

ABSTRACT
One of the proposed strategies for the development of a more efficient HIV-1 vaccine is based on the identification of proteins binding to a paratope of chosen broadly neutralizing antibody (bNAb) that will mimic cognate HIV-1 Env (glyco)protein epitope and could be used as potent immunogens for induction of protective virus-neutralizing antibodies in the immunized individuals. To verify this “non-cognate ligand” concept, we developed a highly complex combinatorial library designed on a scaffold of human myomesin-1 protein domain and selected proteins called Myomedins specifically binding to variable regions of HIV-1 broadly neutralizing antibody 10E8. Immunization of mice with these Myomedin variants elicited the production of HIV-1 Env-specific antibodies. Hyperimmune sera bound to Env pseudotyped viruses and weakly/moderately neutralized 54% of tested clade A, B, C, and AE pseudotyped viruses variants in vitro. These results demonstrate that Myomedin variants have the potential to mimic Env epitopes and could be used as potential HIV-1 vaccine components.
KEYWORDS: protein scaffold, protein mimetics, combinatorial library, Env glycoprotein, broadly neutralizing antibody, HIV vaccine
Introduction
Stimulation of protective immunity by the production of broadly neutralizing antibodies (bNAbs) in immunized individuals remains a challenge in HIV-1 vaccine development. Identification and characterization of highly potent bNAbs cloned from B-cells of elite neutralizers provide a molecular clue for designing new vaccine strategies. The main target of neutralizing antibody on the HIV-1 virus surface is the envelope glycoprotein (Env), a trimer composed of identical gp120/gp41 subunits. Gp120 protrudes from the virus surface whereas gp41 is a transmembrane subunit. Systematic studies of specificity, binding affinity, breadth, and potency led to a clustering of bNAbs into six major groups specific for V2 apex of HIV-1 Env glycoproteins (V2 bNAbs), V3 glycan site (V3 bNAbs), CD4 binding site (CD4 bNAbs), gp120-gp41 interface region (gp120-gp41 bNAbs), membrane-proximal external region (MPER bNAbs), and for so-called “silent face” of gp120 [1–4]. Some of these bNAbs exhibit extraordinary breadth and potency, and efficiently neutralize viral infection of host cells. Thus they represent exclusive candidates for vaccine design and therapy [5–7].
A yet unsolved issue in the current HIV-1 vaccine development is how to target human germline B cells precursors, to stimulate them to mature, and finally to expand to plasma cells secreting high titers of bNAbs which confer protection from virus infection and spreading. Significant progress in this germline B cells-targeting approach has recently been demonstrated. One of the first studies reported that mutation of three glycosylation sites on Env at positions 276, 460, and 463 allowed binding of germline-reverted B cell receptor precursors of two CD4-binding site-specific bNAbs VRC01 and NIH45-46 [8]. The observation was followed by others including immunization with such modified Env antigens. Recently, RC1 immunogen was designed based on the removal of N156 glycan, which induces structural changes of Env allowing better accessibility of antibodies to Env V3 loop base. After immunization of mice, rabbits, and rhesus macaques with RC1, expansion of B cells precursors of V3-type bNAbs was achieved but these Abs did not neutralize HIV-1 strains [9].
A recently introduced general vaccine strategy developed by Steichen et al. [10] is based on deep sequencing human antibody sets allowing identification of B cell precursors with potential to produce bNAbs, most importantly those with dominant HCDR3 contacts. Another example of Env antigen modification targeting germline B cell was reported for the super-site epitope containing N332 [11,12], high-mannose-type glycan, recognized by best-in-class V3-type bNAbs as PGT121, BG18, and 10–1074. By screening the Env mutants recognized by PGT121 and its germline-reverted B cell receptor precursors, and a set of Env mutants containing 5 to 10 mutated amino acids (10MUT, 7MUT, 5MUT, and native Env) was used for sequential immunization of mice, leading to maturation of B cell to finally produce tier-2-neutralizing antibodies resembling the authentic human bNAb such as PGT121 [13,14].
In another approach called signature-based epitope targeted (SET) vaccine design, a big comparative study of neutralizing potency of four groups of bNAbs against a large HIV-1 virus panel allowed the identification of antibody hypervariable region characteristics and virus clade-specific sensitivity. After phylogenetic corrections, this approach led to the identification of a V2-loop epitope signature, which allowed the introduction of V2-consensus guided mutations in a trimeric vaccine that resulted in an increased breadth of bNAbs elicited by immunization of guinea pigs [15]. Several earlier strategies were also reported, focusing on de-glycosylation of antigens by removing the glycan shield from Env glycoprotein [16,17], Env folding- and multimerization-modifying strategies [18,19], and modifying Env glycans toward patterns typical of T cell expression [16,17,20].
To overcome persisting problems with weak production of neutralizing antibodies raised against Env/gp120 and Env-modified glycan-carrying immunogens, we recently proposed a novel strategy that is based on immunization by artificial scaffold proteins mimicking HIV-1 Env epitopes [21]. These protein variants selected from a highly complex albumin-binding domain-derived combinatorial library [22–24] by directed evolution can function as ”protein imprints“ of paratopes of the most-potent HIV-1 bNAbs. In our proof-of-concept study, we demonstrated that variants called VRA017, VRA019, and VRA177 mimicking epitope of VRC01 bNAb elicited virus-neutralizing sera in mice as tested on the panel of pseudotyped HIV-1 viruses on reporter TZM-bl cells. This approach was designated the ”non-cognate ligand“ strategy [25]. However, whether this direction will be a viable solution for HIV-1 vaccine design as a method of reverse vaccinology needs to be investigated.
To verify the concept of ”non-cognate ligand“ strategy (NCLS) for induction of HIV-1 virus-neutralizing antibodies, we targeted another ”super candidate“ bNAb 10E8 that is specific for MPER of gp41 subunit protruding from the viral envelope. In addition to the most potent 10E8, other MPER-binding bNAbs have been identified: 2F5, 4E10, Z13, DH511, and 7H6 [2,6,26,27]. Most of these antibodies preferentially recognize MPER epitopes in CD4-activated HIV-1 Env. Therefore, an efficient bNAb should recognize the cognate MPER epitope in various conformations [28,29]. The MPER bNAbs often exhibit some degree of autoreactivity or polyreactivity but this does not seem to be the case with 10E8 [6,27].
In contrast to previously used three-helix bundle ABD-derived combinatorial library [30–32], here we demonstrate a new loop-randomized scaffold library designed on the structure of domain 10 of human contractile muscle protein myomesin-1. After the screening of randomized library designated as Myomedin library, the best 10E8 binders were identified, produced in prokaryotic expression system, and used as immunogens in experimental mice to stimulate the production of specific antibodies. Env-specificity and virus-neutralizing activity of hyperimmune sera were tested on a panel of 22 pseudotyped HIV-1 viruses of clades A, B, C, D, and AE. We found that 54% of all tested pseudoviruses were weakly/moderately neutralized in vitro and thus, our study further supports the concept of NCLS for HIV-1 immunogen production and underscores NCLS potential in vaccine development.
Materials and methods
Production and crystallization of human myomesin-1 domain 10
Template DNA corresponding to the GenBank BC116183 was obtained from the Source BioScience (Nottingham, UK). Myomesin domain 10 was amplified using primers (forward Myom10-F CATATGAAATCAGAGTTGGCAGTTGAAAT, reverse Myom10-R CAAGAATGGATCAGGAAACAAGGTTAAGGATCC) containing NdeI and BamHI restriction sites for ligation into the pET28b vector. The final protein product contained a 6xHis-tag and thrombin cleavage site at the N-terminus. Myomesin was produced in E. coli BL21 (DE3) cells. Cells grew at 37°C to OD600 of 0.6 in LB (Lysogeny Broth) then the expression of the protein was induced by 1 mM IPTG, and cultivation continued for 4 hours more before the cells were harvested by centrifugation. The protein was purified using Ni-NTA agarose (Qiagen, Germany) under native conditions. Eluted protein fractions were pooled, concentrated, and applied to the size-exclusion chromatography column Superdex 75 10/300 GL (GE Healthcare, UK) with running buffer 20 mM Tris, 50 mM NaCl pH 8.0.
Thermal shift assay
Protein samples (0.1 mg/ml) and 5x Sypro Orange dye (Sigma Aldrich) were added into total volume 25 μl. Using the real-time PCR Detection System CFX96 Touch (Bio-Rad Laboratories), the proteins were incubated in a thermal gradient from 20°C to 80°C at increments of 0.5°C and with 30 s-hold intervals. The degree of protein unfolding was monitored by FRET (fluorescence resonance energy transfer) channel that captured the spectral properties of Sypro Orange unfolded protein complexes (excitation wavelength≈470 nm and emission wavelength≈570 nm). The data were analyzed by CFX Manager software, and the melting temperatures were determined using the first derivative spectra.
Crystallization
The crystallization experiment was performed at 291 K by the hanging drop vapor diffusion method. The solution No. 25 of JCSG+ Suite (Qiagen, Germany) was optimized to 0.1 M citrate/phosphate buffer, pH 5.4, 0.2 M NaCl, 30%(w/v) PEG 3350 with protein concentration 5 mg/mL and ratio of protein to precipitant volume 1:1. A rod crystal was cryoprotected by soaking in mother liquor containing 10% (v/v) PEG 400.
Data collection and processing
The freshly grown crystal with a size of 30 x 30 × 30 μm was vitrified in liquid nitrogen. Diffraction measurement was performed on the BL14.1 beamline at BESSYII, Berlin, Germany, using a Pilatus M6 detector. The XDS program package [33] was used for data processing. The statistics of data collection and processing are summarized in Table S1.
Structure refinement
The structure was solved by molecular replacement using MOLREP [34] with the coordinates of the My10 moiety from the structure with PDB code 3y23 [35] and a sequence identity of 100%. The residues were manually fitted in Coot [36], and the structure was refined to 1.8 Å resolution by the maximum likelihood method including the TLS refinement, as implemented in REFMAC5 [37] giving R and Rfree of 19.8% and 24.4%, respectively (Table S1). The optimized TLS groups were generated using the TLMDS web server [38]. All residues in the refined structure are found in the most favored (97.17%) or additionally allowed (2.82%) regions of the Ramachandran plot.
Myomedin library construction
The combinatorial library we named Myomedin was assembled by a series of three PCR using Phusion High-Fidelity DNA Polymerase (NEB, Massachusetts, USA) using a list of primers and adaptors (see Table S2). 1st PCR (annealing temperature 65°C, 10 cycles) with 100 µM oligonucleotides MYOM-LP_n1F, MYOM-LP_2F, and MYOM-LP_n2R resulted in 147 bp product, which was used in 2nd PCR (annealing temperature 59°C, 10 cycles) with 10 µM oligonucleotides MYOM-LP_1F and MYOM-LP_3R. 3rd PCR with 10 µM oligonucleotides L-for and L-rev was used to complete the Myomedin sequence (333 bp). Two additional PCR steps were done, first using primers JOIN-F and JOIN-R to add ribosome-binding site (RBS), and second using primers T7B and TolAk to add the T7 promoter and a TolA spacer (E. coli str. K-12). TolA spacer template for the last PCR reaction was amplified using primers P7 link and TolA rev from isolated genomic DNA of E. coli. The final PCR product representing linear vector for in vitro transcription and translation (5´-T7 promotor-5´stem loop-RBS-Myomedin-TolAk-3´stem loop-3´) was gel purified.
Antibodies used for selection
Broadly neutralizing human anti-HIV-1 gp41 monoclonal antibody 10E8 (cat#12294) was obtained from the NIH AIDS Reagent Program (Division of AIDS, NIAID, NIH, Germantown, MD). 10E8 IgG was used as a target protein for ribosome display and in ELISA applications (stored as 1 mg/mL aliquoted source stock at −80°C). Human IgG1 with λ light chain (I 5029, purified myeloma protein, Sigma-Aldrich, St. Louis, MO) was chosen as an isotype control for library preselection in ribosome display and as a negative control in ELISA (stored aliquoted as 1 mg/mL source stock at −20°C).
Ribosome display selection
The Myomedin combinatorial library was used for in vitro transcription/translation and ribosome display (RD) selection [21,23]. Three-round RD selections were performed, 96-well Polysorp plates (NUNC, Denmark) were coated by 10E8 IgG1 diluted in coating 100 mM bicarbonate/carbonate solution (pH 9.6) at a concentration according to the adjusted stringency in each round of RD selection procedure: 1st round – 25 µg/mL, 2nd round – 10 µg/mL, and 3rd round – 10 µg/mL. Pre-selection procedure was performed in wells coated with human IgG1 lambda antibody at a constant concentration of 25 µg/mL in each round. Final cDNA after the third round of the selection was amplified by PCR with primers His-Myo-F and JOIN-R. Cleaved PCR product (NcoI, BamHI) was introduced into a pET-28b vector carrying V5 tag sequence downstream of Myomedin cDNA and cloned in E. coli XL1 blue host cells.
Production of Myomedin variants
Myomedin protein variants were produced as 16 kDa recombinant proteins with N-terminal His6 tag and C-terminal V5 tag (His6-Myomedin-V5) in E. coli BL21 (DE3) strain in LB medium containing kanamycin (60 µg/mL), using the same method described earlier. In addition, Myomedin proteins were also produced in E. coli BL21 (DE3) strain containing chaperon plasmid pGRO7 (TaKaRa, Japan) according to the manufacture protocol.
ELISA
Cell lysates of E. coli clones producing Myomedin protein variants were prepared using a sonicator (Misonix 3000). Polysorp plates (NUNC) were coated with 10E8 IgG1, 4E10 IgG1, 2F5 IgG1, VRC01 IgG1 or IgG1 lambda (as an isotype control) at concentration 5 µg/mL in coating buffer (100 mM bicarbonate/carbonate solution, pH 9.6) at 7°C overnight. The plates were washed with PBST solution (PBS buffer containing 0.05% Tween, pH 7.4), and wells were blocked by PBSTB (PBS buffer pH 7.4, containing 0.05% Tween and 1% BSA). Lysate samples (diluted 33x) purified protein variants, and Myomedin-wt (negative control) diluted in PBSTB were applied. Binding of the Myomedin variants was detected using anti-V5 tag – HRP conjugate in PBSTB (1:10,000, Abcam, Cambridge, UK). Results were visualized by the enzymatic reaction of HRP with TMB-Complete 2 substrate (TestLine Clinical Diagnostics s.r.o., Brno, Czech Republic), the reaction was stopped by 2 M sulphuric acid, and absorbance at 450 nm was measured.
Modeling of MLA-10E8 interactions
The structure of studied Myomedin-derived MLA variants was modeled using the MODELER 9v14 suite of programs [39] based on the parental non-mutated myomesin-1 domain 10 structure (PDB ID 3rbs [35], residues 1246–1358). The structure of the 10E8 broadly neutralizing antibody was obtained from the available crystal structures of the HIV-1 gp41 MPER/10E8 complex (PDB ID 5sy8 [40] and 4g6f [41]). The flexible side chain protein-protein global docking was performed using a local copy of the ClusPro server [42,43] docking the MLA variants (as ligands) to the interacting domains of 10E8, chain H residues 1–112 of 5sy8, chain L residues 3–108 of 5sy8, and chain B residues 1–112 of 4g6f, chain D residues 2–108 of 4g6f crystal structure (as the receptor).
Immunization of experimental mice
Female BALB/c mice, 6–8 weeks old, 18–22 g (AnLab, Brno, Czech Republic), were used for all immunization experiments. Animals were housed under standard conditions according to ARRIVE guidelines [44]. The immunization experiments were approved by the Ethics Committee of the Faculty of Medicine and Dentistry (Palacky University, Olomouc, Czech Republic) and the Ministry of Education, Youth and Sports, Czech Republic (MSMT-9487/2019-3). All mice were immunized four times. Pre-immune (naïve) sera were collected before the first immunization. All immunizations were performed by intradermal route with an equal dose of 10 µg of individual Myomedin variant (diluted in 50 µl of DPBS) mixed with 50 µl of Freund´s adjuvant (Sigma Aldrich, St. Louis, MO, USA) per mouse per one immunization.
HIV-1 Env-specific binding antibodies determination
Reactivity of binding antibodies targeting Env was measured by ELISA using pseudotyped viruses prepared for virus neutralization assay (see below). To remove fetal bovine serum pseudotyped viruses were first ultracentrifuged for 3 hours at 50,000 g at 4°C and resuspended in PBS. MaxiSorp ELISA plates (NUNC, Roskilde, Denmark) were coated with pseudotyped viruses diluted in PBS overnight at 4°C. Plates were washed with PBS and blocked with 1% BSA/PBS for 3 hours at room temperature. Mouse sera were serially diluted in blocking buffer in duplicates and incubated overnight at 4°C. Plates were washed with PBS, and bound antibodies targeting Env were determined by incubating with anti-mouse IgG secondary antibody conjugated with horseradish peroxidase diluted in blocking buffer for 3 h. Plates were washed, and the signal was developed with O-phenylenediamine-H2O2 substrate. The reaction was stopped with 1 M sulfuric acid. The absorbance was quantified at 492 nm by ELISA reader.
Competition ELISA
Plates were coated with the selected pseudotyped virus as described in the previous method of Env-specific binding antibodies determination. 10E8 antibody was serially diluted in blocking buffer and applied in duplicates with mouse hyperimmune sera diluted 1:400. Plates were washed, and bound mouse antibodies were detected by rabbit anti-mouse IgG secondary antibody conjugated with horseradish peroxidase diluted in blocking buffer. Signal was developed and measured as described above.
Virus preparation
Pseudotyped viruses were prepared using HEK293/17 cell line grown in 75 cm2 flask at 60–80% confluency co-transfected with 4 µg of plasmid coding Env and 8 µg of plasmid pSG3ΔEnv mixed with 48 µl of transfection reagent FuGene6 (Promega, Madison, WI, USA) in 12 ml of culture medium. After 2 days, produced pseudotyped viruses in culture medium were harvested, filtered, and stored at minus 80°C until used.
Virus neutralization assay
Virus neutralization assay was done as previously described [21,45] using various pseudotyped viruses of clades A, B, C, D, and AE. Murine leukemia virus (MULV)-pseudotyped virus containing the Env of MULV with the same backbone vector as all HIV-1-pseudotyped viruses (pSG3ΔEnv) was used as a negative control. Before the assay, mouse sera were heat-inactivated for 1 hour at 56°C. Sera in duplicates were serially diluted in 100 µl of culture medium in 96-well plates and incubated with 50 µl of pseudotyped viruses at 150,000 RLU for 90 min at 37°C. Then, 10,000 TZM-bl cells stably expressing CD4 receptor, CCR5 and CXCR4 co-receptors, and luciferase and β-galactosidase under control of HIV-1 promotor (NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH) in 50 µl of culture medium were added into each well and incubated for 48 hours at 37°C in 5% CO2 atmosphere. 150 µl of culture medium was removed and 100 µl of lysis buffer containing luciferin (Promega) was added. After 2 minutes, 100 µl of lysate was transferred into a black 96-well plate and luminescence was measured using a HP luminometer.
ELISA coated with CD4-liganded and -unliganded pseudotyped viruses
Nunc Maxisorp ELISA plates were coated with ultracentrifugated pseudotyped viruses in PBS overnight at 4°C. After washing with PBS, plates were blocked with 1% BSA/PBS for 3 hours at room temperature. Then, recombinant human CD4 serially diluted in blocking buffer was added and incubated for 1 hour at 37°C. Selected sera were diluted 1:200 in blocking buffer, added into wells, and incubated for 2 hours at 37°C. After washing, bound antibodies were detected with anti-mouse IgG conjugated with HRP. After washing, signal was developed with OPD, and the reaction was stopped by 1 M sulfuric acid. The absorbance was measured and quantified using ELISA reader. Differences in absorbance of wells with/without added CD4 was determined.
Statistics
Statistical significance of differences between groups was determined using analysis of variance (ANOVA), Kruskal-Wallis test and Dunn´s posttest. All statistical analyses were done by SPSSv.21 statistical packages (IBM Corp., Armonk, NY, USA) or GraphPad Prism 5 Software (GraphPad Software Inc., San Diego, CA, USA).
Results
Human myomesin-1 domain 10 production and structure description
We produced domain 10 of myomesin-1 protein (MYOM1) in E. coli with N-terminal 6xHis-tag and thrombin cleavage site. After purification by Ni-NTA and size-exclusion chromatography, we determined its melting temperature by thermofluor assay to 72°C (Fig S1A, B). Purified protein was also crystallized (Fig. S2), and its X-ray structure was solved to 1.8 Å and deposited to the Protein Data Bank under accession code (PDB ID) 6t3o.
The crystal structure of the myomesin domain (PDB ID 6t3o) is formed by 111 amino acids, which constitute the 2-layer sandwich of the immunoglobulin-like domain with 7 antiparallel β-strands and a terminal α-helix (Figure 1a, b). The domain corresponds to the MY10 part of the myosin filament-linking protein myomesin-1 [35] with 100% identity. Two of the referred structures (PDB codes 2y23 and 3rbs) contain the MY10 entity. Superposition of the Cα atoms from the current myomesin structure and the two previous structures gives a root mean square deviation (RMSD) of 0.81 Å, 0.70 Å respectively (Fig. S3), and showing that the layout of the structure is completely conserved. This demonstrates that myomesin-1 domain 10 maintains structure conformation and stability even when it is separated from the structural context of the myomesin filament.
Figure 1.
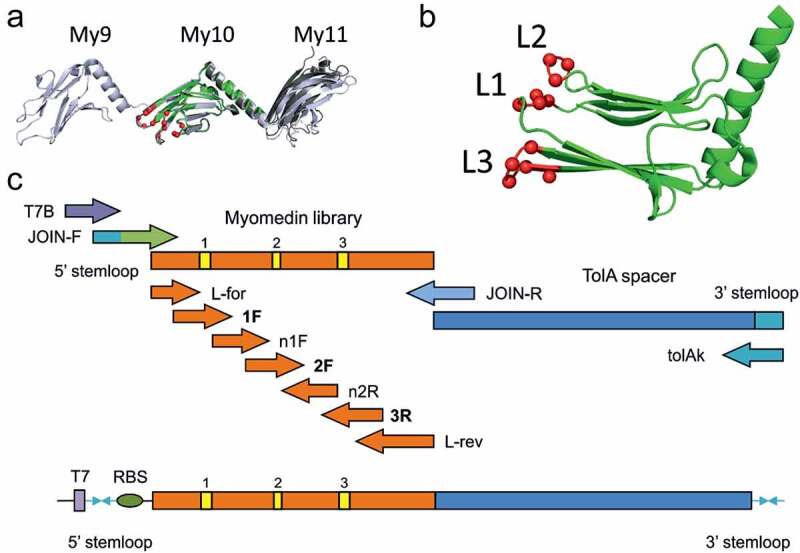
Human myomesin-1 Domain 10, a Loop-type Myomedin library concept. A) The superposition of the crystal structure of the myomesin-1 domain 10 used for randomization (PDB ID 6t3o) to the structures of longer myomesin fragments. The domain 10 is shown in green and the randomized residues indicated as red spheres. The myomesin fragment 10–11 (PDB ID 3rbs) is shown in gray and the 9–10-11 fragment (PDB ID 2y23) is shown in light blue. The Cα RMSD values for the 6t3o domain 10 compared to 2y23 and 3rbs structures are 0.81 Å and 0.70 Å, respectively, showing a high structural similarity of the isolated domain. B) A rotated view on domain 10 is shown in green cartoon representation with the randomized residues indicated as red spheres. C) Diagram of the primers used for the assembly of the Myomedin loop-type combinatorial library via multiple PCR steps
Design of combinatorial library based on scaffold of human myomesin-1 domain
To select a set of myomesin residues suitable for randomization we have performed in silico mutability screening of all amino acid residues. We have used the crystal structure of human myomesin domains 10 and 11 (PDB ID 3rbs) [35] and mutated all residues to all 20 amino acids using the PositionScan routine of FoldX program [46]. According to the predicted change of free energy upon mutation, we have assigned to each residue a mutability score corresponding to several stabilizing substitutions. The score was used for the final selection of candidate residues trying to find a continuous patch of accessible surface residues with high mutability. This procedure led to a selection of the following residues localized in loop regions of myomesin domain 10: E1267 (Loop 1 residue No. 21), K1268 (Loop 1 residue No. 22), L1269 (Loop 1 residue No. 23), S1270 (Loop 1 residue No. 24), R1296 (Loop 2 residue No. 50), N1297 (Loop 2 residue No. 51), T1298 (Loop 2 residue No. 52), D1322 (Loop 3 residue No. 76), G1323 (Loop 3 residue No. 77), K1324 (Loop 3 residue No. 78), A1325 (Loop 3 residue No. 79), and T1326 (Loop 3 residue No. 80), numbered according to the UniProt [47] P52179 record.
Molecular assembly of Myomedin loop-type combinatorial library
In silico prediction of mutable residues defined by a free Gibbs’ energy (ΔΔG) performed for each of 111 amino acid residue of myomesin-1 domain 10 resulted in two possible concepts of randomization. One of them (Figure 1(a,b)) called Myomedin loop-type scaffold library predicted mutability of 4 residues in loop 1 (L1, residues 21–24), 3 residues in loop 2 (L2, residues 50–52), and 5 residues located within loop 3 and the connection to a downstream beta-sheet strain (L3, residues 76–80). To install the designed mutable residues into DNA library strains, a set of 4 forward primers in combination with 3 reverse primers spanning all myomesin-1 domain 10 AA were synthesized (Figure 1c). A supplementary set of helper primers for attachment of ribosome-binding site (RBS), 5´stemloop, 3ʹ-end TolA spacer protein, and 3’stemloop were prepared (Figures 1c and Figures 1a full list of primers in Table S2). To avoid the formation of sulphate bridges in the Myomedin scaffold, we eliminated cysteine and used 19 amino acid trimer-codons (TRIM) for randomization of 12 mutable residues optimized for E. coli expression, thus providing a theoretical library complexity 2 × 1015 Myomedin variants.
Identification of Myomedin variants targeting variable regions of HIV-1 bNAb 10E8
To select protein binders from Myomedin loop-type combinatorial library, a three-round ribosome display was performed on immobilized target 10E8 bNAb with preselection on isotype control – monoclonal myeloma IgG1 antibody with λ light chain (IgG1λ). Thus, both 10E8 bNAb and preselecting antibody are of IgG1 isotype with λ light chain. The final collection of DNA transcripts was cloned into the pET28b vector forming a cDNA library pool. Myomedin protein variants designated as MLA binders, and parental non-mutated control variant called Myomedin wild-type (MyoWT) were expressed in Escherichia coli (E. coli) BL21 (DE3) as 16 kDa fusion protein with C-terminal V5-tag (His6-MLA/MyoWT-V5). Lysates of 180 bacterial clones were analyzed by ELISA for specific binding to 10E8 bNAb. We identified 30 variants representing full-length unique proteins that exhibited preferential binding to 10E8 bNAb in comparison to IgG1λ isotype control. The binding curves of these selected MLA binders confirmed their specificity for 10E8 bNAb and low binding to bovine serum albumin. Based on estimated differences of affinity (Kd values), the group of 8 most promising candidates designated MLA016, MLA024, MLA025, MLA092, MLA093, MLA132, MLA158, and MLA159 (Figure 2, Table 1) were used for further testing their epitope-mimicking potential by immunizations of mice.
Figure 2.
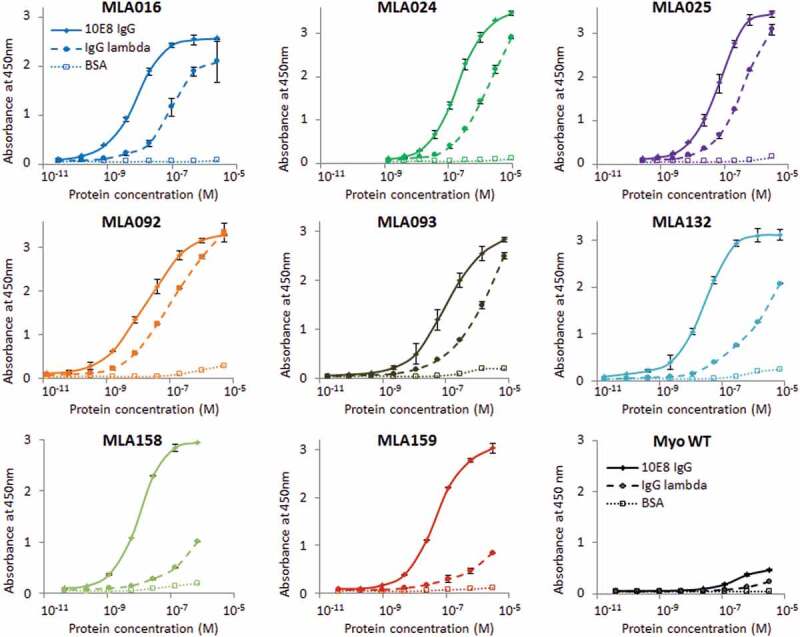
Binding of MLA protein variants to 10E8 bNAb, IgG isotype control and BSA. Myomedin variants of eight selected MLA clones in the form of purified fusion proteins with N-terminal polyhistidine tag and C-terminal V5 tag produced in E.coli BL21 (DE3) were assayed in ELISA, the parental non-randomized Myomedin was used as a negative control. Binding to immobilized 10E8 bNAb (labeled as 10E8 IgG), IgG1λ isotype (labeled as IgG lambda) and BSA was detected by anti-V5 Ab-HRP conjugate. Each point is shown as the mean value of triplicates with standard deviation
Table 1.
Table of MLA sequences. Sequence comparison of the MLA protein variants with parental non-randomized MyoWT protein. Yellow boxes indicate the loop stretches with 12 positions at which the residues of MyoWT were randomized. Other residues of the 111 amino acid Myomedin scaffold protein were not mutated in all shown MLA binding proteins
| 21 | L1 | 24 | 50 | L2 | 52 | 76 | L3 | 80 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MyoWT | Q | A | E | K | L | S | G | N | … | I | D | R | N | T | G | I | … | L | Q | D | G | K | A | T | N | H |
| MLA016 | Q | A | K | A | Q | Q | G | N | … | I | D | I | M | F | G | I | … | L | Q | S | H | H | L | G | N | H |
| MLA024 | Q | A | L | S | V | F | G | N | … | I | D | R | N | T | G | I | … | L | Q | F | M | L | M | M | N | H |
| MLA025 | Q | A | A | T | P | S | G | N | … | I | D | G | H | E | G | I | … | L | Q | V | I | L | I | L | N | H |
| MLA092 | Q | A | E | I | M | W | G | N | … | I | D | P | S | W | G | I | … | L | Q | I | V | T | P | L | N | H |
| MLA093 | Q | A | D | G | S | S | G | N | … | I | D | R | A | N | G | I | … | L | Q | D | F | I | I | W | N | H |
| MLA132 | Q | A | L | L | P | L | G | N | … | I | D | Y | F | W | G | I | … | L | Q | M | W | S | E | - | N | H |
| MLA158 | Q | A | W | M | W | W | G | N | … | I | D | I | T | L | G | I | … | L | Q | L | Y | Y | A | W | N | H |
| MLA159 | Q | A | M | N | L | Y | G | N | … | I | D | Q | A | M | G | I | … | L | Q | M | M | I | E | Y | N | H |
Modeling of MLA interactions with 10E8
To verify the most probable binding areas for all selected MLA binders we performed in silico modeling by docking using two crystal structures of 10E8/Env complex, PDB ID 5sy8 and 4g6f. Although these complexes were described for two different MPER-containing proteins, structures of interacting residues of the IgG chains are nearly identical. We used both these structures to predict the most probable-binding modes for all eight MLA protein variants. Based on available literature data [40,41,48], we defined 3 binding regions for 10E8/gp41 interaction that can be used for evaluation of mimicking potential of each MLA variant. The binding region called Site 1 is formed by MPER epitope residues T676, L679, W680, I682, and K/R683 and corresponding interacting paratope residues Y99, F100a, W100b, Y100e, and P100f of the 10E8 immunoglobulin heavy chain. The second binding region (Site 2) contains gp41 MPER residues N671, W672, and F673 and paratope residues W33, E53, K97, and P100g of the 10E8 heavy chain, and the light chain residue R95B. While sites 1 and 2 involve MPER residues, Site 3 is defined by 10E8 light chain residues L28, R29, S30, H31, Y32, A66, S67, and G68 which were described to be important for the interaction of bNAb 10E8 with lipid membrane components [40]. Results of MLA docking identified three principle binding modes. Myomedin variants MLA 024, 025, 093, 158, and 159 bind almost exclusively via randomized loops of Myomedin scaffold, covering binding Sites 1 and 2 in a vertical orientation. A predicted most probable binding mode for variants 016 and 092 covers Site 1 and 2 as well, but extend to the vicinity of Site 3 in a horizontal orientation. In contrast to all variants, MLA132 prefers horizontal orientation pointing in an opposite direction interacting with Site 1 and 2. Binding modes for all MLA proteins are presented in Figure 3 and Figure S4. Results of docking suggested that all MLA proteins bind preferentially to Site 2 of the 10E8 bNAb paratope. In the case of MLA024 and 093, the interaction surface significantly differs from that of the corresponding gp41 MPER surface, suggesting a worse shape complementarity of these proteins to the 10E8 antibody paratope surface.
Figure 3.
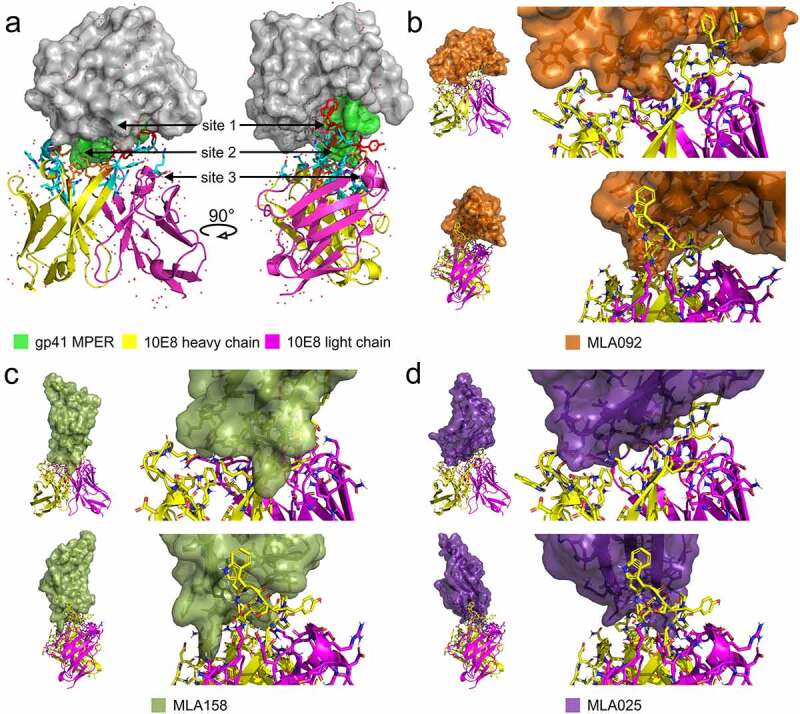
Modeling of MLA-10E8 interaction by docking. A), A crystal structure of the scaffolded HIV-1 Env gp41 MPER/10E8 complex (PDB ID 5sy8) showing the 10E8 antibody heavy chain in yellow cartoon representation, the light chain in magenta, and the gp41/Env MPER as a green surface embedded in a gray helper protein construct. The 10E8 interaction interface is shown as cyan sticks and the positions of the key 10E8 interaction sites are indicated by arrows, Site 1 residues are highlighted as red sticks and Site 2 as orange sticks. B), Predicted binding mode of the MLA092 (orange) to the 10E8 antibody; the upper row of figures shows the whole complex (left) and a detail of the interacting interface (right) in the antibody orientation corresponding to the left-hand side of the panel A (focusing on the interaction site 2). The lower row corresponds to the second antibody orientation (focusing on the interaction site 1). C), Predicted binding mode of the MLA158 (pickle) in the same orientation as in panel B. D), Predicted binding mode of the MLA025 (violet) in the same orientation as in panel B. MLA interface residues within 4 Å of the 10E8 residues are: MLA016 A6, E8, I9, L10, R15, W17, K21, A22, Q23, Q24, H47, I48, D49, I50, M51, F52, E56. MLA024 L21, S22, V23, F24, G25, N26, R50, N51, F76, M77, L78, M79. MLA025 S24, N26, I32, N34, T69, T71, Q73, Q75, I77, L78, L80, H82, T84. MLA092 E7, K11, R15, W17, I22, M23, W24, G25, H47, I48, D49, P50, S51, W52, E56, F58. MLA093 S24, N26, I32, T71, Q73, Q75, F77, I78, W80, H82. MLA132 L22, L24, H47, D49, Y50, F51, W52, E56, M76, W77, S78. MLA158 E3, M18, Q19, A20, W21, M22, W24, N26, L74, L76, Y77, Y78, A79, N81. MLA159 Y24, N26, I32, E35, T71, Q73, M77, I78, E79, Y80, H82. The randomized residues are in bold. The source PyMOL session containing the results for all identified MLA variants is available on 10.5281/zenodo.4650182
Myomedin variants induce serum antibodies recognizing Env glycoproteins on pseudotyped HIV-1 viruses
Following MLA isolation, in vitro and in silico characterization, all eight selected candidates – MLA016, 024, 025, 092, 093, 132, 158, and 159 – were expressed and purified as recombinant proteins, and used for immunization of experimental mice by i.d. route with Freund’s adjuvant by four consecutive antigen injections, according to the schedule presented in Figure 4a. MPER-specific reaction was evaluated on pseudoviruses coated on ELISA panel as the presence of membrane surrounding the gp41 is necessary to MPER epitope. Four pseudoviruses – one Clade C Env (ZM109F) (Figure 4b) and three Clade B Env (AC10.0, QH0692, RHPA) (Figure 4c) were assayed on ELISA. The experiment identified significant binding for MLA025, 092, 093, 158, and 159 at least with one pseudotyped virus (Figure 4(b,c)) when compared with wild-type Myomedin (MyoWT). MyoWT did not elicit detectable Env-pseudovirus-specific serum response.
Figure 4.
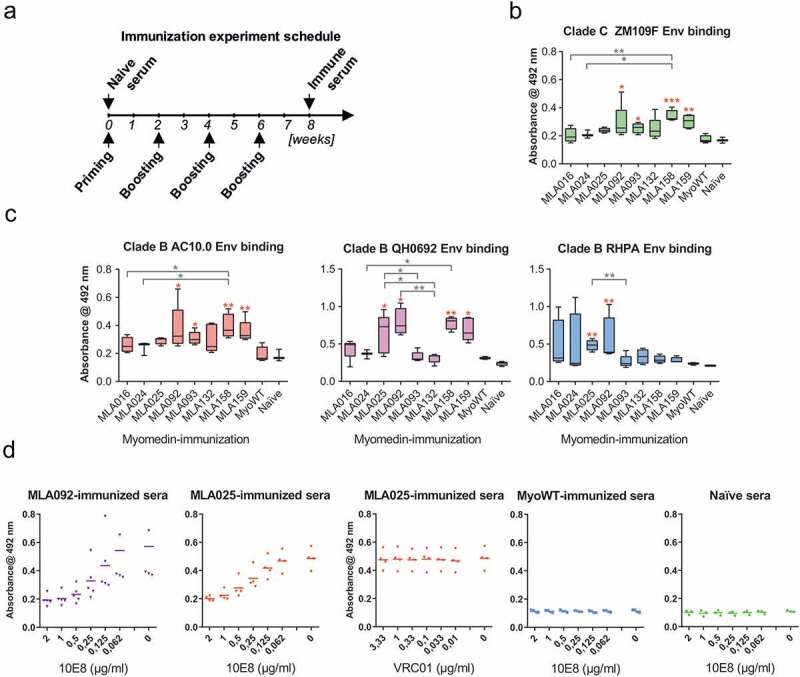
Immunization of experimental mice with a panel of MLA variants induced serum antibodies specifically recognizing HIV-1 Env on the virus surface. A) Mice were immunized by the administration of four doses of individual MLA variants including wild-type (MyoWT). Following immunization, sera were collected and tested in their reactivity with non-replicative HIV-1 viruses pseudotyped with B) Clade C Env (ZM109F) or C) Clade B Env (AC10.0, QH0692, RHPA) coated on ELISA plates. Antibody titers of IgG isotype were measured in ELISA. Statistical comparison was performed by ANOVA Kruskal-Wallis test with Dunn’s posttest (* P < 0.05, ** P < 0.01). The red asterisk indicates a comparison with MyoWT. D) Sera from mice immunized with MLA092 and MLA025 Myomedins were tested for reactivity with RHPA-pseudotyped virus coated on ELISA panel in competition with 10E8 or VRC01. 10E8 serially diluted to achieve final concentrations 2; 1; 0.5; 0.25; 0.125; 0.0625; and 0 μg/mL in blocking buffer was applied with individual MLA092- or MLA025-immunized mouse sera diluted 1:400. After washing, the plates were incubated with rabbit anti-mouse IgG HRP-conjugated antibody, developed with a substrate and O.D. 492 nm was measured. VRC01 antibody was applied in control reaction, as irrelevant antibody, at final 3.33; 1; 0.33; 0.1; 0.033; and 0 μg/mL analogously to 10E8. In separate experiment 10E8 at indicated concentration was applied with individual MyoWT-immunized or naive mouse sera diluted 1:400 as the control (two right panels). All experiments were performed in triplicates. Mean values are indicated by horizontal lines
To further confirm MPER specificity of sera binding, we performed an ELISA competition assay where the binding of sera to selected pseudovirus from Clade B (here RHPA) competed with a serial dilution of 10E8 as an original specific target used for all MLA variants selection (Figure 4d). 10E8 in concentration 2 µg/mL completely inhibited all tested hyperimmune sera reactivity. When irrelevant bNAb was used for competition (here VRC01) no competition was observed even at concentration 3.33 µg/mL. As negative controls, naive sera and Myomedin wild type-immunized mice sera were tested in the same assay, and no binding of sera to RHPA was observed irrespective of titration of 10E8 as a competitor (Figure 4d).
MLA proteins elicit serum antibodies neutralizing the majority of the panel of Clade A, B, C, and AE pseudoviruses
Subsequently, we tested the biological activity of serum antibodies using neutralization assay with the available set of Clade A, B, C, D, and AE pseudoviruses. Due to the limited volume of post boost sera, we tested 22 pseudoviruses, and obtained data were expressed as reciprocal serum dilution resulting in 50% virus neutralization (Figure 5(a,b)). Titration curves are provided for pseudoviruses exhibiting 50% neutralization titers higher than 30 (Figure S5). The most broadly neutralizing serum antibodies were elicited by immunization with MLA variants MLA092 followed by MLA158, MLA132, and MLA025. MLA092 neutralized 54% of tested pseudoviruses at dilution higher than 1:30. MLA092 exhibited also the highest titers of binding antibodies (Figure 4(b,c)). The second most effective Myomedin, MLA158, induced also high titers of binding antibodies as confirmed for three out of four tested pseudoviruses in ELISA (Figure 4(b,c)). Pseudovirus RHPA exhibited binding with sera of MLA025 and MLA092 immunized mice. Myomedin variant MLA132 neutralized 45% of tested pseudoviruses (at dilution higher than 1:30) and exhibited lower relative mean neutralization dilution 42 (Figure 5a), in comparison to MLA158. MLA132-immunized mice exhibited only a modest increase in binding antibodies to all four tested pseudoviruses. Myomedin MLA025 neutralized 40% of pseudoviruses but the mean neutralization dilution for neutralized pseudoviruses was 1:47. All of the tested pseudoviruses were Tier-2 HIV-1 pseudoviruses. This indicates, in agreement with molecular modeling, that screening for 10E8 binding Myomedin library identified MLA variants able to elicit in mice antibodies neutralizing about 50% of tested representative Tier-2 HIV-1 pseudoviruses.
Figure 5.
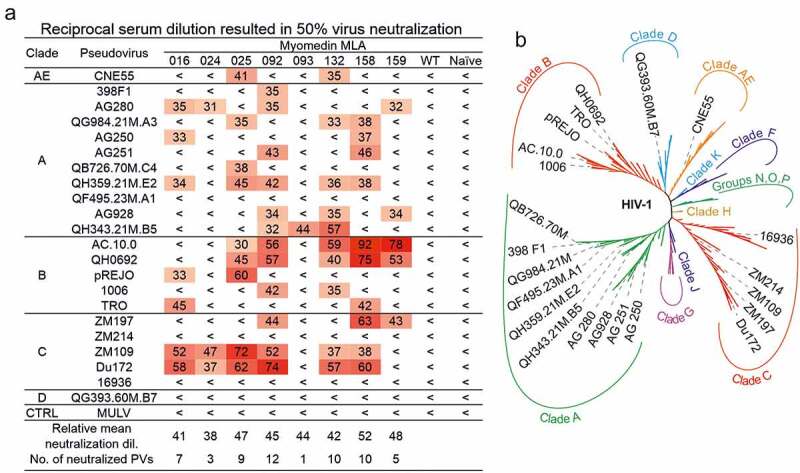
Neutralization activity of MLA-immunized mice sera. A) Values represent the reciprocal serum dilution which resulted in 50% virus neutralization. Relative mean neutralization dilution represents mean values of detected titers among all tested pseudoviruses. No. of neutralized PVs indicates the sum of pseudoviruses neutralized at reciprocal titer higher than 30. Murine leukemia virus (MULV) was used in neutralization assay as a control. Pseudotyped viruses used for analyses were chosen to cover the phylogenetic tree of HIV-1 Env. B) An unrooted phylogenetic tree of 194 representative sequences from HIV-1 2019 Compendium of HIV-1 ENV genes (https://www.hiv.lanl.gov) enriched for 22 HIV-1 ENV sequences used in this analysis. Sequences were processed at EMBL-EBI Simple Phylogeny using neighbor-joining clustering method [68,69]. HIV-1 strains are classified into four groups M, N, O and P. The major group M is further divided into nine genetically distinct subtypes A, B, C, D, AE, G, H, J, F and K. Each subtype contains hybrid viruses – circulating recombinant forms as a result of genetic material combination. For better orientation, only the 22 HIV-1 ENV sequences used in the analysis are visualized. The complete and high-resolution tree is available in figure S6
The HIV-1 Env occurs in a closed, partially open, and open conformation depending on the presence of CD4. Therefore, we tested the dependence of pseudovirus surface-exposed Env interaction with sera of MLA024, MLA092, and MLA158 immunized mice in the presence of soluble CD4. In accord with previous reports, the presence of CD4 was associated with enhanced binding of MLA-immunized mice sera in a dose-dependent manner (see Fig. S10). This indicates that elicited antibodies recognize the MPER epitope similarly to previously reported MPER-specific mAbs [28,49,50].
Discussion
In our previous work, we demonstrated that the three-helix bundle of ABD scaffold can sufficiently mimic a part of the gp120 surface recognized by CD4 site-specific VRC01 bNAb. However, the 46 amino acid ABD scaffold is not sufficient to cover a large binding surface of the MPER region. Instead, we used 111 residues of domain 10 of myomesin-1, which we named Myomedin, that were predicted as a suitable scaffold for the generation of MPER mimicking immunogens. Its benefit lies in human origin, a sufficient rigidity mediated by antiparallel β-sheets and high thermal stability. Several other protein scaffolds have already been developed with extraordinary stability as DARPins [51], Anticalins [52] or OBody [53]. Other scaffolds are also suitable for invivo diagnostics or therapy as Adnectin [54], Affibody [55] or Fynomer [56].
Monoclonal antibodies specific for MPER epitopes identified from B cell of elite HIV-1 neutralizers demonstrated high neutralizing potency and the highest coverage among all HIV-1 bNAbs thanks to high sequence conservation of this Env region. Therefore, stimulation of the production of MPER epitope-specific bNAbs is a priority for the research for HIV-1 eradication [57]. Among naturally developed anti-MPER antibodies, some of them, including 2F5 [58], DH511.2 K3 [59], 4E10 [60], 10E8 [41], and LN01 [61], have exhibited a strong neutralizing potential and serve, therefore, as prototypes and research tools for the design of vaccine components. However, stimulation of high titers of MPER-specific bNAbs in sera of immunized individuals has not been successful so far [62]. Several recombinant gp120/gp41-derived antigens were designed to elicit MPER-specific antibodies but, although specifically recognized by MPER class of bNAbs, they do not elicit serum antibodies with neutralizing potential as described for 10E8 and 4E10 [62–64]. This is mainly due to problems with designing of suitable immunogens resulted from the highly dynamic and, therefore, unknown functional conformation of the gp41 moiety of MPER [57]. Besides, the use of a trimeric envelope protein in the context of phospholipid membrane to mimic MPER is poorly immunogenic compared to other Env epitopes that elicit subtype-specific antibodies without neutralizing activity. Even the strategy based on sequential immunization with recombinant gp41 followed by gp41 on membrane and Env on membrane designated as Incremental, Phased Antigenic Stimulation for Rapid Antibody Maturation was achieving limited efficacy [62].
To overcome current problems with MPER-derived gp41 immunogens, we used our novel strategy (NCLS) stimulating the production of neutralizing antibodies by vaccination employing non-cognate ligands. Recently we have generated a collection of recombinant ligands called VRA proteins targeting gp120-specific bNAb VRC01 that were selected from a highly complex combinatorial library derived from the scaffold of the albumin-binding domain (ABD). We demonstrated that these proteins, when used as immunogens, elicit antibodies neutralizing most of the selected pseudotyped HIV-1 viruses [21]. Here we report on the design of novel protein scaffold Myomedin derived from the human myomesin-1 domain as a robust master structure applicable for combinatorial library design, assembly and generation of ligands for vaccine development. The Myomedin scaffold was chosen mainly due to its high stability, which is related to the general stability of contractile protein domains. The potential advantage of Myomedin-derived immunogens might be also the absence of inherited host reactivity due to natural tolerance of autologous protein myomesin-1, allowing focusing of immune response on randomization-introduced neoantigens.
We applied the NCLS to identify Myomedin variants binding to one of the most prominent MPER-specific bNAb 10E8. This antibody was extensively studied to identify crucial interacting residues located within both the antibody paratope and its cognate MPER epitope [15,40,41]. These studies demonstrated that some of the residues are important for a high-affinity recognition as well as for an observed neutralizing activity, yet the particular contribution varies for each of the interacting residues. Paratope residues Y99, F100a, and W100b were defined as key residues for the neutralization of pseudovirus infection activity [41]. These residues form a part of the Site 1 interaction area together with complementary residues W680 and R/K683 on the MPER epitope. However, for high-affinity binding, residues W672 and especially F673 were shown to be indispensable, as demonstrated by F673A substitution diminishing the binding million-fold [41], and contribute substantially also to the neutralization efficiency. Residues W672 and F673 form a critical point of the Site 2 binding area in this study.
We identified eight gp41/MPER epitope-mimicking candidates that were confirmed to stimulate anti-Env antibody production after the immunization of mice. However, the neutralizing potential tested on a panel of 22 selected pseudoviruses significantly differs among the tested MLA proteins. To gain a rational insight into these differences, we employed molecular modeling approaches. Based on protein-protein docking, we defined 3 principal interaction regions of MLA Myomedins with 10E8 bNAb. Interestingly, in our Myomedins MLA024 and MLA093, insufficient mimicking of F673 residue could explain their low neutralizing activity through the panel of tested pseudoviruses. In contrast to these two variants, all other MLA proteins mimic F673 residue very well, or at least substantially, which is consistent with neutralization activity data. In the case of MLA025, residue F673 is well represented by L80, however, the L82 residue is mimicking W672 only partially, and a similar situation is found for MLA158 involving residues W26 and Y79.
Regarding Site 1, except for MLA093, the interacting surface is well reproduced. Interestingly, variants MLA025 and MLA159 utilize non-randomized H84 residue for mimicking combined W680 and R/K683 residue surfaces. Yet both these protein variants share this interaction mode, they differ in neutralizing potential, probably due to additional interactions across their surface. In the case of MLA158, Site 1 interacting surface forms a deep cavity (Figure 6d lower right) accommodating a key neutralization-supporting paratope residue F100a. This is in a good correlation with the neutralization data, where sera of MLA158 immunized mice show the highest efficiency among all MLA immunized mice (Figure 5a).
Figure 6.
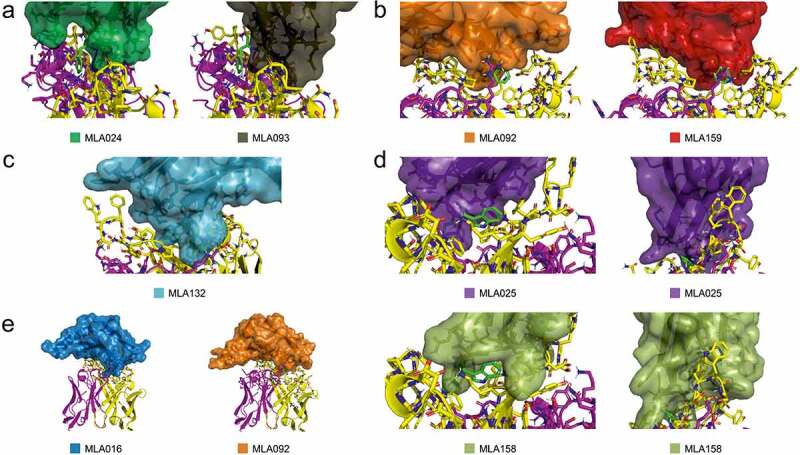
Model-based interpretation of neutralizing potential of all MLA proteins. A) Insufficient mimicking of MPER F673 (green sticks) by MLA variants 024 and 093, B) Partial mimicking of MPER F673 (green sticks) by MLA variants 092 and 159, C) Larger interacting surface within site 2 evolved in MLA132 variant, D) Partial mimicking of MPER W672 (green sticks) by variants MLA025 and MLA158 (left), Comparison of interaction surfaces within site 1 between MLA025 and MLA158, demonstrating the formation of a deep cavity for binding of 10E8 F100a residue (right), E) Potential of variants MLA016 and MLA092 for mimicking of site 3 interaction. As an example, Fig.S11 is summarizing interacting residues of MLA092. For more detailed representation we included the source PyMOL session containing the results for all identified MLA variants available on 10.5281/zenodo.4650182
Myomedin variant MLA092 exhibits the highest breadth in our MLA proteins collection, neutralizing 12 of 22 tested pseudotyped viruses (Figure 5). Results of protein docking suggested that the horizontal-binding mode of this protein variant could contribute to the formation of Site 3 interactions that were described to be responsible for the recognition of membrane components [40]. Available structures 5t80 and 5t85 [40] reveal binding of phospholipids to 10E8 bNAb Site 3, and the negatively charged residue E12 from MLA016 and MLA092 can contribute to the formation of the similar lipid-binding interface in newly elicited antibodies.
Based on the data, we can conclude, that predicted interactions of residues W672, F673, W680, and R/K683 can explain the observed mimicking potential of MLA Myomedins demonstrated by the production of MPER-targeting neutralizing antibodies in immunized mice. This set of residues has recently been assigned as HIV-1 MPER-specific neutralizing antibody signature [48]. It is important to mention that F673 is a key residue recognized not only by 10E8 but also by 4E10, DH511, and LN01 bNAbs [61]. While two former antibodies share the similar MPER-binding mode, LN01 binds MPER in a differently tilted orientation involving a bridging interaction with phospholipids. This combined binding mode resembles the interaction of VRC01/gp120 where water molecules represent a part of the recognized surface, and VRA proteins were found to mimic this combined interface [21].
Motivation to employ Myomedin scaffold for the development of MPER-epitope mimicking protein variants came from the finding that a much larger surface will be necessary to mimic 10E8 bNAb epitope involving key residues of Site 1 as well as Site 2, with possible support by residues of Site 3. In contrast to our previously used albumin-binding domain (ABD)-derived scaffold library providing VRC01-specific VRA binders of 46 amino acid residues [21], Myomedins of 111 residues possess the capacity to evolve a sufficient shape complementarity to the 10E8 antibody paratope required for efficient mimicking potential. In addition, the randomization of 12 mutable residues within three Myomedin scaffold loops increased a theoretical complexity to 2 × 1015 variants in comparison to 11 randomized residues of the ABD-derived NNK-type library, thus providing the complexity 2 × 1014 proteins [22]. As our Myomedin trinucleotide codon-based randomized library eliminated cysteine and stop codons from the TRIM mixture, the formation of covalent dimers was minimized, due to the absence of S-S bridges formation, together with a diminishing of production of truncated variants, thus increasing the efficiency of ribosome display and large scale ELISA screening. As Myomedins are derived from the human protein domain, stimulation of antigenic response targeting only the randomized surface in human immunized individuals is expected to be followed by the production of a more specific antibody repertoire, yet maybe lower than that formed against streptococcal protein G ABD-derived VRA proteins. Sequence comparison between human and mouse DNA coding for myomesin-1 domain 10 revealed only 7 mutations resulting in 93.7% sequence identity. One of them, S/G mutation, is located in the downstream proximity of the loop 1 and another one, S/N, forms a part of the beta-sheet interface, while all others are located in the structure core, thus are expected to be inaccessible for the recognition.
From the collection of tested Myomedin proteins, variants MLA092, MLA158, MLA132 and MLA025 demonstrated the best neutralizing breadth in the panel of tested pseudoviruses. Besides, the level of serum dilution in proteins MLA158, MLA092, MLA159, and MLA025 reached values over 70, suggesting that these variants will be most promising for further development of vaccine immunogens. Interestingly, the level of serum dilution in these Myomedins reached values that were obtained in the case of VRA proteins only for several Clade B/C pseudoviruses by a combinatory immunization scheme consisting of VRA177-TolA priming followed by a booster with VRA017 containing a truncated version of TolA [21]. In addition, Myomedins do not require fusion with TolA/TolS helper proteins for neutralizing antibody production. VRA proteins have not been yet tested for the neutralizing potential using Clade A pseudoviruses, while 7 of 8 Myomedin proteins exhibited virus-neutralizing effect in 9 of 10 pseudoviruses tested on the reporter TZM-bl cells.
Myomedins MLA158, MLA092, MLA025, MLA132, and MLA159 in combination with ABD-derived VRA177 and VRA017 reported earlier could form a unique collection of immunogen candidates available for further testing of potential HIV-1 virus-clades-optimized/targeting vaccine. Indeed, stimulation of VRA-mediated elicitation of CD4bs epitope-neutralizing antibodies in combination with parallel elicitation of MPER-specific bNAbs could increase the neutralizing breadth. It has been recently shown that double, triple or quadruple combinations of monoclonal antibodies exhibited a substantially improved neutralizing breadth in comparison to the particular single ones [65]. In addition, combination therapy employing two potent anti-HIV-1 bNAbs, 3BNC117 and 10–1074, targeting distant Env epitopes, demonstrated a strong treatment efficiency in phase 1b clinical trial [66]. The idea to simultaneously elicit bNAbs against CD4bs, as well as MPER epitopes, using one immunogen mixture of VRA and MLA proteins is also supported by another work. The authors well documented that infection of human individuals by EBV was controlled by a combination of bNAbs targeting non-overlapping epitopes with complementary sensitivity to mutations and concluded that combination of human bNAbs with distinct epitopes suppressed escape mutations, leading to the conclusion that immunotherapy for HBV infection may require combinations of complementary bNAbs [67].
To verify a mimicking potential of MLA protein variants, we tested their binding to other bNAbs. From the group of MPER-targeted bNAbs, we investigated interaction of coated 2F5 and 4E10 antibodies to the MLAs. Our results demonstrate that all tested MLA proteins significantly bind to 4E10 (Fig. S7) in accordance with very similar MPER binding modes as compared to 10E8. In the case of 2F5, which binds MPER epitope more distal to the viral membrane, an absence of MLA158 binding was observed (Fig. S8). In silico analysis of MLA158/10E8 complex suggests that Site 1 residues form the cavity accommodating a key F100a residue, feature not available in 2F5 bNAb. As expected, MPER-unrelated antibody VRC01 exhibited only a minimal binding to all tested MLA variants similar to IgG isotype control (Fig. S9). These combined data support a hypothesis that immunization with MLA proteins can elicit a wider portfolio of neutralizing antibodies than only the parental 10E8.
In this work, we demonstrate the viability of non-cognate ligand strategy for the development of a preventive HIV-1 vaccine. We introduced a new protein scaffold, Myomedin, and documented that highly complex combinatorial library can provide loop-randomized Myomedin variants able to mimic the 10E8 MPER epitope. Even if some of the developed MLA proteins do not possess an ideal mimicking surface involving key residues of Sites 1 and 2, it might be possible to develop MLA-derived variants with improved mimicking potential.
Supplementary Material
Acknowledgments
This work was supported by the Ministry of Education, Youth and Sport of the Czech Republic by OP RDE project CEREBIT No. CZ.02.1.01/0.0/0.0/16_025/0007397, by the Ministry of Health of the Czech Republic via the project of Czech Health Research Council No. 15-32198A, by Institutional Research Concept RVO: 86652036, Palacky University grant LF_UP_2021_015, and by European Regional Development Fund project BIOCEV No. CZ.1.05/1.1.00/02.0109. Authors acknowledged the access to instruments and infrastructure of the project OP RDE FIT No. CZ.02.1.01/0.0/0.0/15_003/0000495 and project CIISB4HEALTH No. CZ.02.1.01/0.0/0.0/16_013/0001776. We thank Linda Malá, Petra Kadlčáková and Josef Šulc for excellent experimental assistance.
Funding Statement
This work was supported by the Ministry of Education, Youth and Sport of the Czech Republic [CZ.02.1.01/0.0/0.0/16_025/0007397]; Institutional Research Concept [RVO: 86652036]; Ministry of Health of the Czech Republic [No. 15-32198A].
Disclosure statement
The authors have no financial conflicts of interest to declare.
Author contributions
P.M. and M.R conceived and designed the study. J.C. conceived structural designing, protein-protein-docking, performed bioinformatics analysis, in silico modeling and designed Myomedin library. H.P., J.Do. and J.Du. designed and performed structural work on myomesin domain. M. K. and L.V. designed collection of primers and adaptors, assembled, and tested the Myomedin DNA library. M.K., V.D.L. and M.M. performed large scale screening of Myomedin library and selection of MLA variants. H.P. performed protein stability analyses. P.K. and M.Kr. performed the immunological analyses. J.M., P.T.K., and M.R. performed immunization experiments. E.V. and L.R.K. engineered and isolated recombinant Env proteins and isolated plasmids for pseudoviruses preparation. P.M., M.R., and J.T. supervised the study. P.M, M.R. and J.C. wrote the manuscript with input from all authors.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].West AP Jr., Scharf L, Scheid JF, et al. Structural insights on the role of antibodies in HIV-1 vaccine and therapy. Cell. 2014. February 13;156(4):633–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Burton DR, Hangartner L.. Broadly neutralizing antibodies to HIV and their role in vaccine design. In: Littman DR, Yokoyama WM, editors. Annual review of immunology. Vol. 34. 2016. pp. 635–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Moore PL. The neutralizing antibody response to the HIV-1 env protein. Curr HIV Res. 2018;16(1):21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Schoofs T, Barnes CO, Suh-Toma N, et al. Broad and potent neutralizing antibodies recognize the silent face of the HIV envelope. Immunity. 2019. June 18;50(6):1513–1529 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Klasse PJ, LaBranche CC, Ketas TJ, et al. Sequential and simultaneous immunization of rabbits with HIV-1 envelope glycoprotein SOSIP.664 trimers from clades A, B and C. PLoS Pathog. 2016. September;12(9):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sok D, Burton DR. Recent progress in broadly neutralizing antibodies to HIV. Nat Immunol. 2018. November;19(11):1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lee JH, Andrabi R, Su CY, et al. A broadly neutralizing antibody targets the dynamic HIV envelope trimer apex via a long, rigidified, and anionic beta-hairpin structure. Immunity. 2017. April 18;46(4):690–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].McGuire AT, Hoot S, Dreyer AM, et al. Engineering HIV envelope protein to activate germline B cell receptors of broadly neutralizing anti-CD4 binding site antibodies. J Exp Med. 2013. April 08;210(4):655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Escolano A, Gristick HB, Abernathy ME, et al. Immunization expands B cells specific to HIV-1 V3 glycan in mice and macaques. Nature. 2019. June;570(7762):468–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Steichen JM, Lin YC, Havenar-Daughton C, et al. A generalized HIV vaccine design strategy for priming of broadly neutralizing antibody responses. Science. 2019. December 6;366(6470):6470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pejchal R, Doores KJ, Walker LM, et al. A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science. 2011. November 25;334(6059):1097–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Behrens A-J, Vasiljevic S, Pritchard LK, et al. Composition and antigenic effects of individual glycan sites of a trimeric HIV-1 envelope glycoprotein. Cell Rep. 2016. March 22;14(11):2695–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Escolano A, Steichen JM, Dosenovic P, et al. Sequential immunization elicits broadly neutralizing anti-HIV-1 antibodies in ig knockin mice. Cell. 2016. September 8;166(6):1445–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Steichen JM, Kulp DW, Tokatlian T, et al. HIV vaccine design to target germline precursors of glycan-dependent broadly neutralizing antibodies. Immunity. 2016. September 20;45(3):483–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bricault CA, Kovacs JM, Badamchi-Zadeh A, et al. Neutralizing antibody responses following long-term vaccination with HIV-1 Env gp140 in guinea pigs. J Virol. 2018. July 1;92(13):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Banerjee K, Michael E, Eggink D, et al. Occluding the mannose moieties on human immunodeficiency virus type 1 gp120 with griffithsin improves the antibody responses to both proteins in Mice. AIDS Res Hum Retroviruses. 2012. February;28(2):206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Banerjee K, Andjelic S, Klasse PJ, et al. Enzymatic removal of mannose moieties can increase the immune response to HIV-1 gp120 in vivo. Virology. 2009. Jun-Jul;389(1–2):108–121. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Raska M, Moldoveanu Z, Novak J, et al. Delivery of DNA HIV-1 vaccine to the liver induces high and long-lasting humoral immune responses. Vaccine. 2008. March 17;26(12):1541–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pugach P, Ozorowski G, Cupo A, et al. A native-like SOSIP.664 trimer based on an HIV-1 subtype B env Gene. J Virol. 2015. March;89(6):3380–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Raska M, Takahashi K, Czernekova L, et al. Glycosylation patterns of HIV-1 gp120 depend on the type of expressing cells and affect antibody recognition. J Biol Chem. 2010. July 2;285(27):20860–20869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kosztyu P, Kuchar M, Cerny J, et al. Proteins mimicking epitope of HIV-1 virus neutralizing antibody induce virus-neutralizing sera in mice. EBioMedicine. 2019. September;47:247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ahmad JN, Li J, Biedermannova L, et al. Novel high-affinity binders of human interferon gamma derived from albumin-binding domain of protein G. Proteins. 2012. March;80(3):774–789. [DOI] [PubMed] [Google Scholar]
- [23].Kuchar M, Vankova L, Petrokova H, et al. Human interleukin-23 receptor antagonists derived from an albumin-binding domain scaffold inhibit IL-23-dependent ex vivo expansion of IL-17-producing T-cells. Proteins. 2014. June;82(6):975–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mareckova L, Petrokova H, Osicka R, et al. Novel binders derived from an albumin-binding domain scaffold targeting human prostate secretory protein 94 (PSP94). Protein Cell. 2015. October;6(10):774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Klasse PJ. Non-cognate ligands of Procrustean paratopes as potential vaccine components. EBioMedicine. 2019. September;47:6–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nelson JD, Brunel FM, Jensen R, et al. An affinity-enhanced neutralizing antibody against the membrane-proximal external region of human immunodeficiency virus type 1 gp41 recognizes an epitope between those of 2F5 and 4E10. J Virol. 2007. April;81(8):4033–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kwong PD, Mascola JR, Nabel GJ. Broadly neutralizing antibodies and the search for an HIV-1 vaccine: the end of the beginning. Nat Rev Immunol. 2013. September;13(9):693–701. [DOI] [PubMed] [Google Scholar]
- [28].Flemming J, Wiesen L, Herschhorn A. Conformation-dependent interactions between HIV-1 envelope glycoproteins and broadly neutralizing antibodies. AIDS Res Hum Retroviruses. 2018. September;34(9):794–803. [DOI] [PubMed] [Google Scholar]
- [29].Wang Q, Finzi A, Sodroski J. The conformational states of the HIV-1 envelope glycoproteins. Trends Microbiol. 2020. August;28(8):655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Krizova L, Kuchar M, Petrokova H, et al. p19-targeted ABD-derived protein variants inhibit IL-23 binding and exert suppressive control over IL-23-stimulated expansion of primary human IL-17+ T-cells. Autoimmunity. 2017. March;50(2):102–113. [DOI] [PubMed] [Google Scholar]
- [31].Hlavnickova M, Kuchar M, Osicka R, et al. ABD-derived protein blockers of human IL-17 receptor A as non-IgG alternatives for modulation of IL-17-dependent pro-inflammatory axis. Int J Mol Sci. 2018. October 9;19(10):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Petrokova H, Masek J, Kuchar M, et al. Targeting human thrombus by liposomes modified with anti-fibrin protein binders. Pharmaceutics. 2019. December 2;11(12):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kabsch W. Xds. Acta Crystallogr., Sect D-Biol Crystallog. 2010. February;66(2):125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr., Sect D-Biol Crystallog. 2010. January;66(1):22–25. [DOI] [PubMed] [Google Scholar]
- [35].Pinotsis N, Chatziefthimiou SD, Berkemeier F, et al. Superhelical architecture of the myosin filament-linking protein myomesin with unusual elastic properties. PLoS Biol. 2012. February;10(2):e1001261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Emsley P, Lohkamp B, Scott WG, et al. Features and development of Coot. Acta Crystallogr., Sect D-Biol Crystallog. 2010. April;66(4):486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997. May 1;53(3):240–255. [DOI] [PubMed] [Google Scholar]
- [38].Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D. 2006. April;62(4):439–450. [DOI] [PubMed] [Google Scholar]
- [39].Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993. December 5;234(3):779–815. [DOI] [PubMed] [Google Scholar]
- [40].Irimia A, Serra AM, Sarkar A, et al. Lipid interactions and angle of approach to the HIV-1 viral membrane of broadly neutralizing antibody 10E8: insights for vaccine and therapeutic design. PLoS Pathog. 2017. February;13(2):e1006212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Huang J, Ofek G, Laub L, et al. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature. 2012. November 15;491(7424):406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kozakov D, Beglov D, Bohnuud T, et al. How good is automated protein docking? Proteins. 2013. December;81(12):2159–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kozakov D, Brenke R, Comeau SR, et al. PIPER: an FFT-based protein docking program with pairwise potentials. Proteins. 2006. November 1;65(2):392–406. [DOI] [PubMed] [Google Scholar]
- [44].Kilkenny C, Browne WJ, Cuthill IC, et al. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010. June 29;8(6):e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Montefiori DC. Evaluating neutralizing antibodies against HIV, SIV, and SHIV in luciferase reporter gene assays. Curr Protoc Immunol. 2005. January;12(12 11). DOI: 10.1002/0471142735.im1211s64. [DOI] [PubMed] [Google Scholar]
- [46].Schymkowitz J, Borg J, Stricher F, et al. The FoldX web server: an online force field. Nucleic Acids Res. 2005. July 1;33:W382–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].UniProt C. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019. January 8;47(D1):D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bricault CA, Yusim K, Seaman MS, et al. HIV-1 neutralizing antibody signatures and application to epitope-targeted vaccine design. Cell Host Microbe. 2019. January 9;25(1):59–72 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Harris AK, Bartesaghi A, Milne JL, et al. HIV-1 envelope glycoprotein trimers display open quaternary conformation when bound to the gp41 membrane-proximal external-region-directed broadly neutralizing antibody Z13e1. J Virol. 2013. June;87(12):7191–7196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Rathinakumar R, Dutta M, Zhu P, et al. Binding of anti-membrane-proximal gp41 monoclonal antibodies to CD4-liganded and -unliganded human immunodeficiency virus type 1 and simian immunodeficiency virus virions. J Virol. 2012. February;86(3):1820–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Binz HK, Amstutz P, Kohl A, et al. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat Biotechnol. 2004. May;22(5):575–582. [DOI] [PubMed] [Google Scholar]
- [52].Gebauer M, Skerra A. Engineered protein scaffolds as next-generation therapeutics. Annu Rev Pharmacol Toxicol. 2020. January;6(60):391–415. [DOI] [PubMed] [Google Scholar]
- [53].Steemson JD, Baake M, Rakonjac J, et al. Tracking molecular recognition at the atomic level with a new protein scaffold based on the OB-fold. PLoS One. 2014;9(1):e86050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Koide A, Bailey CW, Huang X, et al. The fibronectin type III domain as a scaffold for novel binding proteins. J Mol Biol. 1998. December 11;284(4):1141–1151. [DOI] [PubMed] [Google Scholar]
- [55].Nord K, Gunneriusson E, Ringdahl J, et al. Binding proteins selected from combinatorial libraries of an alpha-helical bacterial receptor domain. Nat Biotechnol. 1997. August;15(8):772–777. [DOI] [PubMed] [Google Scholar]
- [56].Bertschinger J, Grabulovski D, Neri D. Selection of single domain binding proteins by covalent DNA display. Protein Eng Des Sel. 2007. February;20(2):57–68. [DOI] [PubMed] [Google Scholar]
- [57].Liu H, Su X, Si L, et al. The development of HIV vaccines targeting gp41 membrane-proximal external region (MPER): challenges and prospects. Protein Cell. 2018. July;9(7):596–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zwick MB, Labrijn AF, Wang M, et al. Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. J Virol. 2001. November;75(22):10892–10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Williams LD, Ofek G, Schatzle S, et al. Potent and broad HIV-neutralizing antibodies in memory B cells and plasma. Sci Immunol. 2017. January 27;2(7):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Muster T, Steindl F, Purtscher M, et al. A conserved neutralizing epitope on Gp41 of human-immunodeficiency-virus type-1. J Virol. 1993. November;67(11):6642–6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pinto D, Fenwick C, Caillat C, et al. Structural basis for broad HIV-1 neutralization by the MPER-specific human broadly neutralizing antibody LN01. Cell Host Microbe. 2019. November 13;26(5):623–637 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Banerjee S, Shi H, Banasik M, et al. Evaluation of a novel multi-immunogen vaccine strategy for targeting 4E10/10E8 neutralizing epitopes on HIV-1 gp41 membrane proximal external region. Virology. 2017. May;505:113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Banerjee S, Shi H, Habte HH, et al. Modulating immunogenic properties of HIV-1 gp41 membrane-proximal external region by destabilizing six-helix bundle structure. Virology. 2016. March;490:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Habte HH, Banerjee S, Shi H, et al. Immunogenic properties of a trimeric gp41-based immunogen containing an exposed membrane-proximal external region. Virology. 2015. December;486:187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kong R, Louder MK, Wagh K, et al. Improving neutralization potency and breadth by combining broadly reactive HIV-1 antibodies targeting major neutralization epitopes. J Virol. 2015. March;89(5):2659–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Mendoza P, Gruell H, Nogueira L, et al. Combination therapy with anti-HIV-1 antibodies maintains viral suppression. Nature. 2018. September 27;561(7724):479-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wang Q, Michailidis E, Yu Y, et al. A combination of human broadly neutralizing antibodies against hepatitis B virus HBsAg with distinct epitopes suppresses escape mutations. Cell Host Microbe. 2020. June 3;28(2):335–349.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Madeira F, Park YM, Lee J, et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019. July 2;47(W1):W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Letunic I, Bork P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019. July 2;47(W1):W256–W259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.