

Abstract
COPD and idiopathic pulmonary fibrosis (IPF) together represent a considerable unmet medical need, and advances in their treatment lag well behind those of other chronic conditions. Both diseases involve maladaptive repair mechanisms leading to progressive and irreversible damage. However, our understanding of the complex underlying disease mechanisms is incomplete; with current diagnostic approaches, COPD and IPF are often discovered at an advanced stage and existing definitions of COPD and IPF can be misleading. To halt or reverse disease progression and achieve lung regeneration, there is a need for earlier identification and treatment of these diseases. A precision medicine approach to treatment is also important, involving the recognition of disease subtypes, or endotypes, according to underlying disease mechanisms, rather than the current “one-size-fits-all” approach. This review is based on discussions at a meeting involving 38 leading global experts in chronic lung disease mechanisms, and describes advances in the understanding of the pathology and molecular mechanisms of COPD and IPF to identify potential targets for reversing disease degeneration and promoting tissue repair and lung regeneration. We also discuss limitations of existing disease measures, technical advances in understanding disease pathology, and novel methods for targeted drug delivery.
Short abstract
Treatment outcomes with COPD and IPF are suboptimal. Better understanding of the diseases, such as targetable repair mechanisms, may generate novel therapies, and earlier diagnosis and treatment is needed to stop or even reverse disease progression. https://bit.ly/2Ga8J1g
Introduction
COPD and idiopathic pulmonary fibrosis (IPF) both represent a substantial unmet clinical need. COPD has become the third leading cause of death globally [1], and IPF has a median survival of ∼3 years after diagnosis, with survival rates comparable to some aggressive cancers [2], and no observed improvement in survival from 2000 to 2012 [3].
In this article, we discuss the current limitations of treatment for COPD and IPF, and potential future strategies with a focus on disease subtypes and lung regeneration and repair. This article is based on discussions at a meeting organised by the authors on 28–29 November 2018 in Gothenburg, Sweden, and supported by AstraZeneca, involving leading global experts in obstructive lung disease who discussed the latest innovations and issues in chronic lung disease (the participants are listed in the Acknowledgements section).
COPD
COPD is a largely preventable and treatable disease characterised by persistent airflow limitation and respiratory symptoms due to chronic inflammation, which causes structural changes, such as fibrosis of the small airways and alveolar wall destruction (emphysema) [1, 4]. Early pathological changes occur in the small airways, with associated inflammation, wall thickening, peribronchiolar fibrosis and loss of terminal and transitional bronchioles and associated vasculature, before the onset of emphysema [5–9]. These early and irreversible destructive events do not initially affect the lung function parameters usually used to define COPD (forced expiratory volume in 1 s (FEV1)/forced vital capacity ratio), making early detection difficult [10]. Smoking is a key risk factor for COPD; however, nonsmokers can also develop COPD (especially in low- and middle-income countries) and many smokers do not develop COPD [11], indicating a role of genetic risk, epigenetics and other environmental factors in its development [12]. The most documented genetic risk factor for COPD is α1-antitrypsin deficiency that represents a specific subtype of COPD (endotype), although different phenotypes exist within this subtype that appear to be caused by variations in other factors, such as tumour necrosis factor (TNF)-α [13]. Autoimmunity and aberrant immunity (suppression of host defence mechanisms and dysfunction of innate immunity) may also contribute to disease progression, especially as the disease advances [14–16].
IPF
IPF is commonly described as a specific form of chronic, progressive, fibrosing, interstitial pneumonia of unknown cause, usually occurring in the elderly [17]. IPF incidence appears to be increasing: the diagnosis of IPF in the UK has increased from approximately 20 per 100 000 patients in 2000 to nearly 40 per 100 000 patients in 2012 [3, 18]. IPF arises from repetitive micro-injuries to the bronchial and alveolar epithelium, which, along with immune system dysregulation [19, 20], results in progressive scarring and the destruction of lung structures [18, 21, 22]. The aetiology of IPF is unknown [3], but smoking is a risk factor and may influence IPF onset [23, 24]. Major genetic risk factors, such as the mucin 5B gene and defective telomerase, have been identified, pointing to future genetic stratification [25, 26]. Of note, pulmonary emphysema and IPF can co-exist in the same patient as a distinct entity termed combined pulmonary fibrosis and emphysema (CPFE) [27]. This condition is characterised by emphysema in the upper lobes and fibrosis in the lower lobes of the lungs [27, 28]. CPFE is estimated to occur in up to 35% of patients with IPF [29].
Limitations of current treatment approaches
Commonly used maintenance treatments in COPD include β2-agonists, anticholinergics, theophylline and corticosteroids [1]. Such treatments are primarily for improving lung function, reducing symptoms and the risk of exacerbations, and improving exercise tolerance and health status. To date, no disease-modifying treatments are available [30]. This is in sharp contrast with other chronic inflammatory-based diseases. In rheumatoid arthritis, for example, scientific advances and early treatment with disease-modifying drugs have resulted in the prevention of disease progression in up to 90% of patients [31].
Treatment options for IPF are even more limited and represent a pressing unmet clinical need [32]. Currently, only two antifibrotic drugs are recommended; namely, pirfenidone and the tyrosine kinase inhibitor nintedanib [17]. However, a recent analysis revealed that 40% of patients with confirmed IPF did not receive antifibrotics, reflecting a possible lack of understanding around the diagnosis and management of the disease, and problems with treatment access [33]. Furthermore, although these treatments may be life-extending [34, 35], potential adverse events could negatively impact quality of life [17], such as gastro-intestinal effects and photosensitivity with pirfenidone, and diarrhoea with nintedanib [36, 37].
Future treatment strategies for COPD and IPF
The marked unmet needs in COPD and IPF therapy highlight the need for new treatment strategies that focus on underlying disease endotypes, regeneration and repair.
A change in mindset is required among pulmonologists, regulators and policymakers to redefine perceptions of COPD and IPF. Current treatments have a “magic bullet” approach, where a single drug is intended to treat all forms of disease. However, both COPD and IPF are heterogeneous diseases with several clinical phenotypes [38] that may reflect multiple but, as of yet, mostly unidentified endotypes (subtypes of disease defined functionally and pathologically by molecular mechanism or treatment response) [39–41]. A move to a “complex subtypes” approach, where precision medicine allows COPD or IPF subtype-specific treatment, could be possible with combinations of interventions. Future treatment strategies may target different aspects of these diseases chronologically, or target several disease mechanisms simultaneously, with subsequent treatment withdrawal upon improvement. For new treatment strategies that focus on underlying disease endotypes, it will be crucial to study COPD and IPF at an early stage before confounding factors, comorbidities and disease progression mask subtle differences.
In the same way, both diseases have been described as “irreversible” [4, 42, 43]. However, lung regeneration, disease reversal and even a cure for COPD and IPF are the ultimate goals in disease management; merely slowing disease progression is important but does not completely address the unmet clinical need [44, 45]. Regeneration efforts could focus on activating the endogenous repair capacity of the lungs, and/or adopting exogenous regeneration through tissue engineering, bio-artificial scaffolds or adding healthy progenitor or stem cells to the lungs [46]. Evidence from retinoic acid studies shows that lung regeneration is feasible, at least in rodent models [47, 48]; however, we need a clear understanding of how endogenous repair processes become dysfunctional in the diseased lungs to identify targets for potential treatment strategies.
New targets for lung regeneration are being identified, but many of these may not be druggable via conventional approaches using either small-molecule inhibitors/activators or systemic antibodies [49]. New modality treatments are being developed, such as approaches using proteolysis-targeting chimaera (PROTAC), inhaled antisense oligonucleotides, gene editing (CRISPR: clustered regular interspaced short palindromic repeats) or exosomes that will allow us to target all pathways of interest (table 1). Although direct delivery to the target organ, in this case the lungs, is possible with inhaled approaches, new methodologies for delivering treatments are also needed.
TABLE 1.
Examples of emerging techniques to deliver therapy in patients with COPD and idiopathic pulmonary fibrosis (IPF) and their limitations
| Technique | Description | Uses and advances made with the technique | Current limitations |
| PROTAC | Proteolysis-targeting chimaera that uses the cell's ubiquitin–proteasome system to target-specific proteins for degradation |
|
|
| CRISPR | Can manipulate gene function through gene deletion, correction or replacement; enhancement of gene expression; base editing |
|
|
| Inhaled antisense oligonucleotides | Single-stranded DNA or RNA (20–21 base pairs) complementary to the target mRNA |
|
|
| Exosomes as delivery systems | A potential delivery system for nucleic acid drugs |
|
|
PROTAC: proteolysis-targeting chimaera; CRISPR: clustered regularly interspaced short palindromic repeats.
Several practical considerations must also be borne in mind. The age and frailty of patient populations with COPD and IPF are likely to be among the greatest challenges to lung regeneration, as well as the existence of comorbid diseases. To achieve significant lung regeneration, it is likely that COPD and IPF treatment will need to be at an earlier stage and in younger patients compared with what currently occurs in clinical practice. This argument is supported by evidence suggesting that early treatment and early smoking cessation have a positive effect on longitudinal lung function and symptoms [57]. COPD is currently diagnosed using spirometry [1], but these changes detected by spirometry occur relatively late in disease progression and are a poor measurement of peripheral airway obstruction in early disease [10]. Redefining COPD based on abnormalities in small airway function, measured using techniques such as magnetic resonance imaging and impulse oscillometry [58, 59], may identify disease earlier than current practises [10, 60] (table 2). Population screening of smokers for COPD could also be a possibility, but screening for IPF less so as it is an uncommon disease and difficult to diagnose [33].
TABLE 2.
Examples of new or emerging techniques for studying COPD and idiopathic pulmonary fibrosis (IPF)
| Technique | Description | Uses and advances made with the technique | Current limitations |
| Micro-CT imaging | High-resolution CT imaging |
|
Performed on ex vivo samples, or explants, rather than on the patient [6, 61, 62] |
| PET | Molecular imaging; most commonly measuring 18F-FDG uptake |
|
Validation of imaging approaches required; changes in lung air, blood and water volumes depending on disease severity can cause variations in signals [63] |
| Gas diffusion MRI | Noble gases such as 3He and 129Xe used to visualise lung structure |
|
Adaption of existing scanners is required [66] |
| SPECT | Radiotracers used to image the lung, where both airways and blood flow can be imaged |
|
|
| IOS | Noninvasive measurement of respiratory mechanics in response to pressure oscillations |
|
|
| OCT | A high-resolution optical imaging method |
|
|
| Multiple-breath nitrogen washout | Noninvasive measurement of residual nitrogen in the airways to detect any abnormalities in gas distribution in the lung |
|
|
| Breathomics | Exhaled breath analysis to detect changes in volatile organic compounds |
|
|
CT: computed tomography; PET: positron emission tomography; 18F-FDG: 18F-2-fluoro-2-deoxy-d-glucose; MRI: magnetic resonance imaging; SPECT: single-photon emission computed tomography; IOS: impulse oscillometry; OCT: optical coherence tomography.
Biomarkers
Biomarkers are central in identifying patient subgroups, phenotypes and endotypes [8, 78, 79]. They are crucial in monitoring and predicting disease progression and predicting responders to treatment [8, 79]. COPD and IPF are highly complex and heterogeneous, and no single biomarker has been identified for clinical applications in either disease [80–82]. Dividing COPD and IPF into endotypes is critical for breaking the diseases down into molecular pathways and disease mechanisms, and for linking molecular mechanisms to clinical features. Treatment targets for specific endotypes could thus be identified and could provide precision treatment to those patients most likely to respond [40, 83]. In the management of cancer, it has long been recognised that genetic mutations can give rise to cancer subtypes that predict prognosis and response to treatment [84]. A similar rationale needs to be applied to COPD and IPF to identify subgroups with distinct disease mechanisms [40, 83].
The Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) and the Genetic Epidemiology of COPD (COPDGene) studies have identified numerous putative biomarkers in COPD. These include protein, cellular and genetic biomarkers associated with COPD characteristics and morbidity (including airflow limitation, emphysema and exacerbation frequency) [85–87]. The analysis of six inflammatory biomarkers (white blood cell count, C-reactive protein, interleukin-6, C-X-C motif chemokine ligand 8, TNF-α and fibrinogen) from patients in ECLIPSE led to the identification of a new COPD phenotype [88].
In IPF, several biomarkers are associated with specific phenotypes [89]. Protein degradation biomarkers and serum biomarkers have been identified that can discriminate between healthy individuals, patients with stable IPF and those with progressive IPF [90]. Four serum proteins have been identified from the metaplastic epithelium that could predict disease progression and mortality; namely, surfactant protein D, matrix metalloproteinase-7, carbohydrate antigen 19–9 and cancer antigen-125 [91]. A gain-of-function variant of the promotor of the mucin 5B (MUC5B) gene is associated with the pathogenesis of IPF [25]; identifying this variation in patients with pre-clinical IPF and targeting MUC5B could enable early diagnosis and prevent the progression of IPF to a state where the lung remodelling is irreversible [25, 92].
Early detection
As we learn more about COPD and IPF endotypes and phenotypes, advances in technology are required to identify these in patients and to allow for the early detection of disease and to monitor disease progression. Reliable tests for small airway function and the ability to quantify disease progression and its links to biomarkers will be essential for advancing our knowledge and for the management of COPD and IPF. Advances in the “-omics” field (for example, genomics, transcriptomics, proteomics, lipidomics and metabolomics) have led to new discoveries and promises to provide insights into endotypes. For instance, it may be possible to use blood samples to detect genomic biomarkers [93], and bioinformatic analysis may identify the activation of particular molecular pathways that could be targeted [94]. Also, breathomics of exhaled breath may help identify COPD phenotypes and provide biomarkers for diagnosis and disease progression [77].
A potential barrier to early, preventative therapies may be that patients who do not feel unwell or who are not experiencing any impact on their quality of life may be reticent about taking medication with associated side-effects. Payers may also be resistant to paying for medication when the current classifications of disease categorise the patient as “at risk” rather than having measurable disease, although it is noteworthy that primary prevention measures do exist in other diseases; for example, the treatment of systemic hypertension and hypercholesterolaemia to prevent cardiovascular diseases [95].
Since late disease is associated with profound structural damage to the lung that is currently irreversible [4, 96], there is great interest in identifying potentially treatable processes much earlier in disease, especially those around regeneration and repair [97–101].
Dysregulated processes presenting opportunities for regeneration and repair
Cellular senescence
Accelerated ageing and senescence are evident in the lungs of patients with COPD and IPF [102, 103]. This can be brought on by DNA damage, mitochondrial dysfunction, telomere shortening, reduced autophagy and stem cell exhaustion, and involves cell cycle arrest and a secretory profile of inflammatory proteins. This is central to lung development and wound repair. In the healthy individual, once wound repair is complete, senescent cells are removed following apoptosis triggered by infiltrating immune phagocytes. However, if senescent cells are not removed, their abnormal secretory profile can lead to pathological tissue changes [104].
Several steps in the senescence pathway could be targeted to halt accelerated ageing and senescence. Cellular senescence is associated with a loss of anti-ageing molecules, such as certain sirtuins and Klotho [105, 106]. The microRNA miR-34a is increased by activation of the phosphoinosite-3-kinase (PI3K)-mammalian target of rapamycin (mTOR) pathway, and downregulates the expression of sirtuin-1 and sirtuin-6; up-regulation of miR-34a in the lungs and cells of patients with COPD results in loss of sirtuin-1 and -6 [106, 107]. miR-570 is also increased in COPD and is activated by p38 mitogen-activated protein kinase, resulting in the downregulation of sirtuin-1 [108]. Inhibition of miR-34a and miR-570 with antagomirs rescues the loss of sirtuin-1 and sirtuin-6, thereby preventing the induction of senescence [106, 108]; the therapeutic administration of antagomirs, possibly via inhaled exosomes [55], could therefore represent a strategy to reverse accelerated ageing [106, 108].
Oxidative stress, via increased reactive oxygen species production or decreased antioxidants, is central in driving senescence in COPD through the activation of the PI3K-mTOR pathway [102, 109]. Reactive oxygen species are potentially generated by the mitochondria in response to cigarette smoke [102, 103]. This results in secretion of a senescence-associated secretory phenotype of inflammatory proteins, which include pro-inflammatory cytokines, chemokines, growth factors and proteases and may account for the low-grade inflammation seen in COPD and the affected patient's systemic circulation [105, 110]. The mTOR inhibitor rapamycin prevents senescence and inhibits components of the senescence-associated secretory phenotype in vitro in pulmonary artery smooth muscle and pulmonary vascular endothelial cells isolated from patients with COPD [109]. The effective dose of rapamycin was low and did not affect cell growth rate, suggesting relatively low doses may be sufficient for a therapeutic effect, thereby reducing potential toxicity.
Multiple cell types are affected by senescence in COPD and IPF, including epithelial, endothelial, fibroblast and smooth muscle cells in COPD [108, 109, 111] and epithelial cells and fibroblasts in IPF [104, 112, 113]. The elimination of senescent cells, or senolysis, is another approach to tackle senescence. Experimental models have shown that the senolytic agents dasatinib and quercetin kill senescent cells and improve lung function [114], and a pilot study of these agents in patients with IPF has shown improvements in physical function with an acceptable safety profile over a 3-week period [115]. The senolytic drug navitoclax (ABT-263) has also been found to reverse pulmonary fibrosis and induce apoptosis in myofibroblasts implicated in fibrosis in animal models [99, 100].
Wnt/β-catenin signalling
The Wnt signalling pathway guides cells to certain fates during lung development and maintains tissue homeostasis in adulthood [116]. Wnt/β-catenin signalling is reduced in mouse models of elastase- and cigarette smoke-induced emphysema, which were attenuated upon Wnt activation with improvements observed in alveolar epithelial structure and function [98]. Cells affected by this pathway include alveolar epithelial type II (ATII) cells in the alveolar epithelium, which have self-renewal properties and rely on Wnt/β-catenin signalling to differentiate to ATI cells in response to alveolar epithelial injury [116]. ATI cells cover the majority of the lung surface area (95–97%) and are responsible for gas exchange, a key lung function [117]. The noncanonical Wnt ligand WNT-5A, which is overexpressed in lungs from animal models of COPD and patients with COPD, antagonises the canonical Wnt/β-catenin signalling pathway resulting in the inhibition of murine lung epithelial cell wound healing and transdifferentiation from ATII to ATI cells in vitro [118]. Lung-specific overexpression of WNT-5A exacerbated the development of emphysema, and prophylactic inhibition of WNT-5A could recuperate alveolar cell function and attenuate lung pathogenesis in COPD animal models [118]. In addition, activation of canonical Wnt/β-catenin signalling with lithium chloride improved alveolar epithelial structure and function in experimental models of emphysema [98]. The canonical Wnt receptor frizzled-4 (FZD4) facilitates ATII to ATI transdifferentiation. FZD4 expression was reduced in patients with COPD, correlating positively with lung function and negatively with smoking (pack-years) [119]. Cigarette smoke directly downregulated FZD4 in vivo and in vitro, thereby preventing Wnt/β-catenin signalling and alveolar tissue repair [119].
Interestingly, activated Wnt/β-catenin signalling in IPF leads to an increase in the Wnt target, WNT-1-inducible signalling protein-1 (WISP1), which in turn induces the expression and secretion of profibrotic mediators, contributing to lung fibrosis [120, 121]. Using antibodies to neutralise WISP1, Königshoff et al. [121] showed reduced pulmonary fibrosis, implicating WISP1 as a potential therapeutic target in IPF.
Several novel approaches to activating and inhibiting Wnt/β-catenin signalling are now in development and look promising with regard to restoring normal lung function in COPD and IPF [97, 122]. These discoveries point to several approaches that could reinstate cell and tissue homeostasis in COPD and IPF.
Stem cell therapy
The basal stem cells (BSCs) in the cartilaginous airways of the lungs are considered to be multipotent lung progenitor cells [123, 124] and drive homeostasis of the normal epithelium and regeneration following injury [123]. Therefore, they could be a potential regeneration target; targeting their proliferation and directing differentiation and stem cell transplantation/bioengineering [123]. In a study of smokers, reductions in the number and function of BSCs were observed in those with COPD compared with those without COPD [125]. Interestingly, low BSC counts in smokers without COPD were associated with lower lung function than in those with high BSC counts [125], which could represent an early pre-diagnostic stage of COPD. However, BSCs isolated from heavy smokers undergoing lung cancer surgery were found to have an increased proliferate potential in vitro compared with never-smokers, whereas ATII cell proliferation decreased [126]. This is in part because BSCs repair damaged DNA by nonhomologous end-joining, which is faster but more error-prone than homologous repair and increases the risk of mutagenesis [126].
Elevated BSCs were observed in the bronchoalveolar lavage fluid from patients with IPF in comparison to healthy individuals [127]. Normally located at the bronchoalveolar duct junction, BSCs were enriched in the alveolar compartment and frequently within fibrotic lesions of patients with IPF [127, 128]. This suggests an unexpected role of BSCs in the pathogenic bronchiolisation of the alveoli in IPF, where bronchial cells appear in this compartment by migration or transdifferentiation [127, 128].
While BSCs theoretically represent an opportunity to reverse COPD- and IPF-associated damage, we need to distinguish between healthy BSCs and those potentially carrying DNA mutations to enhance the positive effects without increasing the risk of pathogenic changes. The interactions between BSCs and immune cells and their role in IPF pathogenesis also need to be understood before BSCs can be considered as therapy.
A population of mesenchymal progenitor cells positive for stage-specific embryonic antigen (SSEA)-4, a cell-surface protein expressed by stem cells, has been identified in the lungs of patients with IPF. These SSEA-4 cells were found to display a pathological gene expression pattern, and their progeny developed a pathological IPF fibroblast phenotype [129]; these cells could be targeted as a therapeutic intervention, although it remains to be seen whether some SSEA-4+ cells are beneficial.
Biological molecules
The vasculature should also not be overlooked when elucidating the mechanisms of lung degeneration and seeking targets for lung regeneration. Retinoic acid is a morphogen that drives tissue regeneration [130, 131] and can induce alveolar regeneration in animal models [47, 48]. In humans, retinoic acid is involved in maintaining the lung microvascular endothelium through up-regulating angiogenesis; in emphysema, expression of the retinoic acid-processing enzyme cytochrome P450 26A1 is elevated in the endothelium, potentially reducing the availability of retinoic acid [132]. It follows that retinoic acid could be a treatment option for lung regeneration; however, early-phase clinical trials of retinoic acid in emphysema have failed to show a clinical benefit [133–135], which underlines the need to understand more about retinoic acid in lung regeneration and whether retinoids can induce lung regeneration.
Hepatocyte growth factor (HGF; also known as scatter factor) promotes airway and bronchoalveolar branching in the developing lung [136, 137], possibly through interaction with vascular endothelial growth factor [138]. HGF also promotes the proliferation and survival of airway epithelial cells [139], plays a role in wound healing [140] and has been shown to improve airspace morphology in emphysema models [139]. Levels of both HGF and vascular endothelial growth factor are reduced in smokers with COPD in comparison to smokers without COPD and nonsmokers, which could contribute to pathogenesis [141], indicating that HGF-enhancing therapy could represent a treatment opportunity for COPD and IPF [142].
Receptor tyrosine kinase pathways have been implicated in aberrant lung remodelling, potentially through growth arrest-specific 6 ligand, TYRO3 protein kinase 3 and Axl [143]. Inhibiting this pathway led to decreased fibrotic responses in vivo and in vitro, suggesting that targeting the receptor tyrosine kinase pathway could be a promising therapeutic strategy [143].
Granulocyte colony-stimulating factor (G-CSF) has been found to be elevated in the lungs of patients with COPD [101]. Interestingly, deletion of G-CSF in a mouse model of COPD led not only to substantially less inflammation and reduced fibrosis in the lung parenchyma and small airways, but it also reduced systemic inflammation and led to improvements in the comorbidities associated with COPD [101], suggesting that G-CSF is a potential therapeutic target in COPD [101].
Role of the extracellular matrix
The extracellular matrix (ECM) plays a central role in guiding cell behaviour and in tissue repair and remodelling. An ex vivo model used bronchial ECM from patients with COPD that was stripped of cells and then repopulated with normal human bronchial cells. The model revealed that the COPD-derived ECM modified the gene expression profile of these healthy cells upon differentiation, altering the activity of mediators involved in regeneration, remodelling, apoptosis, vascularisation and inflammation [144]. Similarly, fibroblasts grown on a stiff matrix resembling a fibrotic ECM, as occurs in IPF, were driven to a myofibroblast phenotype with elevated fibrotic activity, compared with fibroblasts grown on a softer ECM resembling healthy tissue [145]. Such findings emphasise that we need to fully understand the contribution of the ECM in disease and lung regeneration, as the enzymes involved in ECM remodelling could be potential therapeutic targets.
Lessons for the future
Advances in treating obstructive lung diseases such as COPD and IPF have been slow, and improvements in patient outcomes and drug discovery have been poor in respiratory medicine compared with other diseases [146]. Currently, clinical trials require a large number of patients to be assessed over a long period to detect any differences in end-points [146], which could delay results and ultimately slow medical advances. The high costs of such large trials needed to show clinically meaningful effects have discouraged investment in new drug development; many drugs also fail in phase 2 and 3 clinical trials, leading to a rethinking of trial design [146, 147]. Clinical trial design needs to be “smarter”. The design should focus on the biology of the disease and the drug mechanism of action, and end-points should be appropriate for the drug's mechanism of action to establish target engagement; improved proof-of-concept or adaptive trials could help rule out ineffective compounds early on to reduce wasted time and costs [146, 148]. If it is anticipated that a biological or clinical effect of the drug would be observed after a certain time, the trial need only last as long as that period. Also, only the subgroup of patients to whom the drug is targeted should be enrolled, even if this results in a relatively small number of patients. Furthermore, any group of patients responding particularly well to a drug should be closely investigated to understand why.
Recognition of the limitations in current therapies for COPD and IPF, which result in a substantial unmet clinical need, points to possible future treatment strategies. For example, a move to precision medicine as opposed to the “magic bullet” approach could lead to therapeutic advances in these highly complex diseases of varying endotypes. In addition, a change in mindset is needed from considering these diseases to be irreversible, to a focus on early diagnosis when reversibility may be possible (figure 1).
FIGURE 1.
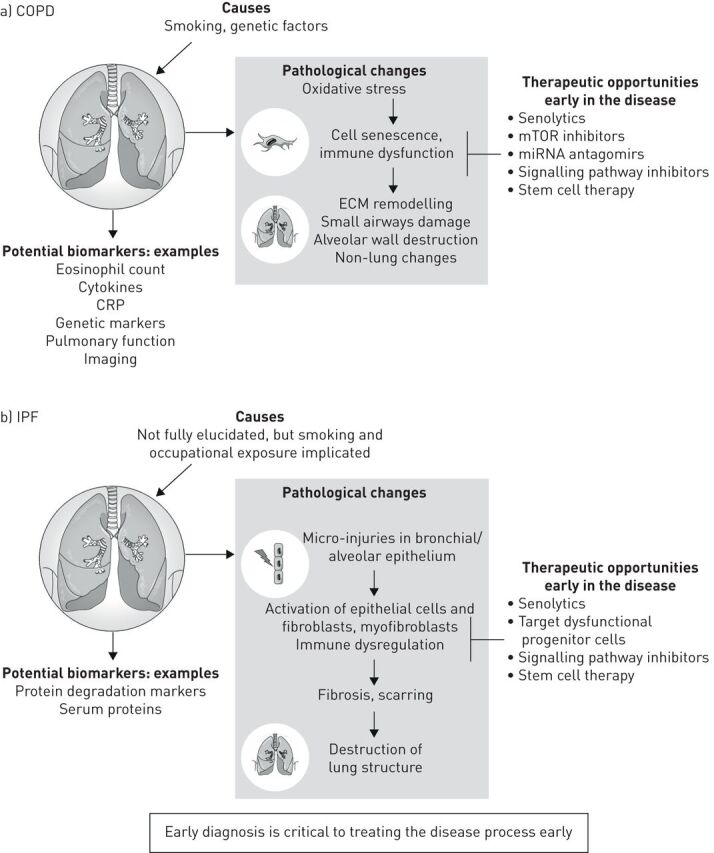
Causes, pathogenesis and opportunities for therapeutic intervention in a) COPD and b) idiopathic pulmonary fibrosis (IPF). CRP: C-reactive protein; ECM: extracellular matrix; miRNA: microRNA; mTOR: mammalian target of rapamycin.
The advances in our knowledge of lung degeneration in COPD and IPF raise further questions. For example, is fibrosis a protective mechanism to prevent peripheral airway destruction? Do terminal bronchioles undergo fibrosis? What is the mechanism for the loss of small airways in early disease? To understand and treat COPD and IPF more effectively, we need clear molecular profiles of the disease; we also need to understand why some areas of the lung are affected and why others are not.
Conclusion
In summary, the considerable body of research into COPD and IPF has yet to translate into improvements in clinical practice. A paradigm shift is required to move the focus to earlier in the disease course, to understand the disease mechanisms more fully, and to measure multiple aspects of the disease. COPD and IPF need to be redefined to better capture the patient populations involved and shift the conceptions about each disease. Novel technologies and the field of “-omics” are providing new insights into COPD and IPF, increasing our ability to predict outcomes and helping to identify new potential therapies to achieve lung regeneration.
Acknowledgements
Meeting attendees: Serge Adnot (Hôpital Henri Mondor & Université Paris Est, Créteil, France), Gary Anderson (University of Melbourne, Melbourne, Australia), Marie-Liesse Asselin-Labat (Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia; Oregon Health and Science University, Portland, OR, USA), Jonathan Baker (Imperial College, London, UK), Peter Barnes (Imperial College, London, UK), Maria Belvisi (AstraZeneca, Gothenburg, Sweden), Francois Xavier Blé (AstraZeneca, Cambridge, UK), Mia Collins (AstraZeneca, Gothenburg, Sweden), James Crapo (National Jewish Health, Denver, CO, USA), Sean Davis (AstraZeneca, Cambridge, UK), Oliver Eickelberg (University of Colorado Denver, Denver, CO, USA), Malin Fagerås (AstraZeneca, Gothenburg, Sweden), Donna Finch (MedImmune Ltd, Cambridge, UK), Hani Gabra (AstraZeneca, Cambridge, UK), Angus Hamblin (AstraZeneca, Gothenburg, Sweden), Petra Hazon (AstraZeneca, Gothenburg, Sweden), Matthew Hind (Royal Brompton Hospital & Imperial College, London, UK), Cory Hogaboam (Cedars-Sinai Medical Center, Los Angeles, CA, USA), Jim Hogg (St Paul's Hospital, Vancouver, Canada), Ellinor Hornberg (AstraZeneca, Gothenburg, Sweden), Rod Hughes (AstraZeneca, Cambridge, UK), Simon Johnson (University of Nottingham, Nottingham, UK), Christina Keen (AstraZeneca, Cambridge, UK), Roland Kolbeck (MedImmune, Gaithersburg, MD, USA), Melanie Königshoff (University of Colorado Denver, Denver, CO, USA), Brian Lipworth (University of Dundee, Dundee, UK), Alex Mackay (AstraZeneca, Gothenburg, Sweden), Toby Maher (Royal Brompton Hospital & Imperial College, London, UK), Anna Malmgren (AstraZeneca, Gothenburg, Sweden), Marc Miravitlles (Hospital Universitari Vall d'Hebron, Barcelona, Spain), Catherine Overed-Sayer (MedImmune Ltd, Cambridge, UK), Edward Piper (AstraZeneca, Cambridge, UK), Adam Platt (AstraZeneca, Cambridge, UK), Ioannis Psallidas (AstraZeneca, Cambridge, UK), Steve Rees (AstraZeneca, Cambridge, UK), Stephen Rennard (AstraZeneca, Cambridge, UK), John Taylor (AstraZeneca, Gothenburg, Sweden), and Jim Wild (University of Sheffield, Sheffield, UK).
Medical writing support (including development of the first draft under author direction) was provided by Sarah Hoyle and Kate Silverthorne (CMC Connect, McCann Health Medical Communications Ltd, Macclesfield, UK) and funded by AstraZeneca (Cambridge, UK), in accordance with Good Publication Practice guidelines.
Footnotes
Provenance: Submitted article, peer reviewed
Conflict of interest: P.J. Barnes reports grants and personal fees from AstraZeneca and Boehringer Ingelheim, and personal fees from Novartis, Teva and Pieris, outside the submitted work.
Conflict of interest: G.P. Anderson served on secondment as Chief Scientist in the Respiratory, Inflammation and Autoimmune division of AstraZeneca in 2016.
Conflict of interest: M. Fagerås is an employee of AstraZeneca.
Conflict of interest: M.G. Belvisi reports personal fees from AstraZeneca, during the conduct of the study.
Support statement: The development of this article was funded by AstraZeneca, Cambridge, UK.
References
- 1.Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease (2020 report). Available from: https://goldcopd.org
- 2.Vancheri C, Failla M, Crimi N, et al. Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J 2010; 35: 496–504. doi: 10.1183/09031936.00077309 [DOI] [PubMed] [Google Scholar]
- 3.Strongman H, Kausar I, Maher TM. Incidence, prevalence, and survival of patients with idiopathic pulmonary fibrosis in the UK. Adv Ther 2018; 35: 724–736. doi: 10.1007/s12325-018-0693-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barnes PJ, Burney PG, Silverman EK, et al. Chronic obstructive pulmonary disease. Nat Rev Dis Primers 2015; 1: 15076. doi: 10.1038/nrdp.2015.76 [DOI] [PubMed] [Google Scholar]
- 5.Hogg JC. Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet 2004; 364: 709–721. doi: 10.1016/S0140-6736(04)16900-6 [DOI] [PubMed] [Google Scholar]
- 6.McDonough JE, Yuan R, Suzuki M, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med 2011; 365: 1567–1575. doi: 10.1056/NEJMoa1106955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koo HK, Vasilescu DM, Booth S, et al. Small airways disease in mild and moderate chronic obstructive pulmonary disease: a cross-sectional study. Lancet Respir Med 2018; 6: 591–602. doi: 10.1016/S2213-2600(18)30196-6 [DOI] [PubMed] [Google Scholar]
- 8.Galbán CJ, Han MK, Boes JL, et al. Computed tomography-based biomarker provides unique signature for diagnosis of COPD phenotypes and disease progression. Nat Med 2012; 18: 1711–1715. doi: 10.1038/nm.2971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Capron T, Bourdin A, Perez T, et al. COPD beyond proximal bronchial obstruction: phenotyping and related tools at the bedside. Eur Respir Rev 2019; 28: 190010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johns DP, Walters JA, Walters EH. Diagnosis and early detection of COPD using spirometry. J Thorac Dis 2014; 6: 1557–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rennard SI, Vestbo J. COPD: the dangerous underestimate of 15%. Lancet 2006; 367: 1216–1219. doi: 10.1016/S0140-6736(06)68516-4 [DOI] [PubMed] [Google Scholar]
- 12.Wain LV, Shrine N, Miller S, et al. Novel insights into the genetics of smoking behaviour, lung function, and chronic obstructive pulmonary disease (UK BiLEVE): a genetic association study in UK Biobank. Lancet Respir Med 2015; 3: 769–781. doi: 10.1016/S2213-2600(15)00283-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wood AM, Simmonds MJ, Bayley DL, et al. The TNFalpha gene relates to clinical phenotype in alpha-1-antitrypsin deficiency. Respir Res 2008; 9: 52. doi: 10.1186/1465-9921-9-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson GP. Advances in understanding COPD. F1000Res 2016; 5: 2392. doi: 10.12688/f1000research.7018.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H, Ying H, Wang S, et al. Imbalance of peripheral blood Th17 and Treg responses in patients with chronic obstructive pulmonary disease. Clin Respir J 2015; 9: 330–341. doi: 10.1111/crj.12147 [DOI] [PubMed] [Google Scholar]
- 16.Donnelly LE, Barnes PJ. Defective phagocytosis in airways disease. Chest 2012; 141: 1055–1062. doi: 10.1378/chest.11-2348 [DOI] [PubMed] [Google Scholar]
- 17.Raghu G, Rochwerg B, Zhang Y, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med 2015; 192: e3–e19. doi: 10.1164/rccm.201506-1063ST [DOI] [PubMed] [Google Scholar]
- 18.Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med 2018; 378: 1811–1823. doi: 10.1056/NEJMra1705751 [DOI] [PubMed] [Google Scholar]
- 19.Noth I, Zhang Y, Ma SF, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med 2013; 1: 309–317. doi: 10.1016/S2213-2600(13)70045-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Misharin AV, Morales-Nebreda L, Reyfman PA, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med 2017; 214: 2387–2404. doi: 10.1084/jem.20162152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011; 378: 1949–1961. doi: 10.1016/S0140-6736(11)60052-4 [DOI] [PubMed] [Google Scholar]
- 22.Fernandez IE, Eickelberg O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet 2012; 380: 680–688. doi: 10.1016/S0140-6736(12)61144-1 [DOI] [PubMed] [Google Scholar]
- 23.Baumgartner KB, Samet JM, Stidley CA, et al. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1997; 155: 242–248. doi: 10.1164/ajrccm.155.1.9001319 [DOI] [PubMed] [Google Scholar]
- 24.Kärkkäinen M, Kettunen HP, Nurmi H, et al. Effect of smoking and comorbidities on survival in idiopathic pulmonary fibrosis. Respir Res 2017; 18: 160. doi: 10.1186/s12931-017-0642-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seibold MA, Wise AL, Speer MC, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med 2011; 364: 1503–1512. doi: 10.1056/NEJMoa1013660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dressen A, Abbas AR, Cabanski C, et al. Analysis of protein-altering variants in telomerase genes and their association with MUC5B common variant status in patients with idiopathic pulmonary fibrosis: a candidate gene sequencing study. Lancet Respir Med 2018; 6: 603–614. doi: 10.1016/S2213-2600(18)30135-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Papaioannou AI, Kostikas K, Manali ED, et al. Combined pulmonary fibrosis and emphysema: the many aspects of a cohabitation contract. Respir Med 2016; 117: 14–26. doi: 10.1016/j.rmed.2016.05.005 [DOI] [PubMed] [Google Scholar]
- 28.Grubstein A, Bendayan D, Schactman I, et al. Concomitant upper-lobe bullous emphysema, lower-lobe interstitial fibrosis and pulmonary hypertension in heavy smokers: report of eight cases and review of the literature. Respir Med 2005; 99: 948–954. doi: 10.1016/j.rmed.2004.12.010 [DOI] [PubMed] [Google Scholar]
- 29.Cottin V. The impact of emphysema in pulmonary fibrosis. Eur Respir Rev 2013; 22: 153–157. doi: 10.1183/09059180.00000813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease (2018 report). Available from: https://goldcopd.org
- 31.Aletaha D, Smolen JS. Diagnosis and management of rheumatoid arthritis: a review. JAMA 2018; 320: 1360–1372. doi: 10.1001/jama.2018.13103 [DOI] [PubMed] [Google Scholar]
- 32.Kolb M, Bonella F, Wollin L. Therapeutic targets in idiopathic pulmonary fibrosis. Respir Med 2017; 131: 49–57. doi: 10.1016/j.rmed.2017.07.062 [DOI] [PubMed] [Google Scholar]
- 33.Maher TM, Molina-Molina M, Russell AM, et al. Unmet needs in the treatment of idiopathic pulmonary fibrosis – insights from patient chart review in five European countries. BMC Pulm Med 2017; 17: 124. doi: 10.1186/s12890-017-0468-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J 2016; 47: 243–253. doi: 10.1183/13993003.00026-2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lancaster L, Crestani B, Hernandez P, et al. Safety and survival data in patients with idiopathic pulmonary fibrosis treated with nintedanib: pooled data from six clinical trials. BMJ Open Respir Res 2019; 6: e000397. doi: 10.1136/bmjresp-2018-000397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roche GmbH. Esbriet® Summary of product characteristics. www.ema.europa.eu/en/documents/product-information/esbriet-epar-product-information_en.pdf Date last updated: 2019; date last accessed: 11 November 2019.
- 37.Boehringer Ingelheim GmbH. Ofev® Summary of product characteristics. www.ema.europa.eu/en/documents/product-information/ofev-epar-product-information_en.pdf. Date last updated: 2019; date last accessed: 11 November 2019.
- 38.Selman M, Martinez FJ, Pardo A. Why does an aging smoker's lung develop idiopathic pulmonary fibrosis and not chronic obstructive pulmonary disease? Am J Respir Crit Care Med 2018; 199: 279–285. doi: 10.1164/rccm.201806-1166PP [DOI] [PubMed] [Google Scholar]
- 39.Anderson GP. Endotyping asthma: new insights into key pathogenic mechanisms in a complex, heterogeneous disease. Lancet 2008; 372: 1107–1119. doi: 10.1016/S0140-6736(08)61452-X [DOI] [PubMed] [Google Scholar]
- 40.Barnes PJ. Inflammatory endotypes in COPD. Allergy 2019; 74: 1249–1256. [DOI] [PubMed] [Google Scholar]
- 41.Vukmirovic M, Kaminski N. Impact of transcriptomics on our understanding of pulmonary fibrosis. Front Med (Lausanne) 2018; 5: 87. doi: 10.3389/fmed.2018.00087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Celli BR, MacNee W. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J 2004; 23: 932–946. doi: 10.1183/09031936.04.00014304 [DOI] [PubMed] [Google Scholar]
- 43.Martinez FJ, Lederer DJ. Focus on idiopathic pulmonary fibrosis: advancing approaches to diagnosis, prognosis, and treatment. Chest 2018; 154: 978–979. doi: 10.1016/j.chest.2018.08.1021 [DOI] [PubMed] [Google Scholar]
- 44.Hess MW, Make B. Back to the future: past, present, and future is COPD360. Chronic Obstr Pulm Dis 2016; 3: 473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spagnolo P, Bonella F, Vasakova M, et al. Current and future therapies for idiopathic pulmonary fibrosis. Pulm Ther 2015; 1: 1–18. doi: 10.1007/s41030-015-0009-4 [DOI] [Google Scholar]
- 46.Ng-Blichfeldt JP, Gosens R, Dean C, et al. Regenerative pharmacology for COPD: breathing new life into old lungs. Thorax 2019; 74: 890–897. doi: 10.1136/thoraxjnl-2018-212630 [DOI] [PubMed] [Google Scholar]
- 47.Hind M, Maden M. Retinoic acid induces alveolar regeneration in the adult mouse lung. Eur Respir J 2004; 23: 20–27. doi: 10.1183/09031936.03.00119103 [DOI] [PubMed] [Google Scholar]
- 48.Maden M. Retinoids have differing efficacies on alveolar regeneration in a dexamethasone-treated mouse. Am J Respir Cell Mol Biol 2006; 35: 260–267. doi: 10.1165/rcmb.2006-0029OC [DOI] [PubMed] [Google Scholar]
- 49.Zhang LM, Zhang J, Zhang Y, et al. Interleukin-18 promotes fibroblast senescence in pulmonary fibrosis through down-regulating Klotho expression. Biomed Pharmacother 2019; 113: 108756. doi: 10.1016/j.biopha.2019.108756 [DOI] [PubMed] [Google Scholar]
- 50.Moon S, Lee BH. Chemically induced cellular proteolysis: an emerging therapeutic strategy for undruggable targets. Mol Cells 2018; 41: 933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zengerle M, Chan KH, Ciulli A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem Biol 2015; 10: 1770–1777. doi: 10.1021/acschembio.5b00216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moses C, Kaur P. Applications of CRISPR systems in respiratory health: entering a new “red pen” era in genome editing. Respirology 2019; 24: 628–637. doi: 10.1111/resp.13527 [DOI] [PubMed] [Google Scholar]
- 53.Lee SH, Kim S, Hur JK. CRISPR and target-specific DNA endonucleases for efficient DNA knock-in in eukaryotic genomes. Mol Cells 2018; 41: 943–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liao W, Dong J, Peh HY, et al. Oligonucleotide therapy for obstructive and restrictive respiratory diseases. Molecules 2017; 22: E139. doi: 10.3390/molecules22010139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ohno S, Kuroda M. Exosome-mediated targeted delivery of miRNAs. Methods Mol Biol 2016; 1448: 261–270. doi: 10.1007/978-1-4939-3753-0_19 [DOI] [PubMed] [Google Scholar]
- 56.Naseri Z, Oskuee RK, Jaafari MR, et al. Exosome-mediated delivery of functionally active miRNA-142-3p inhibitor reduces tumorigenicity of breast cancer in vitro and in vivo. Int J Nanomedicine 2018; 13: 7727–7747. doi: 10.2147/IJN.S182384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Burkes RM, Drummond MB. Initiating drug therapy in early stage chronic obstructive pulmonary disease: does it impact the course and outcome? Curr Opin Pulm Med 2019; 25: 132–137. doi: 10.1097/MCP.0000000000000553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lipworth BJ, Jabbal S. What can we learn about COPD from impulse oscillometry? Respir Med 2018; 139: 106–109. doi: 10.1016/j.rmed.2018.05.004 [DOI] [PubMed] [Google Scholar]
- 59.Fain SB, Korosec FR, Holmes JH, et al. Functional lung imaging using hyperpolarized gas MRI. J Magn Reson Imaging 2007; 25: 910–923. doi: 10.1002/jmri.20876 [DOI] [PubMed] [Google Scholar]
- 60.Lowe KE, Regan EA, Anzueto A, et al. COPDGene® 2019: redefining the diagnosis of chronic obstructive pulmonary disease. Chronic Obstr Pulm Dis 2019; 6: 384–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hoff BA, Pompe E, Galbán S, et al. CT-based local distribution metric improves characterization of COPD. Sci Rep 2017; 7: 2999. doi: 10.1038/s41598-017-02871-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vasilescu DM, Martinez FJ, Marchetti N, et al. Non-invasive imaging biomarker identifies small airway damage in severe COPD. Am J Respir Crit Care Med 2019; 200: 575–581. doi: 10.1164/rccm.201811-2083OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen DL, Cheriyan J, Chilvers ER, et al. Quantification of lung PET images: challenges and opportunities. J Nucl Med 2017; 58: 201–207. doi: 10.2967/jnumed.116.184796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chan HF, Stewart NJ, Parra-Robles J, et al. Whole lung morphometry with 3D multiple b-value hyperpolarized gas MRI and compressed sensing. Magn Reson Med 2017; 77: 1916–1925. doi: 10.1002/mrm.26279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weatherley ND, Stewart NJ, Chan HF, et al. Hyperpolarised xenon magnetic resonance spectroscopy for the longitudinal assessment of changes in gas diffusion in IPF. Thorax 2018; 74: 500–502. doi: 10.1136/thoraxjnl-2018-211851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roos JE, McAdams HP, Kaushik SS, et al. Hyperpolarized gas MR imaging: technique and applications. Magn Reson Imaging Clin N Am 2015; 23: 217–229. doi: 10.1016/j.mric.2015.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bajc M, Chen Y, Wang J, et al. Identifying the heterogeneity of COPD by V/P SPECT: a new tool for improving the diagnosis of parenchymal defects and grading the severity of small airways disease. Int J Chron Obstruct Pulmon Dis 2017; 12: 1579–1587. doi: 10.2147/COPD.S131847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Petersson J, Sánchez-Crespo A, Larsson SA, et al. Physiological imaging of the lung: single-photon-emission computed tomography (SPECT). J Appl Physiol (1985) 2007; 102: 468–476. doi: 10.1152/japplphysiol.00732.2006 [DOI] [PubMed] [Google Scholar]
- 69.Bajc M, Markstad H, Jarenback L, et al. Grading obstructive lung disease using tomographic pulmonary scintigraphy in patients with chronic obstructive pulmonary disease (COPD) and long-term smokers. Ann Nucl Med 2015; 29: 91–99. doi: 10.1007/s12149-014-0913-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pisi R, Aiello M, Zanini A, et al. Small airway dysfunction and flow and volume bronchodilator responsiveness in patients with chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 2015; 10: 1191–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oostveen E, MacLeod D, Lorino H, et al. ERS Task Force on Respiratory Impedance Measurements. The forced oscillation technique in clinical practice: methodology, recommendations and future developments. Eur Respir J 2003; 22: 1026–1041. doi: 10.1183/09031936.03.00089403 [DOI] [PubMed] [Google Scholar]
- 72.Coxson HO, Quiney B, Sin DD, et al. Airway wall thickness assessed using computed tomography and optical coherence tomography. Am J Respir Crit Care Med 2008; 177: 1201–1206. doi: 10.1164/rccm.200712-1776OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen Y, Ding M, Guan WJ, et al. Validation of human small airway measurements using endobronchial optical coherence tomography. Respir Med 2015; 109: 1446–1453. doi: 10.1016/j.rmed.2015.09.006 [DOI] [PubMed] [Google Scholar]
- 74.Robinson PD, Latzin P, Verbanck S, et al. Consensus statement for inert gas washout measurement using multiple- and single- breath tests. Eur Respir J 2013; 41: 507–522. doi: 10.1183/09031936.00069712 [DOI] [PubMed] [Google Scholar]
- 75.Zimmermann SC, Tonga KO, Thamrin C. Dismantling airway disease with the use of new pulmonary function indices. Eur Respir Rev 2019; 28: 180122. doi: 10.1183/16000617.0122-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bell AS, Lawrence PJ, Singh D, et al. Feasibility and challenges of using multiple breath washout in COPD. Int J Chron Obstruct Pulmon Dis 2018; 13: 2113–2119. doi: 10.2147/COPD.S164285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bos LD, Sterk PJ, Fowler SJ. Breathomics in the setting of asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol 2016; 138: 970–976. doi: 10.1016/j.jaci.2016.08.004 [DOI] [PubMed] [Google Scholar]
- 78.Bafadhel M, McKenna S, Terry S, et al. Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med 2011; 184: 662–671. doi: 10.1164/rccm.201104-0597OC [DOI] [PubMed] [Google Scholar]
- 79.Garudadri S, Woodruff PG. Targeting chronic obstructive pulmonary disease phenotypes, endotypes, and biomarkers. Ann Am Thorac Soc 2018; 15: S234–S238. doi: 10.1513/AnnalsATS.201808-533MG [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fuschillo S, Molino A, Stellato C, et al. Blood eosinophils as biomarkers of therapeutic response to chronic obstructive pulmonary disease: still work in progress. Eur J Intern Med 2019; 68: 1–5. doi: 10.1016/j.ejim.2019.07.005 [DOI] [PubMed] [Google Scholar]
- 81.Drakopanagiotakis F, Wujak L, Wygrecka M, et al. Biomarkers in idiopathic pulmonary fibrosis. Matrix Biol 2018; 68–69: 404–421. doi: 10.1016/j.matbio.2018.01.023 [DOI] [PubMed] [Google Scholar]
- 82.Cazzola M, Puxeddu E, Ora J, et al. Evolving concepts in chronic obstructive pulmonary disease blood-based biomarkers. Mol Diagn Ther 2019; 23: 603–614. doi: 10.1007/s40291-019-00413-1 [DOI] [PubMed] [Google Scholar]
- 83.Magnini D, Montemurro G, Iovene B, et al. Idiopathic pulmonary fibrosis: molecular endotypes of fibrosis stratifying existing and emerging therapies. Respiration 2017; 93: 379–395. doi: 10.1159/000475780 [DOI] [PubMed] [Google Scholar]
- 84.Savas S, Liu G. Studying genetic variations in cancer prognosis (and risk): a primer for clinicians. Oncologist 2009; 14: 657–666. doi: 10.1634/theoncologist.2009-0042 [DOI] [PubMed] [Google Scholar]
- 85.Faner R, Tal-Singer R, Riley JH, et al. Lessons from ECLIPSE: a review of COPD biomarkers. Thorax 2014; 69: 666–672. doi: 10.1136/thoraxjnl-2013-204778 [DOI] [PubMed] [Google Scholar]
- 86.Fawzy A, Putcha N, Paulin LM, et al. Association of thrombocytosis with COPD morbidity: the SPIROMICS and COPDGene cohorts. Respir Res 2018; 19: 20. doi: 10.1186/s12931-018-0717-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wells JM, Parker MM, Oster RA, et al. Elevated circulating MMP-9 is linked to increased COPD exacerbation risk in SPIROMICS and COPDGene. JCI Insight 2018; 3: e123614. doi: 10.1172/jci.insight.123614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Agusti A, Edwards LD, Rennard SI, et al. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PLoS One 2012; 7: e37483. doi: 10.1371/journal.pone.0037483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sauleda J, Núñez B, Sala E, et al. Idiopathic pulmonary fibrosis: epidemiology, natural history, phenotypes. Med Sci 2018; 6: 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jenkins RG, Simpson JK, Saini G, et al. Longitudinal change in collagen degradation biomarkers in idiopathic pulmonary fibrosis: an analysis from the prospective, multicentre PROFILE study. Lancet Respir Med 2015; 3: 462–472. doi: 10.1016/S2213-2600(15)00048-X [DOI] [PubMed] [Google Scholar]
- 91.Maher TM, Oballa E, Simpson JK, et al. An epithelial biomarker signature for idiopathic pulmonary fibrosis: an analysis from the multicentre PROFILE cohort study. Lancet Respir Med 2017; 5: 946–955. doi: 10.1016/S2213-2600(17)30430-7 [DOI] [PubMed] [Google Scholar]
- 92.Hancock LA, Hennessy CE, Solomon GM, et al. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat Commun 2018; 9: 5363. doi: 10.1038/s41467-018-07768-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Obeidat M, Nie Y, Fishbane N, et al. Integrative genomics of emphysema-associated genes reveals potential disease biomarkers. Am J Respir Cell Mol Biol 2017; 57: 411–418. doi: 10.1165/rcmb.2016-0284OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang M, Kohler M, Heyder T, et al. Proteomic profiling of lung immune cells reveals dysregulation of phagocytotic pathways in female-dominated molecular COPD phenotype. Respir Res 2018; 19: 39. doi: 10.1186/s12931-017-0699-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Catapano AL, Graham I, De Backer G, et al. 2016 ESC/EAS guidelines for the management of dyslipidaemias. Eur Heart J 2016; 37: 2999–3058. doi: 10.1093/eurheartj/ehw272 [DOI] [PubMed] [Google Scholar]
- 96.Cantor JO, Turino GM. COPD pathogenesis: finding the common in the complex. Chest 2019; 155: 266–271. doi: 10.1016/j.chest.2018.07.030 [DOI] [PubMed] [Google Scholar]
- 97.Huang P, Yan R, Zhang X, et al. Activating Wnt/β-catenin signaling pathway for disease therapy: challenges and opportunities. Pharmacol Ther 2019; 196: 79–90. doi: 10.1016/j.pharmthera.2018.11.008 [DOI] [PubMed] [Google Scholar]
- 98.Kneidinger N, Yildirim AO, Callegari J, et al. Activation of the WNT/β-catenin pathway attenuates experimental emphysema. Am J Respir Crit Care Med 2011; 183: 723–733. doi: 10.1164/rccm.200910-1560OC [DOI] [PubMed] [Google Scholar]
- 99.Pan J, Li D, Xu Y, et al. Inhibition of Bcl-2/xl with ABT-263 selectively kills senescent type II pneumocytes and reverses persistent pulmonary fibrosis induced by ionizing radiation in mice. Int J Radiat Oncol Biol Phys 2017; 99: 353–361. doi: 10.1016/j.ijrobp.2017.02.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lagares D, Santos A, Grasberger PE, et al. Targeted apoptosis of myofibroblasts with the BH3 mimetic ABT-263 reverses established fibrosis. Sci Transl Med 2017; 9: eaal3765. doi: 10.1126/scitranslmed.aal3765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tsantikos E, Lau M, Castelino CM, et al. Granulocyte-CSF links destructive inflammation and comorbidities in obstructive lung disease. J Clin Invest 2018; 128: 2406–2418. doi: 10.1172/JCI98224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mercado N, Ito K, Barnes PJ. Accelerated ageing of the lung in COPD: new concepts. Thorax 2015; 70: 482–489. doi: 10.1136/thoraxjnl-2014-206084 [DOI] [PubMed] [Google Scholar]
- 103.Birch J, Barnes PJ, Passos JF. Mitochondria, telomeres and cell senescence: implications for lung ageing and disease. Pharmacol Ther 2018; 183: 34–49. doi: 10.1016/j.pharmthera.2017.10.005 [DOI] [PubMed] [Google Scholar]
- 104.Waters DW, Blokland KEC, Pathinayake PS, et al. Fibroblast senescence in the pathology of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2018; 315: L162–L172. doi: 10.1152/ajplung.00037.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Barnes PJ, Baker J, Donnelly LE. Cellular senescence as a mechanism and target in chronic lung diseases. Am J Respir Crit Care Med 2019; 200: 556–564. doi: 10.1164/rccm.201810-1975TR [DOI] [PubMed] [Google Scholar]
- 106.Baker JR, Vuppusetty C, Colley T, et al. Oxidative stress dependent microRNA-34a activation via PI3Kalpha reduces the expression of sirtuin-1 and sirtuin-6 in epithelial cells. Sci Rep 2016; 6: 35871. doi: 10.1038/srep35871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nakamaru Y, Vuppusetty C, Wada H, et al. A protein deacetylase SIRT1 is a negative regulator of metalloproteinase-9. FASEB J 2009; 23: 2810–2819. doi: 10.1096/fj.08-125468 [DOI] [PubMed] [Google Scholar]
- 108.Baker JR, Vuppusetty C, Colley T, et al. MicroRNA-570 is a novel regulator of cellular senescence and inflammaging. FASEB J 2019; 33: 1605–1616. doi: 10.1096/fj.201800965R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Houssaini A, Breau M, Kebe K, et al. mTOR pathway activation drives lung cell senescence and emphysema. JCI Insight 2018; 3: e93203. doi: 10.1172/jci.insight.93203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kadota T, Fujita Y, Yoshioka Y, et al. Emerging role of extracellular vesicles as a senescence-associated secretory phenotype: insights into the pathophysiology of lung diseases. Mol Aspects Med 2018; 60: 92–103. doi: 10.1016/j.mam.2017.11.005 [DOI] [PubMed] [Google Scholar]
- 111.Dagouassat M, Gagliolo JM, Chrusciel S, et al. The cyclooxygenase-2-prostaglandin E2 pathway maintains senescence of chronic obstructive pulmonary disease fibroblasts. Am J Respir Crit Care Med 2013; 187: 703–714. doi: 10.1164/rccm.201208-1361OC [DOI] [PubMed] [Google Scholar]
- 112.Yanai H, Shteinberg A, Porat Z, et al. Cellular senescence-like features of lung fibroblasts derived from idiopathic pulmonary fibrosis patients. Aging (Albany NY) 2015; 7: 664–672. doi: 10.18632/aging.100807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Minagawa S, Araya J, Numata T, et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-β-induced senescence of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol 2011; 300: L391–L401. doi: 10.1152/ajplung.00097.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schafer MJ, White TA, Iijima K, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 2017; 8: 14532. doi: 10.1038/ncomms14532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Justice JN, Nambiar AM, Tchkonia T, et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMed 2019; 40: 554–563. doi: 10.1016/j.ebiom.2018.12.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Baarsma HA, Königshoff M. “WNT-er is coming”: WNT signalling in chronic lung diseases. Thorax 2017; 72: 746–759. doi: 10.1136/thoraxjnl-2016-209753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Williams MC. Alveolar type I cells: molecular phenotype and development. Annu Rev Physiol 2003; 65: 669–695. doi: 10.1146/annurev.physiol.65.092101.142446 [DOI] [PubMed] [Google Scholar]
- 118.Baarsma HA, Skronska-Wasek W, Mutze K, et al. Noncanonical WNT-5A signaling impairs endogenous lung repair in COPD. J Exp Med 2017; 214: 143–163. doi: 10.1084/jem.20160675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Skronska-Wasek W, Mutze K, Baarsma HA, et al. Reduced frizzled receptor 4 expression prevents WNT/β-catenin-driven alveolar lung repair in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2017; 196: 172–185. doi: 10.1164/rccm.201605-0904OC [DOI] [PubMed] [Google Scholar]
- 120.Königshoff M, Balsara N, Pfaff EM, et al. Functional Wnt signaling is increased in idiopathic pulmonary fibrosis. PLoS One 2008; 3: e2142. doi: 10.1371/journal.pone.0002142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Königshoff M, Kramer M, Balsara N, et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J Clin Invest 2009; 119: 772–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shi J, Li F, Luo M, et al. Distinct roles of WNT/beta-catenin signaling in the pathogenesis of chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Mediators Inflamm 2017; 2017: 3520581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rock JR, Randell SH, Hogan BL. Airway basal stem cells: a perspective on their roles in epithelial homeostasis and remodeling. Dis Model Mech 2010; 3: 545–556. doi: 10.1242/dmm.006031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hong KU, Reynolds SD, Watkins S, et al. Basal cells are a multipotent progenitor capable of renewing the bronchial epithelium. Am J Pathol 2004; 164: 577–588. doi: 10.1016/S0002-9440(10)63147-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ghosh M, Miller YE, Nakachi I, et al. Exhaustion of airway basal progenitor cells in early and established chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2018; 197: 885–896. doi: 10.1164/rccm.201704-0667OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Weeden CE, Chen Y, Ma SB, et al. Lung basal stem cells rapidly repair DNA damage using the error-prone nonhomologous end-joining pathway. PLoS Biol 2017; 15: e2000731. doi: 10.1371/journal.pbio.2000731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Prasse A, Binder H, Schupp JC, et al. BAL cell gene expression is indicative of outcome and airway basal cell involvement in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2019; 199: 622–630. doi: 10.1164/rccm.201712-2551OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shaykhiev R. Basal-like cells in the BAL fluid: an Echo of regenerative crisis in idiopathic pulmonary fibrosis lungs. Am J Respir Crit Care Med 2019; 199: 555–557. doi: 10.1164/rccm.201808-1557ED [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Xia H, Bodempudi V, Benyumov A, et al. Identification of a cell-of-origin for fibroblasts comprising the fibrotic reticulum in idiopathic pulmonary fibrosis. Am J Pathol 2014; 184: 1369–1383. doi: 10.1016/j.ajpath.2014.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Maden M, Hind M. Retinoic acid, a regeneration-inducing molecule. Dev Dyn 2003; 226: 237–244. doi: 10.1002/dvdy.10222 [DOI] [PubMed] [Google Scholar]
- 131.Piersma AH, Hessel EV, Staal YC. Retinoic acid in developmental toxicology: teratogen, morphogen and biomarker. Reprod Toxicol 2017; 72: 53–61. doi: 10.1016/j.reprotox.2017.05.014 [DOI] [PubMed] [Google Scholar]
- 132.Ng-Blichfeldt JP, Alcada J, Montero MA, et al. Deficient retinoid-driven angiogenesis may contribute to failure of adult human lung regeneration in emphysema. Thorax 2017; 72: 510–521. doi: 10.1136/thoraxjnl-2016-208846 [DOI] [PubMed] [Google Scholar]
- 133.Stolk J, Stockley RA, Stoel BC, et al. Randomised controlled trial for emphysema with a selective agonist of the γ-type retinoic acid receptor. Eur Respir J 2012; 40: 306–312. doi: 10.1183/09031936.00161911 [DOI] [PubMed] [Google Scholar]
- 134.Mao JT, Goldin JG, Dermand J, et al. A pilot study of all-trans-retinoic acid for the treatment of human emphysema. Am J Respir Crit Care Med 2002; 165: 718–723. doi: 10.1164/ajrccm.165.5.2106123 [DOI] [PubMed] [Google Scholar]
- 135.Roth MD, Connett JE, D'Armiento JM, et al. Feasibility of retinoids for the treatment of emphysema study. Chest 2006; 130: 1334–1345. doi: 10.1378/chest.130.5.1334 [DOI] [PubMed] [Google Scholar]
- 136.Ohmichi H, Koshimizu U, Matsumoto K, et al. Hepatocyte growth factor (HGF) acts as a mesenchyme-derived morphogenic factor during fetal lung development. Development 1998; 125: 1315–1324. [DOI] [PubMed] [Google Scholar]
- 137.Kato T, Oka K, Nakamura T, et al. Bronchioalveolar morphogenesis of human bronchial epithelial cells depending upon hepatocyte growth factor. J Cell Mol Med 2015; 19: 2818–2826. doi: 10.1111/jcmm.12672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Seedorf G, Metoxen AJ, Rock R, et al. Hepatocyte growth factor as a downstream mediator of vascular endothelial growth factor-dependent preservation of growth in the developing lung. Am J Physiol Lung Cell Mol Physiol 2016; 310: L1098–L1110. doi: 10.1152/ajplung.00423.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Calvi C, Podowski M, Lopez-Mercado A, et al. Hepatocyte growth factor, a determinant of airspace homeostasis in the murine lung. PLoS Genet 2013; 9: e1003228. doi: 10.1371/journal.pgen.1003228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cahill EF, Kennelly H, Carty F, et al. Hepatocyte growth factor is required for mesenchymal stromal cell protection against bleomycin-induced pulmonary fibrosis. Stem Cells Transl Med 2016; 5: 1307–1318. doi: 10.5966/sctm.2015-0337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kanazawa H, Tochino Y, Asai K, et al. Simultaneous assessment of hepatocyte growth factor and vascular endothelial growth factor in epithelial lining fluid from patients with COPD. Chest 2014; 146: 1159–1165. doi: 10.1378/chest.14-0373 [DOI] [PubMed] [Google Scholar]
- 142.Chakraborty S, Chopra P, Hak A, et al. Hepatocyte growth factor is an attractive target for the treatment of pulmonary fibrosis. Expert Opin Investig Drugs 2013; 22: 499–515. doi: 10.1517/13543784.2013.778972 [DOI] [PubMed] [Google Scholar]
- 143.Espindola MS, Habiel DM, Narayanan R, et al. Targeting of TAM receptors ameliorates fibrotic mechanisms in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2018; 197: 1443–1456. doi: 10.1164/rccm.201707-1519OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hedström U, Hallgren O, Öberg L, et al. Bronchial extracellular matrix from COPD patients induces altered gene expression in repopulated primary human bronchial epithelial cells. Sci Rep 2018; 8: 3502. doi: 10.1038/s41598-018-21727-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Balestrini JL, Chaudhry S, Sarrazy V, et al. The mechanical memory of lung myofibroblasts. Integr Biol (Camb) 2012; 4: 410–421. doi: 10.1039/c2ib00149g [DOI] [PubMed] [Google Scholar]
- 146.Barnes PJ, Bonini S, Seeger W, et al. Barriers to new drug development in respiratory disease. Eur Respir J 2015; 45: 1197–1207. doi: 10.1183/09031936.00007915 [DOI] [PubMed] [Google Scholar]
- 147.Morgan P, Brown DG, Lennard S, et al. Impact of a five-dimensional framework on R&D productivity at AstraZeneca. Nat Rev Drug Discov 2018; 17: 167–181. doi: 10.1038/nrd.2017.244 [DOI] [PubMed] [Google Scholar]
- 148.Medical Research Council. Smarter trials speed up patients’ access to effective treatments. https://mrc.ukri.org/news/browse/smarter-trials-speed-up-patients-access-to-effective-treatments Date last updated: 2018; date last accessed: 21 October 2019.
- 149.Baumgartner KB, Samet JM, Coultas DB, et al. Occupational and environmental risk factors for idiopathic pulmonary fibrosis: a multicenter case-control study. Collaborating Centers. Am J Epidemiol 2000; 152: 307–315. [DOI] [PubMed] [Google Scholar]