

Abstract
The prevalence of COVID-19-associated diabetes is not the result of a single event but of a combination of disease susceptibility associated with chronic illness and COVID-19-specific mechanisms affecting metabolism. Whether a separate entity of post-COVID-19 diabetes, possibly associated with lasting β-cell damage, also exists is not yet clear.
In this Comment, I draw on my experience in treating patients with COVID-19 at Columbia University’s New York Presbyterian Hospital starting in March of 2020, as the pandemic struck New York City. The first wave peaked in mid-April, when our hospital census reached 772 patients with COVID-19, ~280 of whom required invasive assisted mechanical ventilation or extracorporeal membrane oxygenation. From the early days of the pandemic, patients with diabetes were over-represented among the admissions and exhibited an unusually high prevalence of ketoacidosis. Our collective experience as medical faculty at Columbia/New York Presbyterian has been reviewed elsewhere1.
What we know
Recent meta-analyses have confirmed initial observations2 that diabetes is a risk factor for symptomatic SARS-CoV-2 infection and COVID-19-related hospitalization3, escalation of care4, invasive assisted mechanical ventilation5, renal replacement therapy6, cardiac injury7, thromboembolic events8 and death9. My service has reflected these findings: most patients presenting with hyperglycaemia and ketoacidosis had pre-existing, often poorly controlled diabetes (haemoglobin A1c ≥ 10%/13.3 mmol). Although type 2 diabetes prevailed, I saw occasional cases of type 1 diabetes, all in parlous states of control. Previously undiagnosed pre-existing diabetes, on the basis of haemoglobin A1c levels, was common. Excluding pre-existing diabetes can be tricky, particularly in the fraught context of COVID-19 admissions. Patients may have near-normal haemoglobin A1c levels as a result of lifestyle modifications or may have been teetering on the brink of the diagnostic criteria and surpassed the threshold only in the context of SARS-CoV-2 infection. However, patients without clear evidence of pre-existing diabetes may present with stress hyperglycaemia and may not require long-term antidiabetic treatment. Only long-term follow-up can clarify whether COVID-19 accelerates the onset of permanent diabetes to a greater extent than other precipitating factors. For example, community-acquired pneumonia is frequently associated with hyperglycaemia in patients without diabetes, but it can be a harbinger of diabetes, particularly in older people. Thus, hyperglycaemia per se is not unique to COVID-19 (Fig. 1).
Fig. 1 |. Mechanisms of COVID-associated hyperglycaemia and ketoacidosis.
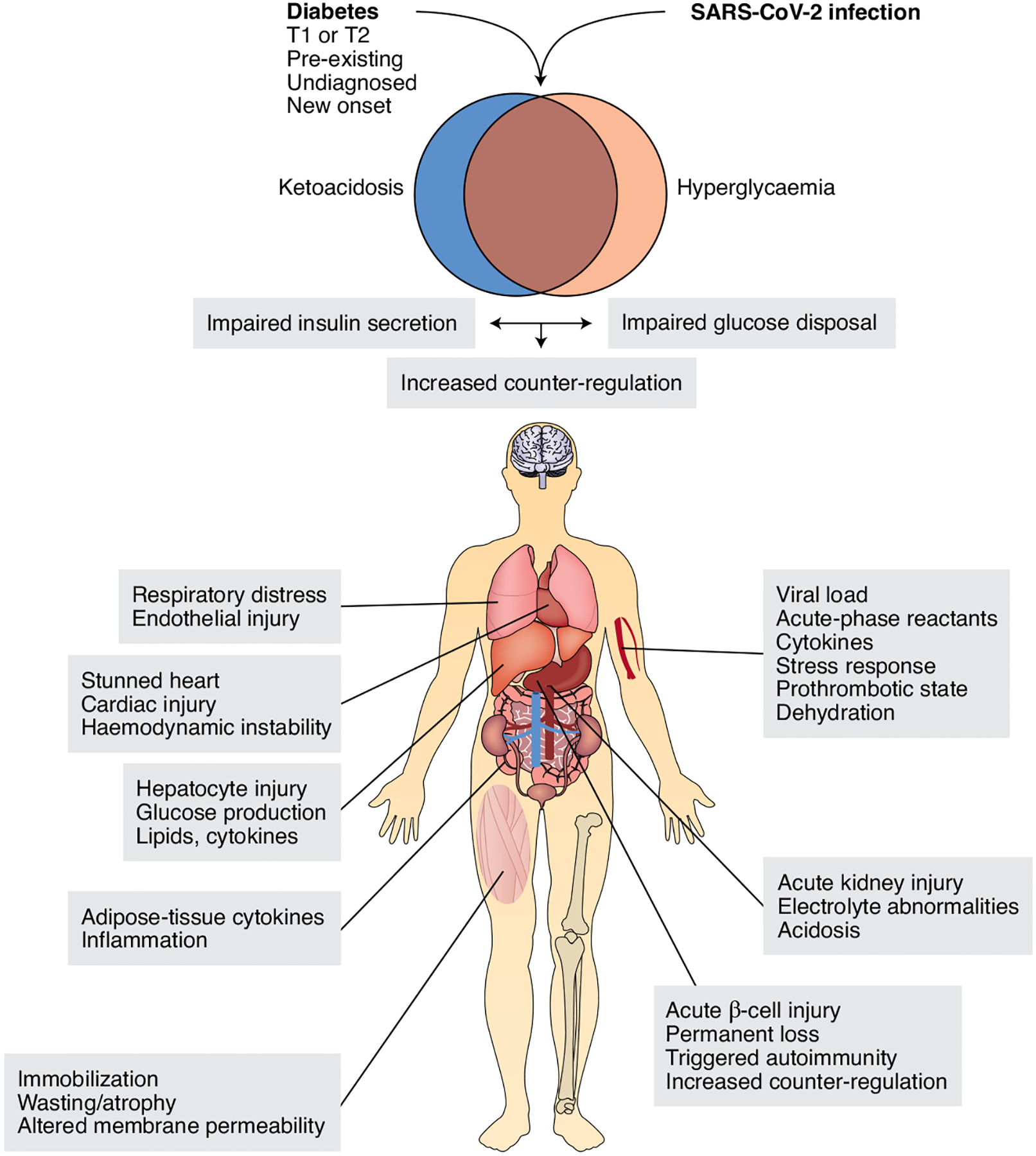
The findings of hyperglycaemia and ketoacidosis in patients with COVID-19 have prompted the question of whether there is underlying diabetes, regardless of whether it was previously recognized. Ketoacidosis can occur independently of hyperglycaemia even in patients who are not being treated with sodium/glucose cotransporter Sglt2 inhibitors. The mechanisms of these metabolic abnormalities involve impaired glucose utilization as well as decreased insulin secretion or increased counter-regulation. Examples of salient pathophysiologic features at the intersection of diabetes, acute intercurrent illness of any kind and COVID-19-specific factors are shown next to each target organ. Depending on the clinical course, these abnormalities may unfold in a rolling fashion rather than all at once.
What we don’t know
The question has been raised as to whether SARS-CoV-2, the pathogenic virus underlying COVID-19, can cause diabetes, among its many effects. But attribution beyond the occasional coincidence remains difficult. The hyperglycaemia in COVID-19 has multiple causes and probably involves impaired glucose disposal as well as diminished insulin secretion (Fig. 1). Any inflammatory state can cause insulin resistance and increase hepatic glucose production through increased counter-regulatory hormones, release of cytokines and lipids, and direct hepatocyte injury. In addition, inflammation can impair peripheral glucose uptake through immobilization, muscle wasting, cytokines and fluid/electrolyte abnormalities that alter membrane permeability. Obesity is another important risk factor for the comorbidities mentioned above10, and local inflammation of adipose tissue can serve as a potential unifying driver of impaired glucose metabolism, inflammation and immune responses (Fig. 1).
Ketoacidosis and COVID-19
The high prevalence of ketoacidosis among patients with COVID-19 and diabetes11 is unusual and has prompted the question of whether the virus might target pancreatic β-cells. We should distinguish between patients with hyperglycaemic versus euglycaemic ketoacidosis. We should also remember that there is scarcely any precedent for viral infections resulting in substantial clinical β-cell damage in the absence of predisposing autoimmunity, and that ketoacidosis is predicated not only on low insulin but also on an altered glucagon/insulin ratio. Thus, the assumption that COVID-19-associated ketoacidosis arises from β-cell damage warrants careful scrutiny. In my view, the cause is likely to be multi-factorial, and β-cell-extrinsic factors are likely to playa larger role than β-cell-intrinsic factors. Among the β-cell-intrinsic factors, in patients with uncontrolled hyperglycaemia, ‘stunned’ β-cells contribute to impaired insulin secretion12. The cytokine storm associated with COVID-19 is also likely to injure β-cells, as is the prothrombotic state, by potentially affecting the islet microvasculature as well as β-cells themselves. The hypothesis that COVID-19 unmasks a ketosis-prone diabetes subtype is attractive but only partly satisfying, because we don’t know what causes the predisposition to ketosis in these patients.
However, many patients with COVID-19 are admitted in a catabolic, ketotic state, exacerbated by a decreased ability to compensate for metabolic acidosis through renal and pulmonary mechanisms, owing to concurrent acute kidney injury with decreased HCO3– reabsorption, as well as impaired pulmonary gas exchange preventing compensatory respiratory alkalosis. In addition, patients are generally volume-depleted as a result of fever and diminished fluid intake, thus exacerbating electrolyte abnormalities. The activation of counter-regulatory responses is likely to further suppress β-cell function. These factors could explain euglycaemic ketoacidosis and are also a reminder that β-cell-extrinsic factors are at least as pathogenetically important as a potential direct hit on β-cells in bringing about hyperglycaemic ketoacidosis.
Pancreatic β-cells and sars-CoV-2
Is COVID-19 capable of inflicting damage on β-cells? We don’t know. Evidence from single-cell RNA surveys available at the outset of the pandemic indicated that the SARS-CoV-2 co-receptor ACE2 is not present in β-cells, whereas the receptor TMPRSS and the newly discovered NRP1 co-receptor are expressed at low levels13. Some surveys of pancreatic islets by immunohistochemistry have yielded results consistent with these findings14,15, whereas others have found SARS-CoV-2 receptors in β-cells16. Could the receptors be induced in β-cells as part of the syndrome? This induction is possible but difficult to demonstrate. In our experience, there is no clear association between diabetic ketoacidosis and readouts of inflammation, such as leukocyte and platelet counts, C-reactive protein, D-dimer and ferritin. However, for the virus alone to cause sufficient loss of β-cell function and impair insulin secretion, the extent of viral entry should be extensive. In addition, if the virus were to cause permanent loss of β-cell function, post-COVID diabetic ketoacidosis should routinely require insulin treatment, which is not the case. Indeed, most instances of ketoacidosis during the acute phase of the illness are mild to moderate. The challenges of treating ketoacidosis in COVID-19 are mostly extrinsic to β-cells: in fluid management, we often must take a narrow path between maintaining organ perfusion and preventing fluid accumulation in the lungs, owing to respiratory distress, stunned heart or outright cardiac injury, and prothrombotic state. Renal tubular damage also leads to protracted and unusual electrolyte abnormalities that can be difficult to manage. In addition, as steroid use to treat COVID-19 has gone mainstream, the recovery of β-cell function is delayed or blunted.
In my view, the focus on whether the virus infects β-cells is too narrow. Regardless of the expression levels of SARS-CoV-2 co-receptors, evidence of viral infections causing permanent β-cell damage is scant17. Stress-induced hyperglycaemia associated with infection is generally transient. Only in patients predisposed to developing type 1 diabetes has an intercurrent viral illness been suspected to deliver the final blow to faltering β-cell function. In this regard, if COVID-19 were in fact to cause permanent insulin-dependent diabetes, SARS-CoV-2 would be unique. Data on SARS1 are inconclusive in this regard18. However, if COVID-19 were to cause β-cell loss in patients with a predisposed autoimmune milieu, we would have seen an excess incidence of type 1 diabetes in association with the pandemic. The limited data in this regard do not bear out this interpretation19. However, we might possibly be undercounting the incidence of type 1 diabetes, because patients tend to avoid treatment at large academic medical centres and are more likely to seek care from primary providers. Recent evidence indicating genetic autoimmune susceptibility to respiratory failure in patients with COVID-19 (ref.20) provides a blueprint for studies addressing the role of type 1 diabetes autoimmunity in COVID-19-associated ketoacidosis. However, whether there are enough patients to conduct such a study is unclear.
What is to be done?
There are multiple reasons why people with diabetes are more susceptible to COVID-19. In addition, because diabetes is a heterogeneous disease, the mechanisms affecting individual patients are also likely to be heterogeneous. We should not dismiss the possibility that SARS-CoV-2 can cause diabetes, but we should not contrive it out of thin air if it is not supported by evidence, either. Studies assessing the in vitro susceptibility of β-cells to SARS-CoV-2 infection should be interpreted cautiously. The ability to bring about an infection in a test tube is not proof that the infection occurs in vivo.
Good glycaemic control is the best way to keep people with diabetes out of COVID-19 wards. We should continue to monitor the possibility that secondary diabetes is a distinct clinical entity within ‘long COVID’, and we should encourage people with diabetes to be vaccinated. Judicious insulin use remains the only efficacious and safe way to treat COVID-19-associated diabetic ketoacidosis, and continuous glucose monitor–based remote monitoring of glycaemia in intensive care units or step-down units to limit provider exposure to virus-shedding patients should be implemented.
Footnotes
Competing interests
D.A. reports no conflicts of interest influencing the content of this Comment. He is a founder, director and chair of the board of advisors of Forkhead BioTherapeutics Corp.
He also serves on the Global Diabetes Advisory Board for Eli Lilly and Co.
References
- 1.Gupta A et al. Nat. Med 26, 1017–1032 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Shang L et al. Arch. Med. Res 51, 700–709 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mantovani A, Byrne CD, Zheng MH & Targher G Nutr. Metab. Cardiovasc. Dis 30, 1236–1248 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hussain S, Baxi H, Chand Jamali M, Nisar N & Hussain MS Diabetes Metab. Syndr 14, 1595–1602 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Figliozzi S et al. Eur. J. Clin. Invest 50, e13362 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Fu EL et al. Clin. Kidney J 13, 550–563 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee MH et al. Diabetes Obes. Metab 23, 287–289 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Piazza G et al. J. Am. Coll. Cardiol 76, 2060–2072 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aggarwal G, Lippi G, Lavie CJ, Henry BM & Sanchis-Gomar FJ Diabetes 12, 851–855 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson MR et al. Ann. Intern. Med 173, 782–790 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J et al. Diabetes Obes. Metab 22, 1935–1941 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferrannini E Cell Metab 11, 349–352 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Veres A et al. Nature 569, 368–373 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coate KC et al. Cell Metab 32, 1028–1040.e4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kusmartseva I et al. Cell Metab 32, 1041–1051.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fignani D et al. Front. Endocrinol 11, 596898 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodriguez-Calvo T, Sabouri S, Anquetil F & von Herrath MG Autoimmun. Rev 15, 964–969 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Yang JK, Lin SS, Ji XJ & Guo LM Acta Diabetol 47, 193–199 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DiMeglio LA, Albanese-O’Neill A, Muñoz CE & Maahs DM Diabetes Care 43, 2631–2634 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Severe Covid-19 GWAS Group N. Engl. J. Med 383, 1522–1534 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]