

Abstract

l-ascorbic acid (AA) or vitamin C is a crucial nutrient needed for optimal health. However, being unable to be synthesized by the body, it is thus necessary to be included in health care products. Moreover, AA is one of the antioxidants that occur naturally, which is used in pharmaceutical and food products as an antioxidant additive. However, AA is vulnerable to environmental settings and undergoes oxidative degradation to dehydroascorbic acid and further to inactive products. Therefore, new research strategies and approaches are required to augment its stability. The objective of this study is to develop and characterize a fiber-reinforced-phospholipid (FRP) matrix-based vehicle, Zeal-AA, for the delivery of AA and optimize the oral bioavailability of the obtained AA powder using an efficacy study by open-label, randomized, single-dose, two-treatment, two-sequence, two-period, two-way crossover. The structural and surface morphologies were analyzed by Fourier transform infrared spectroscopy, transmission electron microscopy, scanning electron microscopy, and differential scanning calorimetry studies. Encapsulation efficiency, mean particle size, size distribution, ζ-potential measurements, and ADMET profiling revealed the potential delivery system for AA. AUC0–t was found to be 55.23 (mg/dL) for Zeal-AA, whereas it was 9.38 (mg/dL) for AA, and Cmax was found to be 6.69 (mg/dL) for Zeal-AA, whereas it was 1.23 (mg/dL) for AA, with a fold difference of bioavailability in terms of AUC found to be 5.9 fold. The results show that a single oral dose of Zeal-AA is capable of rising the AA levels in the body relative to the control up to 24 h.
1. Introduction
l-ascorbic acid (AA or vitamin C) is an essential nutrient and an important bioactive compound due to its potential health benefits. Besides, it is used as an antioxidant additive in the food industry and as an essential vitamin to be acquired from foods; thus, it is a crucial ingredient in health care products. However, its degradability due to different environmental factors, namely, temperature, pH, light, and oxygen, emphasizes its instability in aqueous solutions, which in turn is a major issue during food processing and preparation.1−3 The chief AA degradation pathway in aqueous liquid systems entails its oxidation to dehydroascorbic acid, which quickly degrades itself to 2,3-diketogulonic acid, thus losing its vitamin property.4 Moreover, the oral bioavailability of the AA is less when compared with infusion methods via arteries or veins.5,6 This is particularly important because while seeing the significance of developing orally administered commercial products, the oral bioavailability leads to drug optimization by reaching the bloodstream after oral administration.7 Further, infusion methods have little acceptance and infection transmission risks, discomfort, and phlebitis.8 To overcome the oral bioavailability hitches and therapeutic issues of AA, various strategies such as microencapsulation, using liposomes and nanoparticles, have been adopted.9−11 Among them, phospholipid-based delivery of AA as a drug or supplement, with a naturally slow or regulated pattern of absorption or release, can be accelerated when encapsulated within a liposome.6
Phospholipids, as an appropriate encapsulation medium containing polar as well as nonpolar amphiphilic phospholipid bilayers, can transport both hydrophilic and hydrophobic compounds effectively.12,13 Moreover, lipid molecules that are generally recognized as safe (GRAS) and biodegradable can improve the transcellular transport via transient disruption of cellular lipophilic bilayers and may well progress the paracellular drug transport. Liposomal encapsulation of AA improves the stability and bioavailability and enhances the controlled release within the gastrointestinal tract.14 Nevertheless, conventional liposomal formulations have many drawbacks that affect the therapeutic effect of active biomolecules and are liable to fast “clearance” from the bloodstream.15,16 Phospholipid bilayer membranes of conventional phospholipids are toughly networked with circulating proteins in the bloodstream (opsonization) and taken up by certain macrophages, thereby resulting in a short blood circulation time and swift elimination from blood. Moreover, the physical and chemical attributes of phospholipids can further change due to hydrolysis of the ester bonds (between fatty acids and glycerol), peroxidation of unsaturated acyl chains, and degradation and oxidation of phospholipids.17,18 Thus, these biochemical reactions might alter the quality and diminish the liposomal stability.
In the same way, phospholipids might also undergo aggregation or flocculation and fusion or coalescence, modifying the vesicle size, initiating the drug seepage, or harming the encapsulated drugs. Further, the pharmacokinetics, which concerns the study of the entrance, movement, and changes of the drug in the body and elimination of the drug from the body, inextricably linked to the chemical structure of the drug. Thus, an absorption, distribution, metabolism, excretion, and toxicity (ADMET) analysis on in silico models that link structural changes with changes in response, from which compounds with enhanced properties can be calculated and predicted.19
On this context, our research team developed a stabilized fiber-reinforced-phospholipid (FRP)-based powdered formulation of AA using nanofiber weaving (Zeal) technology with high-pressure homogenization and the spray-drying process to get rid of water. Zeal technology has the utmost potential to shield the functional properties of the active molecules with increased stability and sustained release at a specific period, and thus further improving the bioavailability of active molecules with upgraded healthiness.18 Thus, the purpose of the current study was to develop a powdered form of FRP- vehicle-based AA (Zeal-AA) using the technology to deliver profitably achievable formulation for industrial production. The characterizations of Zeal-AA were carried out by Fourier transform infrared spectroscopy (FTIR), transmission electron microscopy (TEM), scanning electron microscopy (SEM), differential scanning calorimetry (DSC), and thermogravimetric analysis (TGA). The other important objective of this study was to evaluate the effects of Zeal-AA on oral availability by open-label, randomized, single-dose, two-treatment, two-sequence, two-period, two-way crossover.
2. Results and Discussion
2.1. Development of Liposomes
The powder form of AA-loaded FRP vesicles was prepared via Zeal technology to safeguard the functional properties with amplified stability and sustained release of the active molecules at a specific period. Herein, AA is well encapsulated into the liposomal bilayer, and the distinct aqueous space forms the core of the liposomes. During the spray-drying process, the large vesicle liposomes contract to a smaller dimension and form small vesicle liposomes. Then, due to the electrostatic interaction between the phospholipids and the nanofiber, the well-structured turmeric nanofiber (TNF) formed exerts a “fastening effect,” keeping the spherical liposomal organization undamaged. While adding water, the small vesicle bulges and converts into a large vesicle as earlier.
2.2. Morphology by SEM and TEM
The surface morphology and shape of the Zeal-AA powder were studied by SEM and TEM as depicted in Figure 1 below. The Zeal product shows spherical shapes, lacking any aggregation, signifying the stability of the formulation. Similarly, the SEM image of Zeal-AA shows a smooth surface of the liposomal powder with good encapsulation of AA, whereas the normal AA shows the agglomerated and nonspherical form. The TEM images show a highly oriented, circular, liposomal moiety with active ingredient intact, which proves the formation of a stable liposomal product. The TEM images exhibit a distinct dark gray interface inside and a well-rounded transparent phospholipid layer, while the inner portion is seen to be darker, which is ascribed to the fastening effect of TNF.20 The TEM and SEM of Zeal-AA reveal the formation of true liposomal products with a smooth surface and spherical shape without any accumulation, specifying the stability of the formulation.
Figure 1.

Scanning electron microscopy photographs of AA (A) and Zeal-AA (B); transmission electron microscopy of Zeal-AA (C).
2.3. Mean Particle Size, Size Distribution, and ζ-Potential Measurements
The ζ-potential gives an impression of the surface charge, degree of electrostatic repulsion between adjacent particles, colloidal stability, and entrapment efficiency.21 Zeal-AA shows a lower ζ-potential (−40 to 0 mV) when equated to that of AA (−20 to 10 mV) as shown in Figure 2, which is ascribed to the strengthening effect of the specific carriers between the phospholipids and active ingredients. The ζ-potential offers a sign of the stability of Zeal-AA, as the high negative value of the ζ-potential shows that the repulsion between the particles is larger; hence, there will be no propensity for the particles to come together and thus leads to a more stable true liposomal formulation without flocculation.22,23 The particle sizes of Zeal-AA exposed were in the range of 200–700 nm, as shown in Figure 3, in accordance with the results obtained from SEM and TEM.
Figure 2.

ζ-potential of AA (a) and Zeal-AA (b).
Figure 3.

Size distribution curve of Zeal-AA.
2.4. Encapsulation Efficiency (EE)
The encapsulation efficiency of AA onto the phospholipids was examined, for which respectable percentages have been recorded as follows: 83.58 ± 2.18%. These outcomes specify that AA has been successfully assimilated onto the lipid bilayer and has maintained the powder form, through the reinforcement of fibers.
2.5. Fourier Transform Infrared Spectroscopy (FTIR)
The FTIR spectra of AA, TNF, phospholipid, and Zeal-AA are characterized in Figure 4. The −C–O stretching frequency of AA at 1750 cm–1 is present in both AA and Zeal-AA, indicating that AA is well presented in the liposomal matrix in Zeal-AA.11 Similarly, the −O–H stretching of enediol of AA at 1317 cm–1 becomes absent in Zeal-AA, which shows that AA is effectively linked in the liposomal whole matrix11 (Figure 4c). The characteristic peaks of lecithin are present between 900 and 1026 cm–1, attributed to the −P–O and −O–CH3 stretching peaks (Figure 4a), which are still present in the final formulation.24 Similarly, the characteristic peaks of liposomes are present between 2986 and 2806 cm–1 due to the fact that the asymmetric stretching of −CH2 groups decreased considerably in Zeal-AA because of the conjugation of AA in the lipid bilayer.25,26 The peaks at 3283 and 1410 cm–1 are assigned to the stretching vibrations of the −O–H groups in TNF (Figure 4b) and the symmetric bending of −C–H2, respectively.27−29 The deviations can be ascribed to the good encapsulation of AA by FRP inside the liposomal matrix.
Figure 4.
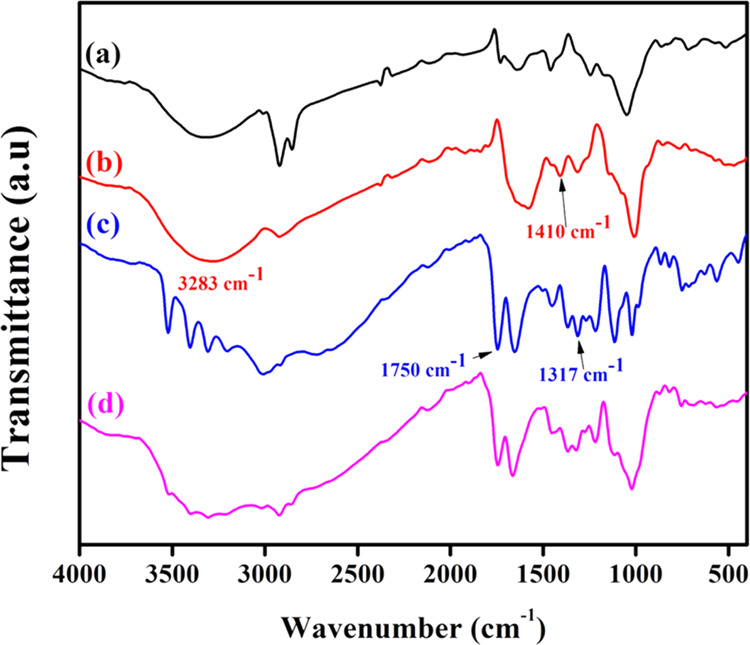
FTIR chromatograms of phospholipid (a), TNF (b), AA (c), and Zeal-AA (d).
2.6. Thermal Analysis by DSC
The interfaces between phospholipids and the encapsulated AA in Zeal-AA can be examined further using DSC. As shown in Figure 5a, the DSC thermogram of AA displays a main endothermic peak at 193 °C, which matches to the crystalline melting point of AA followed by degradation. More importantly, the melting endothermic peak of AA is not present in the thermogram of Zeal-AA (Figure 5b). This reveals the good encapsulation of AA into the nanofiber-stabilized liposomes due to the incorporation of AA into the phospholipids.30 The DSC spectrum of Zeal-AA shows the phospholipid melting point as a minor endothermic peak at 83 °C30,31 and likeness associated with the vaporization of bounded and unbounded water molecules.20 The main exothermic peaks at 193 °C are due to the dissociation of phospholipid–AA cooperative forces (van der Waals forces and H-bonding).30 Similarly, the exothermic peak at 282 °C results during crystallization of the AA matrix with the thermoplastic polymer from the nanofiber, which is well known by the mass reduction of the glycosidic linkage of cellulose and AA degradation.20 The DSC results for FRP show that the stability of FRP is improved due to the incorporation of AA into the lipid bilayer with the fastening effect of the TNF.
Figure 5.
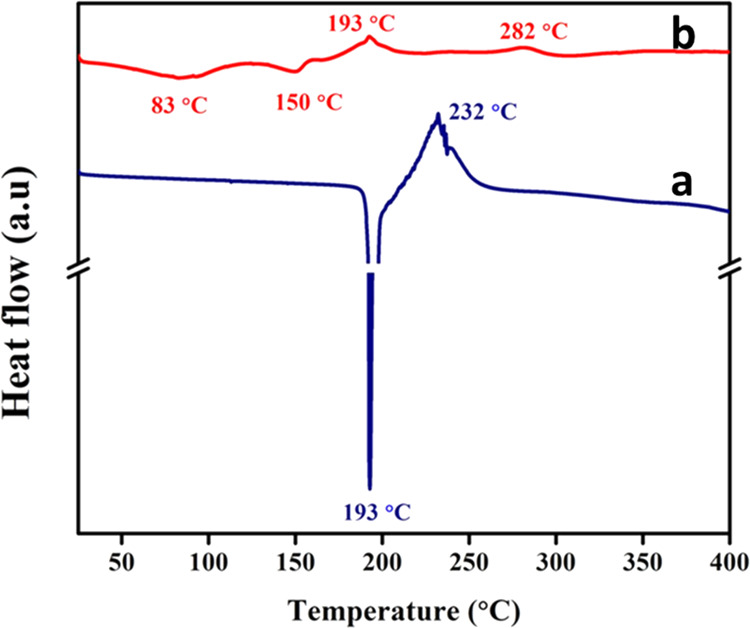
DSC chromatograms of AA (a) and Zeal-AA (b).
2.7. Predicted Pharmacokinetic Properties
To evaluate various ADMET properties, a series of high-grade prediction models were engendered and validated. The results of the in silico study of AA, phospholipid vesicles, and TNF can be seen in Table 1. According to the results, AA and phospholipid vehicles had respectable gastrointestinal absorption (39.154 and 34.698%). Also, the phospholipid vehicle and TNF were foretold to be substrates for P-glycoprotein (P-gp), but not AA. Moreover, the volume of distribution (VDss) was found to be in the order phospholipid < TNF < AA, where phospholipid has the low VDss (−0.134 log L/Kg). Besides, phospholipid was found to be subjected to the metabolism via the CYP3A4 enzyme and none of them were found to be an inhibitor for cytochrome-P450 (CYP-450). Regarding drug clearance, a faster excretion process was found for TNF (1.545 log mL/(min kg)), while the lowest total clearance (both hepatic and renal clearance) was found for AA (0.631 log mL/(min kg)). According to the predicted toxicity results, TNF, phospholipid, and AA were found to be nontoxic (hepatic toxicity), noncarcinogenic (AMES test analyzes the capacity of a compound to induce mutations in DNA), and with no hERG-gene-related cardiotoxicity. Moreover, the prediction for the lowest observed adverse effect level (LOAEL) values of the compounds tested ranged from 3.186 to 4.621 log mg/kg bw/day, which means that they are not included in any toxicity classes.
Table 1. ADMET Studies of AA, Phospholipid Vesicles, and TNF.
| property | AA | phospholipid vesicle | TNF | |
|---|---|---|---|---|
| absorption | intestinal absorption (human) (% absorbed) | 39.154 | 34.698 | 6.412 |
| P-glycoprotein substrate (Yes/No) | No | Yes | Yes | |
| P-glycoprotein I inhibitor (Yes/No) | No | No | No | |
| P-glycoprotein II inhibitor (Yes/No) | No | No | No | |
| distribution | VDss (human) (log L/kg) | 0.218 | –0.134 | 0.203 |
| Fraction unbound (human) (Fu) | 0.825 | 0.376 | 0.629 | |
| metabolism | CYP2D6 substrate (Yes/No) | No | No | No |
| CYP3A4 substrate (Yes/No) | No | Yes | No | |
| CYP2C9 inhibitor (Yes/No) | No | No | No | |
| CYP2D6 inhibitor (Yes/No) | No | No | No | |
| CYP3A4 inhibitor (Yes/No) | No | No | No | |
| excretion | total clearance (log mL/(min/ kg)) | 0.631 | 1.531 | 1.545 |
| renal OCT2 substrate (Yes/No) | No | No | No | |
| toxicity | AMES toxicity (Yes/No) | No | No | No |
| max. tolerated dose (human) (log mg/(kg/day)) | 1.598 | 0.454 | 1.442 | |
| hERG I inhibitor (Yes/No) | No | No | No | |
| hERG II inhibitor (Yes/No) | No | No | No | |
| oral rat chronic toxicity (LOAEL) (log mg/kg bw/day) | 3.186 | 4.064 | 4.621 | |
| hepatotoxicity (Yes/No) | No | No | No | |
2.8. Efficacy of Zeal-AA and Pharmacokinetic Parameters
All of the enrolled research partakers completed the study. The oral administration of Zeal-AA was well accepted, and no adverse events were reported. Baseline circulating concentrations of AA did not diverge between treatments. Circulating concentrations of AA prior to and after administration at different time intervals are presented in Table 2. The effects of the formulation of AA (Zeal-AA) were compared with AA (control). Pharmacokinetic parameters of AA concentrations by mean AUC ±standard deviation (SD), maximum and minimum absorbance for each time interval, and median (all concentrations in mg/dL) for each formulation at different time intervals up to 24 h are also given in Table 2.
Table 2. Plasma Concentrations (mg/dL) of Zeal-AA and Control AA at Different Time Intervals.
| mean ± SD |
minimum |
maximum |
median |
|||||
|---|---|---|---|---|---|---|---|---|
| time (h) | AA | Zeal-AA | AA | Zeal-AA | AA | Zeal-AA | AA | Zeal-AA |
| 0 | 0.027 ± 0.032 | 0.038 ± 0.016 | 0.001 | 0.017 | 0.092 | 0.072 | 0.015 | 0.037 |
| 0.25 | 0.352 ± 0.261 | 1.863 ± 1.080 | 0.010 | 0.284 | 0.753 | 3.383 | 0.340 | 1.871 |
| 0.5 | 0.591 ± 0.395 | 3.086 ± 0.723 | 0.023 | 1.650 | 1.259 | 3.873 | 0.600 | 3.192 |
| 0.75 | 0.667 ± 0.328 | 2.760 ± 0.609 | 0.183 | 1.433 | 1.243 | 3.456 | 0.620 | 2.953 |
| 1 | 0.633 ± 0.396 | 4.010 ± 1.048 | 0.192 | 2.491 | 1.394 | 5.799 | 0.534 | 3.828 |
| 1.5 | 0.480 ± 0.413 | 5.064 ± 1.122 | 0.074 | 3.530 | 1.289 | 6.938 | 0.368 | 4.725 |
| 2 | 0.732 ± 0.328 | 4.897 ± 1.370 | 0.248 | 2.942 | 1.313 | 6.715 | 0.680 | 5.069 |
| 2.5 | 0.867 ± 0.369 | 5.436 ± 1.461 | 0.336 | 3.783 | 1.552 | 8.217 | 0.804 | 5.124 |
| 3 | 1.059 ± 0.442 | 5.067 ± 1.410 | 0.434 | 3.198 | 1.771 | 6.935 | 1.050 | 4.870 |
| 4 | 1.126 ± 0.389 | 5.505 ± 1.466 | 0.345 | 3.086 | 1.471 | 7.866 | 1.239 | 5.585 |
| 5 | 0.782 ± 0.389 | 4.536 ± 1.012 | 0.060 | 3.065 | 1.225 | 6.169 | 0.885 | 4.721 |
| 6 | 0.617 ± 0.373 | 3.807 ± 0.659 | 0.023 | 2.598 | 1.096 | 4.668 | 0.508 | 3.922 |
| 8 | 0.542 ± 0.272 | 3.089 ± 0.988 | 0.021 | 1.823 | 0.846 | 4.565 | 0.611 | 3.118 |
| 10 | 0.362 ± 0.205 | 2.742 ± 0.994 | 0.019 | 1.426 | 0.657 | 4.284 | 0.393 | 2.724 |
| 12 | 0.285 ± 0.261 | 2.742 ± 0.655 | 0.000 | 1.128 | 0.718 | 2.708 | 0.255 | 1.444 |
| 24 | 0.047 ± 0.041 | 0.080 ± 0.026 | 0.002 | 0.029 | 0.120 | 0.114 | 0.046 | 0.086 |
The graphical representations of the mean concentrations of AA and Zeal-AA as a function of time are presented in Figure 6. Multiple AA concentration peaks were detected in the plasma as a function of time for both AA and Zeal-AA. The total average pharmacokinetic variables (mean ± SD) estimated from plasma AA as well as the p-values are given in Table 3. The maximum plasma AA (Cmax) and the AUC for Zeal-AA were roughly 6.69 and 55.6 mg/dL, respectively. In comparison, these values for AA are low, 1.23 and 9.66 mg/dL, respectively. The AUC is the utmost trustworthy measure of the bioavailability, as it quantifies the whole response over time and offers a more precise image of bioavailability.32 The time to reach the maximum plasma concentration (Tmax) is 3–3.5 h for both AA and Zeal-AA, which indicates that the release patterns of AA in both the groups are the same.
Figure 6.
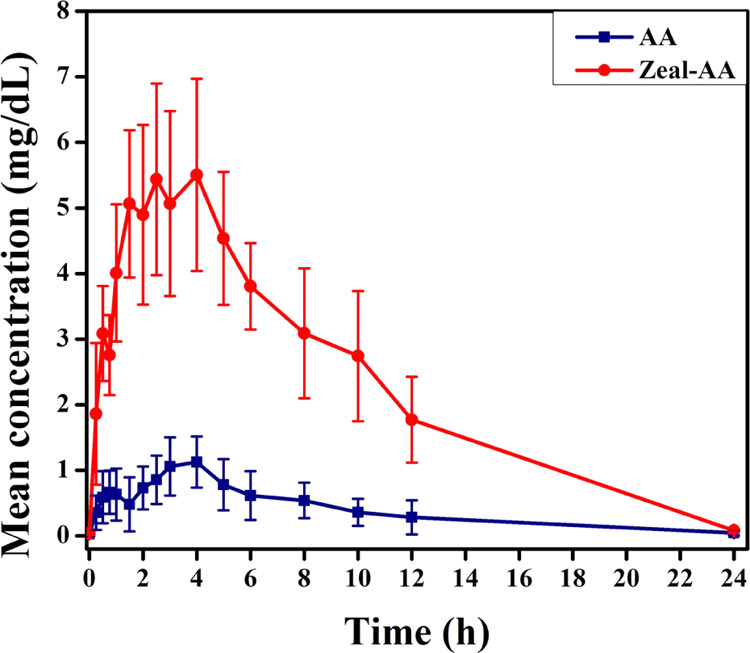
Mean plasma concentrations (mg/dL) of AA (control) compared with Zeal-AA. All of the values stated are mean ± SD.
Table 3. Average PK Variables (mean ± SD) from Plasma Vitamin C of AA and Zeal-AA and the p-Value.
| AA | Zeal-AA | P-value | |
|---|---|---|---|
| AUC0–t (mg·h/dL) | 9.38 ± 4.91 | 55.23 ± 9.39 | <0.0005a |
| AUC0–∞(mg·h/dL) | 9.66 ± 5.02 | 55.61 ± 9.31 | <0.0005a |
| Cmax (mg/dL) | 1.23 ± 0.38 | 6.69 ± 1.00 | <0.0005a |
| Tmax (h) | 3.56 ± 0.62 | 3.06 ± 1.15 | 0.388 |
| T1/2 (h) | 3.14 ± 2.01 | 3.27 ± 0.37 | |
| Kel (h–1) | 0.31 ± 0.18 | 0.21± 0.05 |
Statistically significant.
AA, an essential component of the diet for humans, has three chief roles in human metabolism: as an enzyme cofactor, a chemical reductant, and an antioxidant.33 Thus, an adequate intake is important not only because it is a powerful reducing agent but also because it helps white blood cells in fighting infections and thereby supporting the immune system.34 However, unlike other animals, humans are unable to synthesize AA endogenously, so it is an essential dietary component and has to be obtained from the diet.35 Approximately, 90% of AA in the human diet is obtained from fruits and vegetables as ascorbic and dehydroascorbic acids. Yet, these compounds are sensitive to light and oxygen and may decompose under normal transport and storage conditions, resulting in reduction of the nutritional value of the foodstuffs.36 In addition, the body cannot store AA, and both the elevated oral intake and the intravenous application have certain disadvantages. Thus, as a novel approach, phospholipid-based oral delivery of AA might be a way to overcome these limitations mainly because lipid aggregates can prevent AA degradation in the gastrointestinal tract; thus, a raise of AA in the plasma level is observed after liposomal oral application of AA.37
It is worth mentioning that the absorption of drugs depends on various factors including membrane permeability [indicated by a colon cancer cell line (Caco-2)], intestinal absorption, skin permeability levels, and P-glycoprotein substrates or inhibitors.38 In the current study, AA was loaded in vesicles with fiber-reinforced phospholipids, which form a highly stable lipid bilayer in powdered formulation by fiber core formation. Moreover, the cellulose nanofibers isolated from natural sources have already been reported as drug delivery systems in many studies.39 This fiber core formed after drying the aqueous core thus facilitates accelerated delivery of AA. According to the results, the phospholipid vesicle is predicted to have an appreciable absorption (34.698%), where as a human intestinal absorption parameter, an absorbance of greater than 30% is considered to be highly significant. In addition, P-glycoprotein (P-gp), a member of the ATP-binding transmembrane glycoprotein family and found in cells all over the body, including those lining the intestine, allows pumping of a substrate against a concentration gradient using ATP. The results suggested that phospholipid vesicles being a substrate of the P-gp drug transporter will be transported from the intestine.40
To investigate the physicochemical peculiarities of Zeal-AA and to understand how its characteristics affect its in vivo distribution and behavior of the formulations, morphological studies were carried out. From the morphological analysis carried out by SEM and TEM, the Zeal-AA powder is observed to have a spherical shape, without any aggregation, indicating the stability of the formulation. Moreover, the formulation was uniform in shape and size, and the FRP-based powder exhibited a smooth surface. Similarly, the good encapsulation and formation of a true encapsulated product were visible. Approvingly, the mean percentage of AA encapsulated in the FRP vehicle of 83.5% explains the large aqueous space inside this FRP-based vehicle. This stable core upon drying forms a highly stable fiber core, which thus facilitates accelerated delivery of AA. From the results, it is observed that the bioavailability of Zeal-AA has increased the plasma levels compared with AA. This clearly indicates that a large single oral dose of the FRP-based AA formulation could increase the plasma levels of AA to a maximum of 55 mg/dL. Possibly, the de-encapsulation of the FRP-based vehicle in the plasma offers a sustained release of AA, thereby increasing the plasma AA concentration. Accordingly, a higher VDss (0.218 log L/Kg) predicted for AA than that for phospholipid vesicles (−0.134 log L/Kg) from in silico studies thus further provides a propensity for AA to leave the plasma and move into the extravascular compartments of the body.41 Thus, this increased plasma AA concentration observed in the bioavailability study and thereby increased distribution to other tissues exhibit the effectiveness of FRP-based vehicles as an effective delivery system for AA.
From a research perspective, it is quite interesting to compare the previously published studies on pharmacokinetic responses of the oral dose of liposomal AA to the current study as in Table 4. When we compare the AUC per mg AA and Cmax per mg AA, Zeal-AA shows better results, as shown in Table 4. Davis et al. presented that the oral application of 4 g of liposome-encapsulated AA amplified the plasma concentration up to Cmax/mg (about 0.00082 mg/dL) after 3 h.6 Consistently, another study by Łukawski et al.42 had a Cmax/mg concentration of 0.0016 mg/dL. Promising enough, Zeal-AA achieved a Cmax/mg of 0.045 mg/dL with a low oral dosage of 150 mg, due to the deterrence of AA degradation, while preserving itself inside the FRP-based vehicle. Hickey et al. and Mikirova also studied the efficacy of liposomal AA at high dosages (36 and 25 g, respectively), and the results showed the inefficiency to absorb high dose of AA to the blood even at the liposomal formulations.43,44
Table 4. Comparative Pharmacokinetic Responses to the Oral Dose of Liposomal AA.
Besides, the physical stability of the formulation in a powdered form estimated a negative value for ζ-potential due to electrostatic repulsion between particles, which is larger with no propensity for the particles to come together. This stability of the formulation without flocculation can be ascribed to the strengthening effect of the TNF between the phospholipids and AA. Further, the heat stability was verified by DSC analysis, which indicated the improved stability of AA and further signposted the pharmacokinetic efficacy of the Zeal-AA formulation. This observation is in line with results predicted for the volume of distribution (Vdss), which is an important determinant of pharmacokinetic studies. Metabolism, primarily based on the CYP-P450 enzymes, when analyzed was found to have no effect (on the substrate or inhibitor), meaning that the compounds tend to be metabolized by the body.45
Mechanistically, significant absorption of AA in the presence of phospholipids from the gastrointestinal tract along with the help of P-gp transporter thus might lead to an acceptable AA concentration in plasma. Further, suitable VDss provides major supply to the body tissues. Generally, the possibilities of networking with pharmacological target proteins, namely, receptors, channels, and enzymes, aid a drug to easily diffuse between plasma and tissues in their unbound (free) form. Relevantly, the fraction unbound in plasma for AA (0.825) and FRP vehicle (0.376) forecasted thus promises the effective distribution of AA after achieving the plasma concentration. Further, adequate metabolism as predicted by CYP-P450 enzymes and excretion prevent the accumulation of the drug in the body and hence reduce the toxicity.
3. Conclusions
In brief, this was a comparative oral bioavailability study and characterization of AA encapsulated in FRP- based vehicles on healthy, adult, human participants under fasting conditions. Microscopic images demonstrated the core-type structure, which confirmed the characteristic pattern of phospholipid vesicles of 200–700 nm size. After AA encapsulation, the morphology (spherical) of vesicles remained intact in the presence of turmeric fibers. The encapsulation efficiency (EE%) was 83.58 ± 2.18%, and the results specified that the oral delivery of Zeal-AA showed 5.9 times more bioavailability than that of the normal AA. Zeal-AA confirmed the oral bioavailability of AA by enhancing responses of Cmax, AUC0–t, and AUC0–∞ related to nonliposomal AA. As an illustration, normalization of the data on the basis of Cmax per mg administrated AA indicates that Zeal-AA displays superior oral bioavailability as compared to previous studies.
4. Materials and Methods
4.1. Materials
AA is procured from S. R. Biochem, India, and TNF was provided by Plant Lipids Pvt., Ltd., India. Phospholipids (sunflower lecithin) were bought from Shankar Soya Concepts, India. The standard of AA was obtained from Sigma-Aldrich, India.
4.2. Development of AA-Loaded FRP Vesicles
Phospholipid vesicles reinforced with the turmeric fiber loaded with AA (Zeal-AA) in a powdered form were prepared by Zeal technology. The schematic representation of the preparation is given in Scheme 1. To begin with, 10 g of phospholipids was liquefied in 1 L of Millipore H2O at 80 °C. Into this, 10 g of turmeric fibers was added along with 25 g of modified food starch as a stabilizing agent to preserve the structure of phospholipid vesicles under high shear using a high-speed emulsifier (Homomixer Mark II. 2.5; Primix, Japan). The subsequent mixture was highly sheared continuously, and then AA was added, after which the temperature was brought down to 40 °C. This was then homogenized with a pressure range of 300–500 bars applying an APV 1000 homogenizer (SPX Flow Technology, Germany), and the procedure was carried out three times. The formulation was then dehydrated through the spray-drying (Ohkawara Kakohki Co., Ltd., Japan, model L-8) method with an inlet temperature of 160–170 °C and outlet temperature of 60–70 °C. The final AA-loaded FRP vesicles thus achieved were transferred into vials and retained at room temperature for analyses. AA was analyzed by high-performance liquid chromatography (HPLC),46 and the obtained product was having 28.3% AA content.
Scheme 1. Schematic Representation of the Preparation of Zeal-AA.

4.3. Mean Particle Size, Size Distribution, and ζ-Potential Measurements
The size and ζ-potential (ZP) of AA-loaded FRP vesicles were calculated by the dynamic light scattering method using a Zetasizer Nano ZS instrument (Malvern Instruments, Ltd., Worcestershire, U.K.). The measurements were performed at 25 °C. The intensity was determined at a scattering angle of 90 °C. Then, 1.0 mL of samples was diluted 10-fold with distilled H2O to prevent scattering phenomena.
4.4. Encapsulation Efficiency (EE)
Encapsulation efficiency (EE) was calculated in accordance with the process defined by Parhizkar et al.47 Known amounts of Zeal-AA were dissolved in methanol and agitated to achieve a colloidal solution. The resulting solution was centrifuged for 15 min at 3000 rpm and 5–10 °C. The AA in the supernatant was quantified by HPLC to determine the unencapsulated AA content. Then, the quantity of encapsulated AA and EE (%) were calculated by the equation below.
4.5. Morphological Studies
TEM (JEM-2100, JEOL, Tokyo, Japan) was used to examine the surface morphology of Zeal-AA phospholipid vehicles. The phospholipid suspension was diluted with deionized H2O and positioned on mesh grids. The left-over solution was removed, and the samples were analyzed at 200 kV. The surface morphology of the Zeal-AA samples was also examined using a SEM (Vega3 Tescan, Brno, Czech Republic). Prepared samples were placed in aluminum stubs with double-sided carbon tape and sputter-layered with a fine coating of gold, using a sputter gold coater to eliminate charring, during the analysis. Prepared Zeal-AA samples were scanned using an accelerating voltage of 20 kV.
4.6. Fourier Transform Infrared Spectroscopy (FTIR)
FTIR spectra were recorded using a JASCO ATR-FT/IT-4700 instrument. All of the samples were dried well and analyzed at a scan range of 400–4000 cm–1, with 32 scans per sample using the KBr pellet method.
4.7. Differential Scanning Calorimetry (DSC) Analysis
DSC (Mettler Toledo DSC 822e, Switzerland) was utilized to check the thermal stability of fabricated Zeal-AA. About 10 mg of the sample was placed in hermetically sealed aluminum pans at a heating rate of 10 °C/min within a temperature range of 0–400 °C, under constant purging of nitrogen (40 mL/min), and an empty pan was used as reference.
4.8. In Silico Assay: Prediction of ADME and Toxicity of Compounds
The early ADMET profiling of drug candidates is a crucial component in determining the potential success of drug development. Thus, the properties were determined through the web server pkCSM, which uses graph-based signatures (http://structure. bioc.cam.ac.uk/pkcsm) and performs equally well as or better than several other widely used methods. It is quick and accurate and predicts physically significant descriptors and pharmaceutically relevant properties of organic compounds.48 For the prediction module, SMILES molecular file types were used. Compound IDs (Pubchem) for AA, phospholipids, and TNF were 54670067, 453626, and 16211032, respectively.
4.9. Participants for the Bioavailability Study
Eight male healthy adults, falling in an age group of 21–65 years (both inclusive), were selected for the study. The willingness to follow the protocol requirements was evidenced by written, informed consent. Participants agreed not to use any medication, including vitamins and minerals, during or before the course of this study. The sample size of the study was 8, with 4 participants randomized to each of the two study arms in a double-blinded manner at a 1:1 ratio and received dosing as per randomization code and provided at site by an authorized person independently. During the second period of study, the participants were interchanged for the crossover study after the washout period of 2 weeks.
4.10. Ethics and Approvals
The study was conducted by BioAgile Therapeutics Pvt, Ltd., Bangalore, India. All study-related documents were reviewed by the Independent Ethics Committee and approved. The protocol was registered with Clinical Trials Registry India (clinicaltrials.gov) (CTRI/2019/07/020159) on 12th July 2019. The study was conducted in compliance with current Good Clinical Practices (GCP), Declaration of Helsinki, and applicable regulatory requirements.
4.11. Experimental Design and Overview
The Zeal-AA formulation (500 mg) and dose-normalized control (500 mg that contains 150 mg of AA and 350 mg of food-grade starch) were administered orally to each subject in the sitting posture, with 240 mL of water, as per the randomization schedule, after overnight fasting for at least 10 h. The blood samples were collected for the pharmacokinetic study at 0.25, 0.50, 0.75, 1.00, 1.50, 2.00, 2.50, 3.00, 4.00, 5.00, 6.00, 8.00, 10.00, 12.00, and 24.00 h post dose. Blood was collected in heparin-coated tubes, and collected blood samples were centrifuged at 3500 rpm and 4 °C for 10 min, and the separated plasma was transferred into suitably labeled polypropylene tubes. All plasma samples were stored upright in a freezer set at −20 °C and were analyzed by HPLC.
4.12. Statistical Analysis
Statistical analysis was performed on data using SAS software version 9.4 (SAS Institute Inc.). Arithmetic mean, geometric mean, standard deviation, coefficient of variation, minimum, median, and maximum for all pharmacokinetic parameters were calculated. Pharmacokinetic parameters such as the maximum drug serum concentration (Cmax), the area under the curve calculated for the last measured concentration (AUC0–t), the area under the plasma concentration–time curve extrapolated to infinity (AUC0–∞), and the time at which the Cmax is observed (Tmax) were generated using WinNonlin version 5.0.1 for evaluation. The p-values < 0.05 were considered statistically significant.
Acknowledgments
The authors acknowledge the support from the management of Plant Lipids (P) Ltd., Cochin, India, and BioAgile Therapeutics Private Limited, Bangalore, Karnataka, India, for executing the clinical study. The authors wish to thank Bibin George for the help in make the graphical representation and the cover art.
The authors declare no competing financial interest.
References
- Alvim I. D.; Stein M. A.; Koury I. P.; Dantas F. B. H.; Cruz C. C. V. Comparison between the spray drying and spray chilling microparticles contain ascorbic acid in a baked product application. LWT--Food Sci. Technol. 2016, 65, 689–694. 10.1016/j.lwt.2015.08.049. [DOI] [Google Scholar]
- Fong S. Y. K.; Martins S. M.; Brandl M.; Brandl A. B. Solid phospholipid dispersions for oral delivery of poorly soluble drugs: investigation into celecoxib incorporation and solubility-in vitro permeability enhancement. J. Pharm. Sci. 2016, 105, 11113–11123. 10.1016/S0022-3549(15)00186-0. [DOI] [PubMed] [Google Scholar]
- Matos-Jr F. E.; Sabatino M. D.; Passerini N.; Favaro-T C. S.; Albertini B. Development and characterization of solid lipid microparticles loaded with ascorbic acid and produced by spray congealing. Food Res. Int. 2015, 67, 52–59. 10.1016/j.foodres.2014.11.002. [DOI] [Google Scholar]
- Herbig A. L.; Renard C. M. Factors that impact the stability of vitamin C at intermediate temperatures in a food matrix. Food Chem. 2017, 220, 444–451. 10.1016/j.foodchem.2016.10.012. [DOI] [PubMed] [Google Scholar]
- Padayatty S. J.; Sun H.; Wang Y.; Riordan H. D.; Hewitt S. M.; Katz A.; Wesley R. A.; Levine M. vitamin C pharmacokinetics: implications for oral and intravenous use. Ann. Intern. Med. 2004, 140, 533–537. 10.7326/0003-4819-140-7-200404060-00010. [DOI] [PubMed] [Google Scholar]
- Davis J. L.; Paris H. L.; Beals J. W.; Binns S. E.; Giordano G. R.; Scalzo R. L.; Schweder M. M.; Blair E.; Bell C. Liposomal-encapsulated ascorbic acid: Influence on vitamin C bioavailability and capacity to protect against ischemia-reperfusion injury. Nutr. Metab. Insights. 2016, 9, NMI.S39764 10.4137/NMI.S39764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner J. V.; Agatonovic-Kustrin S.. In Silico Prediction of Oral Bioavailability. In Comprehensive Medicinal Chemistry II; 1st ed.; Taylor J. B.; Triggle D. J., Eds.; Elsevier: Netherlands, 2007; pp 699–724. [Google Scholar]
- Parhizkar E.; Rashedinia M.; Karimi M.; Alipour S. Design and development of vitamin C-encapsulated proliposome with improved in-vitro and ex-vivo antioxidant efficacy. J. Microencapsulation 2018, 35, 301–311. 10.1080/02652048.2018.1477845. [DOI] [PubMed] [Google Scholar]
- Comunian T. A.; Abbaspourrad A.; Favaro-T C. S.; Weitz D. A. Fabrication of solid lipid microcapsules containing ascorbic acid using a microfluidic technique. Food Chem. 2014, 152, 271–275. 10.1016/j.foodchem.2013.11.149. [DOI] [PubMed] [Google Scholar]
- Farhang B.; Kakuda Y.; Corredig M. Encapsulation of ascorbic acid in liposomes prepared with milk fat globule membrane-derived phospholipids. Dairy Sci. Technol. 2012, 92, 353–366. 10.1007/s13594-012-0072-7. [DOI] [Google Scholar]
- Chakraborty A.; Jana N. R. Vitamin C-Conjugated nanoparticle protects cells from oxidative stress at low doses but induces oxidative stress and cell death at high doses. ACS Appl. Mater. Interfaces 2017, 9, 41807–41817. 10.1021/acsami.7b16055. [DOI] [PubMed] [Google Scholar]
- Emami S.; Azadmard-Damirchi S.; Peighambardoust S. H.; Valizadeh H.; Hesari J. Liposomes as carrier vehicles for functional compounds in food sector. J. Exp. Nanosci. 2016, 11, 737–759. 10.1080/17458080.2016.1148273. [DOI] [Google Scholar]
- Toniazzo T.; Berbel I. F.; Cho S.; Fávaro-T C. S.; Moraes I. C. F.; Pinho S. C. β-carotene-loaded liposome dispersions stabilized with xanthan and guar gums: Physico-chemical stability and feasibility of application in yogurt. LWT--Food Sci. Technol. 2014, 59, 1265–1273. 10.1016/j.lwt.2014.05.021. [DOI] [Google Scholar]
- Anthony A. A.; Mumuni A. M.; Philip F.. Lipid Nanoparticulate Drug Delivery Systems: A Revolution in Dosage Form Design and Development Builders. In Recent Advances in Novel Drug Carrier Systems; 1st ed.; Sezer A. D., Ed.; In Tech: Croatia, 2012; pp 107–140. [Google Scholar]
- Gabizon A.; Horowitz A. T.; Goren D.; Tzemach D.; Shmeeda H.; Zalipsky S. In vivo fate of folate-targeted polyethylene-glycol liposomes in tumor-bearing mice. Clin. Cancer Res. 2003, 9, 6551–6559. [PubMed] [Google Scholar]
- Gabizon A.; Tzemach D.; Mak L.; Bronstein M.; Horowitz A. T. Dose dependency of pharmacokinetics and therapeutic efficacy of pegylated liposomal doxorubicin (DOXIL) in murine models. J. Drug Targeting. 2002, 10, 539–548. 10.1080/1061186021000072447. [DOI] [PubMed] [Google Scholar]
- Gabizon A.; Shmeeda H.; Barenholz Y. Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies. Clin. Pharmacokinet. 2003, 42, 419–436. 10.2165/00003088-200342050-00002. [DOI] [PubMed] [Google Scholar]
- Gopi S.; Amalraj A.; Jacob J.; Kalarikkal N.; Thomas S.; Guo Q. Preparation, characterization and in vitro study of liposomal curcumin powder by cost effective nanofiber weaving technology. New J. Chem. 2018, 42, 5117–5127. 10.1039/C7NJ05029A. [DOI] [Google Scholar]
- Davis A. M.; Riley R. J. Predictive ADMET studies, the challenges and the opportunities. Curr. Opin. Chem. Biol. 2004, 8, 378–386. 10.1016/j.cbpa.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Amalraj A.; Jacob J.; Varma K.; Gopi S. Preparation and characterization of liposomal β-Caryophyllene (Rephyll) by nanofiber weaving technology and its effects on delayed onset muscle soreness (DOMS) in humans: A randomized, double-blinded, crossover-designed, and placebo-controlled study. ACS Omega 2020, 5, 24045–24056. 10.1021/acsomega.0c03456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raval N.; Maheshwari R.; Kalyane D.; Youngren-Ortiz S. R.; Chougule M. B.; Tekade R. K.. IMportance of Physicochemical Characterization of Nanoparticles in Pharmaceutical Product Development Basic Fundamentals of Drug Delivery; 1st ed.; Tekade R. K., Ed.; Academic Press: London, 2019; pp 369–400. [Google Scholar]
- Samimi S.; Maghsoudnia N.; Eftekhari R. B.; Dorkoosh F.. Lipid-Based Nanoparticles for Drug Delivery Systems. In Characterization and Biology of Nanomaterials for Drug Delivery; 1st ed.; Mohapatra S.; Ranjan R.; Dasgupta N.; Mishra R. K.; Thomas S., Eds.; Elsevier: Netherlands, 2019; pp 47–76. [Google Scholar]
- Joseph E.; Singhvi G.. Multifunctional Nanocrystals for Cancer Therapy: A Potential Nanocarrier. In Nanomaterials for Drug Delivery and Therapy; 1st ed.;Grumezescu A. M., Ed.; Elsevier: Netherlands, 2019; pp 91–116. [Google Scholar]
- Prasad R.; Yadav A. S.; Gorain M.; Chauhan D. S.; Kundu G. C.; Srivastava R.; Selvaraj K. Graphene oxide supported liposomes as red emissive theranostics for phototriggered tissue visualization and tumor regression. ACS Appl. Bio. Mater. 2019, 2, 3312–3320. 10.1021/acsabm.9b00335. [DOI] [PubMed] [Google Scholar]
- Deygen I. M.; Seidl C.; Kölmel D. K.; Bednarek C.; Heissler S.; Kudryashova E. V.; Brase S.; Schepers U. Novel prodrug of doxorubicin modified by stearoylspermine encapsulated into PEG-chitosan-stabilized liposomes. Langmuir 2016, 32, 10861–10869. 10.1021/acs.langmuir.6b01023. [DOI] [PubMed] [Google Scholar]
- Vaghasiya K.; Sharma A.; Kumar K.; Ray E.; Adlakha S.; Katare O. P.; Hota S. K.; Verma R. K. Heparin-encapsulated metered-dose topical “nano-spray gel” liposomal formulation ensures rapid on-site management of frostbite injury by inflammatory cytokines scavenging. ACS Biomater. Sci. Eng. 2019, 5, 6617–6631. 10.1021/acsbiomaterials.9b01486. [DOI] [PubMed] [Google Scholar]
- Jacob J.; Peter G.; Thomas S.; Haponiuk J. T.; Gopi S. Chitosan and polyvinyl alcohol nanocomposites with cellulose nanofibers from ginger rhizomes and its antimicrobial activities. Int. J. Biol. Macromol. 2019, 129, 370–376. 10.1016/j.ijbiomac.2019.02.052. [DOI] [PubMed] [Google Scholar]
- Yao J.; Chen S.; Chen Y.; Wang B.; Pei Q.; Wang H. Macrofibers with high mechanical performance based on aligned bacterial cellulose nanofibers. ACS Appl. Mater. Interfaces 2017, 9, 20330–20339. 10.1021/acsami.6b14650. [DOI] [PubMed] [Google Scholar]
- Jacob J.; Haponiuk J. T.; Thomas S.; Peter G.; Gopi S. Use of ginger nanofibers for the preparation of cellulose nanocomposites and their antimicrobial activities. Fibers 2018, 6, 79 10.3390/fib6040079. [DOI] [Google Scholar]
- Huang Z.; Brennana C. S.; Zhao H.; Liu J.; Guan W.; Mohan M. S.; Stipkovits L.; Zheng H.; Kulasiri D. Fabrication and assessment of milk phospholipid-complexed antioxidant phytosomes with vitamin C and E: A comparison with liposomes. Food Chem. 2020, 324, 126837 10.1016/j.foodchem.2020.126837. [DOI] [PubMed] [Google Scholar]
- Djekic L.; Krajisnik D.; Micic Z.; Calija B. Formulation and physicochemical characterization of hydrogels with 18b-glycyrrhetinic acid/phospholipid complex phytosomes. J. Drug Delivery Sci. Technol. 2016, 35, 81–90. 10.1016/j.jddst.2016.06.008. [DOI] [Google Scholar]
- Gopi S.; Jacob J.; Varma K.; Jude S.; Amalraj A.; Arundhathy C. A.; George R.; Sreeraj T. R.; Divya C.; Kunnumakkara A. B.; Stohs S. J. Comparative oral absorption of curcumin in a natural turmeric matrix with two other curcumin formulations: An open-label parallel-arm study. Phytother. Res. 2017, 31, 1883–1891. 10.1002/ptr.5931. [DOI] [PubMed] [Google Scholar]
- Hays N. P.; Roberts S. B.. Aging – Nutritional Aspects. In Encyclopedia of Food Sciences and Nutrition,; 2 nd ed.; Caballero B., Ed.; Academic Press:2003; pp 81–87. [Google Scholar]
- Casas C.8.6 - Vitamins. In Analysis of Cosmetic Products; 1st ed.; Salvador A.; Chisvert A., Eds.; Elsevier, 2007; pp 364–379. [Google Scholar]
- Li Y.; Schellhorn H. E. New developments and novel therapeutic perspectives for vitamin C. J. Nutr. 2007, 137, 2171–2184. 10.1093/jn/137.10.2171. [DOI] [PubMed] [Google Scholar]
- Zee J. A.; Carmichael L.; Codère D.; Poirier D.; Fournier M. Effect of storage conditions on the stability of vitamin C in various fruits and vegetables produced and consumed in Quebec. J. Food Compos. Anal. 1991, 4, 77–86. 10.1016/0889-1575(91)90050-G. [DOI] [Google Scholar]
- Prantl L.; Eigenberger A.; Gehmert S.; Haerteis S.; Aung T.; Rachel R.; Jung E. M.; Felthaus O. Enhanced resorption of liposomal packed vitamin C monitored by ultrasound. J. Clin. Med. 2020, 9, 1616 10.3390/jcm9061616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y.; Zhang J.; Hu C. Q.; Zhang X.; Ma B.; Zhang P. In silico ADME and toxicity prediction of ceftazidime and its impurities. Front. Pharmacol. 2019, 10, 434 10.3389/fphar.2019.00434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob J.; Haponiuk J. T.; Thomas S.; Gopi S. Biopolymer based nanomaterials in drug delivery systems: a review. Mater. Today Chem. 2018, 9, 43–55. 10.1016/j.mtchem.2018.05.002. [DOI] [Google Scholar]
- Lagorce D.; Douguet D.; Miteva M. A.; Villoutriex B. O. Computational analysis of calculated physicochemical and ADMET properties of protein-protein interaction inhibitors. Sci. Rep. 2017, 7, 46277 10.1038/srep46277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo F.; Bentzien J.; Berellini G.; Muegge I. In silico models of human pk parameters. prediction of volume of distribution using an extensive data set and a reduced number of parameters. J. Pharm. Sci. 2021, 110, 500–509. 10.1016/j.xphs.2020.08.023. [DOI] [PubMed] [Google Scholar]
- Łukawski M.; Dałek P.; Borowik T.; Foryś A.; Langner M.; Witkiewicz W.; Przybyło M. New oral liposomal vitamin C formulation: Properties and bioavailability. J. Liposome Res. 2020, 30, 227–234. 10.1080/08982104.2019.1630642. [DOI] [PubMed] [Google Scholar]
- Hickey S.; Roberts H. J.; Miller N. J. Pharmacokinetics of oral vitamin C. J. Nutr. Environ. Med. 2008, 17, 169–177. 10.1080/13590840802305423. [DOI] [Google Scholar]
- Mikirova N. A. Ascorbic acid and dehydroascorbic acid concentrations in plasma and peripheral blood mononuclear cells after oral liposomal-encapsulated or intravenous ascorbic acid delivery. J. Orthomol. Med. 2017, 32, 1–9. [Google Scholar]
- Tyzack J. D.; Kirchmair J. Computational methods and tools to predict cytochrome P450 metabolism for drug discovery. Chem. Biol. Drug Des. 2019, 93, 377–386. 10.1111/cbdd.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille L.; Hoffer L. J. A simple method for plasma total vitamin C analysis suitable for routine clinical laboratory use. Nutr. J. 2015, 15, 40 10.1186/s12937-016-0158-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parhizkar E.; Rashedinia M.; Karimi M.; Alipour S. Design and development of vitamin C encapsulated proliposome with improved in-vitro and ex-vivo antioxidant efficacy. J. Microencapsulation 2018, 35, 301–311. 10.1080/02652048.2018.1477845. [DOI] [PubMed] [Google Scholar]
- Pires D. E. V.; Blundell T. L.; Ascher D. B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]