

Abstract
Introduction
Mucopolysaccharidosis type IV A (MPS IVA) or Morquio A syndrome is an autosomal recessive lysosomal storage disease caused by GALNS gene mutations that lead to a deficiency of the N-acetylgalactosamine-6-sulfate sulfatase enzyme and the accumulation of two glycosaminoglycans in cell lysosomes, namely, chondroitin and keratan sulfate.
Objective
To present two female patients with Morquio A syndrome in their late adult years (over 50 years of age) with a classical phenotype, treated with enzyme replacement therapy; and to present a summary of the natural history and the characteristics of the disease, and the benefit of comprehensive management.
Materials and methods
Descriptive clinical study before and after the treatment with enzyme replacement therapy as part of the comprehensive management of MPS IVA.
Results
Enzyme replacement therapy with elosulfase alfa was effective, with an adequate safety profile in these two patients, showing evidence of sustained improvement in terms of endurance and gait patterns.
Conclusion
We present two cases of MPS IVA, with longer survival than reported previously in classical phenotypes associated with this disease condition. There is a paucity of reports of similar cases in the literature. We believe that the clinical heterogeneity of the disease manifesting with the classical phenotype, together with comprehensive management, have played a role in the survival of these two patients. Therapy with elosulfase alfa as part of comprehensive management has been crucial; we suspect a clinical response and infer a better quality of life and reduced burden for the caregiver, supporting its use in older patients.
Keywords: Mucopolysaccharidosis IV A, Morquio A syndrome, Enzyme replacement therapy
Highlights
-
•
Case report of two patients with MPS IVA with exceptional survival for this type of disease condition.
-
•
Two factors are associated with survival: the clinical heterogeneity (attenuated phenotype) and comprehensive management.
-
•
Enzyme replacement therapy has been a crucial contributor to survival in these patients.
1. Introduction
Mucopolysaccharidosis type IV (MPS IV), also known as Morquio - Brailsford disease or syndrome (OMIM # 253000), was described concurrently in 1929 by Luis Morquio, pediatrician from Uruguay, in 4 children in Montevideo, [1] and by radiologist James Brailsford in Birmingham (England). [2] It is a rare autosomal recessive disease with a point prevalence ranging between 1/599,000 live births in United Kingdom and 1/926,000 live births in Australia. [3] There are two forms of MPS IV, type A and type B, depending on the enzyme involved. In MPS IVA, the deficient lysosomal enzyme is N-acetylgalactosamine-6-sulfate sulfatase (GALNS). This deficiency results in reduced or absent catabolism of two glycosaminoglycans (GAG), namely, chondroitin sulfate (CS) and keratan sulfate (KS), which are preferentially localized in cartilage and cornea. GALNS is a homodimeric glycoprotein formed by two monomers, each of them with two domains [4]. Both domain 1 (aminoacids 28 to 379) as well as domain 2 (aminoacids 380 to 481) define parts of the active site. [5] (Fig. 1).
Fig. 1.
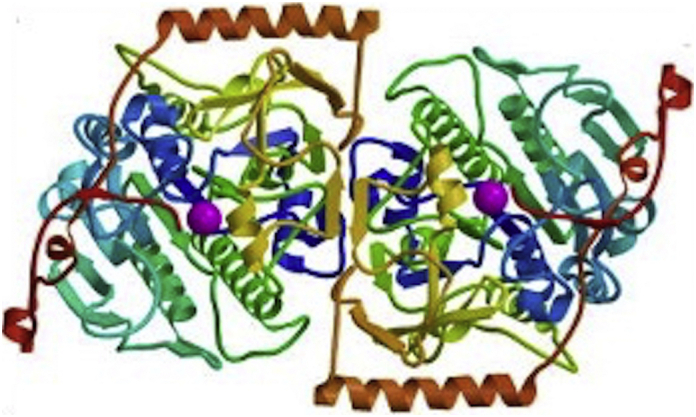
3D structure of GALNS.
Taken from: Rivera-Colón Y, Schutsky EK, Kita AZ, Garman SC. The structure of human GALNS reveals the molecular basis for MPS IVA. J Mol Biol. 2012 Nov 9;423 [5]:736–51. doi: https://doi.org/10.1016/j.jmb.2012.08.020. Epub 2012 Aug 29. PubMed PMID: 22940367; PubMed Central PMCID: PMC3472114.
The GALNS encoding gene is located on chromosome 16q24.3. It consists of 2339 nucleotides and contains 14 exons (50 Kb) with a single splicing product: a 1566 base pair messenger ribonucleic acid (mRNA) encoding a 522-aminoacid protein precursor. [6] Following a 21-aminoacid N-terminal signal peptide splitting and glycosylation, it turns into subunits of 40 and 15 kDa, forming a mature active enzyme. [[7], [8], [9]] Molecular analysis in different population groups has shown that, currently, there are more than 277 mutations, [10] the most frequent being missense and nonsense mutations. [[10], [11], [12]] Three missense mutations are described as the most frequent in the world population: p.R386C, p.G301C and p.I113F, but due to the great allele heterogeneity described, these mutations account for only 20% of all the alleles analyzed [10,13].
In a study conducted in 2018 in the Colombian population, the pathogenic p.Gly301Cys variant was reported in 51.6% of the alleles analyzed. [14,15] These results are consistent with a previous description in 1997, with a reported prevalence of 68.4% for the p.G301C mutation, considered as a founder mutation in the Colombian population [16]. At present, this mutation has also been reported in the Moroccan, Portuguese, French, Brazilian and Spanish populations, being more frequent in the latter (20%). The homozygous p.G301C mutation is related to a severe phenotype of the disease, given that it disrupts the hydrophobic core of the enzyme, affecting its folding but not its secondary structure (Fig. 2).
Fig. 2.

Mutational spectrum of the GALNS gene. Aminoacids corresponding to the active site of the GALNS enzyme are marked as (*): p.Asp39, p.Asp40, p.Cys79/dihydroxy alanine, p.Arg83, p.Tyr108, p.Lys140,p.His142, p.His236, p.Asp288, p.Asn289, and p.Lys310. Taken from Amelia Morrone et al., 2014 and Rivera-Colon et al., 2012.
This wide allelic heterogeneity in GALNS mutations is what supports the broad spectrum of clinical phenotypes observed in patients with MPS IVA. A genotype-phenotype correlation has been reported in some groups of patients, [11,[13], [14], [15], [16], [17], [18], [19], [20], [21], [22], [23], [24], [25], [26]] making it even more important to insist on early diagnosis followed by prompt initiation of treatment, in order to improve prognosis.
The clinical expression in MPS IVA depends to a large extent on GALNS residual activity and the accumulation and location of the storage material in the tissues. GAG accumulation occurs mainly in cartilage and its extracellular matrix (ECM), interrupting cell function and metabolism in cartilage and disrupting cartilage and bone development, with failed ossification, growth imbalance and progressive systemic skeletal dysplasia. [18] In MPS IVA, the heterogeneity and distribution of the storage material in different tissues results in a broad spectrum of symptoms as well as phenotype variations, leading to multi-system involvement with a chronic and progressive course of the disease. To date, the majority of reported phenotypes generally appear to be more severe, affecting 68.4% of patients, while 15.1% are classified as intermediate and 9.8% as mild. [11]
Patients usually do not present symptoms at birth, although some may have clubfoot or limited hip abduction. Initial signs become evident by one year of age, with the development of thoracolumbar hump or dorsal kyphosis, clearly noticeable in the sitting position. Stunting is observed by the second year of life, characterized by a short trunk in relation to limb length. In 60% of patients, findings include bone deformities (genu valgum, kyphosis, pectum carinatum), low stature, altered gait, instability due to joint laxity, and impaired articular motion. All patients with MPS IVA have dental abnormalities. Corneal opacity is a frequent in older patients and it is a finding which must not be missed. Hepatosplenomegaly is uncommon. Significant central nervous system manifestations of GAG are not evident, and the intellectual quotient appears to be preserved. Psychomotor development is normal, although it could be impaired by the presence of sensorineural or conductive hearing loss. Ambulation may be difficult because of the genu valgum deformity. It is important during early childhood to look for signs of spinal canal compression (rapid gait decline or pyramidal signs in the lower limbs).
It is estimated that the majority of patients with MPS IVA do not live beyond the second decade of life; however, patients with mild manifestations may reach 50–60 years of age. [27] In the fourth or fifth decade of life, significant thoracic deformities impair lung and heart function, shortening life expectancy. [11,19,28]
Most patients exhibit the classical phenotype described above, but others may have a late-progression phenotype with atypical signs such as stiffness and hip pain. [11]
Given the progressive nature of MPS IV A, early diagnosis is of crucial importance if management outcomes are to be optimized. Patients generally seek help due to a series of progressive multisystemic clinical and/or radiographic findings. [11,20] Definitive diagnosis is based on the metabolic workup, using a qualitative and/or quantitative GAG measurement in the urine and dry blood spot and/or leukocyte/fibroblast enzyme activity tests showing reduced GALNS activity. [11,29] Enzyme activity testing in leukocytes or fibroblasts is recommended not only when screening is positive or uncertain, but also when it is negative and clinical and radiological findings suggest MPS IVA. When enzyme activity testing in fibroblasts or leukocytes is inconclusive, the test must be repeated or molecular genetic testing must be carried out. Given that the diseases is autosomal recessive, enzyme detection of carriers among the patient's family members is carried out by means of molecular testing once the mutation in the index case has been identified. In cases of inconclusive enzyme test results, two pathogenic mutations must be identified in separate alleles in order to arrive at the definitive diagnosis. Reaching the right diagnosis is essential given the current availability of enzyme replacement therapy (ERT) for MPS IVA.
Elosulfase alfa is a recombinant form of the human N-acetylgalactosamine-6-sulfatase (rhGALNS) obtained by means of recombinant DNA technology from Chinese hamster ovarian cell cultures. [22] This molecule contains an oligosaccharide with a mannose 6-phosphate terminal which reaches the lysosome when taken up by the body, triggering the correct catabolism of chondroitin sulfate and keratan sulphate, thus avoiding their accumulation in the body as well as the complications of the disease. It is administered intravenously on a weekly basis at a dose of 2 mg/kg/week. [4,5] This molecule has been available since 2014, when approved by the European Medicine Agencies (EMA) and the Food and Drug Administration (FDA), for the treatment of mucopolysaccharidosis IVA; [20,23,24] in Colombia it was approved on March 2015. [30] Before it became available, the treatment for these patients was limited to palliative care based on symptom management. [4] Elosulfase alfa is a recognized therapy for patients with MPS IVA, and has been shown to stabilize KS in the urine, with ambulation improvement in the 6-min walk test (6MWT), improvement in the 3-min stair climb test (3MSCT), lung function tests, muscle tone and pain in these patients. [25,26]. Long-term follow-up in adult patients has shown that therapy with elosulfase alfa is associated with greater walking endurance and improved performance in activities of daily living. [31] Other treatments available are hematopoietic stem cell transplantation (HSCT) [[32], [33], [34]] and gene therapy [35]; both treatment options have reported on the literature cases in which follow up shows stabilization of the disease and a positive impact on natural history.
2. Methodology
Two clinical cases are reported and the presentation, management, follow-up and course of the disease are analyzed. A review of the topic was also conducted using the Medline database and the PUBMED, SCIENCEDIRECT and SCIELO specific search engines, in order to support the findings of the study.
3. Results
3.1. Index case 1
A 62-year-old female patient born in El Tambo (Cauca) to a G11P11A0 mother in her fifth pregnancy. Both parents were of caucano ancestry. Two siblings of the index case (female and male who died at 19 and 20 years of age, respectively) with a similar non-elucidated clinical picture (Fig. 3).
Fig. 3.

Family pedigree showing four affected individuals and two index cases; two affected individuals with the same clinical phenotype died at 19 and 20 years of age.
The patient has had difficulty walking since she was three years of age, with progressive abnormal leg position, chest deformity and growth delay. She lost her ability to walk when she was 54 (2014). Dyspnea and other persistent respiratory symptoms added to repeated pneumonia appeared in early childhood. There was also progressive loss of hearing after 5 years of age, with impaired verbal communication, as well as progressive loss of visual acuity.
The clinical genetic assessment conducted at 56 years of age showed a patient weighing 90 kg with a height of 94 cm, macrocrania, coarse features, flat nasal bridge, wide mouth, thick lips, bilateral cataracts, poor quality dental enamel with multiple cavities, severe kyphoscoliosis, short neck, short chest, increased antero-posterior diameter (pectum carinatum), horizontalized ribs, grade I/IV systolic murmur of mitral focus, no enlarged viscera, hyperlaxity of the hip joint with severe shortening of the 4 limbs, genu valgum and bilateral flat foot (Fig. 4, Fig. 5). Intellectual quotient is normal.
Fig. 4.
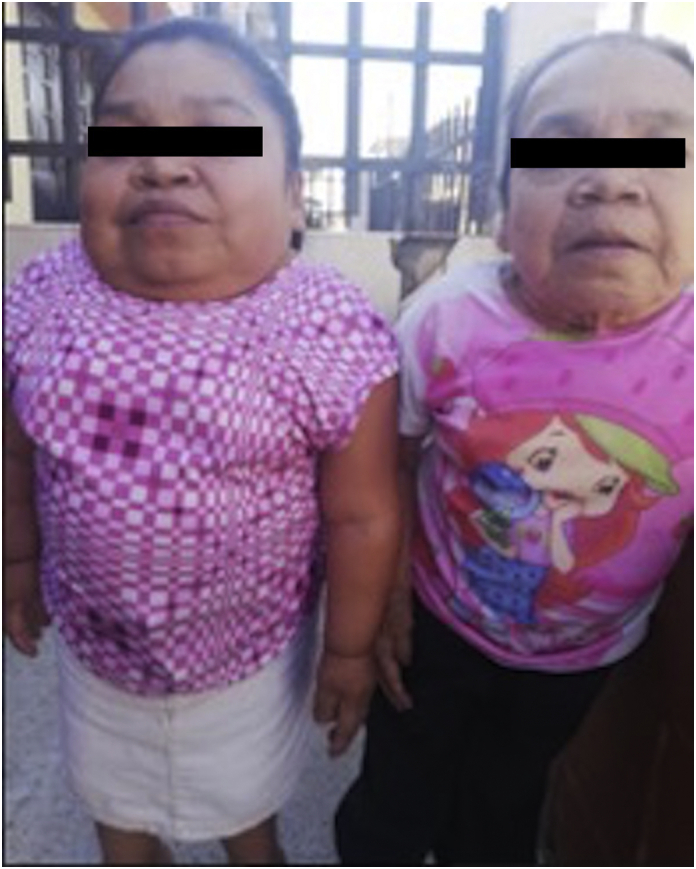
Body characteristics of the two cases showing disharmonic dwarfism with short trunk in relation to the lower limbs, overall shortening of the limbs with brachydactyly, bell-shaped chest, prominent sternum, kyphosis. Picture obtained after informed consent from the patients.
Fig. 5.
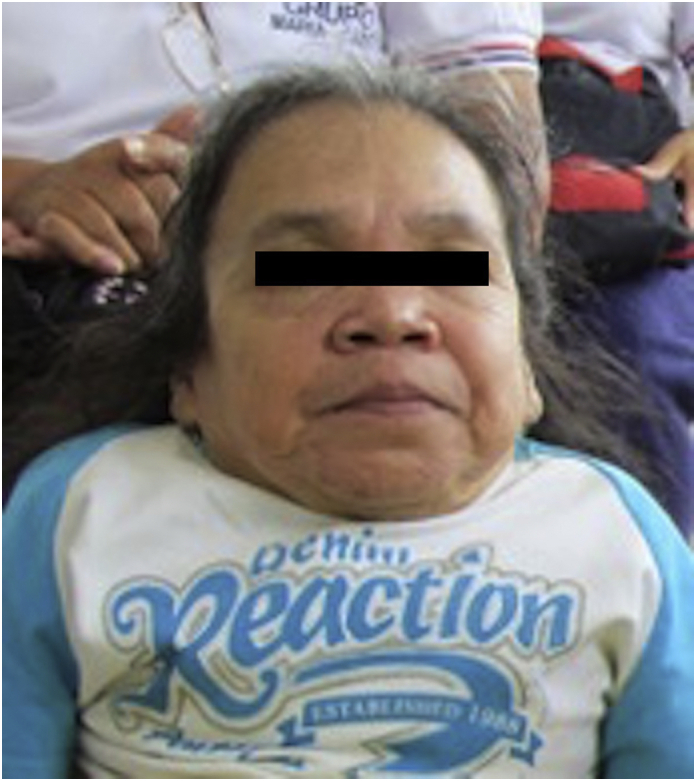
Facial characteristics of the index case. Evidence of broad forehead, macrostomia, square jaw, flat nasal bridge and short neck. Picture obtained after informed consent from the patient.
Auditory evoked potentials were consistent with severe bilateral profound sensorineural hearing loss in the left ear.
Fig. 6 shows the findings on the spinal x-ray. The film of the hip and lower limbs shows coxa valga, flattening of the femoral head, metacarpal hypoplasia with widening of both metacarpal and proximal and distal phalangeal metaphysis; reduced amplitude of metacarpal and intercarpophalangeal spaces, with joint surface sclerosis; bilateral distal radio-ulnar dislocation with ulnar variance, and interphalangeal soft tissue edema; hypoplasia of the first and fourth metatarsals with widening of proximal and distal metatarsal metaphyses; reduced amplitude in tarsal joint spaces and soft tissue edema.
Fig. 6.

Spinal radiograph: evidence of deviation secondary to vertebral body deformities with shortened height and anterior wedging, odontoid hypoplasia and platyspondylia.
Echocardiography shows evidence of dilated cardiomyopathy, moderate left ventricular dilation with severe concentric hypertrophy (left ventricular mass index [LVMI] 123 g/m2), sigmoid septum with dyssynchronic motion, no segmental contractility defects and 55% left ventricular ejection fraction (LVEF). Type I diastolic dysfunction, moderate mitral regurgitation, mild biatrial dilation, left atrial volume 29 ml, floppy atrial septum, mild aortic root dilation. Pulmonary artery systolic pressure (PASP) could not be measured due to tricuspid gradient. Spirometry could not be performed due to technical limitations for conducting the test with non-reproducible stress and short expiration time. However, there is no clinical evidence of lung function decline. Abdominal ultrasound does not show abnormalities associated with the disease. Patient receives integral management with physical, occupational, respiratory and rehabilitation therapies.
Based on these findings, the clinical diagnosis suggestive of MPS IV was made at 52 years of age. Mucopolysaccharide electrophoresis showed migration patterns corresponding to keratan and chondroitin sulfate, with positive GAG quantification in the urine. Table 1 shows the results of the enzymatic activity tests.
Table 1.
GALNS enzymatic activity in the index case.
| Betagalactosidase | 201.7 | 80–260 nmol/mg protein/H | Leukocytes |
|---|---|---|---|
| Galactose-6-sulfate sulfatase | 0.0 | Range in controls (N = 50): (2–35.9) | Leukocytes |
| Arylsulfatase B | 290.2 | 115–226 nmol/mg protein/H | Leukocytes |
NOTE: Enzymatic values are consistent with MPS IVA.
GALNS sequencing showed two pathogenic mutations described below:
-
•
Position chr16:88.904.105 Variation T > G p.Asn164Thr Heterozygocity
-
•
Position chr16:88.898.507 Variation C > A p.Gly301Cys Heterozygocity
Based on these findings, enzyme replacement therapy with elosulfase alfa was initiated in August 2016 with a weekly continuous intravenous dose of 2 mg/kg of body weight, leading to an improvement of the respiratory component in the form of absence of respiratory exacerbations, improvement in dyspnea and gait recovery (in 2017) after a little more than 12 months of therapy. At present, the subjective measurements reports a toleration in 500-m unsupported walk, with lower pain and fatigue levels during exercise. No objective measurements could be performed. The patient has not had any drug-related adverse events. She is receiving comprehensive management involving cardiology, pneumology, orthopedics, pain clinic, respiratory, physical and occupational therapy, physiatry and psychological rehabilitation, with trimestral evaluations by different specialties.
3.2. Index case 2
A 50-year-old patient coming from El Tambo (Cauca), sister of the first patient described above.
The onset of the disease at 7 years of age manifested with bone deformities and initial diagnosis of stunting (dwarfism), associated with progressive decline of visual and hearing acuity and intermittent ear ache, asymptomatic umbilical hernia, and difficulty walking due to severe genu valgum and articular hyperlaxity. At 9 years of age, she underwent bilateral knee osteotomy, with the development of debilitating pain of the left knee.
At 42 years of age, the patient complained of dyspnea with mild exertion and repeated bronchopulmonary episodes. These findings led to the diagnosis of chronic respiratory failure due to severe restrictive lung disease. At present, the patient is in a wheelchair and is oxygen-dependent, with non-productive cough, allergic rhinitis, bronchial hyperresponsiveness, and obstructive sleep apnea. She has severe bilateral hearing loss and has been fitted with hearing aids. Additional diagnoses include obesity, mixed hyperlipidemia, swallowing disorder and tricompartmental arthrosis of the left knee. Dilated cardiomyopathy was found on echocardiography. There is no evidence visceral enlargement. A clinical genetics assessment was performed for the first time at 44 years of age.
Findings on physical examination were: weight 25.6 kg, size 91 cm, grade I obesity, macrocrania, coarse features, prognathism, bilateral nasal pterygium, poor quality dental enamel, wide mouth, short neck, depressed nasal bridge, anteverted nostrils, malar hypoplasia, chest with increased AP diameter kyphoscoliosis, scant bibasal rhonchi, liver contour at 4 cm, global limb shortening with trunk/limb disproportion, severe genu valgum, brachydactyly, flexion contractures, bilateral flat foot (See Fig. 7).
Fig. 7.
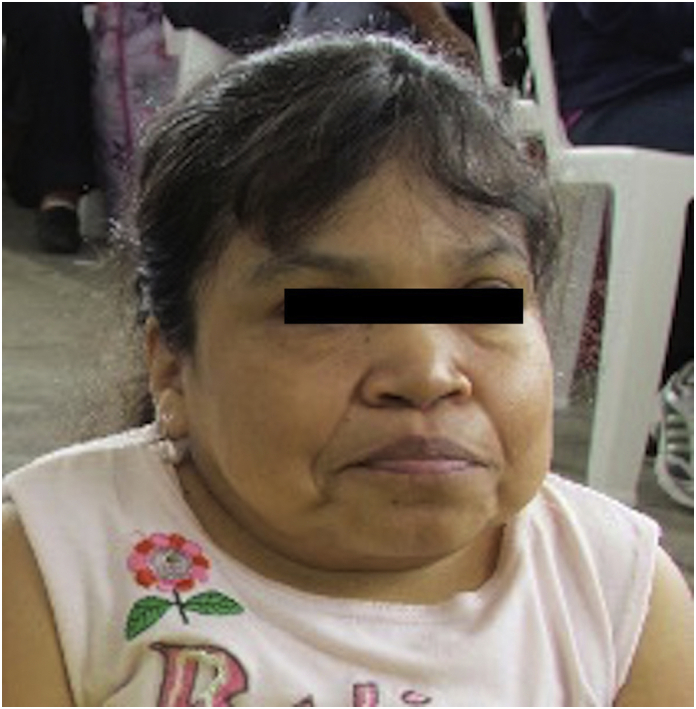
Facial characteristics of the index case's sibling: There is evidence of prognatism, macrostomia, flat nasal bridge, corneal opacity and short neck. Picture obtained after informed consent from the patient.
Cardiac imaging studies showed dilated cardiomyopathy with mild concentric hypertrophy (LVMI: 108 g/m2), sigmoid septum with dyssynchronic motion, LVEF 60%, type I diastolic dysfunction, mild mitral regurgitation, 30 ml of indexed left atrial volume, mild aortic root dilation, PASP 32 mmHg. The electrocardiogram showed sinus tachycardia and criteria for right ventricular hypertrophy. A Holter test performed in 2017 revealed ventricular and supraventricular extrasystoles.
The chest x-ray showed bilateral parahilar symmetrical reticular interstitial infiltrates; congestive pulmonary hilum, with solid nodular image in the left perilingular region and well-defined contours and edges; global cardiomegaly, widened costal arch deformities, and generalized abnormal bone tissue density (osteopenia) (Fig. 8).
Fig. 8.

Chest x-ray with bilateral parahilar symmetrical reticular interstitial infiltrates; congestive pulmonary hilum, with solid nodular image in the left perilingular region and well-defined contours and edges; global cardiomegaly, widened costal arch deformities, and generalized abnormal bone tissue density (osteopenia).
Tuberculin test was negative. Multiple atelectasis were seen on high resolution computed tomography (CT) scan. Respiratory home therapy was ordered. Audiometry showed severe bilateral sensory hearing loss.
Pelvis and hip radiographs showed bilateral coxofemoral joint morphological and structural changes, with femoral head destruction, osteochondral osteophytic reactions around the acetabular regions and preservation of greater and lesser femoral condyles, and diffuse bone mineralization alteration due to osteopenia. The full-length radiograph of the lower limbs showed bilateral genu valgum, with 12 and 40 degrees of right and left tibiofemoral angles, respectively; estimated 11 mm shortening of the left lower limb and diffuse alteration of bone mineralization due to osteopenia. Both hips showed loss of their spherical morphology with acetabular verticalization, suggesting advanced osteoarthrosis with dislocation. Marked arthritis of the tibiofemoral and ankle joints. CT and magnetic resonance imaging (MRI) of the left knee showed subchondral bone erosion with loss of bone substance in the femoral condyles and tibial plateaus, and synovial hypertrophy. Severe osteoarthrosis, complete rupture of the lateral meniscus and incomplete medial meniscal rupture, grade II injury of the anterior cruciate ligament, with pre-patellar bursitis. Patient have been on physical, occupational, respiratory and rehabilitation therapies, with periodic steroid infiltrations on her knee as part of pain management, along with analgesics and anti-inflammatory drugs.
Based on these findings and the family history, clinical genetics came to the diagnosis of MPS IVA.
Electrophoresis for urine mucopolysaccharides showed positive GAG migration patterns corresponding to keratan and chondroitin sulfate, with positive glycosaminoglycan quantification. Table 2 illustrates enzymatic activity results.
Table 2.
GALNS enzymatic activity. NOTE: Enzymatic values are consistent with MPS IVA.
| Betagalactosidase | 81.6 | 80–260 nmol/mg protein/H | Leukocytes |
|---|---|---|---|
| Galactose-6-sulfate sulfatase | 0.07 | Range in controls (N = 50): (2–35.9) | Leukocytes |
| Arylsulfatase B | 268.3 | 115–226 nmol/mg protein/H | Leukocytes |
GALNS sequencing was performed, showing evidence of two pathogenic mutations, as described below:
-
•
Position chr16:88.904.105 Variation T > G p.Asn164Thr Heterozygous
-
•
Position chr16:88.898.507 Variation C > A p.Gly301Cys Heterozygous
Based on these findings, enzyme replacement therapy with elosulfase alfa was initiated in August 2016, at a weekly continuous intravenous dose of 2 mg/Kg, with improvement of symptoms. Within 18 months of the start of treatment, the patient was able to leave the wheelchair and start progressive walks, initially with support, and then independently. The current 6-min walk test shows a distance of 208 m, i.e., 37.11% of the predicted estimated value. Consequently, an objective positive impact of the enzyme replacement therapy on quality of life is observed, with potential improvement in the burden for the caregiver and in the quality of life for the family. This impact may be considered clinically relevant.
The patient has not had any drug-related adverse events and she is receiving comprehensive management with the participation of other specialties (cardiology, pneumology, orthopedics, pain clinic, respiratory, physical and occupational therapy, psychology, physiatry and rehabilitation).
4. Discussion
We describe the clinical presentation, paraclinical findings, course and response to treatment in two patients with late diagnosis of the classical form of MPS IV A. Two older patients have been previously reported in the literature (ages 70 and 74), these patients shared clinical characteristics with our case report (short statue, abnormal gait, skeletal manifestations, airway obstruction and hearing lost), with an earlier diagnosis (age 4) and independent walking until later in life. [36] The analysis of the clinical findings is shown in Table 3. It is striking to see how two sisters with the same molecular findings may present subtle phenotypical variations which point to clinical heterogeneity and heterogenous distribution of the material stored in the different tissues in MPS IVA, with a broad spectrum of symptoms and multisystemic compromise. [11] In the molecular analysis, both patients have heterozygous mutations in p.G301C and p.N164T. The p.G301C mutation is associated with a severe disease phenotype because it alters the hydrophobic core of the enzyme, affecting its folding, but not its secondary structure. On the other hand, the p.N164T mutation found in the two patients with the classical phenotype has been reported in indeterminate phenotypes in the literature [37] and creates a change in the protein with a surface interruption, preventing the adequate formation of hydrogen bridges, specifically in domain 1. It is suggested that this mutation must be classified as indeterminate, considering that it is found in patients with both the severe and the attenuated phenotypes. [24]
Table 3.
Clinical characteristics of the patients.
| Clinical characteristic | Index case 1 | Index case sister |
|---|---|---|
| Very short stature | + | + |
| Odontoid hypoplasia | + | + |
| Pectum carinatum | + | + |
| Kyphoscolosis | + | + |
| Deformities of wrists, elbows and shoulders | + | + |
| Joint laxity | + | − |
| Femoral head flattening and scant femoral development | + | + |
| Genu valgum | + | + |
| Altered gait. Difficulty walking | + | + |
| Flattened facial features | + | − |
| Prognathism | + | + |
| Wide mouth | + | + |
| Dental diastemas and poor enamel quality | + | + |
| Diffuse corneal opacity | + | − |
| Hearing loss | + | + |
| Valve dysfunction | + | + |
| Myocardial infiltration | + | + |
| Obstructive sleep apnea | + | − |
| Obstructive and restrictive pulmonary disease | + | + |
| Umbilical hernia | + | − |
| Hepatomegaly | − | + |
| Cervical instability | + | + |
Up until only a few years ago, the treatment of MPS IVA was primarily limited to symptomatic support, symptom-based pharmacological management, physical therapy, surgery and rehabilitation. The first recombinant therapy (elosulfase alfa) was approved by the FDA on February 14, 2014, and on February 20 of the same year, the European Medicines Agency recommended its approval for the same indication.
Although enzyme replacement therapy has been shown to be of greater benefit in the pediatric population, this case report supports its use and benefit in older patients. The treatment with elosulfase alfa has been shown to have a favorable safety profile in these types of patients, and we suspect it has had a favorable outcome. Therapy has been associated with sustained endurance improvement in the 6-min walk test (6MWT) and in the 3-min stair climb test (3MSCT); it also results in improvements in respiratory parameters in populations of 57 years of age (MOR 004 and 005 Study). [[27], [28], [29],38] The two patients presented here, were able to recover their ability to walk and experienced improvement in pain and fatigue. One patient had 37.11% of the predicted estimated value in the 6MWT and her sister reported subjective improvement (500-m unsupported walk) so that one can suspect a clinical response and infer a better quality of life and reduced burden for the caregiver. These findings are consistent with reports in the literature regarding the positive impact of enzyme replacement therapy on lysosomal diseases, and support the importance of early initiation following diagnostic confirmation. [31] It is also worth highlighting the importance of multidisciplinary management and its impact during follow-up.
5. Conclusion
Mucopolysaccharidosis IVA is a rare disease associated with a high morbidity and mortality burden. There is limited knowledge of the disease at the present time. This fact, associated with a heterogenous clinical presentation, may explain underdiagnosis or late diagnosis of the disease. Consequently, in case of clinical suspicion, radiographic, biochemical and molecular testing is required in order to define diagnosis, prognosis and accurate early treatment of the disease so as to diminish complications and improve the quality of life of these patients.
In the two patients reported in this paper, enzyme replacement therapy with elosulfase alfa had an adequate safety profile; we suspect a clinical response and infer a better quality of life and reduced burden for the caregiver, supporting its use in older patients.
Ethical considerations
The two patients described in this case report gave their informed consent for the publication of their cases and the use of the photographic material.
Editorial coordination
Integralis HGS (Doctors Daniel Rodríguez and María Stella Salazar).
Article submission and verification statement
The authors certify that this article has not been submitted simultaneously to another journal or published previously in any form, and is not under consideration for submission. Additionally, publication was approved by all the authors and this article shall not be published in the same form in any other journal or in digital form without the consent of the copyright owner.
Authors' contributions
All the authors participated in the study design, data collection and/or patient recruitment, analysis of the results and drafting of the final manuscript.
Funding
Financial support for the retainer of a medical editor received from BioMarin Colombia Ltda. The sponsor did not play any role in the study design, data collection, data interpretation or drafting of the report.
Declaration of Competing Interest
None.
Acknowledgments
None.
References
- 1.Chudley A.E., Chakravorty C. Genetic landmarks through philately: Luís Morquio 1867-1935. Clin. Genet. 2002;62(6):438–439. doi: 10.1034/j.1399-0004.2002.620603.x. [DOI] [PubMed] [Google Scholar]
- 2.Brailsford J.F. Chondro osteo dystrophy. Roentgenographic and clinical features of a child with dislocation of vertebrae. Clin. Orthop. Relat. Res. 1976;114:4–9. doi: 10.1259/0007-1285-4-38-83. [DOI] [PubMed] [Google Scholar]
- 3.Leadley R.M., Lang S., Misso K., Bekkering T., Ross J., Akiyama T. A systematic review of the prevalence of Morquio a syndrome: challenges for study reporting in rare diseases. J Rare Dis. 2014;9:1–17. doi: 10.1186/s13023-014-0173-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rivera-Colón Y., Schutsky E.K., Kita A.Z., Garman S.C. The structure of human GALNS reveals the molecular basis for mucopolysaccharidosis IV a. J. Mol. Biol. 2012;423(5):736–751. doi: 10.1016/j.jmb.2012.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sukegawa K. Biochemical and structural analysis of missense mutations in N-acetylgalactosamine-6-sulfate sulfatase causing mucopolysaccharidosis IVA phenotypes. Hum. Mol. Genet. 2000;9(9):12. doi: 10.1093/hmg/9.9.1283. -83–90. [DOI] [PubMed] [Google Scholar]
- 6.Tomatsu S., Montaño A.M., Nishioka T., Gutierrez M.A., Peña O.M., Trandafirescu G.G. Mutation and polymorphism spectrum of the GALNS gene in mucopolysaccharidosis IVA (Morquio A) Hum. Mutat. 2005;26(6):500–512. doi: 10.1002/humu.20257. [DOI] [PubMed] [Google Scholar]
- 7.Montaño A.M., Kaitila I., Sukegawa K., Tomatsu S., Kato Z., Nakamura H. Mucopolysaccharidosis IVA: characterization of a common mutation found in Finnish patients with attenuated phenotype. Hum. Genet. 2003;113(2):162–169. doi: 10.1007/s00439-003-0959-8. [DOI] [PubMed] [Google Scholar]
- 8.Tomatsu S., Fukuda S., Masue M., Sukegawa K., Fukao T., Yamagishi A. Morquio disease: isolation, characterization and expression of full-length cDNA for human N-acetylgalactosamine-6-sulfate sulfatase. Biochem. Biophys. Res. Commun. 1991;181(2):677–683. doi: 10.1016/0006-291X(91)91244-7. [DOI] [PubMed] [Google Scholar]
- 9.Dorfman A., Arbogast B., Matalon R. The enzymic defects in Morquio and Maroteaux-Lamy syndrome. Adv. Exp. Med. Biol. 1976;68:251–276. doi: 10.1007/978-1-4684-7735-1_18. [DOI] [PubMed] [Google Scholar]
- 10.Morrone A., Caciotti A., Atwood R., Davidson K., Du C., Francis-Lyon P. Morquio a syndrome-associated mutations: a review of alterations in the GALNS gene and a new locus-specific database. Hum. Mutat. 2014;35(11):1271–1279. doi: 10.1002/humu.22635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hendriksz C.J., Harmatz P., Beck M., Jones S., Wood T., Lachman R. Review of clinical presentation and diagnosis of mucopolysaccharidosis IVA. Mol. Genet. Metab. 2013;110(1–2):54–64. doi: 10.1016/j.ymgme.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomatsu S., Dieter T., Schwartz I.V., Sarmient P., Giugliani R., Barrera L.A. Identification of a common mutation in mucopolysaccharidosis IVA: correlation among genotype, phenotype, and keratan sulfate. J. Hum. Genet. 2004;49(9):490–494. doi: 10.1007/s10038-004-0178-8. [DOI] [PubMed] [Google Scholar]
- 13.Pajares S., Alcalde C., Couce M.L., Del Toro M., González-Meneses A., Guillén E. Molecular analysis of mucopolysaccharidosis IVA (Morquio A) in Spain. Mol. Genet. Metab. 2012;106(2):196–201. doi: 10.1016/j.ymgme.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 14.Tapiero-Rodriguez S.M., Guio J.C.A., Porras-Hurtado G.L., García N., Solano M., Pachajoa H. Determination of genotypic and clinical characteristics of Colombian patients with mucopolysaccharidosis IVA. Appl. Clin. Genet. 2018;11:45–57. doi: 10.2147/TACG.S141881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moreno Giraldo L.J., Escudero Rodríguez Á.M., Sánchez Gómez A., Satizabal Soto J.M. Clinical and molecular characteristics of colombian patients with mucopolysaccharidosis IVA, and description of a new galns gene mutation. Mol Genet Metab Reports. 2018;16:53–56. doi: 10.1016/j.ymgmr.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kato Z., Fukuda S., Tomatsu S., Vega H., Yasunaga T., Yamagishi A. A novel common missense mutation G301C in the N-acetylgalactosamine-6-sulfate sulfatase gene in mucopolysaccharidosis IVA. Hum. Genet. 1997;101(1):97–101. doi: 10.1007/s004390050594. [DOI] [PubMed] [Google Scholar]
- 17.Tomatsu S., Dieter T., Schwartz I.V., Sarmient P., Giugliani R., Barrera L.A. Identification of a common mutation in mucopolysaccharidosis IVA: correlation among genotype, phenotype, and keratan sulfate. J. Hum. Genet. 2004 doi: 10.1007/s10038-004-0178-8. [DOI] [PubMed] [Google Scholar]
- 18.Peracha H., Sawamoto K., Averill L., Kecskemethy H., Theroux M., Thacker M. Molecular genetics and metabolism special edition: diagnosis and prognosis of Mucopolysaccharidosis IVA. Mol. Genet. Metab. 2018;125:18–37. doi: 10.1016/j.ymgme.2018.05.004.Molecular. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan S., Sawamoto K., Mackenzie W.G., Theroux M.C., Pizarro C., Mason R.W. Mucopolysaccharidosis IVA and glycosaminoglycans. Mol. Genet. Metab. 2017;120:78–95. doi: 10.1016/j.ymgme.2016.11.007.Mucopolysaccharidosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tomatsu S., Yasuda E., Patel P., Ruhnke K., Shimada T., Alméciga-díaz C.J. Morquio a syndrome: diagnosis and current and future therapies. Pediatr. Endocrinol. Rev. 2014;12(01):141–151. [PMC free article] [PubMed] [Google Scholar]
- 21.Tomatsu S., Fukuda S., Cooper A., Wraith J.E., Rezvi G.M.M., Yamagishi A. Mucopolysaccharidosis IVA: identification of a common missense mutation I113F in the N-acetylgalactosamine-6-sulfate sulfatase gene. Am. J. Hum. Genet. 1995;57(3):556–563. [PMC free article] [PubMed] [Google Scholar]
- 22.BioMarin Pharmaceutical Inc. Vimizin. N-actilgalactosamina-6-6sulfatasa recombinante humana . 2014. Elosulfasa alfa. Inser [Internet] [Internet] p. 12. [Google Scholar]
- 23.Food and Drug Administration . 2014. Vimizim (elosulfase alpha) injection, For Intravenous Use: US Prescribing Information. [Google Scholar]
- 24.Sanford M., Lo J.H. Elosulfase alfa: First global approval. Drugs. 2014 doi: 10.1007/s40265-014-0210-z. [DOI] [PubMed] [Google Scholar]
- 25.Regier D.S., Tanpaiboon P. Role of elosulfase alfa in mucopolysaccharidosis IVA. Appl. Clin. Genet. 2016;9:67–74. doi: 10.2147/TACG.S69080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hendriksz C.J., Giugliani R., Harmatz P., Mengel E., Guffon N., Valayannopoulos V. Multi-domain impact of elosufase alfa in Morquio a syndrome in the pivotal phase III trial. Mol. Genet. Metab. 2015;114(2):178–185. doi: 10.1016/j.ymgme.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 27.Lavery C., Hendriksz C. Mortality in patients with Morquio syndrome a. J. Inherit. Metab. Dis. 2015;15:59–66. doi: 10.1007/8904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Politei J., Schenone A.B., Guelbert N., Fainboim A., Szlago M. Enfermedad de Morquio ( mucopolisacaridosis IV-A ): aspectos clínicos, diagnósticos y nuevo tratamiento con terapia de reemplazo enzimático. Arch. Argent. Pediatr. 2015;113(4):359–364. doi: 10.5546/aap.2015.359. [DOI] [PubMed] [Google Scholar]
- 29.Hendriksz C.J., Berger K.I., Giugliani R., Harmatz P., Kampmann C., Mackenzie W.G. International Guidelines for the Management and Treatment of Morquio A Syndrome. Am J Med Genet A. 2014;167(A (1)):11–25. doi: 10.1002/ajmg.a.36833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ministerior de salud y protección Social, INVIMA. COMISIÓN REVISORA SALA ESPECIALIZADA DE MEDICAMENTOS Y PRODUCTOS BIOLÓGICOS. ACTA No. 2015:08. [Google Scholar]
- 31.Hughes D., Giugliani R., Guffon N., Jones S.A., Mengel K.E., Parini R. Clinical outcomes in a subpopulation of adults with Morquio a syndrome: results from a long-term extension study of elosulfase alfa. Orphanet J Rare Dis. 2017 doi: 10.1186/s13023-017-0634-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor M., Khan S., Stapleton M., Wang J., Chen J., Wynn R. Hematopoietic stem cell transplantation for mucopolysaccharidoses; past, present, and future. Biol Blood Marrow Transpl. 2019;25(7):e226–e246. doi: 10.1016/j.bbmt.2019.02.012.Hematopoietic. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yabe H., Tanaka A., Chinen Y., Kato S., Yasuda E., Shintaku H. Hematopoietic stem cell transplantation for Morquio a syndrome. Mol. Genet. Metab. 2016;117(2):84–94. doi: 10.1016/j.ymgme.2015.09.011.Hematopoietic. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chinen Y., Higa T., Tomatsu S., Suzuki Y., Orii T., Hyakuna N. Long-term therapeutic efficacy of allogenic bone marrow transplantation in a patient with mucopolysaccharidosis IVA. Mol Genet Metab Reports [Internet]. 2014;1(1):31–41. doi: 10.1016/j.ymgmr.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hodges B.L., Cheng S.H. Cell and gene-based therapies for the lysosomal storage diseases. Curr Gene Ther. 2006;6:227–241. doi: 10.2174/156652306776359522. [DOI] [PubMed] [Google Scholar]
- 36.Peretz R.H., Flora C.H., Adams D.J. Natural history of the oldest known females with mucopolysaccharidosis type IVA (Morquio A syndrome) Am J Med Genet Part A. 2020;182(6):1491–1495. doi: 10.1002/ajmg.a.61566. [DOI] [PubMed] [Google Scholar]
- 37.Montaño A.M., Sukegawa K., Kato Z., Carrozzo R., Di Natale P., Christensen E. Effect of “attenuated” mutations in mucopolysaccharidosis IVA on molecular phenotypes of N-acetylgalactosamine-6-sulfate sulfatase. J. Inherit. Metab. Dis. 2007;30(5):758–767. doi: 10.1007/s10545-007-0702-z. [DOI] [PubMed] [Google Scholar]
- 38.Akyol M.U., Alden T.D., Amartino H., Ashworth J., Belani K., Berger K.I. Recommendations for the management of MPS IVA: systematic evidence- a nd consensus-based guidance. Orphanet J Rare Dis. 2019;14(1):1–21. doi: 10.1186/s13023-019-1074-9. [DOI] [PMC free article] [PubMed] [Google Scholar]