalized multivariate linear mixed model.43 The expression of HS2ST1 and two other HS glycotransferases genes were shown to be decreased in the conjunctiva of patients with dry eyes.44 This could play a role in the down-regulation of Notch signaling and pathophysiology of dry eye disease. Notch receptors are glycosylated cell surface receptors. Interestingly, expression of HS2ST1 and other enzymes involved in HS and chondroitin sulphate chains biosynthesis increases through human B cell differentiation from CD27+ memory B cells to bone marrow plasma cells. Furthermore, increased HS2ST1 expression in multiple myeloma cells (malignant plasma cells) has been associated with a good prognosis.45 However, whether HS2ST1 plays a role in vascular inflammation remains unknown.
No DE genes encoded in the BN Chr 2 middle fragment could be associated with the greater BP under HSD observed in S2Bb rats compared to the S2Ba and SS rats. However, one of the two candidates, Gja5 that encodes Cx40 remains interesting as its expression seems to be regulated by HSD and tended to be higher in S2Bb rats. Cx40 is expressed in vascular cells, renal renin-producing cells, atrial cardiomyocytes and in the cardiac conduction system.46,47 Gja5 has been shown to play a role in flow-driven arteriogenesis. Gja5 knockout reduced femoral artery occlusion-induced formation of collateral arterial network.46 Germline and somatic mutations causing loss- or gain-of-function have been linked to atrial fibrillation (reviewed in48). Interestingly, it has also been shown that a single nucleotide polymorphism in the Cx40 promoter, the minor allele −44A, was associated with hypertension in men.47,49 However, whether Cx40 plays a role in salt-induced BP elevation remains to be determined.
Limitations
This study was designed to profile gene expression by RNA-seq in aorta of SS, S2Ba and S2Bb rats fed a NSD or HSD. Whether similar changes in gene expression exist in other vascular beds or tissues needs to be determined in a future study. This study was not designed to compare the effects of NSD and HSD on BP, aorta structure, vascular inflammation and gene expression. It was designed to identify genes regulating vascular inflammation under a NSD or HSD. The study, however, failed to identify DE genes encoded in the introgressed BN Chr 2 middle portion associated to a greater BP under HSD.
Perspectives
This study using an unbiased approach has identified two differentially expressed genes encoded within introgressed BN Chr 2 distal fragment, Enpep and Hs2st1, which were associated with reduced vascular inflammation under NSD and increased vascular inflammation under HSD, respectively. Understanding how changes in expression of these genes affects vascular inflammation may reveal new therapeutic targets to reduce vascular inflammation and end-organ damage in hypertension. Interestingly, blocking brain ENPEP is under investigation as an anti-hypertensive treatment in hypertensive patients.
Supplementary Material
Novelty and Significance.
What Is New?
We discovered that two differently expressed genes encoded within introgressed Brown Norway chromosome 2 distal portion, down-regulated Enpep and up-regulated Hs2st1, were associated with reduced and increased vascular inflammation under normal- and high-salt diet, respectively.
What Is Relevant?
Understanding the mechanisms of vascular inflammation may allow discovery of novel treatments in order to reduce end-organ damage in hypertension.
Summary
Down-regulated Enpep was associated with reduced vascular inflammation under a normal salt diet and up-regulated Hs2st1 was associated with enhanced vascular inflammation under a high salt diet. Understanding how vascular inflammation was affected by these genes under these different conditions of salt intake may reveal new therapeutic targets to reduce end-organ damage in hypertension.
Acknowledgments
We are grateful to laboratory technicians Adriana Cristina Ene, Guillem Colell Dinarès, and Isabelle Miguel, the Lady Davis Institute for Medical Research Imaging and Phenotyping core facility and the Sainte-Justine University Hospital Integrated Centre for Pediatric Clinical Genomic sequencing platform core facility for excellent technical support. This research was enabled in part by the computing infrastructure provided Calcul Québec (http://www.calculquebec.ca/) and Compute Canada (www.computecanada.ca).
Sources of Funding
This research was supported by Canadian Institutes of Health Research (CIHR) grant 123465 and CIHR First Pilot Foundation Grant 143348, a Tier 1 Canada Research Chair (CRC) on Hypertension and Vascular Research by the CRC Government of Canada/CIHR Program, and by the Canada Fund for Innovation, all to ELS; by a Terry Fox New Frontiers Program grant 217967 to DS; by National Institutes of Health (NIH) grant U01HL066579 to AEK and by a fellowships to OB (Société québécoise d’hypertension artérielle [SQHA]), and SO (CIHR Canada Graduate Scholarship-Master’s scholarship).
Footnotes
DISCLOSURES
The authors declared no conflict of interest.
References
- 1.Caillon A, Paradis P, Schiffrin EL. Role of immune cells in hypertension. Br J Pharmacol. 2019;176:1818–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ehret GB, Ferreira T, Chasman DI, et al. The genetics of blood pressure regulation and its target organs from association studies in 342,415 individuals. Nat Genet. 2016;48:1171–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kunert MP, Drenjancevic-Peric I, Dwinell MR, Lombard JH, Cowley AW Jr., Greene AS, Kwitek AE, Jacob HJ. Consomic strategies to localize genomic regions related to vascular reactivity in the Dahl salt-sensitive rat. Physiol Genomics. 2006;26:218–225. [DOI] [PubMed] [Google Scholar]
- 4.Alemayehu A, Breen L, Krenova D, Printz MP. Reciprocal rat chromosome 2 congenic strains reveal contrasting blood pressure and heart rate QTL. Physiol Genomics. 2002;10:199–210. [DOI] [PubMed] [Google Scholar]
- 5.Deng AY, Dene H, Rapp JP. Mapping of a quantitative trait locus for blood pressure on rat chromosome 2. J Clin Invest. 1994;94:431–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dutil J, Eliopoulos V, Marchand EL, Devlin AM, Tremblay J, Prithiviraj K, Hamet P, Migneault A, deBlois D, Deng AY. A quantitative trait locus for aortic smooth muscle cell number acting independently of blood pressure: implicating the angiotensin receptor AT1B gene as a candidate. Physiol Genomics. 2005;21:362–369. [DOI] [PubMed] [Google Scholar]
- 7.Garrett MR, Rapp JP. Multiple blood pressure QTL on rat Chromosome 2 defined by congenic Dahl rats. Mamm Genome. 2002;13:41–44. [DOI] [PubMed] [Google Scholar]
- 8.Viel EC, Lemarie CA, Benkirane K, Paradis P, Schiffrin EL. Immune regulation and vascular inflammation in genetic hypertension. Am J Physiol Heart Circ Physiol. 2010;298:H938–944. [DOI] [PubMed] [Google Scholar]
- 9.Cowley AW Jr., Liang M, Roman RJ, Greene AS, Jacob HJ. Consomic rat model systems for physiological genomics. Acta Physiol Scand. 2004;181:585–592. [DOI] [PubMed] [Google Scholar]
- 10.Cowley AW Jr., Roman RJ, Jacob HJ. Application of chromosomal substitution techniques in gene-function discovery. J Physiol. 2004;554:46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lagrange D, Fournie GJ. Generation of congenic and consomic rat strains. Methods Mol Biol. 2010;597:243–266. [DOI] [PubMed] [Google Scholar]
- 12.Mattson DL, Dwinell MR, Greene AS, Kwitek AE, Roman RJ, Jacob HJ, Cowley AW Jr. Chromosome substitution reveals the genetic basis of Dahl salt-sensitive hypertension and renal disease. Am J Physiol Renal Physiol. 2008;295:F837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wiessner JH, Garrett MR, Roman RJ, Mandel NS. Dissecting the genetic basis of kidney tubule response to hyperoxaluria using chromosome substitution strains. Am J Physiol Renal Physiol. 2009;297:F301–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forman JP, Fisher ND, Pollak MR, Cox DG, Tonna S, Curhan GC. Renin-angiotensin system polymorphisms and risk of hypertension: influence of environmental factors. J Clin Hypertens (Greenwich). 2008;10:459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kato N, Takeuchi F, Tabara Y, et al. Meta-analysis of genome-wide association studies identifies common variants associated with blood pressure variation in east Asians. Nat Genet. 2011;43:531–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Surendran P, Drenos F, Young R, et al. Trans-ancestry meta-analyses identify rare and common variants associated with blood pressure and hypertension. Nat Genet. 2016;48:1151–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitsui T, Nomura S, Okada M, Ohno Y, Kobayashi H, Nakashima Y, Murata Y, Takeuchi M, Kuno N, Nagasaka T, J OW, Cooper MD, Mizutani S. Hypertension and angiotensin II hypersensitivity in aminopeptidase A-deficient mice. Mol Med. 2003;9:57–62. [PMC free article] [PubMed] [Google Scholar]
- 18.Wu Q, Lahti JM, Air GM, Burrows PD, Cooper MD. Molecular cloning of the murine BP-1/6C3 antigen: a member of the zinc-dependent metallopeptidase family. Proc Natl Acad Sci U S A. 1990;87:993–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li L, Wu Q, Wang J, Bucy RP, Cooper MD. Widespread tissue distribution of aminopeptidase A, an evolutionarily conserved ectoenzyme recognized by the BP-1 antibody. Tissue Antigens. 1993;42:488–496. [DOI] [PubMed] [Google Scholar]
- 20.Gao J, Marc Y, Iturrioz X, Leroux V, Balavoine F, Llorens-Cortes C. A new strategy for treating hypertension by blocking the activity of the brain renin-angiotensin system with aminopeptidase A inhibitors. Clin Sci (Lond). 2014;127:135–148. [DOI] [PubMed] [Google Scholar]
- 21.Mizutani S, Ishii M, Hattori A, Nomura S, Numaguchi Y, Tsujimoto M, Kobayshi H, Murohara T, Wright JW. New insights into the importance of aminopeptidase A in hypertension. Heart Fail Rev. 2008;13:273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishii M, Hattori A, Numaguchi Y, Tsujimoto M, Ishiura S, Kobayashi H, Murohara T, Wright JW, Mizutani S. The effect of recombinant aminopeptidase A on hypertension in spontaneously hypertensive rats: its effect in comparison with candesartan. Horm Metab Res. 2008;40:887–891. [DOI] [PubMed] [Google Scholar]
- 23.Nomura M, Nomura S, Mitsui T, Suzuki M, Kobayashi H, Ito T, Itakura A, Kikkawa F, Mizutani S. Possible involvement of aminopeptidase A in hypertension and renal damage in Dahl salt-sensitive rats. Am J Hypertens. 2005;18:538–543. [DOI] [PubMed] [Google Scholar]
- 24.Llorens-Cortes C, Touyz RM. Evolution of a New Class of Antihypertensive Drugs: Targeting the Brain Renin-Angiotensin System. Hypertension. 2020;75:6–15. [DOI] [PubMed] [Google Scholar]
- 25.Reaux A, Iturrioz X, Vazeux G, Fournie-Zaluski MC, David C, Roques BP, Corvol P, Llorens-Cortes C. Aminopeptidase A, which generates one of the main effector peptides of the brain renin-angiotensin system, angiotensin III, has a key role in central control of arterial blood pressure. Biochem Soc Trans. 2000;28:435–440. [PubMed] [Google Scholar]
- 26.Marc Y, Gao J, Balavoine F, Michaud A, Roques BP, Llorens-Cortes C. Central antihypertensive effects of orally active aminopeptidase A inhibitors in spontaneously hypertensive rats. Hypertension. 2012;60:411–418. [DOI] [PubMed] [Google Scholar]
- 27.Marc Y, Hmazzou R, Balavoine F, Flahault A, Llorens-Cortes C. Central antihypertensive effects of chronic treatment with RB150: an orally active aminopeptidase A inhibitor in deoxycorticosterone acetate-salt rats. J Hypertens. 2018;36:641–650. [DOI] [PubMed] [Google Scholar]
- 28.Ahmari N, Santisteban MM, Miller DR, Geis NM, Larkin R, Redler T, Denson H, Khoshbouei H, Baekey DM, Raizada MK, Zubcevic J. Elevated bone marrow sympathetic drive precedes systemic inflammation in angiotensin II hypertension. Am J Physiol Heart Circ Physiol. 2019;317:H279–H289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carnevale D, Perrotta M, Pallante F, Fardella V, Iacobucci R, Fardella S, Carnevale L, Carnevale R, De Lucia M, Cifelli G, Lembo G. A cholinergic-sympathetic pathway primes immunity in hypertension and mediates brain-to-spleen communication. Nat Commun. 2016;7:13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alanazi AZ, Clark MA. Angiotensin III Induces JAK2/STAT3 Leading to IL-6 Production in Rat Vascular Smooth Muscle Cells. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed]
- 31.Alanazi AZ, Clark MA. Angiotensin III induces p38 Mitogen-activated protein kinase leading to proliferation of vascular smooth muscle cells. Pharmacol Rep. 2020;72:246–253. [DOI] [PubMed] [Google Scholar]
- 32.Alanazi AZ, Clark MA. Effects of angiotensin III on c-Jun N terminal kinase in Wistar and hypertensive rat vascular smooth muscle cells. Peptides. 2020;123:170204. [DOI] [PubMed] [Google Scholar]
- 33.Suhre K, Shin SY, Petersen AK, et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature. 2011;477:54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. [DOI] [PubMed] [Google Scholar]
- 35.Li JP, Kusche-Gullberg M. Heparan Sulfate: Biosynthesis, Structure, and Function. Int Rev Cell Mol Biol. 2016;325:215–273. [DOI] [PubMed] [Google Scholar]
- 36.Wilson VA, Gallagher JT, Merry CL. Heparan sulfate 2-O-sulfotransferase (Hs2st) and mouse development. Glycoconj J. 2002;19:347–354. [DOI] [PubMed] [Google Scholar]
- 37.Bullock SL, Fletcher JM, Beddington RS, Wilson VA. Renal agenesis in mice homozygous for a gene trap mutation in the gene encoding heparan sulfate 2-sulfotransferase. Genes Dev. 1998;12:1894–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Axelsson J, Xu D, Kang BN, Nussbacher JK, Handel TM, Ley K, Sriramarao P, Esko JD. Inactivation of heparan sulfate 2-O-sulfotransferase accentuates neutrophil infiltration during acute inflammation in mice. Blood. 2012;120:1742–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adhikari N, Basi DL, Townsend D, Rusch M, Mariash A, Mullegama S, Watson A, Larson J, Tan S, Lerman B, Esko JD, Selleck SB, Hall JL. Heparan sulfate Ndst1 regulates vascular smooth muscle cell proliferation, vessel size and vascular remodeling. J Mol Cell Cardiol. 2010;49:287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turnbull JE, Fernig DG, Ke Y, Wilkinson MC, Gallagher JT. Identification of the basic fibroblast growth factor binding sequence in fibroblast heparan sulfate. J Biol Chem. 1992;267:10337–10341. [PubMed] [Google Scholar]
- 41.Li J, Shworak NW, Simons M. Increased responsiveness of hypoxic endothelial cells to FGF2 is mediated by HIF-1alpha-dependent regulation of enzymes involved in synthesis of heparan sulfate FGF2-binding sites. J Cell Sci. 2002;115:1951–1959. [DOI] [PubMed] [Google Scholar]
- 42.Kinsella MG, Irvin C, Reidy MA, Wight TN. Removal of heparan sulfate by heparinase treatment inhibits FGF-2-dependent smooth muscle cell proliferation in injured rat carotid arteries. Atherosclerosis. 2004;175:51–57. [DOI] [PubMed] [Google Scholar]
- 43.Liu J, Huang J, Ma S. Penalized multivariate linear mixed model for longitudinal genome-wide association studies. BMC Proc. 2014;8:S73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mantelli F, Schaffer L, Dana R, Head SR, Argueso P. Glycogene expression in conjunctiva of patients with dry eye: downregulation of Notch signaling. Invest Ophthalmol Vis Sci. 2009;50:2666–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bret C, Hose D, Reme T, Sprynski AC, Mahtouk K, Schved JF, Quittet P, Rossi JF, Goldschmidt H, Klein B. Expression of genes encoding for proteins involved in heparan sulphate and chondroitin sulphate chain synthesis and modification in normal and malignant plasma cells. Br J Haematol. 2009;145:350–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buschmann I, Pries A, Styp-Rekowska B, et al. Pulsatile shear and Gja5 modulate arterial identity and remodeling events during flow-driven arteriogenesis. Development. 2010;137:2187–2196. [DOI] [PubMed] [Google Scholar]
- 47.Schmidt K, Kaiser FJ, Erdmann J, Wit C. Two polymorphisms in the Cx40 promoter are associated with hypertension and left ventricular hypertrophy preferentially in men. Clin Exp Hypertens. 2015;37:580–586. [DOI] [PubMed] [Google Scholar]
- 48.Bai D Atrial fibrillation-linked GJA5/connexin40 mutants impaired gap junctions via different mechanisms. FEBS Lett. 2014;588:1238–1243. [DOI] [PubMed] [Google Scholar]
- 49.Firouzi M, Kok B, Spiering W, Busjahn A, Bezzina CR, Ruijter JM, Koeleman BP, Schipper M, Groenewegen WA, Jongsma HJ, de Leeuw PW. Polymorphisms in human connexin40 gene promoter are associated with increased risk of hypertension in men. J Hypertens. 2006;24:325–330. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.