

Abstract
Bacterial low-copy-number plasmids require partition (par) systems to ensure their stable inheritance by daughter cells. In general, these systems consist of three components: a centromeric DNA sequence, a centromere-binding protein and a nucleotide hydrolase that polymerizes and functions as a motor. Type III systems, however, segregate plasmids using three proteins: the FtsZ/tubulin-like GTPase TubZ, the centromere-binding protein TubR and the MerR-like transcriptional regulator TubY. Although the TubZ filament is sufficient to transport the TubR-centromere complex in vitro, TubY is still necessary for the stable maintenance of the plasmid. TubY contains an N-terminal DNA-binding helix-turn-helix motif and a C-terminal coiled-coil followed by a cluster of lysine residues. This study determined the crystal structure of the C-terminal domain of TubY from the Bacillus cereus pXO1-like plasmid and showed that it forms a tetrameric parallel four-helix bundle that differs from the typical MerR family proteins with a dimeric anti-parallel coiled-coil. Biochemical analyses revealed that the C-terminal tail with the conserved lysine cluster helps TubY to stably associate with the TubR-centromere complex as well as to nonspecifically bind DNA. Furthermore, this C-terminal tail forms an amphipathic helix in the presence of lipids but must oligomerize to localize the protein to the membrane in vivo. Taken together, these data suggest that TubY is a component of the nucleoprotein complex within the partitioning machinery, and that lipid membranes act as mediators of type III systems.
Keywords: TubZ, crystal structure, bacteria, plasmid partitioning, DNA segregation, segrosome, MerR, amphipathic helix, plasmid, DNA binding protein, cytoskeleton, plasmid partioning
Accurate DNA segregation is essential for transmission of genetic information to daughter cells. The well-studied process of eukaryotic DNA segregation involves the mitotic spindle and kinetochores, which are composed of multiple protein complexes. By contrast, much less is known about prokaryotic DNA segregation.
Partition systems (par) for low-copy-number plasmids are thought to be the best model for studying bacterial DNA segregation, as only three components are required: a centromere DNA site, a centromere-binding protein (CBP) and an ATP or GTP hydrolase (NTPase; 1, 2). In general, par systems are classified into three types based on the nature of the NTPases that provide the driving force for plasmid segregation. Type I and II partition systems employ a Walker-type ATPase ParA and an actin-like ATPase ParM, respectively, whereas type III systems use an FtsZ/tubulin-like GTPase TubZ. The behaviors of these noncanonical motor proteins are distinct from one another in the cell, implying that the molecular mechanisms of the three systems are dissimilar (2, 3). Plasmids are delivered to daughter cells by these NTPases, whereas the CBPs function as adaptors between the centromere and NTPases. Unlike the NTPases, the sequence homology of the CBPs is fairly low even within the same partition system (4). Hence, the centromere sequences are not conserved but consist mostly of multiple tandem repeats. These repeats contribute to formation of the segrosome, a higher-order nucleoprotein complex containing the CBPs and centromere (reviewed in ref. 5). The segrosome activates the NTPases to achieve partitioning of the plasmid. The type I ATPase ParA nonspecifically associates with nucleoid DNA, and when the segrosome interacts with the ParA on the nucleoid, it stimulates the ATPase activity of ParA, leading to dissociation of ParA from the nucleoid (6). Subsequently the segrosome diffuses and associates with neighboring ParA, which results in a directional movement of the plasmid. In type II systems, the ParM ATPase forms a bipolar spindle whose ends are stabilized by binding to the segrosome and polymerization of ParM forces the segrosome to the cell poles, causing the plasmids to segregate to the daughter cells (7–9). The segrosome of type III systems associates with the minus-ends of TubZ filaments and is pulled by filaments while they treadmill (10).
In all segregation systems, three partitioning factors of the centromere and the proteins are coded within a par operon, but in some cases, proteins encoded outside par are required for efficient partition. For instance, the centromere region of the P1 plasmid in the type I system contains the binding site for IHF (integration host factor) as well as the CBP (11, 12). IHF can bend DNA by ∼180°, which allows the CBP to associate with the centromere to form a segrosome (13, 14). Another example is TubY from type III partition systems. TubY is a DNA-binding protein with a putative helix-turn-helix (HTH) motif that functions as a transcriptional activator of the tubRZ operon (15). More importantly, TubY modulates the interaction between the segrosome and the TubZ filaments (16); however, it remains unclear how TubY is involved in plasmid segregation.
The type III par system was first identified in virulent Bacillus species (17, 18). The centromere tubC is localized upstream of the tandemly arranged tubR and tubZ genes and consists of several direct or inverted repeats, providing multiple binding sites for TubR, the CBP (16, 18–21). TubR binding leads to segrosome formation as well as transcriptional repression of tubRZ. TubY is not encoded inside the tubRZ operon, but its locus is in the same vicinity (16). Stable maintenance of the plasmid requires TubY, which strongly suggests that tubY and tubRZ form a regulon for plasmid partitioning (15). Notably in this regard, tubY from virulent Bacillus species is located upstream of tubRZ, whereas tubY from Clostridium botulinum is downstream of tubRZ, and the transcription direction of tubY is not conserved among type III systems (16). These observations raise the question of how much diversity, both structurally and functionally, type III systems possess.
Here, I report the crystal structure of the TubY C-terminal coiled-coil domain of the pXO1-like plasmid pBc10987 from Bacillus cereus. The structure contains a tetrameric four-helix bundle that enables the N-terminal HTH motif to bind DNA. In addition, I found that the amphipathic C-terminal tail of TubY contributes not only to nonspecific DNA binding but also to association with lipid membranes. These data suggest that TubY may function as a modulator of segrosome formation and localization in the cell.
Results
TubY binds upstream of the tubRZ operon in pBc10987
TubY, a putative MerR family transcriptional regulator, is encoded upstream of the tubRZ operon in pBc10987 (Fig. 1A). In type III par systems, although tubY genes are located in the vicinity of tubRZ, they are not likely to be transcribed as part of the same transcriptional unit (15, 16). Because the tubY promoter is flanked by tubRZ, I examined the binding of TubR or TubY of pBc10987 to the region of DNA between tubY and tubR. DNA fragments from the promoter regions were radioactively labeled and examined for binding of His-tag fused TubR or TubY by pulldown assays. TubY preferentially bound to the pro1 region (65171–65320 nt) carrying the putative promoter -35 and -10 elements of tubRZ, but not to the TubR-binding region pro2 (65321–65577; 21).
Figure 1.

The tubYRZ regulon. A, Gene organization of the tubYRZ regulon in the plasmids from different Bacillus species (pBc10987 from Bacillus cereus, pBtoxis from Bacillus thuringiensis and pBsph from Bacillus sphaericus), along with the regulons in the pCLG2 plasmid and prophage c-st from Clostridium botulinum. Type III partitioning systems containing tubR, tubZ and tubY are listed. Transcription directions of tubY (gray), tubR (white) and tubZ (black) are shown as arrows. DNA fragments used in this study are shown in the box. Binding of TubR and TubY, determined by the pulldown assay, is indicated. B, Domain organization of Bacillus cereus TubY. Mutations critical for DNA binding are labeled. Constructs used in this study are shown in the box. C, EMSA analysis of BcY and its mutants (BcYN and BcYC) binding to the pro12 region. Protein concentration is 1 μm. Reactions were analyzed by electrophoresis using a 6% polyacrylamide gels.
Full-length TubY from B. cereus (BcY) was mostly expressed as an insoluble protein, similar to the one previously reported for C. botulinum TubY (16). Therefore, denatured BcY was refolded and purified to homogeneity. During purification, BcY was proteolytically sensitive: specifically, it was degraded into two peptides, indicating that it possesses structurally flexible regions. To identify the domain structure of BcY, the digested fragments were analyzed by MS and determined to be BcYN and BcYCΔ (Fig. 1B). Sequence analyses revealed that BcYN and BcYCΔ contain the HTH motif and coiled-coil domains, respectively. The conserved C-terminal tail was susceptible to degradation, presumably because of the intrinsic flexibility of the clusters of lysine and phenylalanine residues (see below). In analysis of DNA binding by BcYN and BcYCΔ with the tail (BcYC) using electrophoretic mobility shift assays (EMSA), only BcY bound pro12 (65171–65577 nt), indicating that both domains are required for DNA binding (Fig. 1C).
BcY is a tetramer
Although the primary sequence of the C terminus is not tightly conserved among the TubY proteins encoded by the plasmids or phage, it is predicted to form a coiled-coil (16; Fig. 2A). I determined the crystal structure of BcYCΔ (Fig. 1B, Fig. 2B and Table 1). The asymmetric unit contains four BcYCΔ monomers, which forms a tetrameric four-helix bundle with dimensions 90 Å × 20 Å × 20 Å. These four monomers are parallel and related by a pseudo-2-fold axis, and the root-mean-square deviation (rmsd) of Cα atoms (residues 112–174 of chain A and B versus C and D) is 0.37 Å (Fig. 2C). The BcYCΔ structure reveals that the hydrophobic core of the four-helix bundle is mostly aliphatic (Fig. 2A, B). Because of sequence similarity at the N terminus, TubY is a member of the MerR family (16). The MerR family is a group of metal-dependent transcriptional regulators which typically have a HTH motif at the N terminus followed by a coiled-coil region (23; Fig. S1). The coiled-coil region of the MerR proteins forms an anti-parallel dimer that functions as a metal sensing domain: metals are coordinated by conserved histidine or cysteine residues, which induce a conformational change and activate transcription. Another MerR family protein, TnrA, is a fairly small dimeric protein with a unique winged-HTH structure (24). Regarding BcY, BcYN is monomeric (Fig. S2) and incapable of binding to DNA (Fig. 1C), whereas BcYC is a tetrameric parallel coiled-coil with no cysteine or histidine residues. On the basis of these findings, I predict that TubY activates the transcription of tubRZ in a different manner than MerR or TnrA.
Figure 2.

Crystal structure of the tetramerization domain of BcY. A, Sequence alignment of the C-terminal domains in TubY listed in Fig. 1A: BcYC (NCBI Reference Sequence ID: WP_000564666; pBt158 from pBtoxis (WP_086402658); Bsph_p187 from pBsph (WP_069511974); and CLG_A0046 (ACT33709) and CST188 (YP_398618) from pCLG2 and c-st, respectively. The α-helix observed in the crystal structure is highlighted in yellow. The C-terminal amphipathic tail is shown in cyan. Residues involved in the hydrophobic core are indicated by green arrows. Conserved residues in the C-terminal tail are indicated by asterisks. B, Tetramerization domain of BcY. Residues forming the hydrophobic core are shown as sticks and labeled. Each monomer is shown in a different color. The N and C termini are labeled. C, Top view of the tetramerization domain. The orientation in (C) is related to that in (B) by a 90° rotation about a vertical axis. The 2-fold axis is indicated.
Table 1.
Crystallographic statistics
| Data Collection | |
| BcYCΔ (Se-MAD) | |
| Beamline | PF-NW12A |
| Space group | P1 |
| Unit cell dimensions, Å, ° |
a = 27.1, b = 40.3, c = 80.7 α = 102.0, β = 94.2, γ = 98.5 |
| Wavelength, Å | 0.9791 |
| Data range, Å | 30 – 2.6 |
| Completenessc, % | 98.0 (95.1) |
| Redundancyc | 3.9 (3.6) |
| I/σ(I)c | 27.2 (4.3) |
| Rmergea,c | 0.064 (0.330) |
| Refinement | |
| Resolution range, Å | 26.2 – 2.6 |
| No. reflections | 9865 |
| Rcryst (Rfree)b,c | 0.224 (0.277) |
| RMSD: bond length, Å | 0.0075 |
| RMSD: bond angle, ° | 1.52 |
| B factors, Å2: protein | 63.9 |
| B factors, Å2: water | 63.5 |
| PDB ID code | 7C7Y |
aRmerge = ∑ |Iobs − |/∑ Iobs, where Iobs is the intensity measurement and is the mean intensity for multiply recorded reflections (22).
bRcryst and Rfree = ∑ ‖Fobs| - |Fcalc‖/|Fobs| for reflections in the working and test sets, respectively. The R-free value was calculated using a randomly selected 5% of the data set that was omitted through all stages of refinement.
cNumbers in parentheses refer to statistics for the highest shell of data.
BcY associates with the TubR-centromere complex
To analyze the contribution of each domain to DNA binding, I constructed two BcY mutants: BcYmut, which possesses two mutations (R28A and K47A) in the HTH motif, and BcYΔ, which lacks the C-terminal tail (Fig. 1B). Curiously, both mutants were expressed in soluble form in E. coli, but were purified in the same manner as WT BcY. CD (CD) spectroscopy confirmed that these refolded proteins, as well as soluble BcYmut and BcYΔ, were correctly folded (Fig. S3).
EMSA analysis of BcY with the pro12 DNA fragment revealed complex formation, consistent with the results of the pulldown assay (Fig. 1A and Fig. 3A). The R28 and K47 residues are the putative DNA-binding sites predicted from sequence alignment of the MerR family proteins with BcYN (Fig. S1). These residues were simultaneously mutated to alanine. As predicted, the mutations abolished the DNA-binding activity of BcY, indicating that BcYN is critical for DNA binding but must oligomerize with BcYC to form a protein-DNA complex (Fig. 1C and Fig. 3A). When BcYΔ was mixed with pro12, it retarded the mobility of pro12, but the complex migrated faster than the BcY-pro12 complex, implying that the tail region of BcY contributes a nonspecific interaction with DNA (Fig. 3A).
Figure 3.
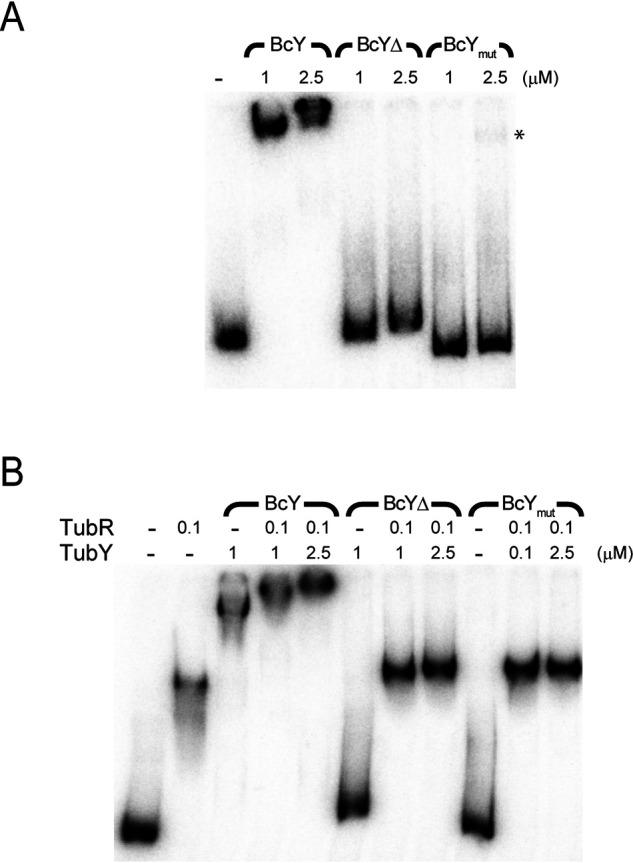
DNA-binding analysis of BcY and its mutants. A, EMSA analysis of binding of BcY and its mutants BcYΔ and BcYmut to the pro12 region. The amounts of BcY (in µM) are indicated above the lanes. Asterisk indicates very weak binding of BcYmut to pro12. Reactions were analyzed by electrophoresis on 4% polyacrylamide gels. B, EMSA analysis of BcY and TubR with pro12.
Next, to examine binding of BcY and TubR to pro12, TubR was preincubated with pro12 and then BcY was added (Fig. 3B). EMSA analysis revealed that the TubR-pro12 complex band was further supershifted in the presence of BcY, indicating that TubR and BcY bind their own binding sites in pro12. When BcYmut or BcYΔ was added to the mixture of pro12 and TubR, the bands were slightly retarded relative to TubR alone, implying that the BcY mutants may associate with the centromere-bound TubR. These EMSA analyses demonstrate that both the HTH motif and the C-terminal tail of BcY are required for stable segrosome formation.
TubR and BcY binding to pro2 was then examined whether BcY can associate with TubR in complex with pro2 which lacks BcY-binding sites (Fig. 1A and Fig. 4A). Although BcY did not bind pro2, the TubR-pro2 complex migrated slower in the presence of BcY compared with TubR-pro2 alone, which suggests that BcY is involved in segrosome formation. Hydroxyl radical footprinting analysis of pro2 supported these results (Fig. 4B and Fig. S4): BcY generated hypersensitive patterns across multiple sites, indicating that BcY nonspecifically interacts with DNA and might influence DNA structure. In the presence of both TubR and BcY, the footprinting patterns showed that the TubR-binding region was highly protected by BcY at TubR concentrations above 50 nm. Thus, TubR and BcY form a supramolecular complex at the centromeric DNA site, and BcY association is presumably achieved via the BcYCΔ region because the BcY mutants, BcYmut and BcYΔ, only weakly associate with the segrosome (Fig. 3B).
Figure 4.

Centromere binding of TubR and BcY. A, EMSA analysis of TubR and BcY binding to the pro2 region. TubR binds pro2, whereas BcY does not. Addition of both TubR and BcY results in a supershifted pro2 band, indicating that BcY binds to the centromere and TubR complex. Reactions were analyzed by electrophoresis using a 6% polyacrylamide gel. B, Hydroxyl radical footprinting analysis of the tubRZ promoter region. The amounts of TubR and BcY (in nm) are indicated above the lane. The numbers on the right-hand side show the location on the pBc10987 plasmid. Gray bars on the left-hand side indicate the regions containing the TubR-binding sites (21). Hyper-sensitive sites are marked by red asterisks. At higher concentrations of TubR (> 50 nm; red bracket), BcY protects the TubR-binding region (65460–65530 nt).
BcY binds lipid membranes in vitro
The C-terminal tail of the recombinant BcY caused low solubility of the protein. Sequence analysis reveals that this C-terminal tail is conserved not only in TubY of the plasmids or phages from virulent Bacillus species or Clostridium botulinum, but also in chromosomal TubY from some Clostridia (Fig. 2A and Fig. S5). This amphipathic tail is a characteristic of many peripheral membrane proteins, including MinD, MreB and SepF (25–28). Analysis of the C-terminal sequence of BcY (BcYtail) using the amphipathic helix prediction software AmphipaSeek indicated that the very C-terminal end is likely to form a helix (Fig. 5A; 29). Such amphipathic helical structures are thought to interact with lipids directly: the residues on the hydrophobic surface of the helix insert into the lipid bilayer, whereas the cationic residues on the polar surface associate with the head groups of anionic phospholipids.
Figure 5.

BcY C-terminal tail binds phospholipids. A, Helical wheel representation of the amphipathic helix in BcYtail. Residue numbers are labeled. Hydrophobic residues are highlighted in black. The peptide sequence used in the experiment is given at the bottom: the amphipathic helix region shown in the helical wheel is highlighted in gray (residues 192–199). Hydrophobic residues are shown in a larger font size. B, Far-UV circular dichroic spectra of the BcYtail peptide in the absence (dotted line) or presence (solid line) of phospholipids (pl). C, Co-sedimentation of BcY and BcYΔ with phospholipids. BcY co-sediments with phospholipid vesicles, whereas BcYΔ does not.
A synthetic peptide of BcYtail was examined for direct binding to lipid membranes by CD spectroscopy (Fig. 1B and Fig. 5B). In the absence of lipids, the BcYtail peptide yielded a spectrum with a single minimum at 200 nm, typical of an unstructured peptide and consistent with previous studies of MinD (25). When lipid membranes were mixed with the peptide, significant spectral changes were observed, with a minimum of 206 nm and peak intensity at 228 nm. This spectral change indicates that the BcYtail peptide interacts directly with lipid membranes to yield an α-helical conformation.
To determine whether BcYtail is critical for lipid binding, lipid pelleting assays were performed with BcY or BcYΔ (Fig. 5C). BcY co-sedimented with phospholipids, whereas BcYΔ did not, confirming that the BcYtail region is required for interaction with lipid membranes.
The BcY C terminus is a membrane targeting domain
To investigate further whether the C-terminal domain of BcY acts as a membrane targeting motif, BcY and its mutants were fused to the C terminus of GFP and their localization was analyzed in vivo (Fig. 6). These experiments were performed in Bacillus subtilis because transformation of B. cereus cells was not successful. As expected, GFP-BcY exhibited its characteristic distribution around the cell periphery, suggesting that BcY associates with membranes. A similar peripheral localization pattern was observed when GFP-BcYC was introduced, which indicated that the HTH motif is not required for the membrane localization.
Figure 6.
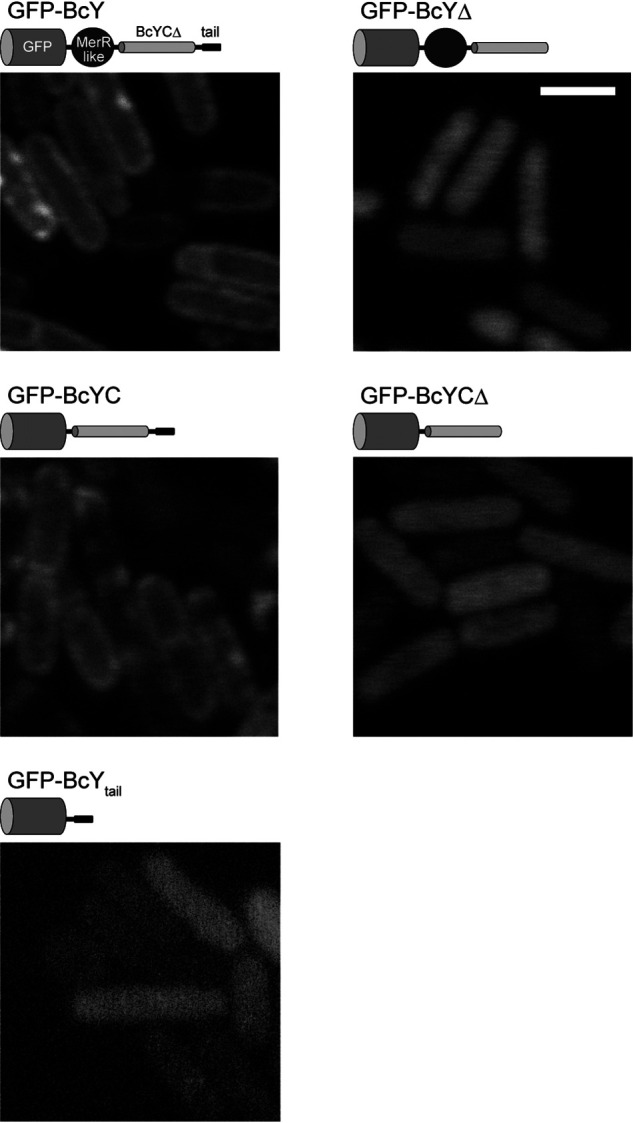
BcY localization in B. subtilis cells. Fluorescence micrographs of BcY and its mutants (BcYΔ, BcYC, BcYCΔ, BcYtail) fused to GFP in B. subtilis cells. Cartoons of each construct are shown above the micrographs. Scale bar, 2 μm.
Next, to determine the importance of the amphipathic tail in membrane localization of BcY, BcYΔ or BcYCΔ fused to GFP was analyzed. Removal of the C-terminal 19 residues abolished the peripheral localization pattern, and the mutants were uniformly distributed in the cytoplasm. These results suggest that the BcYtail region is essential for membrane localization of BcY. In addition, BcYtail was fused to GFP to investigate whether the BcYtail peptide itself is capable of targeting the cell membrane. GFP-BcYtail was not localized to the cell periphery, indicating that a single BcYtail peptide is not sufficient for the membrane attachment, and that tetramerization by the BcYCΔ region increases the affinity of BcYtail for lipid membranes.
Discussion
TubY, a putative MerR family protein, is considered to be an integral element of plasmid partitioning in type III par systems. This study shows that B. cereus TubY is a novel membrane-binding transcription factor and segrosome component by binding to the TubR-centromere complex, suggesting that the cell membrane is involved in the process of type III systems.
The crystal structure of the BcY C terminus revealed that the coiled-coil region is a parallel tetramerization domain, demonstrating that BcY is a unique tetrameric MerR-like transcriptional regulator. The results of this study show that oligomerization enables efficient binding of BcY to DNA. Moreover, the C-terminal tail, which contains basic residues, promotes the interaction with DNA nonspecifically and may change the DNA conformation, as suggested by the footprinting experiment.
The domain organization and DNA-binding function of BcY are analogous to those of tumor suppressor p53. p53 is a tetrameric transcription factor in which an anti-parallel α-helical tetramerization domain is flanked by two distinct DNA-binding regions (reviewed in ref. 30). The central domain binds DNA in a sequence-specific manner, whereas the C-terminal region is a short, unstructured tail of lysine residues that nonspecifically interacts with DNA and can slide along DNA when it is linked to the tetramerization domain. Given the analogy between p53 and BcY, the central DNA-binding domain of p53 corresponds to BcYN which specifically binds pro1. Although the current study has not demonstrated the sliding ability of BcY along DNA, the data indicate that several lysine residues in the flexible amphipathic tail of BcY are responsible for nonspecific DNA binding. The lysine-rich C-terminal tail of p53 is also responsible for the recruitment of protein cofactors and can adopt a helical conformation when interacting with DNA or some of the cofactors (30–32). Although shorter than the p53 tail, BcYtail may also allow BcY to engage various partners in different ways to regulate transcription and plasmid partitioning by changing the conformation of BcYtail.
Previously, tubY and tubRZ were shown to form a regulon whose gene products work cooperatively (15, 16). The EMSA analyses in this study showed that BcY associates with the TubR-DNA complex to form a supramolecular structure, implying that BcY is a component of the segrosome. Although the stoichiometry and affinity between the segrosome and BcY remain undetermined, full-length BcY is required for the stable complex. In particular, the BcYCΔ domain seems critical for complex assembly, in which BcYtail plays a supportive role. It should be noted that the cooperativity between TubR and TubY varies among species: tubC of pBsph is composed of three blocks of repeated sequences, and when TubY binds its recognition site, TubR is somehow removed from one of the blocks (15). The association of TubY with TubR and tubC may induce structural rearrangement of the ternary complex. Further molecular analysis is required to dissect the process of segrosome formation involving BcY.
Structural analyses of TubR-tubC complexes have been performed for four species (19, 21, 33). The results of these studies revealed that the nucleoprotein complexes from pBtoxis and pBc10987 have an extended filament structure (19, 21), whereas the others possess a rather rigid ring form (19, 33), indicating that the structural rearrangement might occur to function as a segrosome. TubR-tubC of pBtoxis is sufficient for tracking the minus-end of the TubZ filament, but it remains to be determined how the filaments drop off the plasmid to the daughter cells (10). TubR-tubC of pBtoxis has been shown to interact with the flexible C-terminal tail of TubZ (34). The TubZ tail of pBc10987 and pBtoxis, which is critical for TubZ assembly, possesses clusters of basic residues (34–36) and is somewhat similar to the amphipathic tail of BcY. In this study, I demonstrated that BcY directly associates with TubR-tubC. In type III plasmid partitioning, TubR-tubC, which is pulled by the TubZ filament, may abandon the filament and preferentially associate with TubY. Previous analysis of C. botulinum TubY showed that TubY, together with TubR-tubC, induces disassembly of the TubZ polymers (16). If this is a common mechanism in type III systems, the observation implies either that the quaternary complex between TubZ, TubY and TubR-tubC is highly unstable or that the binding site of TubY does not coincide with that of the TubZ filament in TubR-tubC. Both possibilities result in unloading of the plasmid, and thus depolymerization of the filaments, as TubR-tubC stabilizes the minus-end of the filament (10). Ultimately, the C-terminal tail of the cytoskeletal proteins may use a tail-mimicking mechanism that enables interplay between the filament and the associated proteins, as observed between tubulin and EB1 at microtubule plus-ends (37).
BcYtail interacts with lipid membranes as well. The amphipathic helix is seen in many peripheral membrane proteins, but barely found in transcriptional regulators. One of the exceptions is the yeast transcriptional repressor Opi1, which uses the amphipathic helix to occasionally target lipid membranes by altering its affinity for phosphatidic acid (38, 39). The amphipathic helices are suggested to sense membrane curvature or recognize specific lipids (38, 40, 41). In the case of TubY, the sequence analysis indicates that the amphipathic tail is conserved in TubY (Fig. 2A and Fig. S5). Although exact functions of the amphipathic tail are unknown, TubY may play a critical role in the localization of the segrosome and TubZ filaments. It should be noted that, in some Clostridia, TubY and TubZ but not TubR are encoded on the chromosome (16). Because chromosome segregation of Clostridium seems to rely on Soj (ParA) and Spo0J (ParB), TubZ and TubY may not be involved in DNA segregation (16, 42). In either case, because the chromosome-encoded TubY proteins possess a putative amphipathic helix (Fig. S5), TubY presumably functions as a modulator of TubZ filaments.
Based on the observation that TubZ filaments from B. thuringiensis seem to treadmill at the cell periphery, it has been speculated that the cell membrane is involved in detachment of the segrosome from the filaments (34, 43). These findings suggest that TubY and lipid membranes act as mediators in type III partition systems (Fig. 7). The TubR-tubC complex transported by the treadmilling TubZ filament encounters TubY at the membrane, detaches itself from the minus-end of the polymer, and associates with TubY, ultimately leading to plasmid partition. In this context, TubY is a key component of the partition machinery that serves to release the segrosome at a certain location within the cell.
Figure 7.

Model for TubY-mediated plasmid release. Hypothetical model for TubY-mediated plasmid release. In the absence of TubY, the plasmid is transported by the TubZ filament (10). When the segrosome at the minus-end of the filament approaches TubY attached to the cell membrane (left), TubY may dissociate from the membrane to interact with the TubR-centromere complex (right). TubY induces dissociation of the TubR-plasmid complex from the TubZ filament (16), delivering the plasmid to the daughter cell.
Experimental Procedures
Protein preparation
The tubY gene of pBc10987 was cloned in-frame into pET28a using NdeI and NotI with an N-terminal histidine tag and an additional tobacco etch virus (TEV) protease recognition sequence. TubY and its mutants (BcYΔ: residues 1–182; BcYmut: BcY with R28A and K47A mutations; BcYN: 1–93; BcYC: 107–201) were expressed in BL21(DE3). Cells were suspended in a buffer containing 20 mm Tris (pH 8.0), 0.1 M NaCl, 5 mm β-mercaptoethanol and 3 M urea and lysed by ultra-sonication. Then the histidine-tagged BcY or its mutants were isolated using HisTrap (GE Healthcare). Denatured proteins were refolded by dialysis at 4 °C in a stepwise fashion (1, 0.5 and 0 M urea). After cleavage of the tag by the histidine-tagged TEV protease, the protein was loaded onto a HiTrap Q and Superdex 75 (GE Healthcare). CD spectroscopy confirmed that the purified proteins had secondary structures, and that the mutations of R28A and K47A did not affect folding. The gene encoding the TubY C-terminal domain (BcYCΔ; residues 107–182) was cloned into pET21. The protein was expressed in a soluble form in BL21(DE3) and purified from the bacterial extract by precipitation with 60% saturated ammonium sulfate, followed by HiTrap SP cation exchange chromatography (GE Healthcare). Selenomethionine-substituted BcYCΔ was expressed in BL21(DE3) by inhibition of the methionine biosynthetic pathway and purified as for the native protein (44). TubR was purified as reported previously (21).
Crystallization, data collection, and structure determination
Se-substituted BcYCΔ protein was concentrated to 1.4 mm in a buffer containing 10 mm Tris (pH 8.0), 0.1 M NaCl and 3 mm DTT. Crystals were grown at 20 °C by sitting-drop vapor diffusion at 20 °C with a reservoir solution containing 0.1 M HEPES (pH 7.5), 0.2 M MgCl2 and 30% PEG400, and then frozen in a nitrogen stream at 95K.
All diffraction data were collected at the Photon Factory (Tsukuba, Japan). Diffraction images were processed with HKL2000 (22, 44). Initial phases were obtained by Se-SAD with the Phenix program AutoSol (45; Table 1). After automatic model building, the remaining residues were built manually in Coot (46). The structures were refined using Refmac (47). All figures were generated using PyMOL (48).
DNA binding
DNA fragments of interest were generated by PCR with [5′-32P]-labeled primers. To identify the binding region of TubY or TubR, His-tag fused proteins were immobilized on Ni-NTA agarose beads (Qiagen). Bound proteins were mixed with a radioactive probe in 10 mm Tris-HCl (pH 8.0), 0.1 M KCl, 1 mm MgCl2, 0.05 mg/ml salmon sperm DNA and 0.1 mg/ml BSA, and incubated on ice for 30 min. Beads were washed with buffer containing 10 mm Tris-HCl (pH 8.0), 0.1 M KCl and 0.05% Tween20. The remaining radioactivity was counted in a scintillation counter. The protocol used for EMSA was described previously (21). Experiments were performed in triplicate.
Hydroxyl radical footprinting
Hydroxyl radical footprinting was carried out as described previously (21, 49). Binding of TubR and BcY to the pro2 region (nt 65321–65577) was analyzed by end-labeling the DNA fragment with [γ-32P]ATP. For footprinting experiments, pro2 was cloned into pCR2.1 (Thermo Scientific) and digested with a restriction enzyme to create a single-end labeled DNA probe.
Liposome pelleting assay
Phosphatidylethanolamine (PE) and phosphatidylglycerol (PG) from chicken egg were purchased from Sigma and prepared as described previously (41). PE and PG were mixed at a ratio of 7:3. Dried lipids were solubilized in 20 mm HEPES (pH 7.0) and 0.1 M NaCl, and sonicated for 20 min until the solution became clear.
For pelleting experiments, 0.5 mg/ml BcY or BcYΔ and 2.5 mg/ml liposomes were mixed and incubated for 10 min at 20 °C. The mixture was then centrifuged using a Beckman TLA100.3 rotor at 80,000 rpm for 20 min. Pellets were analyzed by SDS-PAGE with Coomassie Brilliant Blue staining. Experiments were performed in triplicate.
CD measurements
The CD spectra of the peptide encompassing the C-terminal tail of BcY (BcYtail; residues 183–201; CS Bio) were collected between 195 and 260 nm at 20 °C using a JASCO J-720W spectropolarimeter with a 0.1 cm pathlength quartz cuvette. Samples at a concentration of 0.1 mg/ml in the presence or absence of 2.5 mg/ml phospholipids in a buffer containing 20 mm HEPES (pH 7.0) and 0.1 M NaCl were scanned four times with a step size of 0.5 nm and averaged.
Fluorescence microscopy
The genes encoding tubY and its mutants were inserted into the N-terminal GFP fusion vector pSG1729 using the BamHI and XhoI sites and transformed into B. subtilis 168 as described previously (50). Overnight cultures were diluted 1:100 in fresh LB supplemented with 250 μg/ml spectinomycin and grown to exponential phase at 30 °C. Expression of GFP-fused protein was induced with 0.75% xylose. Cells were washed three times with PBS, immobilized on microscope slides covered with 1% agarose in PBS and visualized under a confocal laser scanning microscope (Leica TCS SP8, Leica Microsystems).
Data availability
Crystallographic data and coordinates were deposited in Protein Data Bank with the accession number of 7C7Y.
Supplementary Material
Acknowledgments
I thank the members of the beamline staff at NW12A of the Photon Factory (Tsukuba, Japan) for help with data collection. I also thank Miyuki Katano for help with fluorescence microscopy; Tetsuro Kokubo for helpful discussion; and Hirofumi Yoshikawa for providing pSG1729 and B. subtilis 168. This work was supported by JSPS KAKENHI Grant Numbers JP15H01326 and JP19K05774.
This article contains supporting information.
Author contributions—IH conceptualization; IH data curation; IH formal analysis; IH funding acquisition; IH investigation; IH writing-original draft; IH project administration; IH writing-review and editing.
Funding and additional information—This work was supported by MEXT, Japan Society for the Promotion of Science (JSPS) Grants 15H01326 and 19K05774 (to I. H.).
Conflict of interest—The author declares that he has no conflicts of interest with the contents of this article.
- CBP
- centromere-binding protein
- HTH
- helix-turn-helix
- EMSA
- electrophoretic mobility shift assays.
References
- 1. Ebersbach G., and Gerdes K. (2005) Plasmid segregation mechanisms. Annu. Rev. Genet. 39, 453–479 10.1146/annurev.genet.38.072902.091252 [DOI] [PubMed] [Google Scholar]
- 2. Baxter J. C., and Funnell B. E. (2014) Plasmid Partition Mechanisms. Microbiol. Spectr. 2, [DOI] [PubMed] [Google Scholar]
- 3. Brooks A. C., and Hwang L. C. (2017) Reconstitutions of plasmid partition systems and their mechanisms. Plasmid 91, 37–41 10.1016/j.plasmid.2017.03.004 [DOI] [PubMed] [Google Scholar]
- 4. Hayes F., and Barillà D. (2006) The bacterial segrosome: a dynamic nucleoprotein machine for DNA trafficking and segregation. Nat Rev Microbiol. 4, 133–143 10.1038/nrmicro1342 [DOI] [PubMed] [Google Scholar]
- 5. Oliva M. A. (2016) Segrosome Complex Formation during DNA Trafficking in Bacterial Cell Division. Front. Mol. Biosci. 3, 51 10.3389/fmolb.2016.00051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vecchiarelli A. G., Neuman K. C., and Mizuuchi K. (2014) A propagating ATPase gradient drives transport of surface-confined cellular cargo. Proc. Natl. Acad. Sci. U.S.A. 111, 4880–4885 10.1073/pnas.1401025111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Møller-Jensen J., Borch J., Dam M., Jensen R. B., Roepstorff P., and Gerdes K. (2003) Bacterial mitosis: ParM of plasmid R1 moves plasmid DNA by an actin-like insertional polymerization mechanism. Mol. Cell 12, 1477–1487 10.1016/S1097-2765(03)00451-9 [DOI] [PubMed] [Google Scholar]
- 8. Gayathri P., Fujii T., Møller-Jensen J., van den Ent F., Namba K., and Löwe J. (2012) A bipolar spindle of antiparallel ParM filaments drives bacterial plasmid segregation. Science 338, 1334–1337 10.1126/science.1229091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bharat T. A., Murshudov G. N., Sachse C., and Löwe J. (2015) Structures of actin-like ParM filaments show architecture of plasmid-segregating spindles. Nature 523, 106–110 10.1038/nature14356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fink G., and Löwe J. (2015) Reconstitution of a prokaryotic minus end-tracking system using TubRC centromeric complexes and tubulin-like protein TubZ filaments. Proc. Natl. Acad. Sci. U.S.A. 112, E1845–1850 10.1073/pnas.1423746112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davis M. A., and Austin S. J. (1988) Recognition of the P1 plasmid centromere analog involves binding of the ParB protein and is modified by a specific host factor. EMBO J. 7, 1881–1888 10.1002/j.1460-2075.1988.tb03021.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Funnell B. E. (1988) Participation of Escherichia coli integration host factor in the P1 plasmid partition system. Proc. Natl. Acad. Sci. U.S.A. 85, 6657–6661 10.1073/pnas.85.18.6657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rice P. A., Yang S., Mizuuchi K., and Nash H. A. (1996) Crystal structure of an IHF-DNA complex: a protein-induced DNA U-turn. Cell 87, 1295–1306 10.1016/S0092-8674(00)81824-3 [DOI] [PubMed] [Google Scholar]
- 14. Schumacher M. A., and Funnell B. E. (2005) Structures of ParB bound to DNA reveal mechanism of partition complex formation. Nature 438, 516–519 10.1038/nature04149 [DOI] [PubMed] [Google Scholar]
- 15. Ge Y., Zhao N., Hu X., Shi T., Cai Q., and Yuan Z. (2014) A novel transcriptional activator, tubX, is required for the stability of Bacillus sphaericus mosquitocidal plasmid pBsph. J. Bacteriol. 196, 4304–4314 10.1128/JB.01855-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oliva M. A., Martin-Galiano A. J., Sakaguchi Y., and Andreu J. M. (2012) Tubulin homolog TubZ in a phage-encoded partition system. Proc. Natl. Acad. Sci. U.S.A. 109, 7711–7716 10.1073/pnas.1121546109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tinsley E., and Khan S. A. (2006) A novel FtsZ-like protein is involved in replication of the anthrax toxin-encoding pXO1 plasmid in Bacillus anthracis. J. Bacteriol. 188, 2829–2835 10.1128/JB.188.8.2829-2835.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tang M., Bideshi D. K., Park H. W., and Federici B. A. (2006) Minireplicon from pBtoxis of Bacillus thuringiensis subsp. israelensis. Appl. Environ. Microbiol. 72, 6948–6954 10.1128/AEM.00976-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aylett C. H., and Löwe J. (2012) Superstructure of the centromeric complex of TubZRC plasmid partitioning systems. Proc. Natl. Acad. Sci. U.S.A. 109, 16522–18527 10.1073/pnas.1210899109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ge Y., Hu X., Zhao N., Shi T., Cai Q., and Yuan Z. (2014) A new tubRZ operon involved in the maintenance of the Bacillus sphaericus mosquitocidal plasmid pBsph. Microbiology 160, 1112–1124 10.1099/mic.0.075465-0 [DOI] [PubMed] [Google Scholar]
- 21. Hayashi I., Oda T., Sato M., and Fuchigami S. (2018) Cooperative DNA Binding of the Plasmid Partitioning Protein TubR from the Bacillus cereus pXO1 Plasmid. J. Mol. Biol. 430, 5015–5028 10.1016/j.jmb.2018.11.001 [DOI] [PubMed] [Google Scholar]
- 22. Otwinowski Z., and Minor W. (1997) Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 23. Brown N. L., Stoyanov J. V., Kidd S. P., and Hobman J. L. (2003) The MerR family of transcriptional regulators. FEMS Microbiol. Rev. 27, 145–163 10.1016/S0168-6445(03)00051-2 [DOI] [PubMed] [Google Scholar]
- 24. Schumacher M. A., Chinnam N. B., Cuthbert B., Tonthat N. K., and Whitfill T. (2015) Structures of regulatory machinery reveal novel molecular mechanisms controlling B. subtilis nitrogen homeostasis. Genes Dev. 29, 451–464 10.1101/gad.254714.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Szeto T. H., Rowland S. L., Rothfield L. I., and King G. F. (2002) Membrane localization of MinD is mediated by a C-terminal motif that is conserved across eubacteria, archaea, and chloroplasts. Proc. Natl. Acad. Sci. U.S.A. 99, 15693–15698 10.1073/pnas.232590599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hu Z., and Lutkenhaus J. (2003) A conserved sequence at the C-terminus of MinD is required for binding to the membrane and targeting MinC to the septum. Mol. Microbiol. 47, 345–355 10.1046/j.1365-2958.2003.03321.x [DOI] [PubMed] [Google Scholar]
- 27. Salje J., van den Ent F., de Boer P., and Löwe J. (2011) Direct membrane binding by bacterial actin MreB. Mol. Cell 43, 478–487 10.1016/j.molcel.2011.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Duman R., Ishikawa S., Celik I., Strahl H., Ogasawara N., Troc P., Löwe J., and Hamoen L. W. (2013) Structural and genetic analyses reveal the protein SepF as a new membrane anchor for the Z ring. Proc. Natl. Acad. Sci. U.S.A. 110, E4601–4610 10.1073/pnas.1313978110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Combet C., Blanchet C., Geourjon C., and Deléage G. (2000) NPS@: network protein sequence analysis. Trends Biochem. Sci. 25, 147–150 10.1016/S0968-0004(99)01540-6 [DOI] [PubMed] [Google Scholar]
- 30. Laptenko O., Tong D. R., Manfredi J., and Prives C. (2016) The Tail That Wags the Dog: How the Disordered C-Terminal Domain Controls the Transcriptional Activities of the p53 Tumor-Suppressor Protein. Trends Biochem. Sci. 41, 1022–1034 10.1016/j.tibs.2016.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Friedler A., Veprintsev D. B., Freund S. M., von Glos K. I., and Fersht A. R. (2005) Modulation of binding of DNA to the C-terminal domain of p53 by acetylation. Structure 13, 629–636 10.1016/j.str.2005.01.020 [DOI] [PubMed] [Google Scholar]
- 32. Rustandi R. R., Baldisseri D. M., and Weber D. J. (2000) Structure of the negative regulatory domain of p53 bound to S100B(betabeta). Nat. Struct. Biol. 7, 570–574 [DOI] [PubMed] [Google Scholar]
- 33. Martín-García B., Martín-González A., Carrasco C., Hernández-Arriaga A. M., Ruíz-Quero R., Díaz-Orejas R., Aicart-Ramos C., Moreno-Herrero F., and Oliva M. A. (2018) The TubR-centromere complex adopts a double-ring segrosome structure in Type III partition systems. Nucleic Acids Res. 46, 5704–5716 10.1093/nar/gky370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ni L., Xu W., Kumaraswami M., and Schumacher M. A. (2010) Plasmid protein TubR uses a distinct mode of HTH-DNA binding and recruits the prokaryotic tubulin homolog TubZ to effect DNA partition. Proc. Natl. Acad. Sci. U.S.A. 107, 11763–11768 10.1073/pnas.1003817107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hoshino S., and Hayashi I. (2012) Filament formation of the FtsZ/tubulin-like protein TubZ from the Bacillus cereus pXO1 plasmid. J. Biol. Chem. 287, 32103–32112 10.1074/jbc.M112.373803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Montabana E. A., and Agard D. A. (2014) Bacterial tubulin TubZ-Bt transitions between a two-stranded intermediate and a four-stranded filament upon GTP hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 111, 3407–3412 10.1073/pnas.1318339111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lansbergen G., and Akhmanova A. (2006) Microtubule plus end: a hub of cellular activities. Traffic 7, 499–507 10.1111/j.1600-0854.2006.00400.x [DOI] [PubMed] [Google Scholar]
- 38. Hofbauer H. F., Gecht M., Fischer S. C., Seybert A., Frangakis A. S., Stelzer E., Covino R., Hummer G., and Ernst R. (2018) The molecular recognition of phosphatidic acid by an amphipathic helix in Opi1. J. Cell Biol. 217, 3109–3126 10.1083/jcb.201802027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Giménez-Andrés M., Čopič A., and Antonny B. (2018) The many faces of amphipathic helices. Biomolecules 8, 45 10.3390/biom8030045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Drin G., Casella J. F., Gautier R., Boehmer T., Schwartz T. U., and Antonny B. (2007) A general amphipathic alpha-helical motif for sensing membrane curvature. Nat. Struct. Mol. Biol., 14, 138–146 10.1038/nsmb1194 [DOI] [PubMed] [Google Scholar]
- 41. Szeto T. H., Rowland S. L., Habrukowich C. L., and King G. F. (2003) The MinD membrane targeting sequence is a transplantable lipid-binding helix. J. Biol. Chem. 278, 40050–40056 10.1074/jbc.M306876200 [DOI] [PubMed] [Google Scholar]
- 42. Lee P. S., and Grossman A. D. (2006) The chromosome partitioning proteins Soj (ParA) and Spo0J (ParB) contribute to accurate chromosome partitioning, separation of replicated sister origins, and regulation of replication initiation in Bacillus subtilis. Mol. Microbiol. 60, 853–869 10.1111/j.1365-2958.2006.05140.x [DOI] [PubMed] [Google Scholar]
- 43. Larsen R. A., Cusumano C., Fujioka A., Lim-Fong G., Patterson P., and Pogliano J. (2007) Treadmilling of a prokaryotic tubulin-like protein, TubZ, required for plasmid stability in Bacillus thuringiensis. Genes Dev. 21, 1340–1352 10.1101/gad.1546107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Van Duyne G. D., Standaert R. F., Karplus P. A., Schreiber S. L., and Clardy J. (1993) Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 229, 105–124 10.1006/jmbi.1993.1012 [DOI] [PubMed] [Google Scholar]
- 45. Terwilliger T. C., Adams P. D., Read R. J., McCoy A. J., Moriarty N. W., Grosse-Kunstleve R. W., Afonine P. V., Zwart P. H., and Hung L. W. (2009) Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr. D Biol. Crystallogr. 65, 582–601 10.1107/S0907444909012098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 47. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 48. DeLano W. L. (2002) The PyMOL Molecular Graphics System, Version 1.2b2 Schrödinger, LLC. [Google Scholar]
- 49. Jain S. S., and Tullius T. D. (2008) Footprinting protein-DNA complexes using the hydroxyl radical. Nat. Protoc. 3, 1092–1100 10.1038/nprot.2008.72 [DOI] [PubMed] [Google Scholar]
- 50. Lewis P. J., and Marston A. L. (1999) GFP vectors for controlled expression and dual labelling of protein fusions in Bacillus subtilis. Gene 227, 101–110 10.1016/S0378-1119(98)00580-0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Crystallographic data and coordinates were deposited in Protein Data Bank with the accession number of 7C7Y.