

Abstract
Background and aims:
Treprostinil is a prostacyclin analog used to treat pulmonary arterial hypertension. Dosing is empiric and based on tolerability. Adverse effects are common and can affect treatment persistence. Pharmacogenomic variants that may affect treprostinil metabolism and transport have not been well-characterized. We aimed to investigate the pharmacogenomic sources of variability in treatment persistence and dosing.
Methods:
Patients were prospectively recruited from an IRB approved biobank registry at a single pulmonary hypertension center. A cohort of patients who received oral treprostinil were screened for participation. Pharmacogenomic analysis was for variants in CYP2C8, CYP2C9, and ABCC4. A retrospective review was conducted for demographics, clinical status, dosing, and response. Fisher’s exact test was used for categorical data and Kruskal–Wallis test or Wilcoxon rank sum were used for continuous data.
Results:
A total of 15 patients received oral treprostinil and were consented. Their median age was 53 years, 73% were female, and 93% were White. The median total daily dose was 22.5 mg (13.5, 41) at last clinical observation. 40% of patients discontinued treatment with a majority due to adverse effects. Approximately 27% of patients had a loss-of-function variant in CYP2C8 (*1/*3 or *1/*4), whereas 47% of patients had a loss-of-function variant in CYP2C9 (*1/*2, *1/*3, or *2/*2). Minor allele frequencies for ABCC4 (rs1751034 and rs3742106) were 0.17 and 0.43, respectively. Survival analysis showed that increased CYP2C9 activity score was associated with decreased risk for treatment discontinuation [hazard ratio (HR): 0.13; 95% confidence interval (CI): 0.02, 0.91; p = 0.04]. Genetic variants were not significantly associated with dosing.
Conclusion:
Genetic variants responsible for the metabolism and transport of oral treprostinil were common. Increased CYP2C9 activity score was associated with decreased risk for treatment discontinuation. However, dosing was not associated with genetic variants in metabolizing enzymes for treprostinil. Our findings suggest significant variability in treatment persistence to oral treprostinil, with pharmacogenomics being a potentially important contributor.
The reviews of this paper are available via the supplemental material section.
Keywords: personalized medicine, pharmacogenomics, pulmonary arterial hypertension, treprostinil
Introduction
Pulmonary arterial hypertension (PAH) is a progressive disease characterized by increases in pulmonary vascular resistance, right ventricular failure, and death if left untreated.1 Despite a rapid expansion of treatments approved over the last two decades, long-term outcomes remain unsatisfactory.2 Prostacyclins are a mainstay of treatment for patients with more advanced symptoms of PAH.3 Treprostinil is a prostacyclin analog approved for the treatment of PAH and available in several different routes of administration, including subcutaneous, intravenous, inhalation, and oral.4 Oral treprostinil diolamine (Orenitram®) was the first oral prostacyclin analog to be approved by the FDA in 2013.5
Prostacyclin treatment response is highly individualized and predictors of response are not well-understood.6 Current treatment paradigms are empiric and iterative, and primarily based on indices of PAH disease severity through risk stratification.3 The heterogeneity of prostacyclin response is illustrated through prior reports of epoprostenol, which included the identification of some patients being described as ‘super responders.’6 Less is known about treprostinil, including the oral formulation, which has the advantage of not requiring an indwelling catheter and may be appropriate for select patients who are eligible for transition from parenteral treprostinil.3,7 Clinical studies have also shown significant variability in dosing and treatment response with oral treprostinil.7–10 Treatment persistence and higher doses improve clinical outcome with oral treprostinil, but patients may develop intolerable adverse effects or require transition to infusion or inhaled prostacyclins from the oral formulation.10–13 Understanding which patients would be most likely to gain benefit and least likely to experience toxicity from prostacyclin analogs underscores the importance of a precision medicine approach to PAH care.14,15
Pharmacogenomics is the study of how genetic variations affect medication response through the correlation of gene expression or single nucleotide polymorphisms with efficacy and toxicity.16 Genomic studies in PAH are emerging and are being increasingly used to improve our knowledge of the disease process, although few studies have evaluated pharmacogenomics in this population.17,18 Pharmacogenomic variants which affect prostacyclin metabolism and transport have not been well-characterized. The primary enzyme pathways that metabolize treprostinil are the hepatic cytochrome P-450 (CYP) isoenzyme system (CYP2C8 and CYP2C9) and these are known to be polymorphic.5,19 In fact, pharmacogenomic variants of CYP2C9 (*2 and *3 alleles) with known functional consequences (e.g. reduced or no function alleles leading to intermediate or poor metabolizer phenotypes) have reported population frequencies of 35% in Caucasians.19,20 In addition to metabolic pathways for treprostinil, transporters also have the potential to influence medication response. ATP-binding cassette (ABC) transporters are transmembrane proteins that use ATP for the active transport of various drugs, thereby leading to their elimination. Multidrug resistance protein 4 (MRP4), encoded by ABCC4, is among the most common transporters, is highly polymorphic, and is of interest with regard to its role as a potential transporter of treprostinil. MRP4 may also have a role in cardiovascular homeostasis and platelet function.21
The primary aim of our study was to evaluate the effect of pharmacogenomics as a source of variability in treatment persistence and dosing with oral treprostinil. The secondary aim was to characterize whether an association exists between predicted metabolizer status and treprostinil drug concentrations.
Methods
Study design
This was an investigator-initiated, proof-of-concept, pilot study of oral treprostinil dosing and clinical response variability as a function of predicted metabolizer status based on pharmacogenomic variants. In addition, we explored trends between predicted metabolizer status and treprostinil concentrations. Study participants were recruited from an Institutional Review Board (IRB) approved biobank registry of patients with pulmonary hypertension at UPMC Presbyterian Hospital in Pittsburgh, PA. All patients with PAH (World Health Organization Group 1) aged 18 years and older who were prescribed oral treprostinil since FDA approval (20 December 2013) through 30 June 2019 were screened for participation and had the opportunity to enroll. The target population included both those who were newly initiated on oral treprostinil (de novo), and those who transitioned from other treprostinil formulations (inhaled, intravenous, and subcutaneous).
The University of Pittsburgh IRB approved the consent and study protocol. This was in accordance with the ethical standards of the Helsinki Declaration. All study participants provided written informed consent.
Clinical data were extracted from the electronic health record to capture the following variables: age, sex, race, weight, PAH etiology, World Health Organization (WHO) functional class, background PAH therapy, de novo oral treprostinil use or transition from another treprostinil formulation, total daily dose of treprostinil at last date of clinical observation (last visit with PAH provider closest to the end of the study period), dosing frequency, adverse effects, and persistence with treatment. Treatment persistence was defined as maintenance on therapy without discontinuation. Participants who remained on treatment at the last date of clinical follow-up were presumed to have maintained on therapy for analysis purposes. For instances of treatment discontinuation before the end of observation, the reason(s) were also collected.
Pharmacogenomic analysis
Genomic DNA was isolated from whole blood or buffy coat using Gentra Puregene Blood Kit according to the manufacturer (Qiagen, Germantown, MD). DNA was quantified using Nanoquant and an Infinite® 200 PRO plate reader (Tecan, Switzerland). Variants in CYP2C8, CYP2C9, and ABCC4 were detected by TaqMan® allelic discrimination assay (Thermo-Fisher Scientific, Waltham, MA). Variants tested for in CYP2C8 were the *3 (rs10509681, rs11572080) and *4 (rs1058930) alleles which have an expected frequency of 10–20% in the Caucasian population. CYP2C9 variants tested were the *2 (rs1799853, decreased function) and *3 (rs1057910, no function) alleles which have an expected frequency of 15% in Caucasians. Finally, variants in ABCC4 included rs1751034 and rs3742106, which have expected minor allele frequencies of 20% and 40%, respectively.22 Predicted phenotypes for CYP2C9 were based on diplotype to activity score mappings by the Clinical Pharmacogenetics Implementation Consortium (CPIC) where *1 = 1, *2 = 0.5, and *3 = 0.20 Predicted phenotypes for CYP2C8 were determined to be intermediate or poor metabolizer status when one or two decreased or loss-of-function alleles were detected, respectively. For both CYP2C8 and CYP2C9, in the complete absence of variants tested, participants were assigned normal metabolizer status (*1/*1 genotype).
Pharmacokinetic analysis
For the oral treprostinil pharmacokinetic analysis, we assumed steady state concentrations with three-times daily dosing and slow absorption with a time to maximum concentration (Tmax) of approximately 4–6 h.23 One 4 ml blood sample was collected from each research participant at a time point that was approximated to be at or near the anticipated maximum concentration (Cmax). Blood samples were collected in K3 EDTA tubes and kept immediately in ice. These were then centrifuged as soon as possible at 25°C for 10 min at 2500 g. Plasma was placed in polypropylene cryovials and stored at −80°C until analysis. A validated HPLC-MS-MS assay was used to measure treprostinil concentrations in the plasma. The coefficient of variation of the assay is less than 10%.
Sample size and statistical analysis
This was a pilot study designed to evaluate a cohort of all patients treated with oral treprostinil at our institution since its FDA approval. Statistical analyses were performed with R version 3.6.2 (R Core Team, Vienna Austria). Continuous data are presented as mean and standard deviation or median and interquartile range. Categorical data were evaluated with Fisher’s Exact test. Continuous data were evaluated by Kruskal–Wallis or Wilcoxon Rank Sum tests. Genetic variants were included in models according to an additive (e.g. AA versus Aa versus aa) inheritance pattern or based on established genotype to phenotype definitions (e.g. metabolizer status, activity score). The primary endpoints were oral treprostinil treatment persistence and dose as a function of genotype and predicted phenotype. Total daily dose was evaluated by Kruskal–Wallis and dosing frequency assessed by Wilcoxon Rank Sum to compare all patients with no variants detected versus at least one variant detected. Treatment persistence was evaluated with survival analysis. Time to treprostinil discontinuation was defined as the date of follow-up when treatment was discontinued. Cases were censored at the last date of follow-up. The overall time to treprostinil discontinuation based on activity score was compared using Cox regression. The secondary aim was to explore trends between predicted metabolizer status and treprostinil concentrations. The Kruskal–Wallis test was used to test differences between phenotype and dose-normalized concentrations as a function of each gene.
Results
Patient and treatment characteristics
A total of 15 adult patients received oral treprostinil and consented to study participation. Baseline demographic and treatment characteristics of study participants are summarized in Table 1. The study population was predominantly females who were of middle age. The majority of the population was Caucasian. Nearly 80% of patients had either PAH associated with connective tissue disease or idiopathic PAH. Nearly all patients had WHO functional class II or III symptoms at the time of oral treprostinil initiation. The majority of patients were on background therapy with a phosphodiesterase type-5 inhibitor and/or an endothelin receptor antagonist. Nine of the 15 patients were on both classes of these medications. The cohort was approximately evenly split between those who were prostacyclin naïve and those who transitioned from other treprostinil formulations. Variability in oral treprostinil dosing was evident, with a more than four-fold difference in total daily dose achieved between patients with a median of 22.5 mg (13.5, 41) at last clinical observation. There were also notable differences in dose frequency, including both three- and four-times daily dosing at last follow-up.24 Patient demographics and treatment characteristics by treatment persistence are summarized in Table 2.
Table 1.
Baseline demographic and treatment characteristics.
| Characteristic (n = 15) | |
|---|---|
| Demographics | |
| Age‡ (years) | 53 (42, 61) |
| Female sex (%) | 73 |
| Caucasian race (%) | 93 |
| PAH etiology, severity, and background treatment | |
| PAH associated with connective tissue disease (%) | 43 |
| Idiopathic PAH (%) | 36 |
| WHO functional class II or III (%) | 87 |
| Background PDE5i therapy (%) | 93 |
| Background ERA therapy (%) | 67 |
| Prostacyclin naïve (%) | 47 |
| Oral Treprostinil Dosing Information | |
| Transition from other treprostinil formulation (%) | 53 |
| Total daily dose‡ (mg) | 22.5 (13.5, 43.1) |
| Three-times daily dose frequency (%) | 67 |
| Four-times daily dose frequency (%) | 33 |
Median, interquartile range.
ERA, endothelin receptor antagonist; PAH, pulmonary arterial hypertension; PDE5i, phosphodiesterase type-5 inhibitor; WHO, World Health Organization.
Table 2.
Baseline demographics and treatment characteristics by phenotype.
| Characteristic (n = 15) | Treatment persistence (n = 9) | Treatment discontinuation (n = 6) |
|---|---|---|
| Age‡ (years) | 57 (50, 61) | 41 (36, 55) |
| Female sex (%) | 67 | 83 |
| PAH associated with connective tissue disease (%) | 56 | 17 |
| Idiopathic PAH (%) | 33 | 50 |
| Background PDE5i therapy (%) | 100 | 83 |
| Background ERA therapy (%) | 56 | 83 |
| Prostacyclin naïve (%) | 56 | 33 |
| Transition from other treprostinil formulation (%) | 44 | 67 |
| Oral treprostinil dose at last follow-up (mg) | 16 (13.5, 42) | 28.8 (15.8, 41.9) |
Median, interquartile range.
ERA, endothelin receptor antagonist; PAH, pulmonary arterial hypertension; PDE5i, phosphodiesterase type-5 inhibitor.
A total of 40% of patients (6/15) discontinued treatment, the majority (5/6) of whom experienced intolerable adverse effects. The median time to discontinuation was 13 months. The reasons for discontinuation for these five patients included: gastrointestinal adverse effects (2/6), gastrointestinal adverse effects and headache (1/6), flushing (1/6), and non-specific complaints (1/6). Of these five patients who discontinued oral treprostinil due to adverse effects, three were switched to subcutaneous treprostinil and two were switched to selexipag. There was one patient who discontinued oral treprostinil due to clinical worsening (progressive dyspnea, orthopnea, and paroxysmal nocturnal dyspnea over several months), then subsequently was switched to intravenous treprostinil and later underwent lung transplantation. The median total daily dose for the six patients who discontinued oral treprostinil was 28.8 mg at last clinical follow-up (Table 2). Each of these patients received three-times daily dosing, with the exception of one who received four-times daily dosing.
Pharmacogenomic analysis
Approximately 27% of patients had a variant in CYP2C8 (*1/*3 or *1/*4), whereas 47% of patients had a variant in CYP2C9 (*1/*2, *1/*3, or *2/*2). Minor allele frequencies for ABCC4 (rs1751034 and rs3742106) were 0.17 and 0.43, respectively. The total daily dose and status (discontinued or still taking) at last follow-up across all genes and variants are shown in Figure 1. Survival analysis (Figure 2) showed that increased CYP2C9 activity score was associated with decreased risk for treatment discontinuation (HR: 0.13; 95% CI: 0.02, 0.91; p = 0.04). Most patients (83%) who had at least one decreased or no function allele in CYP2C9 discontinued treatment due to adverse events, which limited the available ‘final’ dosing available for comparison. For ABCC4 rs1751034, all samples had at least one variant present which precluded further statistical analyses for dosing differences. There were no significant differences in total daily dose detected for CYP2C9 (p = 0.82), CYP2C8 (p = 0.15), or ABCC4 rs3742106 (p = 0.51). Similarly, no significant differences in dosing frequency were found for CYP2C9 (p = 0.39), CYP2C8 (p = 0.50), and ABCC4 rs3742106 (p = 0.21). Although genetic variants were not significantly associated with daily dosing or dose frequency, the single lowest total daily dose was reported in the patient with CYP2C9 *2/*2 who would be a predicted intermediate metabolizer and in the lowest CYP2C9 activity score (1) group observed in this cohort.20 A similar trend was observed between alleles predicting decreased CYP2C8 metabolizer status (*3, *4) and dosing, although this did not meet statistical significance. No apparent trend was seen between ABCC4 and dosing. Figure 3 shows treprostinil total daily dose as a function of predicted metabolizer status for CYP2C8 and CYP2C9, but no significant differences were observed (p = 0.15 and p = 0.26, respectively).
Figure 1.

Treprostinil dosing and pharmacogenomic variants.
Total daily dose defined as the date of last clinical observation.
For patients who discontinued therapy, total daily dose was determined on date of discontinuation.
TRE, treprostinil.
Figure 2.

Cox regression for probability of treprostinil continuation.
Date of treatment discontinuation was defined as the date of follow-up in which treatment was discontinued. Subjects were censored at the last date of follow up during which therapy was still active.
AS, activity score; TRE, treprostinil.
Figure 3.
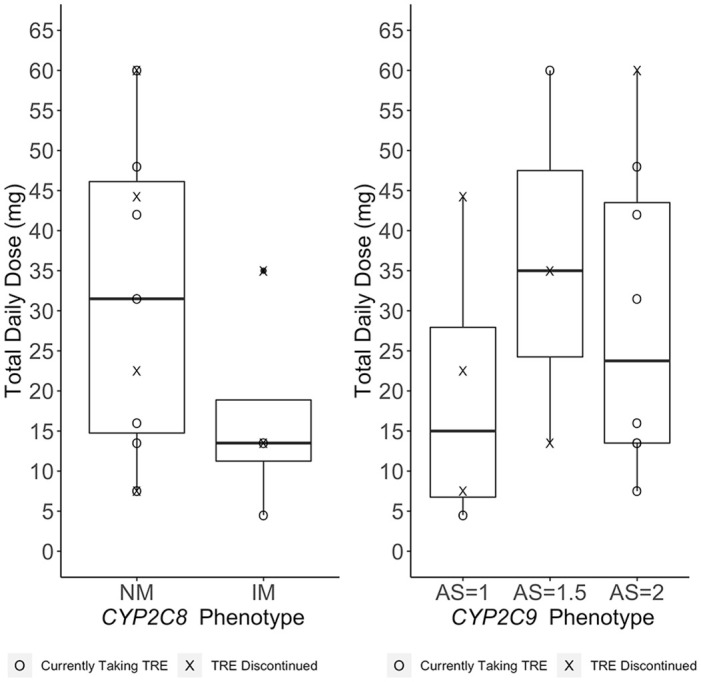
Treprostinil dosing and predicted metabolizer status.
Total daily dose defined as the date of last clinical observation.
For patients who discontinued therapy, total daily dose was determined on date of discontinuation.
AS, activity score; IM, intermediate metabolizer; NM, normal metabolizer; TRE, treprostinil.
Pharmacokinetic analysis
A total of nine patients whose treatment with oral treprostinil persisted during the study follow-up period consented to venipuncture and provided blood samples for determination of treprostinil concentration. The median concentration was 2.86 ng/ml (2.17 ng/ml, 3.46 ng/ml) and the median sampling time post-dose was 4.25 h (3.4–5.75 h with one at 1.75 h). It is known that Cmax after oral administration is around 4–6 h.5 Therefore, the measured concentrations can be considered to be close to Cmax values. The median total daily dose for these patients was 16 mg at last clinical observation. The median duration of treatment was approximately 48 months. Five of these patients received three-times daily dosing, with the remainder dosed at four-times daily dosing. Treprostinil Cmax concentrations were linear across the range of total daily doses (Figure 4). A significant positive correlation was found between total daily dose and treprostinil Cmax concentration (Figure 4; Spearman rank correlation coefficient = 0.79; p < 0.05). Figure 5 illustrates the dose normalized Cmax concentrations for treprostinil as a function of predicted metabolizer status for CYP2C8 and CYP2C9. No significant differences were found between the CYP2C8 and CYP2C9 phenotypes and treprostinil concentrations (p = 0.38 and p = 0.77, respectively).
Figure 4.

Treprostinil concentrations and total daily dosing.
Figure 5.
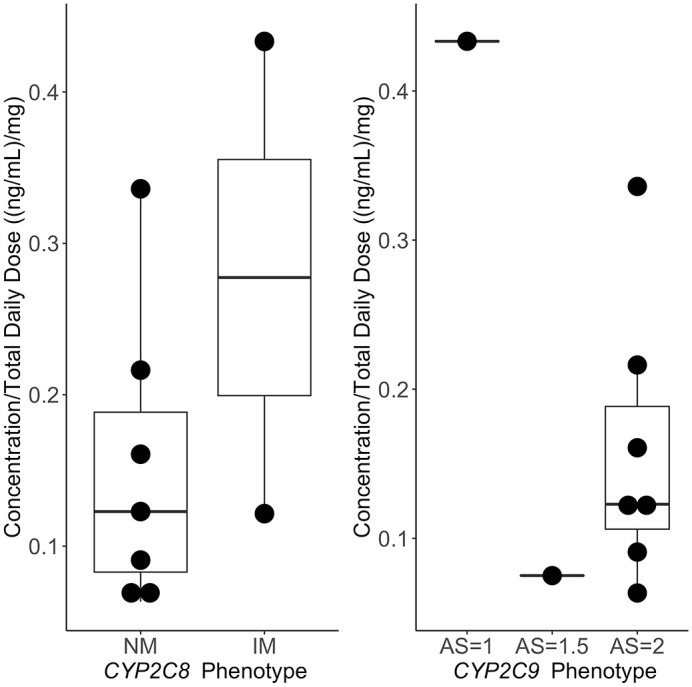
Dose-normalized treprostinil concentrations and predicted metabolizer status.
AS, activity score; IM, intermediate metabolizer; NM, normal metabolizer.
Discussion
Our main finding is that pharmacogenomics appears to be an important driver of variability in treatment persistence to oral treprostinil. Study participants with an estimated CYP2C9 activity score of 2 were 13% as likely to discontinue therapy compared to those with an activity score of 1. This assessment occurred at approximately 48 months after treatment initiation. Interestingly, we did not observe a significant association between predicted CYP2C8 activity and treprostinil persistence. However, markers for CYP2C9 *2 and CYP2C8 *3 are in linkage disequilibrium (tend to co-occur), making it difficult to ascertain which gene is ultimately responsible for these findings.25 Both CYP2C8 and CYP2C9 are responsible for the metabolism of treprostinil, even though the drug is metabolized to a lesser extent by CYP2C9.5 A trend was apparent for each gene where patients with alleles predicting decreased activity were the ones who generally received lower oral treprostinil doses. This was particularly noteworthy for the patient who was predicted to be a CYP2C9 intermediate metabolizer with the lowest activity score, and who received the lowest oral treprostinil dose in this study. However, these overall differences in dosing based on allele and predicted phenotype were not significant and may be related to insufficient power. Finally, no significant findings were seen between the ABCC4 transporter gene variation and dosing.
Registry data in the modern era indicates that PAH mortality remains high, even with rapid advances in medical care over the past two decades.26–28 Current treatments are based primarily on markers of disease severity, with parenteral therapies reserved for those with advanced disease, and oral and inhaled therapies often indicated for those with milder disease.3,29 In aggregate, the effect of PAH medications on the historical benchmark of 6-minute walk distance improvement has been relatively modest.17,30 Predicting individual response to a certain medication is typically not possible, therefore, a trial-and-error approach is the current standard of practice.18 Individual medication and/or formulation selection within a class is often left to prescriber preference and patient convenience. For those patients who have a poor response by way of adverse effects or lack of benefit, the time lost to inadequate treatment leads to further disease progression.1,2
Matching an individual patient to the medication likely to be most effective is the promise of precision medicine.31 This concept is not new to PAH, as a notable example includes the use of calcium channel blockers for patients with a positive vasoreactivity test.32 Significant interindividual variability in response to different PAH medications is clinically apparent and suggests a role for genetic differences in treatment response, disease severity, or both. Endothelin receptor antagonists (ERAs) provide a compelling use case. For example, women and Caucasians may have a better response to ERAs than men and non-Caucasians.33 Another example includes the association of a decreased function allele CYP2C9*2 with bosentan-induced liver injury.34 In addition, polymorphisms in the endothelin-1 pathway have been shown to be predictive of treatment response.35 Integrating pharmacogenomic data could enhance the efficiency of appropriate clinical treatment and address the vexing challenge in PAH care of selecting the most appropriate treatment for an individual patient. Furthermore, identifying genes of significance to PAH medications could help to enrich future studies by including those with an excellent response (i.e. ‘super responders’).6 This would offer advantages by maximizing therapeutic benefit for study participants while saving time and resources for those conducting and sponsoring such trials.36
The advancement of pharmacogenomic approaches to PAH management was highlighted as one of the key opportunities by the National Heart, Lung, and Blood Institute and will be a high priority for ongoing and future research.14,17 Our study adds to the ongoing efforts to research personalized medicine approaches in PAH by providing important clues to understanding variability in medication response. Our work is innovative because it has evaluated the extent to which pharmacogenomics effects variability in response to oral treprostinil and describes associations with drug concentrations. We determined treprostinil plasma concentrations for 9 out of 15 patients in the study cohort. Treprostinil Cmax concentrations were positively correlated across the dosing range and included patients who received up to a total daily dose of 60 mg. Dose-normalized concentrations as a function of predicted metabolizer status showed a pattern of higher drug concentrations among patients who were intermediate or poor versus normal metabolizers; however, these findings were not significant due to the small number of subjects in each group. This finding could also be the result of titration of the medication to effect over time. It is possible that any apparent difference in metabolism earlier in therapy would be accounted for by dose adjustments throughout longer-term treatment. Patients who discontinued therapy earlier in treatment due to adverse effects did not have an opportunity for blood sample collection to determine drug concentrations. Therefore, this pharmacokinetic aim should be further explored in future studies.
Our pilot data should prompt further investigations of pharmacogenomic associations with treatment response in a broader population of patients with PAH including those treated with other prostacyclins and non-prostacyclins. The prostacyclin use case would be of particular interest considering the multiple formulations available in practice and recent study showing improvement in PAH disease progression with oral treprostinil.10 The significance of any pharmacogenomic associations with treatment persistence, adverse effects, and dosing should also be evaluated in the context of whether the prostacyclin was de novo or transitioned from a different formulation. This could help to discern whether there is a treatment interaction between prostacyclin exposure and pharmacogenomics.
Limitations
The main limitations of our study are that it was single center study and that our sample size was limited. While all participants at our center who had received oral treprostinil were enrolled, the relative number of patients on this particular treatment was relatively low. Therefore, the study was likely underpowered to detect differences in treatment response and dosing based on predicted metabolizer status and reduced function alleles in CYP2C8 and CYP2C9. Nearly all study participants with CYP2C9 decreased function allele(s) ultimately discontinued oral treprostinil during follow-up; therefore, the final dosing assessment could not be determined. Nonetheless, this underscores one of the primary findings of the study, which is that patients carrying CYP2C9 decreased function alleles were significantly associated with lower treatment persistence.
In addition, Cox regression is a semi-parametric test, and it is reasonable to suggest that the low sample size in this study precludes optimal use of parametric tests. However, the assumptions of the Cox model were upheld in our analysis (Schoenfeld test; p = 0.63). We also captured several clinical variables of interest which could have influenced medication response (e.g. age, sex, PAH etiology, concomitant PAH therapy) and reported differences between those who remained on oral treprostinil and those who discontinued treatment (Table 2). Patients who discontinued treprostinil were younger and were less likely to have PAH associated with connective tissue disease. Patients who did not persist on treatment during follow-up also reached numerically higher median doses than those who continued treatment. However, other clinical variables were not able to be collected (i.e. right ventricular function, biomarkers, and hemodynamics) and could not be compared between groups. We also did not collect certain interacting medications that significantly interact with treprostinil (e.g. rifampin and gemfibrozil), although these are uncommonly used within our practice. Sample size also precluded controlling for clinical variables when ascertaining potential genetic differences underlying treatment response. With regards to the pharmacokinetics aim of the study, we were only able to obtain a single blood sample for each participant. This resulted from the study design itself, which relied on patient recruitment from the clinic environment. Therefore, a more robust study evaluating multiple samples per participant would be needed to fully characterize any associations between drug concentrations and pharmacogenomics. We were also unable to collect blood samples from the patients who had already discontinued oral treprostinil at the time of consent for this aim.
Conclusion
Genetic variants responsible for the metabolism of oral treprostinil were common. An increased CYP2C9 activity score was associated with decreased risk for treatment discontinuation. However, dosing was not associated with genetic variants in the major metabolizing enzymes for treprostinil. Our findings suggest significant variability in treatment persistence to oral treprostinil with pharmacogenomics being a potentially important contributor. Future studies should explore the effect of pharmacogenomics on PAH medication dosing and response in larger patient cohorts.
Supplemental Material
Supplemental material, sj-pdf-1-tar-10.1177_17534666211013688 for A pilot study of oral treprostinil pharmacogenomics and treatment persistence in patients with pulmonary arterial hypertension by James C. Coons, Karryn Crisamore, Solomon Adams, Ashley Modany, Marc A. Simon, Wenchen Zhao, Imam H. Shaik, Raman Venkataramanan and Philip E. Empey in Therapeutic Advances in Respiratory Disease
Supplemental material, sj-pdf-2-tar-10.1177_17534666211013688 for A pilot study of oral treprostinil pharmacogenomics and treatment persistence in patients with pulmonary arterial hypertension by James C. Coons, Karryn Crisamore, Solomon Adams, Ashley Modany, Marc A. Simon, Wenchen Zhao, Imam H. Shaik, Raman Venkataramanan and Philip E. Empey in Therapeutic Advances in Respiratory Disease
Supplemental material, sj-pdf-3-tar-10.1177_17534666211013688 for A pilot study of oral treprostinil pharmacogenomics and treatment persistence in patients with pulmonary arterial hypertension by James C. Coons, Karryn Crisamore, Solomon Adams, Ashley Modany, Marc A. Simon, Wenchen Zhao, Imam H. Shaik, Raman Venkataramanan and Philip E. Empey in Therapeutic Advances in Respiratory Disease
Acknowledgments
United Therapeutics (the manufacturer of oral treprostinil) supported the conduct of the study at UPMC by its investigator-initiated-study mechanisms. The authors acknowledge the PittPharmacy (University of Pittsburgh School of Pharmacy) Pharmacogenomics (PGx) Center of Excellence, The Clinical Pharmacokinetics Laboratory (University of Pittsburgh School of Pharmacy), The Institute for Precision Medicine, a partnership of the University of Pittsburgh and UPMC, the UPMC Heart and Vascular Institute, and the UPMC Comprehensive Pulmonary Hypertension Program. The authors also acknowledge Yingze Zhang, PhD (Director, Translational Research Core Laboratory and Cardiology Biobank, Department of Pulmonary, Allergy and Critical Care Medicine, University of Pittsburgh Medical Center) for her contributions to sample processing and biobanking, and Yassmin Al Aaraj, MD, MPH (Clinical Research Coordinator, Pittsburgh Heart, Lung, Blood, and Vascular Medicine Institute, Division of Cardiology, Department of Medicine, University of Pittsburgh Medical Center) for her contributions to sample collection.
Footnotes
Contributorship: Each author contributed to the conception and design of the manuscript. In addition, each author contributed to the acquisition, analysis, and interpretation of the data presented in the manuscript. James C. Coons drafted the manuscript, and all authors critically revised the content accordingly. Each author also gave final approval of the manuscript and agrees to be accountable for all aspects of work ensuring the integrity and accuracy of the content.
Conflict of interest statement: James C. Coons has received investigator-initiated grant funding from United Therapeutics and research support from Pfizer, Inc.
Karryn Crisamore has no relevant disclosures.
Solomon Adams has no relevant disclosures.
Ashley Modany has no relevant disclosures.
Marc A. Simon has received research support from Novartis and Aadi, and has received consulting fees from United Therapeutics, Actelion, Complexa, Gilead, Acceleron.
Wenchen Zhao has no relevant disclosures.
Imam H Shaik has no relevant disclosures.
Raman Venkataramanan has received grant funding from United Therapeutics.
Philip Empey has no relevant disclosures.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors disclose that this work was supported through the investigator-initiated-funding mechanisms at United Therapeutics in contract with the University of Pittsburgh (Coons), the National Center for Advancing Translational Sciences of the National Institutes of Health (award number TL1TR001858-04; Crisamore), the American Foundation for Pharmaceutical Education (Crisamore), and NIH (grant numbers PO1HL103455, R01AG058659, and UL1TR001857; Simon). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Ethical approval: All ethical procedures were followed in accordance with the conduct of this research and the preparation of this manuscript. All patient identifiers have been removed.
ORCID iD: James C. Coons  https://orcid.org/0000-0002-7193-3751
https://orcid.org/0000-0002-7193-3751
Supplemental material: The reviews of this paper are available via the supplemental material section.
Contributor Information
James C. Coons, University of Pittsburgh School of Pharmacy, Clinical Pharmacist, Cardiology, UPMC Presbyterian Hospital, Salk Hall, Room 727, 3501 Terrace Street, Pittsburgh, PA 15261, USA.
Karryn Crisamore, Department of Pharmaceutical Sciences, Center for Clinical Pharmaceutical Sciences, University of Pittsburgh School of Pharmacy, Pittsburgh, PA, USA.
Solomon Adams, Shenandoah University, Winchester, VA, USA.
Ashley Modany, UPMC Presbyterian Hospital, Pittsburgh, PA, USA.
Marc A. Simon, Bioengineering, and Clinical Translational Science, Department of Medicine/Division of Cardiology, Vascular Medicine Institute, University of Pittsburgh, Pittsburgh, PA, USA Heart and Vascular Institute, Heart Failure Research, University of Pittsburgh Medical Center, Pittsburgh, PA, USA.
Wenchen Zhao, Department of Pharmaceutical Sciences, University of Pittsburgh School of Pharmacy, Pittsburgh, PA, USA.
Imam H. Shaik, Department of Pharmaceutical Sciences, University of Pittsburgh School of Pharmacy, Pittsburgh, PA, USA
Raman Venkataramanan, Department of Pharmaceutical Sciences, University of Pittsburgh School of Pharmacy, Pittsburgh, PA, USA.
Philip E. Empey, Pharmacogenomics Center of Excellence, Institute for Personalized Medicine, Department of Pharmacy and Therapeutics, Center for Clinical Pharmaceutical Sciences and the Clinical and Translational Science Institute, University of Pittsburgh School of Pharmacy, Pittsburgh, PA, USA
References
- 1. Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med 2004; 351: 1425–1436. [DOI] [PubMed] [Google Scholar]
- 2. Gomberg-Maitland M, Glassner-Kolmin C, Watson S, et al. Survival in pulmonary arterial hypertension patients awaiting lung transplantation. J Heart Lung Transplant 2013; 32: 1179–1186. [DOI] [PubMed] [Google Scholar]
- 3. Galie N, Channick RN, Frantz RP, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J 2019; 53: 1801889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Coons JC, Pogue K, Kolodziej AR, et al. Pulmonary arterial hypertension: a pharmacotherapeutic update. Curr Cardiol Rep 2019; 21: 141. [DOI] [PubMed] [Google Scholar]
- 5. United Therapeutics Corporation. Orenitram (treprostinil) [package insert]. Research Triangle Park, NC: United Therapeutics Corporation, 2017. [Google Scholar]
- 6. Halliday SJ, Hemnes AR. Identifying “super responders” in pulmonary arterial hypertension. Pulm Circ 2017; 7: 300–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chakinala MM, Feldman JP, Rischard F, et al. Transition from parenteral to oral treprostinil in pulmonary arterial hypertension. J Heart Lung Transplant 2017; 36: 193–201. [DOI] [PubMed] [Google Scholar]
- 8. Tapson VF, Torres F, Kermeen F, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): a randomized controlled trial. Chest 2012; 142: 1383–1390. [DOI] [PubMed] [Google Scholar]
- 9. Tapson VF, Jing ZC, Xu KF, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients receiving background endothelin receptor antagonist and phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C2 study): a randomized controlled trial. Chest 2013; 144: 952–958. [DOI] [PubMed] [Google Scholar]
- 10. White RJ, Jerjes-Sanchez C, Bohns Meyer GM, et al. Combination therapy with oral treprostinil for pulmonary arterial hypertension: a double-blind, placebo-controlled study. Am J Respir Crit Care Med 2020; 201: 707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chin KM, Ruggiero R, Bartolome S, et al. Long-term therapy with oral treprostinil in pulmonary arterial hypertension failed to lead to improvement in important physiologic measures: results from a single center. Pulm Circ 2015; 5: 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Coons JC, Miller T. Extended-release oral treprostinil in the management of pulmonary arterial hypertension: clinical evidence and experience. Ther Adv Respir Dis 2018; 12: 1753466618766490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ramani G, Cassady S, Shen E, et al. Novel dose–response analyses of treprostinil in pulmonary arterial hypertension and its effects on six-minute walk distance and hospitalizations. Pulm Circ 2020; 10: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Erzurum S, Rounds SI, Stevens T. et al. Strategic plan for lung vascular research: an NHLBI-ORDR workshop report. Am J Respir Crit Care Med 2010; 182: 1554–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pereira NL, Weinshilboum RM. Cardiovascular pharmacogenomics and individualized drug therapy. Nat Rev Cardiol 2009; 6: 632–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Evans WE, McLeod HL. Pharmacogenomics–drug disposition, drug targets, and side effects. N Engl J Med 2003; 348: 538–549. [DOI] [PubMed] [Google Scholar]
- 17. Newman JH, Rich S, Abman SH. et al. Enhancing insights into pulmonary vascular disease through a precision medicine approach. A joint NHLBI-Cardiovascular Medical Research and Education Fund Workshop Report. Am J Respir Crit Care Med 2017; 195: 1661–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Austin ED, West J, Loyd JE, et al. Translational advances in the field of pulmonary hypertension molecular medicine of pulmonary arterial hypertension. From population genetics to precision medicine and gene editing. Am J Respir Crit Care Med 2017; 195: 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee CR, Goldstein JA, Pieper JA. Cytochrome P450 2C9 polymorphisms: a comprehensive review of the in-vitro and human data. Pharmacogenetics 2002; 12: 251–263. [DOI] [PubMed] [Google Scholar]
- 20. Karnes JH, Rettie AE, Somogyi AA, et al. Clinical pharmacogenetics implementation consortium (CPIC) guideline for CYP2C9 and HLA-B genotypes and phenytoin dosing: 2020 update. Clin Pharmacol Ther 2021; 109: 302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Belleville-Rolland T, Sassi Y, Decouture B, et al. MRP4 (ABCC4) as a potential pharmacologic target for cardiovascular disease. Pharmacol Res 2016; 107: 381–389. [DOI] [PubMed] [Google Scholar]
- 22. Karczewski KJ, Francioli LC, Tiao G, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019.531210. DOI: 10.1101/531210. [DOI] [Google Scholar]
- 23. Jones A, Wang-Smith L, Pham T, et al. Pharmacokinetics of 3 times a day dosing of oral treprostinil in healthy volunteers. J Cardiovasc Pharmacol 2014; 63: 227–232. [DOI] [PubMed] [Google Scholar]
- 24. Coons JC, Bunner C, Ishizawar DC, et al. Impact of four times daily dosing of oral treprostinil on tolerability and daily dose achieved in pulmonary hypertension. Pulm Circ 2018; 8: 2045893217744512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yasar U, Lundgren S, Eliasson E, et al. Linkage between the CYP2C8 and CYP2C9 genetic polymorphisms. Biochem Biophys Res Commun 2002; 299: 25–28. [DOI] [PubMed] [Google Scholar]
- 26. Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010; 122: 156–163. [DOI] [PubMed] [Google Scholar]
- 27. Benza RL, Miller DP, Gomberg-Maitland M, et al. Predicting survival in pulmonary arterial hypertension: insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management (REVEAL). Circulation 2010; 122: 164–172. [DOI] [PubMed] [Google Scholar]
- 28. Benza RL, Gomberg-Maitland M, Elliott CG, et al. Predicting survival in patients with pulmonary arterial hypertension: the REVEAL risk score calculator 2.0 and comparison with ESC/ERS-based risk assessment strategies. Chest 2019; 156: 323–337. [DOI] [PubMed] [Google Scholar]
- 29. Galie N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 2016; 37: 67–119. [DOI] [PubMed] [Google Scholar]
- 30. Peacock AJ, Naeije R, Galie N, et al. End-points and clinical trial design in pulmonary arterial hypertension: have we made progress? Eur Respir J 2009; 34: 231–242. [DOI] [PubMed] [Google Scholar]
- 31. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med 2015; 372: 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rich S, Brundage BH. High-dose calcium channel-blocking therapy for primary pulmonary hypertension: evidence for long-term reduction in pulmonary arterial pressure and regression of right ventricular hypertrophy. Circulation 1987; 76: 135–141. [DOI] [PubMed] [Google Scholar]
- 33. Gabler NB, French B, Strom BL, et al. Race and sex differences in response to endothelin receptor antagonists for pulmonary arterial hypertension. Chest 2012; 141: 20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Markova SM, De Marco T, Bendjilali N, et al. Association of CYP2C9*2 with bosentan-induced liver injury. Clin Pharmacol Ther 2013; 94: 678–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Benza RL, Gomberg-Maitland M, Demarco T, et al. Endothelin-1 pathway polymorphisms and outcomes in pulmonary arterial hypertension. Am J Respir Crit Care Med 2015; 192: 1345–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pencina MJ, Peterson ED. Moving from clinical trials to precision medicine: the role for predictive modeling. JAMA 2016; 315: 1713–1714. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-pdf-1-tar-10.1177_17534666211013688 for A pilot study of oral treprostinil pharmacogenomics and treatment persistence in patients with pulmonary arterial hypertension by James C. Coons, Karryn Crisamore, Solomon Adams, Ashley Modany, Marc A. Simon, Wenchen Zhao, Imam H. Shaik, Raman Venkataramanan and Philip E. Empey in Therapeutic Advances in Respiratory Disease
Supplemental material, sj-pdf-2-tar-10.1177_17534666211013688 for A pilot study of oral treprostinil pharmacogenomics and treatment persistence in patients with pulmonary arterial hypertension by James C. Coons, Karryn Crisamore, Solomon Adams, Ashley Modany, Marc A. Simon, Wenchen Zhao, Imam H. Shaik, Raman Venkataramanan and Philip E. Empey in Therapeutic Advances in Respiratory Disease
Supplemental material, sj-pdf-3-tar-10.1177_17534666211013688 for A pilot study of oral treprostinil pharmacogenomics and treatment persistence in patients with pulmonary arterial hypertension by James C. Coons, Karryn Crisamore, Solomon Adams, Ashley Modany, Marc A. Simon, Wenchen Zhao, Imam H. Shaik, Raman Venkataramanan and Philip E. Empey in Therapeutic Advances in Respiratory Disease