

Abstract
There is a growing awareness of the underlying power of catalytic reactions in water that is not limited to innate sustainability alone. Some Type III reactions are catalytically accelerated without dissolution of reactants and are occasionally highly selective, as shown by comparison with the corresponding reactions run in organic solvents or under solvent-free conditions. Such catalysts are highly diversified, including hydrophilic, lipophilic, and even solid catalysts. In this Outlook, we highlight the impressive characteristics of illustrative catalysis that is exerted despite the immiscibility of the substrates and reveal the intrinsic benefits of these enigmatic reactions for synthetic organic chemistry, albeit with many details remaining unclear. We hope that this brief introduction to the expanding field of synthetic organic “aquachemistry” will inspire organic chemists to use the platform to invent new transformations.
Short abstract
The counterintuitive and underexplored catalysis that is exerted at the reactant(s)−water interface is discussed. Illustrative examples are presented to highlight the benefits of the approach to synthetic organic chemistry.
Introduction
Modern synthetic organic chemistry relies heavily on the use of vast amounts of organic solvents to perform many aspects of both laboratory synthesis and industrial production. Solvents are used to mediate heat dissipation and to bring the reactants together in a uniform phase to enhance the rate of those chemical reactions that are governed by the collision frequency between reactants. This implicit view that the reactants should be dissolved, however, perpetuates a belief that a chemical reaction does not take place unless the reactants are in solution, a view that can be traced back to a quote ascribed to Aristotle.1 In recent years, growing concerns over the environmental impact resulting from the accumulated use of solvents in chemical production have driven the streamlining of synthetic methodologies in line with the principle of green sustainability. Driven in part by these external stimuli, the use of water as a reaction medium without the aid of organic cosolvents has emerged as a preferred remedy2 in addition to solvent-free mechanochemical techniques that have recently seen a resurgent,3 albeit seldom adopted, approach in organic synthesis. Both approaches, although old-fashioned, transcend the traditional paradigm of “solution chemistry” and obviate the need for bulk dissolution of reactants. The fledgling field of organic chemistry was intimately bound up with the use of water as a reaction medium; venerable examples include Wöhler’s urea synthesis in 1828,4 self-condensation of acetone in 1838,5 Kolbe electrolysis in 1847,6 and even Robinson’s landmark synthesis of tropinone in 1917.7 The historical links between organic chemistry and aqueous environments, however, lapsed into obscurity, largely in the context of flourishing organometallic chemistry that typically forces us to operate in rigorously anhydrous environments. Not until the rediscovery of rate acceleration in water by Breslow,8 subsequently termed the “on-water” effect,9 did organic chemists dare to again manipulate chemical reactions in the presence of water. Even today, the apodictic benefits of embracing sustainability notwithstanding, organic chemists are generally reluctant to harness water as a reaction medium.
A common approach that is used to sidestep an immiscibility issue is appending surfactants to partition lipophilic organic compounds. It was reported that the amount of organic waste that is produced can be reduced tremendously by the use of 10–2 to 10–4 M surfactant solutions instead of 100% organic solvents.10 Extensive efforts to realize the maximum effect of surfactants even under dilute conditions (especially catalytic amounts) resulted in the advent of designed nonionic surfactants11 and surfactant-combined catalysts such as metal dodecylsulfates12 or amphiphilic ligands.13 In parallel, the use of dilute solutions of these surfactants has enabled multifarious catalytic reactions including asymmetric synthesis to be performed without the assistance of organic solvents.
Apart from surfactant-based approaches, surfactant-free catalytic reactions in water, “aquachemistry”, have come to be recognized as an offbeat yet useful tool in synthetic organic chemistry. One may perceive this as a biomimetic regime because water is a peerless medium in which nature furnishes complex molecules. The biosynthetic process is critically dependent on water: it regulates reaction kinetics, selectivity, substrate binding, and conformation. In addition, in vivo reactions are often not reproducible based on conventional organic chemistry that has been developed in organic solvents, which does encourage us to explore this straightforward, albeit counterintuitive, approach. For example, the reactants of in vivo reactions are not necessarily water-soluble, for example, 25-hydroxylation of practically water-insoluble14 cholecalciferol (vitamin D3) in human liver (Scheme 1) and enzymatic cellulose hydrolysis.
Scheme 1. Example of an Enzymatic Reaction of a Water-Insoluble Compound.

For all that, one may be bewildered by the absence of reliable methods to leverage this counterintuitive chemistry in organic synthesis, especially when one would like to transform water-immiscible reactants. To stimulate discussions, such surfactant-free catalytic reactions using water as a reaction medium can be roughly classified into three types (Figure 1).15 The physicochemical properties of catalysts determine whether they are Type IIIa, IIIb, or IIIc, pushing aside supramolecular catalysts such as enzymes, abzymes, capsules, cyclodextrins, and hydrogels. Type IIIa reactions involve water-soluble catalysts, IIIb reactions involve lipophilic catalysts that are miscible with reactants, and IIIc reactions correspond to reactions in which neither reactants nor catalysts are water-soluble and are immiscible with each other. Above all, Type IIIc reactions are highly counterintuitive, and it is difficult to anticipate positive outcomes because the reactions take place where neither reactants nor catalysts are miscible with water.
Figure 1.

Schematic images of Type III reactions with catalysts (no cosolvents, no amphiphilic molecules). Yellow shapes represent droplets consisting of reactant(s); capital C indicates a soluble catalyst (blue: soluble in water, red: lipophilic and soluble in suspended droplets), and purple hexagons indicate an insoluble solid catalyst.
One feasible advantage of this chemistry is rate acceleration as is often the case with catalyst-free reactions. Although catalysts often alter the mechanism of reactions in substantial ways, the catalytic activation of reactants is compatible with accelerative effects at the surface of the water droplets. Moreover, Type IIIa–c reactions sometimes allow unusual selectivity that differs from that in organic solvents. In this context, asymmetric synthesis without the use of cosolvents or surfactants is particularly challenging because the precise and subtle interactions essential for stereodifferentiation may be disrupted by water acting as both a hydrogen-bond donor and acceptor; existing methods that address these issues are thus still few. An early form of a catalytic asymmetric Type III reaction emerged in the early 2000s16,17 with a catalyst that was likely dissolved in suspended droplets (Type IIIb).18 Although Type IIIb reactions catalyzed by proline derivatives were subsequently reported in 2006,19 these examples are not regarded as following the “on-water” mechanism.15 This is because the effect of water is concluded to be the suppression of the formation of key intermediates within the cycle as well as oxazolidinones,20 as is often quoted in various discussions.21
Type III reactions may be characterized by an expedient isolation of organic compounds pursuant to green and sustainable chemistry. The postreaction mixtures can separate into two or three phases, thereby avoiding the need for extractive workup. In particular, Type IIIc reactions are ideal because centrifugation of the postreaction mixtures would allow their separation into water, an organic phase including products, and a solid catalyst (vide infra). On the other hand, one may be concerned that the innate heterogeneity of Type III reactions is a deterrent for applications at manufacture scale. Although there has been indeed no application to an industrial process, Novaltis’s adoption of a catalyst-free “on-water” process en route to the anticonvulsant rufinamide22 and many successes of gram-scale Type III reactions using catalysts including unreported results will lower the barrier for process chemists to embrace “aquachemical” approaches. In addition, many chemical mixing tools including the venturi tube, ball miller, and microfluidic devices may help drive future success. When scaled up, “aquachemical” exothermic processes become safer and more selective because of an excellent heat capacity of water.
We herein discuss illustrative examples to showcase their intrinsic values in synthetic organic chemistry that complement techniques using organic solvents. We also summarize the challenges involved in better understanding the seemingly capricious catalysis that is not exerted in analogous homogeneous reactions performed in organic solvents.
Vignettes of Synthetic Organic “Aquachemistry”
It is important that both reactants and catalyst are embedded in the surface of the water droplets for efficient Type III reactions. The involvement of interfacial water molecules in the reaction mechanism is anticipated to underlie the water-induced enhancement of the reactivity and to determine selectivity control.23 Indeed, the pronounced role of interfacial water molecules is often seen in enzymatic catalysis.
Hydrogen-Bonding and Water-Induced Hydrophobic Amplification
The water-mediated hydrogen-bonding network is found in thymidylate synthase to reduce the energy necessary to reach the intermediate.24 Syrén and co-workers adopted an enzyme engineering strategy for accelerated catalysis in which water bridges between the productive transition state and the protein backbone were redesigned to stabilize the transition state.25 Although noncovalent hydrogen-bond organocatalysis is a priori not amenable to Type III reactions, a noteworthy example of epoxide hydrolases that detoxify living cells by epoxide hydration supports the viability of this catalysis. In the transition-state model, two tyrosine residues act through hydrogen bonds to activate the epoxide, with a water molecule activated by the nearby histidine and aspartic acid residues. The role of the two tyrosine residues was confirmed using the double mutant, which showed no detectable catalytic activity, and by inhibition studies with ureas.26
The concept of highly enantioselective noncovalent organocatalysis was reported by Song and co-workers (Table 1),27 wherein the use of brine provided a remarkable rate acceleration over organic solvents while retaining high levels of enantioselectivity. A significant decline in the level of conversion in saturated LiClO4 solution is the same observation as catalyst-free Type III reactions.28 Therefore, the authors attribute the beneficial role of water to the hydrophobic hydration effect. The benefits of adopting “aquachemistry” are evidenced by the successful 1,4-addition of dithiomalonate with less reactive β,β-disubstituted nitroalkenes with no conversion in organic solvents.29 Notably, these Type IIIb protocols are amenable to multigram-scale synthesis, and the catalyst can be quantitatively recovered as a solid by filtration from postreaction mixtures after addition of methylcyclohexane.27
Table 1. Water-Accelerated Type IIIb Noncovalent Hydrogen-Bond Catalysis.

| entry | medium | conv (%)a |
|---|---|---|
| 1 | brine | >99 (92)b |
| 2 | LiClO4 in H2O (sat.) | 14 |
| 3 | MeOH | 34 |
| 4 | CH2Cl2 | 52 |
| 5 | THF | 21 |
| 6 | toluene | 17 |
| 7 | CH3CN, 1,4-dioxane | <10 |
Calculated based on crude 1H NMR analysis.
Enantiomeric excess (% ee).
[Cp*MCl2]2-Catalyzed C(sp2)–H Bond Activation
The contribution of water to C(sp2)–H bond activation with the assistance of carboxylates has been underpinned by several Type III metal-catalyzed cross-coupling reactions,30 in which water facilitates the production of both coordinatively unsaturated metal species and a water-bound metal species.31 The involvement of interfacial water molecules in a concerted metalation–deprotonation (CMD) mechanism was proposed for a rhodium-catalyzed Type IIIb reaction (Table 2).32 The authors confirmed that a quantitative amount of rhodacycle A was formed when [Cp*RhCl2]2 was mixed with 2-phenylpyridine in water at an elevated temperature and that the rhodacycle A was able to catalyze the reaction (entry 8) as the active species. Given that general rhodacycle formation via C–H activation commences with the cleavage of dimeric [Cp*RhCl2]2 to form a reactive monomeric [Cp*RhCl(OAc)] in the presence of NaOAc,33 it is considered that water substitutes for the acetate ion. The negative results observed in the presence of catalytic amounts of water under homogeneous conditions (entries 5 and 6) strongly suggest the involvement of interfacial water molecules, as depicted in transition state B. Less than 50% conversion even with a longer reaction time in DCE (entry 9) suggests that half the amount of 2-phenylpyridine may act as a base. Within the catalytic cycles, rhodacycle A can undergo hydration to form an aqua complex or monohydroxide, leading to a water-promoted mechanism involving intermolecular hydrogen bonding.
Table 2. Water-Involved Type IIIb Rhodacycle Formation/N-Boc Amidation in Water.

| entry | medium | conv (%)a |
|---|---|---|
| 1 | H2O | 88 |
| 2 | DCE, PhMe, CH3CN, THF, DMSO, iPrOH | <5 |
| 3 | hexane | 8 |
| 4 | DMF | 11 |
| 5 | DCE/H2O (0.2, 1.0, or 10 equiv) | <5 |
| 6 | iPrOH/H2O (10 equiv) | 8 |
| 7 | –(neat) | 25 (16 h) |
| 8b | H2O | 83 |
| 9b | DCE | 46 (48)c |
Calculated based on crude 1H NMR analysis.
Rhodacycle A was used instead of [Cp*RhCl2]2 as a catalyst.
For 12 h.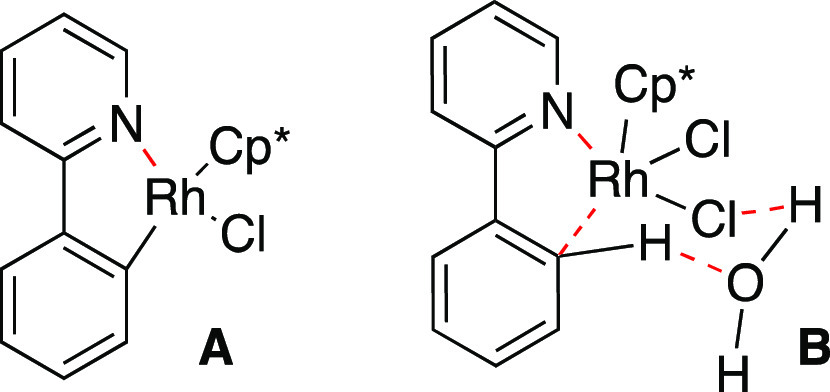
Rare water-accelerated iridium(III) catalysis for aldehyde C–H activation with the assistance of carboxylic acid was reported. The annulation between salicylaldehyde and α-diazoester proceeded efficiently in water, allowing access to chromane synthesis (Table 3).34 Incubation of the catalyst with salicylaldehyde in D2O resulted in around 17% deuterium exchange, indicating the reversibility of aldehyde C–H activation. The use of a tert-butyl diazoester resulted in a product switch induced by water and led to one-pot annulation–decarboxylation (entries 6 and 7). The reaction can be carried out on a gram scale.
Table 3. Water-Promoted Type IIIb Annulation of Salicylaldehyde with α-Diazo Carbonyl Compound.

| entry | R | medium | yield (%) |
|---|---|---|---|
| 1 | Et | H2O | 90 (75)a |
| 2 | Et | MeOH | (68)a |
| 3 | Et | CHCl3 | (40)a |
| 4 | Et | DCE | (70)a |
| 5 | Et | TCE | (55)a |
| 6b | tBu | H2O | 86c |
| 7b | tBu | DCE | 53 |
AcOH was added instead of PivOH.
100 °C, 8 h.
A chromanone was obtained.
Metal-Bound Water Molecules
Metal-bound water molecules form much stronger hydrogen bonds than bulk water molecules,35 thereby participating in intramolecular hydrogen bonds within the first hydration shell36 and sometimes conferring a pronounced Brønsted acidity37 that governs the aqua-hydroxo equilibrium. The resulting hydroxo metal complexes are expected to be promising Lewis acid/Brønsted base hybrid catalysts for bond-forming reactions. Because they have been infrequently found in catalyst structures, their catalytic versatility may have been overlooked in organic synthesis. Considering that calculations revealed that the surrounding water molecules would supply hydrogen bonds to stabilize the hydrolyzed metal cations in water,38 the polarization of adjacent cluster water molecules would enhance the stability of metal cations in M···OH species through hydrogen bonding. A tantalizing catalysis of redox-inert hafnium(IV) was found for the oxy-functionalization of active methylene compounds in water (Table 4).39 During the reaction, chlorine dioxide, a long-lived paramagnetic gaseous free radical, was generated, and it is considered to be a bona fide oxidant of this reaction. The reaction using TfOH instead of Hf(OTf)4 suffered from low levels of conversion (<2%), suggesting a distinction between the role of hafnium(IV) and that of a Brønsted acid. The significant retardation of the reaction in both ethanol and aqueous acetonitrile, which can both solubilize sodium chlorite (entries 2 and 3), indicates the importance of water. Given that the structure of anhydrous Hf(OTf)4 was reported to be built up of tetrameric [Hf4(OH)8(OTf)8],40 it may adopt an exceptionally kinetically stable octahydroxohexadecaaquatetrahafnium (hafnyl) ion [Hf4(OH)8(OH2)16]8+ in aqueous media. The presumable involvement of tetrameric hafnyl implies the pivotal role of hydroxo bridges or metal-bound water molecules in the activation of chlorine dioxide.
Table 4. Water-Promoted Hafnium(IV)-Catalyzed Oxyfunctionalization of Active Methylene Compounds in Water.

| entry | medium | yield (%)a |
|---|---|---|
| 1 | H2O | 89 |
| 2 | EtOH | 49 (48 h) |
| 3 | CH3CN/H2O = 2/1 | 52 |
Calculated based on crude 1H NMR analysis.
These examples indicate that although water is involved in the mechanism, it is not involved in the selectivity determination. The hydroxo scandium complex can be considered as an active species in thia-Michael addition in the presence of pyridine (Table 5).41 Notably, the reaction run in organic solvents or aqueous cosolvents suffered from decreased enantioselectivity. Considering the hydrolysis constant of Sc3+ (pKh = 4.3), 4-fold amounts of pyridine were sufficient to form Sc(OH)2+ along with polymeric hydroxy-bridged Sc2(OH)24+. The observation of a positive nonlinear effect also supports the formation of a heterooligomeric complex. The catalyst loading can be reduced to 1 mol % without loss of enantioselectivity.
Table 5. Influence of Water in Enantioselective Thia-Michael Addition.

| entry | medium | yield (%) | ee (%)a |
|---|---|---|---|
| 1 | H2O | 84 (91)b | 91 (91)b |
| 2 | CH2Cl2 | 93 | 28 |
| 3 | THF | 91 | 31 |
| 4 | EtOH | 88 | 63 |
| 5 | PhMe | 82 | 75 |
| 6 | THF/H2O = 9/1 | 54 | 64 |
| 7 | EtOH/H2O = 9/1 | 90 | 59 |
Determined using HPLC analysis.
At 1 mol % catalyst loading.
The immense role played by water in determining enantiocontrol was also revealed for palladium-catalyzed indole C–H functionalization via the putative σ-indolylpalladium intermediate (Table 6).42 Notably, substrates reacted in a highly enantioselective manner in water despite being immiscible (Type IIIa reaction). The reaction suffered from a significant reduction of enantioselectivity when run either in organic solvents or under solvent-free conditions. The incubation of the catalyst with indole in water plays a prominent role in achieving a high level of stereocontrol. Incubation under anhydrous conditions led to a significant decrease in enantioselectivity (61% ee) even though the reaction was performed in water. The rapid, quantitative, and exclusive deuterium exchange at the C3 position of indole in D2O and contrasting lack of a corresponding D incorporation in organic solvents denote the efficient electrophilic palladation that is triggered by the aqua complex. The feasible electrophilic activation of a carbonyl or soft π-bond would provoke the C3-alkylation of indole in aprotic solvents without forming the σ-indolylpalladium intermediate. It is noted that the addition of even small amounts of organic solvents disabled stereoselective catalysis because the aqua complex preferentially undergoes water exchange reaction with these organic solvents with consequent loss of performance.
Table 6. Water-Controlled Enantioselectivity Switch in Type IIIa Indole C–H Functionalizationa.

| entry | medium | yield (%) | ee (%)b |
|---|---|---|---|
| 1 | H2O | 97 | 90 |
| 2 | nhexane | 95 | 31 |
| 3 | CH2Cl2 | 80 | 34 |
| 4 | toluene | 94 | 16 |
| 5 | THF | 92 | 1 |
| 6 | Et2O | 72 | 0 |
| 7 | EtOAc | 77 | 1 |
| 8 | DMSO | NR | |
| 9 | acetone | 44 | 1 |
| 10 | MeCN | 90 | 1 |
| 11 | MeOH | 91 | 2 |
| 12 | –(neat) | 95 | 12 |
NR = no reaction.
Determined using HPLC analysis.
Type IIIc Reactions
Although counterintuitive and difficult to predict, water upregulates catalytic activity in some Type IIIc reactions in which the substrates, catalyst, and water are immiscible with one another. The pivotal role of water is clear for the enantioselective β-borylation of chalcone (Table 7).43 Notably, in this case, all the components involved are solid, and all are practically insoluble in water. The reaction, therefore, proceeds heterogeneously on the surface of chirally modified Cu(OH)2 (entry 1), whereas the reaction did not proceed at all in typical organic solvents in which both substrates are soluble (entries 2–8). The reaction proceeded sluggishly in alcohols with a quite low enantioselectivity (entries 9 and 10) and failed to give the desired adduct under neat (solvent-free) conditions (entry 11). In addition, the inclusion of aqueous cosolvents resulted in a significant decrease in enantioselectivity (entries 12 and 13). A simple filtration after the first run confirmed the heterogeneity of the active form of the catalyst; the Cu content in the filtrate was less than the detection limit of the ICP equipment (5 ppb), and the filtrate did not show any catalytic activity. Extensive investigations identified water-soluble Cu(OAc)2 as an alternative to water-insoluble Cu(OH)2, affording the product with slightly higher enantioselectivity. Notable is the exceptionally high turnover with the Cu(OH)2 catalyst, which reaches up to 43 200 h–1 at 0.005 mol % catalyst loading in a gram-scale reaction, the highest value in β-borylation reported to date. In addition, the Type IIIc conditions were applicable to β-borylation of water-sensitive α,β-unsaturated imines with perfect enantioselectivity.44 Unusual Type IIIc catalysis using Cu(0) powder for enantioselective β-borylation is also noteworthy.45
Table 7. Water-Enabled Type IIIc Asymmetric β-Borylationa.

| entry | medium | yield (%) | ee (%)b |
|---|---|---|---|
| 1 | H2O | 83 | 81 |
| 2 | toluene | NR | |
| 3 | CH2Cl2 | NR | |
| 4 | DMF | NR | |
| 5 | DMSO | NR | |
| 6 | THF | NR | |
| 7 | Et2O | NR | |
| 8 | MeCN | NR | |
| 9 | EtOH | 1 | |
| 10 | MeOH | 17 | 29 |
| 11 | –(neat) | NR | |
| 12 | H2O/THF = 1/4 | 77 | 79 |
| 13 | H2O/MeOH = 1/1 | 82 | 49 |
NR = no reaction.
Determined using HPLC analysis.
Because Cu(OH)2 possesses a layered structure where the hydroxide ligands in the plane are either doubly or triply bridging, a hydroxide-bridged multicopper complex is presumed as an active species. The mode of Type IIIc reactions based upon this architecture allowed unprecedented 1,6-borylation in a highly enantioselective manner (Scheme 2).46 Considering that the copper is expected to activate the carbonyl group of cyclic dienones and to mount boron on their γ-position with high enantioselectivity, a monomeric copper complex is sterically not feasible. Indeed, cyclic dienones underwent a remarkable switch of regioselectivity from 1,6- to common 1,4-borylation in the presence of water-soluble monomeric Cu(OAc)2.
Scheme 2. Uncommon Asymmetric 1,6-Borylation under Type IIIc Conditions.

Enantioselective β-silylation was enabled by acicular purplish crystals comprising Cu(acac)2 and a chiral 2,2′-bipyridine ligand under Type IIIc conditions (Table 8).47 In striking contrast to the Type IIIc reaction, the use of organic solvents, alcohols, and even aqueous organic cosolvents gave inferior results in both yield and enantioselectivity. Notably, the reaction did not proceed at all in most cases including solvent-free conditions. The centrifugation of the reaction tube after the first run allowed separation of the reaction mixture into aqueous, organic, and solid phases, which delineates the technical advantage of Type IIIc reactions over conventional chemistry. A sharp increase in enantioselectivity correlated with a decrease in solubility of the catalyst is insightful (entries 10–12). The success of this chemistry may be built upon higher aggregation states associated with the insoluble catalyst. The advantage of the reactions lies in the application to enantioselective β-silylation of β-nitrostyrenes that remains still unrivaled (Scheme 3).
Table 8. Water-Enabled Type IIIc Asymmetric β-Silylationa.

| entry | medium | yield (%) | ee (%)b |
|---|---|---|---|
| 1 | H2O | 92 | 93 |
| 2 | toluene | NR | |
| 3 | CH2Cl2 | NR | |
| 4 | DMSO | NR | |
| 5 | THF | NR | |
| 6 | Et2O | NR | |
| 7 | EtOH | 4 | 0 |
| 8 | MeOH | 22 | 31 |
| 9 | –(neat) | NR | |
| 10 | H2O/THF = 1:4 | 73 | 6 |
| 11 | H2O/THF = 1:1 | 75 | 6 |
| 12 | H2O/THF = 4:1 | 88 | 72 |
| 13 | H2O/MeOH = 1:4 | 76 | 37 |
NR = no reaction.
Determined using HPLC analysis.
Scheme 3. Type IIIc Asymmetric β-Silylation of β-Nitrostyrene.

New Frontiers and Challenges in Synthetic Organic “Aquachemistry”
Since the 18th century, the position of water in organic chemistry has changed too vertiginously. Marginalized once with organometallic chemistry flourishing, water has recently been spotlighted as a green reaction medium to realize clean chemical processes with an aid of surfactants or cosolvents. The early observation of “on-water” acceleration united synthetic chemists with aqueous media, and surfactant-based approaches are prospering to fulfill our synthetic needs. Aside from this, catalytic reactions in water that rely on neither cosolvents nor surfactants have been sporadically reported especially in the past decade. As amazing as it is, the “aquachemistry”49 enables us to do more with less. As evidenced by chirality amplification in confined water cages,48 water-induced hydrophobic amplification may result in enhanced enantioselectivity. Assembled with hydrogen-bonding catalysis, the hydrophobic hydration effect may provide new strategies to find out new catalytic reactions. Metal-bound water molecules form much stronger hydrogen bonds than bulk water molecules and may confer a pronounced Brønsted acidity. Corresponding hydroxo metal complexes formed after deprotonation may serve as Lewis acid/Brønsted base hybrids. They prove their superior worth in reactions involving CMD, radical activation, and σ-metathesis. Although the role of water remains elusive in many aspects, the examples outlined herein represent a broad perspective of synthetic organic “aquachemistry” that may ameliorate some problems that have plagued researchers so far or allow the invention of new reactions.
The pluripotent power of synthetic organic “aquachemistry” can be further elaborated. For instance, in organic electrochemistry, aqueous electrolytes have the advantages of high ionic conductivity and noncorrosiveness compared with organic electrolytes, in addition to the clear general benefits of using water. Representative electrochemical Type III reactions include Kolbe electrolysis6 and Zn-mediated Barbier-type electrochemical allylation.50 Ackermann and co-workers reported an illuminating Type IIIa reaction under electrochemical conditions in a simple undivided cell with a platinum cathode and a reticulated vitreous carbon (RVC) anode.51 Although the oxidation potential of Co(OAc)2 is lower than that of the starting benzamide, in methanol competitive C–H oxygenation takes place. Unlike catalyst-free Type III reactions, the combination between synthetic organic electro- and aquachemistry is still in its infancy. Despite its narrow potential window, electrochemical Type III reactions using catalysts have the potential to enable transformations that exhibit the unique characteristics of both electrochemistry and “aquachemistry.”
Table 9. Water-Promoted Type IIIa Electrochemical Alkyne Annulation.

| entry | medium | yield (%) |
|---|---|---|
| 1 | H2O | 64 |
| 2 | HFIP | 19 |
| 3 | TFE | 50 |
| 4 | DMSO | 31 |
| 5 | MeOH | 64 (17)a |
The yield of the C–H oxygenated product.
By and large, the further development of synthetic organic “aquachemistry” requires that entire data sets need to be examined. In-depth mechanistic and kinetic studies are imperative to elucidate the role of interfacial water molecules in catalytically accelerated Type III reactions and to theorize synthetic organic “aquachemistry”, a field that still has a long way to go. The main reason for the lack of kinetic data in these reactions stems from the technical difficulties involved in establishing reproducible control of overall interfacial areas, surface to volume ratios, and droplet sizes in the reaction mixtures. Given that the physicochemical properties of droplets are highly dependent upon the nature of the components, including viscosity and melting point, they need to be determined as a time-dependent dynamic parameter. On a laboratory scale, these parameters are also governed by both the stirring method and the size of the reaction flask. Recent progress has leveraged the development of interface-sensitive spectroscopic techniques. Adoption of the microfluidic technique enabled the influence of the water surface on catalyst-free Type III reactions to be quantified52 and was even used to analyze the effect of droplet size on the enantioselectivity in a cinchonine-catalyzed Type IIIb reaction.48 Nevertheless, a biphasic system is required to generate a precisely defined monodisperse water phase. Although electrospray ionization techniques have also been used in kinetic studies of catalyst-free Type III reactions,53 microdroplet chemistry is distinct from Type III reactions.
Direct analysis in real time mass spectrometry (DART-MS) methods with isotopically labeled indicators has emerged recently as a complementary way to analyze catalytically accelerated Type III reactions directly and semiquantitatively.54 DART-MS is based on atmospheric-pressure, soft, and robust ionization pathways, thereby allowing versatile applications that are not possible with electrospray ionization. The use of isotopically labeled indicator can significantly improve the reproducibility of the measurements by compensating for signal fluctuation and matrix effect that is usually ineluctable for ambient ionization techniques.55 Nevertheless, this analytical method presupposes an even distribution of the components in the reaction mixture, including isotope effects on the diffusion process, thereby suffering from poor reproducibility for catalytically accelerated Type III reactions, especially when the starting material or product is solid. Hence, the development of a quantitative analysis technique that does not need an aliquot of the sample is coveted to deepen understanding of “aquachemistry.”
Quantum mechanics/molecular mechanics (QM/MM) approaches are often used to understand catalyst-free Type III reactions, and a few examples of organocatalytic reactions have been reported.56 The difficulty of calculating catalytic reactions lies in the fact that the structural fluctuation of a catalyst and its surroundings in aqueous environments, because of hydration, significantly multiplies the number of transition states en route to the desired product. Although the artificial force-induced reaction (AFIR) method enabled the challenging systematic sampling of transition states to determine the stereochemical information on the reactions performed in aqueous media,57 there has been no QM study on catalytically accelerated Type III reactions at the interface. This is because it is too computationally expensive to calculate reaction pathways involving interfacial water molecules. Thus, theoretical advances that significantly reduce the calculation costs are required.
In sharp contrast to conventional organic reactions in which the reactants are in solution, the bizarre nature of catalytically accelerated Type III reactions has so far thwarted attempts to completely elucidate their details using existing theory and techniques. Nevertheless, we believe that the very elusive nature of the underlying processes that are inherent to this chemistry belies the great potential for reaction invention and the expansion of organic synthesis in new, vibrant, and creative directions.
Acknowledgments
This work was supported by a Grant-in-Aid for Science Research (JP15H05698 to S.K. and JP19H05288 and JP20K15272 to T.K.) from the Japan Society for the Promotion of Science (JSPS).
The authors declare no competing financial interest.
References
- “The substances do not react unless fluid or if dissolved”, quoted by Aristotle (384–322 BCE).
- a Cortes-Clerget M.; Yu J.; Kincaid J. R. A.; Walde P.; Gallou F.; Lipshutz B. H. Water as the reaction medium in organic chemistry: from our worst enemy to our best friend. Chem. Sci. 2021, 12, 4237. 10.1039/D0SC06000C. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhou F.; Hearne Z.; Li C.-J. Water–the greenest solvent overall. Curr. Opin. Green Sustain. Chem. 2019, 18, 118–123. 10.1016/j.cogsc.2019.05.004. [DOI] [Google Scholar]; c Kitanosono T.; Masuda K.; Xu P.; Kobayashi S. Catalytic organic reactions in water toward sustainable society. Chem. Rev. 2018, 118, 679–746. 10.1021/acs.chemrev.7b00417. [DOI] [PubMed] [Google Scholar]; and references cited therein.
- Recent authoritative reviews, see:; a Andersen J.; Mack J. Mechanochemistry and organic synthesis: from mystical to practical. Green Chem. 2018, 20, 1435–1443. 10.1039/C7GC03797J. [DOI] [Google Scholar]; b Tan D.; Friščić T. Mechanochemistry for organic chemists: an update. Eur. J. Org. Chem. 2018, 2018, 18–33. 10.1002/ejoc.201700961. [DOI] [Google Scholar]; c Do J.-L.; Friščić T. Mechanochemistry: a force of synthesis. ACS Cent. Sci. 2017, 3, 13–19. 10.1021/acscentsci.6b00277. [DOI] [PMC free article] [PubMed] [Google Scholar]; and references cited therein.
- Wöhler F. Ueber künstliche Bildung des Harnstoffs. Ann. Phys. 1828, 87, 253–256. 10.1002/andp.18280870206. [DOI] [Google Scholar]
- a Kane R. Ueber eine aus dem Essiggeist entspringende Reihe von Verbindungen. Ann. Phys. 1838, 120, 473–494. 10.1002/andp.18381200711. [DOI] [Google Scholar]; b Kane R. Ueber den Essiggeist und einige davon abgeleitete Verbindungen. J. Prakt. Chem. 1838, 15, 129–155. 10.1002/prac.18380150112. [DOI] [Google Scholar]
- Kolbe J. Beobachtungen über die oxydirende Wirkung des Sauerstoffs, wenn derselbe mit Hülfe einer elektrischen Säule entwickelt wird. J. Prakt. Chem. 1847, 41, 137–139. 10.1002/prac.18470410118. [DOI] [Google Scholar]
- Robinson R. LXIII. A synthesis of tropinone. J. Chem. Soc., Trans. 1917, 111, 762–768. 10.1039/CT9171100762. [DOI] [Google Scholar]
- Rideout D. C.; Breslow R. Hydrophobic acceleration of Diels-Alder reactions. J. Am. Chem. Soc. 1980, 102, 7816–7817. 10.1021/ja00546a048. [DOI] [Google Scholar]
- Narayan S.; Muldoon J.; Finn M. G.; Fokin V. V.; Kolb H. C.; Sharpless K. B. “On water”: unique reactivity of organic compounds in aqueous suspension. Angew. Chem., Int. Ed. 2005, 44, 3275–3279. 10.1002/anie.200462883. [DOI] [PubMed] [Google Scholar]
- Pharr D. Y. Green analytical chemistry–the use of surfactants as a replacement of organic solvents in spectroscopy. Phys. Sci. Rev. 2017, 2, 1–25. 10.1515/psr-2017-0006. [DOI] [Google Scholar]
- Recent authoritative reviews, see:; a Lorenzetto T.; Berton G.; Fabris F.; Scarso A. Recent designer surfactants for catalysis in water. Catal. Sci. Technol. 2020, 10, 4492–4502. 10.1039/D0CY01062F. [DOI] [Google Scholar]; b Lipshutz B. H.; Ghorai S.; Cortes-Clerget M. The hydrophobic effect applied to organic synthesis: recent synthetic chemistry “in water.. Chem. - Eur. J. 2018, 24, 6672–6695. 10.1002/chem.201705499. [DOI] [PubMed] [Google Scholar]; and references cited therein.
- Recent authoritative reviews, see:; a Singh R.; Bhardwaj D.; Ganaie S. A.; Singh A. Lewis acid surfactant combined (LASC) catalyst as a versatile heterogeneous catalyst in various organic transformations. Mini-Rev. Org. Chem. 2020, 17, 124–140. 10.2174/1570193X16666181228112313. [DOI] [Google Scholar]; b Kobayashi S.; Manabe K. Development of novel Lewis acid catalysts for selective organic reactions in aqueous media. Acc. Chem. Res. 2002, 35, 209–217. 10.1021/ar000145a. [DOI] [PubMed] [Google Scholar]; and references cited therein.
- a Kitanosono T.; Kobayashi S. Toward chemistry-based design of the simplest metalloenzyme-like catalyst that works efficiently in water. Chem. - Asian J. 2015, 10, 133–138. 10.1002/asia.201403004. [DOI] [PubMed] [Google Scholar]; b Chen Z. L.; Lin L. L.; Wang M.; Liu X. H.; Feng X. M. Asymmetric synthesis of trans-β-lactams by a Kinugasa reaction on water. Chem. - Eur. J. 2013, 19, 7561–7567. 10.1002/chem.201204373. [DOI] [PubMed] [Google Scholar]; c Li J. H.; Tang Y. F.; Wang Q. W.; Li X. F.; Cun L. F.; Zhang X. M.; Zhu J.; Li L. C.; Deng J. G. Chiral surfactant-type catalyst for asymmetric reduction of aliphatic ketones in water. J. Am. Chem. Soc. 2012, 134, 18522–18525. 10.1021/ja308357y. [DOI] [PubMed] [Google Scholar]
- The solubility of cholecalciferol is estimated to be 13 ng/L (34 pM): https://pubchem.ncbi.nlm.nih.gov/compound/5280795#section=Solubility&fullscreen=true.
- Kitanosono T.; Kobayashi S. Reactions in water involving the “on-water” mechanism. Chem. - Eur. J. 2020, 26, 9408–9429. 10.1002/chem.201905482. [DOI] [PubMed] [Google Scholar]
- Kobayashi S.; Kakumoto K.; Mori Y.; Manabe K. Chiral Lewis acid-catalyzed enantioselective Michael reactions in water. Isr. J. Chem. 2001, 41, 247–249. 10.1560/6GRQ-YRVV-6KU3-RHGX. [DOI] [Google Scholar]
- Wei C.; Li C.-J. Enantioselective direct-addition of terminal alkynes to imines catalyzed by copper(I)pybox complex in water and in toluene. J. Am. Chem. Soc. 2002, 124, 5638–5639. 10.1021/ja026007t. [DOI] [PubMed] [Google Scholar]
- Some examples of catalytic asymmetric Diels–Alder and hydrogenation reactions in the 1990s are Type Ia reactions performed homogeneously. For the earliest reports, see:; a Otto S.; Boccaletti G.; Engberts J. B. F. N. A chiral Lewis-acid-catalyzed Diels-Alder reaction. water-enhanced enantioselectivity. J. Am. Chem. Soc. 1998, 120, 4238–4239. 10.1021/ja972772+. [DOI] [Google Scholar]; b Wan K. T.; Davis M. E. Asymmetric hydrogenation in water by a rhodium complex of sulfonated 2,2’bis(diphenylphosphino)-1,1’-binaphthyl (binap). J. Chem. Soc., Chem. Commun. 1993, 74, 1262–1264. 10.1039/C39930001262. [DOI] [Google Scholar]
- a Hayashi Y.; Sumiya T.; Takahashi J.; Gotoh H.; Urushima T.; Shoji M. Highly diastereo- and enantioselective direct aldol reactions in water. Angew. Chem., Int. Ed. 2006, 45, 958–961. 10.1002/anie.200502488. [DOI] [PubMed] [Google Scholar]; Long alkyl chain-tethered proline:; b Mase N.; Nakai Y.; Ohara N.; Yoda H.; Takabe K.; Tanaka F.; Barbas C. F. III Organocatalytic direct asymmetric aldol reactions in water. J. Am. Chem. Soc. 2006, 128, 734–735. 10.1021/ja0573312. [DOI] [PubMed] [Google Scholar]; c Hayashi Y.; Aratake S.; Okano T.; Takahashi J.; Sumiya T.; Shoji M. Combined Proline-Surfactant Organocatalyst for the Highly diastereo- and enantioselective aqueous direct cross-aldol reaction of aldehydes. Angew. Chem., Int. Ed. 2006, 45, 5527–5529. 10.1002/anie.200601156. [DOI] [PubMed] [Google Scholar]; Representative reviews:; d Yamashita Y.; Yasukawa T.; Yoo W.-J.; Kitanosono T.; Kobayashi S. Catalytic enantioselective aldol reactions. Chem. Soc. Rev. 2018, 47, 4388–4480. 10.1039/C7CS00824D. [DOI] [PubMed] [Google Scholar]; e Mlynarski J.; Paradowska J. Catalytic asymmetric aldol reactions in aqueous media. Chem. Soc. Rev. 2008, 37, 1502–1511. 10.1039/b710577k. [DOI] [PubMed] [Google Scholar]
- a Zotova N.; Franzke A.; Armstrong A.; Blackmond D. G. Clarification of the role of water in proline-mediated aldol reactions. J. Am. Chem. Soc. 2007, 129, 15100–15101. 10.1021/ja0738881. [DOI] [PubMed] [Google Scholar]; b Blackmond D. G.; Armstrong A.; Coombe V.; Wells A. Water in organocatalytic processes: debunking the myths. Angew. Chem., Int. Ed. 2007, 46, 3798–3800. 10.1002/anie.200604952. [DOI] [PubMed] [Google Scholar]
- a Hayashi Y. In water or in the presence of water?. Angew. Chem., Int. Ed. 2006, 45, 8103–8104. 10.1002/anie.200603378. [DOI] [PubMed] [Google Scholar]; b Brogan A. P.; Dickerson T. J.; Janda K. D. Enamine-based aldol organocatalysis in water: are they really ″all wet″?. Angew. Chem., Int. Ed. 2006, 45, 8100–8102. 10.1002/anie.200601392. [DOI] [PubMed] [Google Scholar]
- Portmann P.Process for preparing 1-substituted 4-cyano-1,2,3-triazoles. Patent WO 9802423, 1998.
- Ruiz-Lopez M. F.; Francisco J. S.; Martins-Costa M. T. C.; Anglada J. M. Molecular reactions at aqueous interfaces. Nature Rev. Chem. 2020, 4, 459–475. 10.1038/s41570-020-0203-2. [DOI] [PubMed] [Google Scholar]
- Sage C. R.; Rutenber E. E.; Stout T. J.; Stroud R. M. An essential role for water in an enzyme reaction mechanism: the crystal structure of the thymidylate synthase mutant E58Q. Biochemistry 1996, 35, 16270–16281. 10.1021/bi961269r. [DOI] [PubMed] [Google Scholar]
- Hendil-Forssell P.; Martinelle M.; Syrén P.-O. Exploring water as building bricks in enzyme engineering. Chem. Commun. 2015, 51, 17221–17224. 10.1039/C5CC07162C. [DOI] [PubMed] [Google Scholar]
- Yamada T.; Morisseau C.; Maxwell J. E.; Argiriadi M. A.; Christianson D. W.; Hammock B. D. Biochemical evidence for the involvement of tyrosine in epoxide activation during the catalytic cycle of epoxide hydrolase. J. Biol. Chem. 2000, 275, 23082–23088. 10.1074/jbc.M001464200. [DOI] [PubMed] [Google Scholar]
- a Bae H. Y.; Song C. E. Unprecedented hydrophobic amplification in noncovalent organocatalysis “on water”: hydrophobic chiral squaramide catalyzed Michael addition of malonates to nitroalkenes. ACS Catal. 2015, 5, 3613–3619. 10.1021/acscatal.5b00685. [DOI] [Google Scholar]; b Bae H. Y.; Some S.; Oh J. S.; Lee Y. S.; Song C. E. Hydrogen bonding mediated enantioselective organocatalysis in brine: significant rate acceleration and enhanced stereoselectivity in enantioselective Michael addition reactions of 1,3-dicarbonyls to β-nitroolefins. Chem. Commun. 2011, 47, 9621–9623. 10.1039/c1cc13637b. [DOI] [PubMed] [Google Scholar]
- Breslow R. Determining the geometries of transition states by use of antihydrophobic additives in water. Acc. Chem. Res. 2004, 37, 471–478. 10.1021/ar040001m. [DOI] [PubMed] [Google Scholar]
- Sim J. H.; Song C. E. Water-enabled catalytic asymmetric Michael reactions of unreactive nitroalkenes: one-pot synthesis of chiral GABA-analogs with all-carbon quaternary stereogenic centers. Angew. Chem., Int. Ed. 2017, 56, 1835–1839. 10.1002/anie.201611466. [DOI] [PubMed] [Google Scholar]
- Li B.; Dixneuf P. H. sp2 C–H bond activation in water and catalytic cross-coupling reactions. Chem. Soc. Rev. 2013, 42, 5744–5767. 10.1039/c3cs60020c. [DOI] [PubMed] [Google Scholar]; and references cited therein.
- Ferrer-Flegeau E.; Bruneau C.; Dixneuf P. H.; Jutand A. Autocatalysis for C–H bond activation by ruthenium(II) complexes in catalytic arylation of functional arenes. J. Am. Chem. Soc. 2011, 133, 10161–10170. 10.1021/ja201462n. [DOI] [PubMed] [Google Scholar]
- Ali M. A.; Yao X.; Sun H.; Lu H. [RhCp*Cl2]2-catalyzed directed N-Boc amidation of arenes “on water”. Org. Lett. 2015, 17, 1513–1516. 10.1021/acs.orglett.5b00392. [DOI] [PubMed] [Google Scholar]
- Li J.; Hu W.; Peng Y.; Zhang Y.; Li J.; Zheng W. Theoretical study on iridacycle and rhodacycle formation via C–H activation of phenyl imines. Organometallics 2014, 33, 2150–2159. 10.1021/om400832c. [DOI] [Google Scholar]
- Debbarma S.; Sk M. R.; Modak B.; Maji M. S. On-water Cp*Ir(III)-catalyzed C–H functionalization for the synthesis of chromones through annulation of salicylaldehydes with diazo-ketones. J. Org. Chem. 2019, 84, 6207–6216. 10.1021/acs.joc.9b00418. [DOI] [PubMed] [Google Scholar]
- Andrić J. M.; Janjić G. V.; Ninković D. B.; Zarić S. D. The influence of water molecule coordination to a metal ion on water hydrogen bonds. Phys. Chem. Chem. Phys. 2012, 14, 10896–10898. 10.1039/c2cp41125c. [DOI] [PubMed] [Google Scholar]
- Ye Y.; Ball N. D.; Kampf J. W.; Sanford M. S. Oxidation of a cyclometalated Pd(II) dimer with “CF3+”: formation and reactivity of a catalytically competent monomeric Pd(IV) aquo complex. J. Am. Chem. Soc. 2010, 132, 14682–14687. 10.1021/ja107780w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara E.; Fujii A.; Sodeoka M. Enantioselective addition of enol silyl ethers to imines catalyzed by palladium complexes: a novel way to optically active acylalanine derivatives. J. Am. Chem. Soc. 1998, 120, 2474–2475. 10.1021/ja973962n. [DOI] [Google Scholar]
- Robertazzi A.; Platts J. A. Hydrogen bonding, solvation, and hydrolysis of cisplatin: A theoretical study. J. Comput. Chem. 2004, 25, 1060–1067. 10.1002/jcc.20038. [DOI] [PubMed] [Google Scholar]
- Kitanosono T.; Tani S.; Kobayashi S. Oxyfunctionalization of active methylene compounds using sodium chlorite in water. Asian J. Org. Chem. 2018, 7, 350–354. 10.1002/ajoc.201700646. [DOI] [Google Scholar]
- Hagfeldt C.; Kessler V.; Persson I. Structure of the hydrated, hydrolysed and solvated zirconium(IV) and hafnium(IV) ions in water and aprotic oxygen donor solvents. A crystallographic, EXAFS spectroscopic and large angle X-ray scattering study. Dalton Trans. 2004, 2142–2151. 10.1039/B402804J. [DOI] [PubMed] [Google Scholar]
- a Kitanosono T.; Sakai M.; Ueno M.; Kobayashi S. Chiral-Sc catalyzed asymmetric Michael addition/protonation of thiols with enones in water. Org. Biomol. Chem. 2012, 10, 7134–7147. 10.1039/c2ob26264a. [DOI] [PubMed] [Google Scholar]; b Bonollo S.; Lanari D.; Pizzo F.; Vaccaro L. Sc(III)-catalyzed enantioselective addition of thiols to α,β-unsaturated ketones in neutral water. Org. Lett. 2011, 13, 2150–2152. 10.1021/ol200379r. [DOI] [PubMed] [Google Scholar]; c Ueno M.; Kitanosono T.; Sakai M.; Kobayashi S. Chiral Sc-catalyzed asymmetric Michael reactions of thiols with enones in water. Org. Biomol. Chem. 2011, 9, 3619–3621. 10.1039/c1ob05424d. [DOI] [PubMed] [Google Scholar]
- Kitanosono T.; Hisada T.; Yamashita Y.; Kobayashi S. Hydrogen-bonding-assisted cationic aqua palladium(II) complex enables highly efficient asymmetric reactions in water. Angew. Chem., Int. Ed. 2021, 60, 3407. 10.1002/anie.202009989. [DOI] [PubMed] [Google Scholar]
- a Kitanosono T.; Xu P.; Kobayashi S. Heterogeneous versus homogeneous copper(II) catalysis in enantioselective conjugate-addition reactions of boron in water. Chem. - Asian J. 2014, 9, 179–188. 10.1002/asia.201300997. [DOI] [PubMed] [Google Scholar]; b Kobayashi S.; Xu P.; Endo T.; Ueno M.; Kitanosono T. Chiral copper(II)-catalyzed enantioselective boron conjugate additions to α,β-unsaturated carbonyl compounds in water. Angew. Chem., Int. Ed. 2012, 51, 12763–12766. 10.1002/anie.201207343. [DOI] [PubMed] [Google Scholar]
- Kitanosono T.; Xu P.; Isshiki S.; Zhu L.; Kobayashi S. Cu (II)-catalyzed asymmetric boron conjugate addition to α,β-unsaturated imines in water. Chem. Commun. 2014, 50, 9336–9339. 10.1039/C4CC04062G. [DOI] [PubMed] [Google Scholar]
- Kitanosono T.; Kobayashi S. Asymmetric boron conjugate additions to enones in water catalyzed by copper(0). Asian J. Org. Chem. 2013, 2, 961–966. 10.1002/ajoc.201300201. [DOI] [Google Scholar]
- Kitanosono T.; Xu P.; Kobayashi S. Heterogeneous and homogeneous chiral Cu(II) catalysis in water: enantioselective boron conjugate additions to dienones and dienoesters. Chem. Commun. 2013, 49, 8184–8186. 10.1039/c3cc44324h. [DOI] [PubMed] [Google Scholar]
- Kitanosono T.; Zhu L.; Liu C.; Xu P.; Kobayashi S. An insoluble copper(II) acetylacetonate-chiral bipyridine complex that catalyzes asymmetric silyl conjugate addition in water. J. Am. Chem. Soc. 2015, 137, 15422–15425. 10.1021/jacs.5b11418. [DOI] [PubMed] [Google Scholar]
- Song C. E.; Park S. J.; Hwang I.-S.; Jung M. J.; Shim S. Y.; Bae H. Y.; Jung J. Y. Hydrophobic chirality amplification in confined water cages. Nat. Commun. 2019, 10, 851. 10.1038/s41467-019-08792-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intrinsically, Type Ib reactions are supposed to be categorized into “aquachemistry.”
- Huang J.-M.; Dong Y. Zn-mediated electrochemical allylation of aldehydes in aqueous ammonia. Chem. Commun. 2009, 3943–3945. 10.1039/b905553c. [DOI] [PubMed] [Google Scholar]
- Tian C.; Massignan L.; Meyer T. H.; Ackermann L. Electrochemical C–H/N–H activation by water-tolerant cobalt catalysis at room temperature. Angew. Chem., Int. Ed. 2018, 57, 2383–2387. 10.1002/anie.201712647. [DOI] [PubMed] [Google Scholar]
- a Guo D.; Zhu D.; Zhou X.; Zheng B. Accelerating the “on water” reaction: by organic–water interface or by hydrodynamic effects?. Langmuir 2015, 31, 13759–13763. 10.1021/acs.langmuir.5b04031. [DOI] [PubMed] [Google Scholar]; b Mellouli S.; Bousekkine L.; Theberge A. B.; Huck W. T. S. Investigation of “on water” conditions using a biphasic fluidic platform. Angew. Chem., Int. Ed. 2012, 51, 7981–7984. 10.1002/anie.201200575. [DOI] [PubMed] [Google Scholar]
- Bain R. M.; Sathyamoorthi S.; Zare R. N. On-droplet” chemistry: the cycloaddition of diethyl azodicarboxylate and quadricyclane. Angew. Chem., Int. Ed. 2017, 56, 15083–15087. 10.1002/anie.201708413. [DOI] [PubMed] [Google Scholar]
- Masuda K.; Kobayashi S. Direct and quantitative monitoring of catalytic organic reactions under heterogeneous conditions using direct analysis in real time mass spectrometry. Chem. Sci. 2020, 11, 5105–5112. 10.1039/D0SC00021C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bridoux M.; Malandain H.; Leprince F.; Progent F.; Machuron-Mandard X. Quantitative analysis of phosphoric acid esters in aqueous samples by isotope dilution stir-bar sorptive extraction combined with direct analysis in real time (DART)-orbitrap mass spectrometry. Anal. Chim. Acta 2015, 869, 1–10. 10.1016/j.aca.2015.01.010. [DOI] [PubMed] [Google Scholar]; b Hajslova J.; Cajka T.; Vaclavik L. Challenging applications offered by direct analysis in real time (DART) in food-quality and safety analysis. TrAC, Trends Anal. Chem. 2011, 30, 204–218. 10.1016/j.trac.2010.11.001. [DOI] [Google Scholar]
- a Yang L.; Zhao J.; Yang X.; Chen M.; Xue Y. Effects of solvents on the DACBO-catalyzed vinylogous Henry reaction of isatin with 3,5-dimethyl-4-nitroisoxazole “on-water” and in solution from QM/MM MC simulations. RSC Adv. 2019, 9, 4932–4941. 10.1039/C9RA00082H. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhao J.; He F.; Zhang F.; Yang X.; Tian Z.; Xue Y. The role of water in the catalyst-free aldol reaction of water-insoluble N-methyl-2,4-thiazolidinedione with N-methylisatin from QM/MM Monte Carlo simulations. ChemPhysChem 2017, 18, 2123–2131. 10.1002/cphc.201700423. [DOI] [PubMed] [Google Scholar]; c Zhang J.; Yang Y. I.; Yang L.; Gao Y. Q. Dynamics and kinetics study of “in-water” chemical reactions by enhanced sampling of reactive trajectories. J. Phys. Chem. B 2015, 119, 14505–14514. 10.1021/acs.jpcb.5b08690. [DOI] [PubMed] [Google Scholar]; d Acevedo O.; Jorgensen W. L. CP2K: Atomistic simulations of condensed matter systems. WIREs Comput. Mol. Sci. 2014, 4, 422–435. 10.1002/wcms.1180. [DOI] [PMC free article] [PubMed] [Google Scholar]; and references cited therein.
- a Sameera W. M. C.; Hatanaka M.; Kitanosono T.; Kobayashi S.; Morokuma K. The mechanism of iron(II)-catalyzed asymmetric Mukaiyama aldol reaction in aqueous media: density functional theory and artificial force-induced reaction study. J. Am. Chem. Soc. 2015, 137, 11085–11094. 10.1021/jacs.5b05835. [DOI] [PubMed] [Google Scholar]; b Hatanaka M.; Morokuma K. Role of water in Mukaiyama-aldol reaction catalyzed by lanthanide Lewis acid: a computational study. J. Am. Chem. Soc. 2013, 135, 13972–13979. 10.1021/ja407357c. [DOI] [PubMed] [Google Scholar]; c Hatanaka M.; Maeda S.; Morokuma K. Sampling of transition states for predicting diastereoselectivity using automated search method–aqueous lanthanide-catalyzed Mukaiyama aldol reaction. J. Chem. Theory Comput. 2013, 9, 2882–2886. 10.1021/ct4002637. [DOI] [PubMed] [Google Scholar]