

Abstract
Mutations of SPINT2, the gene encoding the integral membrane, Kunitz-type serine inhibitor HAI-2, primarily affect the intestine, while sparing many other HAI-2-expressing tissues, causing sodium loss in patients with syndromic congenital sodium diarrhea. The membrane-bound serine protease prostasin was previously identified as a HAI-2 target protease in intestinal tissues but not in the skin. In both tissues, the highly related inhibitor HAI-1 is, however, the default inhibitor for prostasin and the type 2 transmembrane serine protease matriptase. This cell-type selective functional linkage may contribute to the organ-selective damage associated with SPINT 2 mutations. To this end, the impact of HAI-2 deletion on matriptase and prostasin proteolysis was, here, compared using Caco-2 human colorectal adenocarcinoma cells and HaCaT human keratinocytes. Greatly enhanced prostasin proteolytic activity with a prolonged half-life and significant depletion of HAI-1 monomer were observed with HAI-2 loss in Caco-2 cells but not HaCaT cells. The constitutive, high level prostasin zymogen activation observed in Caco-2 cells, but not in HaCaT cells, also contributes to the excessive prostasin proteolytic activity caused by HAI-2 loss. HAI-2 deletion also caused increased matriptase zymogen activation, likely as an indirect result of increased prostasin proteolysis. This increase in activated matriptase, however, only had a negligible role in depletion of HAI-1 monomer. Our study suggests that the constitutive, high level of prostasin zymogen activation and the cell-type selective functional relationship between HAI-2 and prostasin renders Caco-2 cells more susceptible than HaCaT cells to the loss of HAI-2, causing a severe imbalance favoring prostasin proteolysis.
Introduction
Hepatocyte growth factor (HGF) activator inhibitor (HAI)-2, an integral membrane, Kunitz-type, serine protease inhibitor, is broadly expressed in organ systems with epithelial components (1,2). Despite this widespread expression, the defects caused by the mutations of serine peptidase inhibitor, Kunitz type 2 (SPINT2), which encodes HAI-2, are primarily focused on the gastrointestinal (GI) tract in patients with syndromic congenital sodium diarrhea [SCSD (3)]. These organ-selective defects suggest that there may be organ-selective regulation and functions of HAI-2. The SPINT2 mutations identified in SCSD patients either result in the loss of protein expression or reportedly cause a reduction or the loss of HAI-2 protease inhibitory activity (3). Dysregulated proteolytic activity of HAI-2 target proteases has, therefore, been proposed to contribute to the disease. HAI-2 contains two Kunitz domains, which are structurally similar to aprotinin, the prototype for this protease inhibitor family. The specificity of these serine protease inhibitors is determined by the sequence of the canonical protease binding loop within the Kunitz domains (4,5). The reactive center loop (P1–P1′) site of both the HAI-2 Kunitz domains are Arg-48 and Arg-143, and therefore the target protease(s) of HAI-2 are serine proteases with trypsin-like activity (1). The blood-borne serine protease HGF activator was initially used as a ‘bait’ protease to identify, purify and characterize HAI-2 as well as HAI-1 serine peptidase inhibitor, Kunitz type 1 (SPINT1) which is closely related to HAI-2 in terms of the overall protein structure and its specificity against serine proteases (1,6). The type 2 transmembrane serine protease matriptase and the membrane-associated serine protease prostasin are, however, the two most prominent physiological target proteases for HAI-1 and HAI-2 (7–12).
The structurally complementary match between the serine protease domains and the Kunitz domains provides the biochemical basis for the potency and specificity of the HAIs as inhibitors of matriptase and prostasin (13–16). It is, however, the tissue distribution and subcellular localization that determine whether this biochemical relationship is further expanded and established as cellular and physiological relationships. For example, HAI-1 can serve as an endogenous inhibitor for both matriptase and prostasin in human skin due to its co-localization with matriptase at the intercellular junctions in the keratinocytes of the basal and spinous layers and its co-localization with prostasin at an organelle-like structure, the appearance of which is dynamically altered along with epidermal differentiation (17–19). In contrast, although HAI-2 is co-expressed with matriptase and prostasin to some extent over the course of epidermal differentiation, the intracellular localization of HAI-2 results in its not being an endogenous inhibitor for either matriptase or prostasin in human skin (17). This differential functional relationship has been also demonstrated in HaCaT keratinocytes. When these human keratinocytes are induced to activate matriptase, HAI-1, but not HAI-2, rapidly inhibits both serine proteases via the formation of stable enzyme-inhibitor complexes (20). Since the formation of enzyme-inhibitor complexes has also been observed in a variety of other epithelial/carcinoma cell systems, the functional relationship between HAI-1 and these two serine proteases is likely ubiquitous (21). HAI-2, in contrast, exhibits marked cell-type selectivity with respect to its role in the regulation of matriptase and prostasin activity. For example, HAI-2-mediated matriptase inhibition has been observed in some breast cancer cells but not in 184A1N4 human mammary epithelial cells. This differential role likely results from the co-localization of HAI-2 and matriptase on cell surface of breast cancer cells but not the mammary epithelial cells (22). Furthermore, HAI-2 has been shown to be the primary inhibitor of matriptase in some neoplastic B-cells, in which HAI-1 is not expressed (2), although the HAI-2 is not effective for the control of the shed extracellular active matriptase because of the predominantly intracellular localization of the HAI-2 (23).
Cell-type selective, HAI-2-mediated protease inhibition has also been observed for prostasin. Prostasin-HAI-2 complex have been detected along with prostasin-HAI-1 complexes in human intestinal tissues lysates (24). Furthermore, the functional linkage between HAI-2 and prostasin is also supported by the co-localization of these proteins near the terminal webs underneath the microvilli in these tissues (24). The dual mechanism underlying the control of prostasin proteolysis by HAI-1 and HAI-2 has been also seen in Caco-2 colorectal adenocarcinoma cells, in which prostasin-HAI-1 complexes and prostasin-HAI-2 complexes are present, along with matriptase-HAI-1 complex (24). The identification of prostasin as the cell-type selective HAI-2 target protease could help explain why intestinal tissues are particularly susceptible to HAI-2 mutations, since such mutations might result in elevated prostasin proteolytic activity in the GI tract thereby contributing to the pathogenesis of the SCSD. Since prostasin proteolysis is also under regulation by HAI-1, which also serves as the primary inhibitor for matriptase, a much more complicated protease network system is present in intestinal tissues than in other epithelial counterparts. To further complicate this already complicated relationship, matriptase can serve as an upstream activator for prostasin (20), and prostasin deficiency causes reduced matriptase zymogen activation in some model systems (25,26). In the current study, we set out to determine the differential impact of HAI-2 loss on the matriptase and prostasin proteolysis status in Caco-2 colorectal adenocarcinoma cells versus HaCaT keratinocytes with the goal of better understanding the molecular and cellular mechanism underlying the organ-selective defects caused by SPINT2 mutations.
Results
HAI-2 loss causes an increase in prostasin zymogen activation and prolongs active prostasin in Caco-2
Given the differential role of HAI-2 in the control of prostasin proteolysis in Caco-2 colorectal adenocarcinoma cells versus that in HaCaT keratinocytes, we postulated that Caco-2 cells might differently respond to the loss of HAI-2 than the HaCaT cells, particularly in terms of the proteolysis status of prostasin and possibly matriptase. Thus, we compared the impact of targeted HAI-2 deletion on prostasin and matriptase proteolysis in both cell lines. Two different HAI-2 knockout (KO) Caco-2 variants were generated using clustered regularly interspaced short palindromic repeats (CRISPR) technology. The lack of HAI-2 protein expression in the two knockout variants (HAI-2 KO) was verified by western blot analysis (Fig. 1A). Mature HAI-2 is synthesized as glycoproteins with complex type N-glycosylation (22,27). HAI-2 monomer was, therefore, detected as very diffuse bands in the parental cells (Fig. 1A, lane 1). In addition, a proportion of HAI-2 can form, and has previously been detected as stable enzyme-inhibitor complexes with prostasin in the parental cells [(24); Fig. 1A, lane 1]. Neither the monomer nor complexes were detected in the two KO variants (Fig. 1A, lanes 2 and 3). Examining the cellular protein composition of Caco-2 parental and the two KO variants further demonstrated that HAI-2 deficiency did not appear to cause significant changes in global protein expression (Fig. 1B). The intensity of cellular proteins loaded was similar among the three lines and served here as the loading control for the western blot analysis.
Figure 1 .

Targeted deletion of HAI-2 does not alter global cellular protein profile in Caco-2 colorectal adenocarcinoma cells. HAI-2, encoded by SPINT2, was deleted in a targeted fashion by CRISPR in Caco-2 colorectal adenocarcinoma cells. Lysates were prepared from parental Caco-2 cells (Parental, lane 1) and two representative clones (KO1 and KO2, lanes 2 and 3) and equal amounts of protein were analyzed by western blot for HAI-2 protein expression using the mAb DC16 (A) and by Coomassie Blue staining for the protein profile and as a loading control (B). HAI-2 monomer and HAI-2 complexes are indicated.
The impact of targeted HAI-2 deletion on prostasin proteolysis was investigated next. Analyses of prostasin species by immunoblot revealed that HAI-2 loss increased prostasin proteolysis (Fig. 2). The increase resulted not only from the higher cellular levels of prostasin detected in the two KO variants compared with the parental cells but also from an increase in prostasin zymogen activation and an increase in the lifespan of free active prostasin. The increase in zymogen activation can be appreciated by 1) a significant decrease in the levels of prostasin zymogen in the two HAI-2 KO variants compared with the parental cells and 2) as a result, a significant increase in the ratio of activated prostasin relative to prostasin zymogen (Fig. 2A). In addition to the zymogen and complexed prostasin forms, a 35 kDa species was observed in the HAI-2 KO variants (Fig. 2A, compare lanes 2 and 3 with lane 1, for active prostasin). This 35 kDa species has previously been shown to be enzymatically active prostasin by virtue of its ability to form a stable complex with HAI-1 and cleave an optimal prostasin fluorogenic substrate (24). It is worth noting that the sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) in this study was performed under non-reducing and non-boiled conditions, under which conditions, active prostasin and zymogen prostasin contain the same number of amino acid residues. The localized conformational changes associated with the cleavage of zymogen activation apparently results in a relatively relaxed conformation for active prostasin versus a much more compacted one for the zymogen counterpart. Thus, active prostasin exhibits a slower migration rate on SDS-PAGE. This larger size by SDS-PAGE is contradictory to the general perception that active prostasin is smaller than zymogen prostasin. This is true only when SDS-PAGE is run under reducing and boiled conditions. Since the light-chain is very small in this enzyme, with only 12 amino acid residues, it is not easy to see the difference in size between active versus zymogen prostasin by western blot under reducing and boiled conditions.
Figure 2 .

Targeted HAI-2 deletion causes increased prostasin proteolysis with increased lifespan of active prostasin in Caco-2 cells. (A) Lysates were prepared from parental Caco-2 cells (Parental, lane 1) and two representative clones (KO1 and KO2, lanes 2 and 3), and equal amounts of protein were analyzed by western blot for prostasin expression using the mAb YL11. (B) through (F) The HAI-2 KO1 Caco-2 cells were engineered to express HAI-2 (C, D; HAI-2 Teton) or HAI-1 (E, F; HAI-1 Teton) or with empty vector as a control for doxycycline treatment (B; LVX-control) in a doxycycline-inducible manner. The Caco-2 variants were treated with increasing concentrations of doxycycline, as indicated, to induce expression of the HAIs. Lysates were prepared from these cells, and equal amounts of protein were analyzed by immunoblot for prostasin species using the prostasin mAb YL11 (B, D and F), for HAI-2 using the HAI-2 mAb CD16 (C) and for HAI-1 using the HAI-1 mAb M19 (E). The prostasin species, including the complexes with the HAIs (Prostasin Complexes with HAI-1 or HAI-2), enzymatically active prostasin (Active Prostasin) and the zymogen form (Zymogen Prostasin) are indicated. HAI-2 monomer and HAI-1 monomer are also indicated.
The appearance of active prostasin in the two KO variants suggests a prolonged lifespan for enzymatically active prostasin as a result of HAI-2 loss. Since prostasin is also under tight inhibitory control by HAI-1 in Caco-2 cells, the appearance of active prostasin indicates that endogenous HAI-1 protein might not be sufficiently abundant enough to compensate for the loss of HAI-2 for rapid prostasin inhibition. Alternatively, it is possible that prostasin is targeted to two different subcellular compartments, in which HAI-1 and HAI-2 independently control prostasin activity. As a result, targeted HAI-2 deletion would lead to the generation of free active prostasin, regardless of the level of endogenous HAI-1.
The HAI-2 KO1 cells were next engineered to express HAI-1 or HAI-2 in a doxycycline-dependent manner to determine the dynamic relationship between HAI expression and the roles of the HAIs in the regulation of prostasin proteolysis in the intestinal epithelial cells. We were particularly interested in assessing the impact on the generation of free active prostasin in a relatively quantitative manner. In the control cells transduced with an empty virus (LVX, Lily virus X), addition of increasing amounts of doxycycline up to 5 μg/ml did not affect the status of prostasin with the vast majority of the protein found in activated forms, including complexes with HAI-1 and the enzymatically active form (Fig. 2B). These results demonstrate that doxycycline is relatively inert with regard to the endpoints we are studying and demonstrate feasibility of using this model system. Treatment of cells transduced with the Teton-HAI-2 virus with increasing concentrations of doxycycline resulted in an increasing level of the HAI-2 protein expression from very low level (requiring longer exposure to see the signal) to very high levels at 0.5 and 5 μg/ml (Fig. 2C). Increasing expression of HAI-1 was also induced by increasing concentrations of doxycycline in cells transduced with the Teton-HAI-1 virus, in a fashion similarly to the HAI-2 cells (Fig. 2E). Coincident with the very low level of the HAIs induced by treatment with 0.02 μg/ml doxycycline, the level of enzymatically active prostasin began to decrease (Fig. 2D and F, lanes 2, active prostasin). This decrease in active prostasin became more obvious at 0.1 μg/ml doxycycline (Fig. 2D and F, lanes 3, active prostasin). Along with the significant decrease in active prostasin produced by HAI expression, prostasin zymogen began to appear (Fig. 2D and F, lanes 3, zymogen prostasin). The residual enzymatically active prostasin was further reduced to essentially zero in those cells with very high-level exogenous expression of HAI-2 and also HAI-1, where prostasin zymogen completely replaced active prostasin (Fig. 2D and F, lanes 4 and 5). The process of prostasin zymogen activation remains very active even in the face of very high-level expression of the HAIs, as demonstrated by the high levels of activated prostasin complexes with HAI-2 and HAI-1 detected (Fig. 2D and F, lanes 4 and 5). These data suggest that the prolonged life of active prostasin observed in the knockout cells is the result of the severe imbalance between protease and protease inhibitor caused by the HAI-2 deletion, which can be reverted by re-expression of not only HAI-2 but also HAI-1. Some interesting differences between HAI-1 and HAI-2 in the regulation of prostasin were, however, observed. High-level exogenous HAI-1 prevented the degradation of 100 kDa prostasin-HAI-1 complex into the 70 kDa form (Fig. 2F, lanes 4 and 5). High-level expression of HAI-2 reduced the level of prostasin-HAI-1 complexes (Fig. 2D, lanes 4 and 5). Given that the high-level expression of the HAIs under these conditions is likely to be significantly higher than the nature level of endogenous HAI expression in Caco-2 cells, the physiologic relevance of these differences remains unclear.
Targeted deletion of HAI-2 causes an increase in matriptase zymogen activation in Caco-2 cells
The impact of targeted HAI-2 deletion on matriptase proteolysis was investigated next (Fig. 3). In contrast to prostasin, which is predominantly cell-associated, matriptase can be shed into the extracellular milieu in amounts, particularly following matriptase zymogen activation (28). Matriptase is synthesized as a 94 kDa full-length form, which undergoes N-terminal processing to become mature matriptase zymogen. Mature matriptase zymogen contains two fragments, C-terminal 70 kDa (amino acids 150-885) and N-terminal 25 kDa (amino acids 1–149). The matriptase monoclonal antibody (mAb) used in this study is targeted against the 70 kDa C-terminal fragment. Matriptase zymogen undergoes autoactivation to become active form, which is rapidly inhibited by binding with HAI-1 to form a 120 kDa matriptase-HAI-1 complex. Shedding of matriptase requires proteolytic cleavage at Arg-186, and this cleavage generates shed matriptase with a slightly smaller size than its cell-associated counterpart—by 37 amino acid residues (Fig. 3A, bands f, in lanes 2, 4 and 6). Shedding of the 120 kDa matriptase-HAI-1 complex also requires the proteolytic cleavage of HAI-1. HAI-1 can be shed by cleavage at two different sites, generating 50 and 40 kDa fragments. The former when bound with activated matriptase forms the 110 kDa complex and the latter forms the 95-Da complex (Fig. 3A, bands d and e, in lanes 2, 4 and 6). Matriptase species were, therefore, analyzed by immunoblot in cell lysates and conditioned medium prepared from the cells (Fig. 3A). There were six dominant matriptase species detected, three of which were cell-associated, including the 120 kDa matriptase-HAI-1 complex, the 94 kDa full-length matriptase and 70 kDa matriptase zymogen (Fig. 3A, bands a, b and c, respectively, in lanes 1, 3 and 5). Since the cell lysates and conditioned media were prepared with same final volume, the ratio of activated matriptase (bands a, d and e) to total matriptase (bands a, b, c, d, e and f) can easily be estimated by densitometry. In the parental cells, ~40% of the total matriptase was in the activated form (Fig. 3A, right). Enhanced matriptase zymogen activation was observed in both HAI-2 KO cells with increased ratios of 68.3 and 66.2%, respectively (Fig. 3A, right).
Figure 3 .

Targeted HAI-2 deletion causes an increase in matriptase zymogen activation in Caco-2 cells. (A) The parental (Paren., lanes 1 and 2), HAI-2 KO1 (KO1, lane 3 and 4) and HAI-2 KO2 (KO2, lanes 5 and 6) Caco-2 cells were grown in serum-free medium for 24 h. Lysates were prepared from the three cell lines in a standard volume, and conditioned media were collected and concentrated to the same volumes as that of cell lysates. The protein concentration in the lysates prepared from the parental cells (lane 1), the KO1 cells (lane 3) and KO2 cells (lane 5) were determined and a volume of each lysate containing the same amount of protein was prepared for electrophoresis. That same volume of each of the corresponding conditioned media (lanes 2, 4 and 6, respectively) was also prepared for electrophoresis. The samples of lysate and conditioned media were then analyzed by western blot for matriptase species using the mAb M24. (B) and (C) The HAI-2 Teton cells (B) and HAI-1 Teton cells (C) were treated with increasing concentrations of doxycycline, as indicated, to induce expression of the HAIs. Cell lysates and conditioned media were prepared, as described above. As before, equivalent volumes of lysate and concentrated conditioned media (based on the protein concentration in the lysates), were analyzed by western blot for matriptase levels using the M24 mAb. The matriptase species, including 120 kDa matriptase-HAI-1 complex (band a), full-length matriptase (band b), cell-associated 70 kDa matriptase (band c), shed 110 kDa matriptase-HAI-1 complex (band d), shed 95 kDa matriptase-HAI-1 complex (band e) and shed 70 kDa matriptase (band f), are indicated. The ratio of activated/total matriptase was calculated by a + d + e/a + b + c + d + e + f.
The increased matriptase zymogen activation caused by HAI-2 deletion was reversed by the exogenous re-expression of HAI-2. A steady decrease in the ratio of activated to total matriptase was observed along with a rapid increase in HAI-2 expression induced by increasing concentrations of doxycycline (Fig. 3B). Similarly, a dose-dependent decrease in the ratio was also observed with increased exogenous expression of HAI-1 (Fig. 3C). Thus, elevation of either of the HAIs can revert the increased matriptase zymogen activation caused by HAI-2 loss. Furthermore, when very high-level HAI expression was induced, matriptase zymogen activation and shedding were further suppressed to a level lower than observed in the parental cells. It seems that the HAIs can regulate matriptase proteolytic activity not only via suppression of matriptase enzymatic activity but also by preventing matriptase zymogen activation and shedding, although the HAI expression levels needed for the suppression of the zymogen activation are far higher that the nature level.
There are, however, some subtle differences between HAI-2 and HAI-1. For example, the HAIs differ with respect to their impact on the shedding of the 120 kDa matriptase-HAI-1 complex. Although the 120 kDa matriptase-HAI-1 complex can be shed as 110 and 95 kDa forms, the 95 kDa form is the major species shed by the parental cells (Fig. 3A, lane 2). Both the 110 and 95 kDa complexes were shed at similar levels by the two HAI-2 KO cells (Fig. 3A, lanes 4 and 6). Re-expression of HAI-2 reverted this altered matriptase-HAI-1 complex shedding pattern (Fig. 3B, comparing lane 4 with lane 2 for the bands d and e). In contrast, exogenous expression of HAI-1 reduced shedding of the 95 kDa form much more effective than that of the 110 kDa form (Fig. 3C, comparing lane 6 with lanes 2 and 4, for bands d and e).
HAI-2 loss results in the almost complete depletion of HAI-1 monomer in Caco-2
As the primary protease inhibitor for matriptase and prostasin, HAI-1 is detected either as a monomer, the form that is capable of inhibiting active matriptase and prostasin, or as a complex with activated matriptase or prostasin. Once it is in a complex, HAI-1 loses the ability to serve as a potential protease inhibitor due to Kunitz domain 1 already being occupied by an activated molecule of matriptase or prostasin. As shown in Figure 4A (lane 1), the HAI-1 species, were detected either in 120 kDa complexes with activated matriptase or 100 kDa complexes with activated prostasin (Fig. 4A, lane 1, band a′ and a, respectively). The majority of the HAI-1 was, however, detected as the 60 kDa monomer (Fig. 4A, lane 1, band d). From our previous studies we know that several HAI-1 degradation products are frequently detected, including species of 50-, 40- and 25 kDa, all of which have lost the transmembrane domain but retain the intact Kunitz domain 1 (7,8). Thus, they can be detected in complexes with activated matriptase and activated prostasin. Because activated matriptase is rapidly shed via proteolytic cleavage at Arg-186, these matriptase-HAI-1 complexes containing the shorter forms of HAI-1 have been seen in body fluids and the conditioned medium of cell cultures. In contrast, the vast majority of activated prostasin-HAI-1 complexes remain cell-associated because the membrane anchorage mechanisms of prostasin, either with the transmembrane domain or GPI-anchor, remain intact. It is worth noting that although active prostasin or activated prostasin in complexes with the HAIs are eventually shed into body fluids, the shedding of prostasin occurs at a much slower rate than that for matriptase. Thus, cell-associated prostasin-HAI-1 complexes have been detected as several smaller forms in addition to the intact 100 kDa form. For example, a prostasin-HAI-1 complex was seen on the top of the monomer HAI-1 in the parental Caco-2 cells (Fig. 4A, lane 1). It is worth noting that the complex is likely located on the basolateral plasma membrane (29). Targeted deletion of HAI-2 appears to have a significant impact on HAI-1 (Fig. 4A, lanes 2 and 3). First, the level of the 120 kDa matriptase-HAI-1 complex appeared to be decreased (Fig. 4A, lane 2 and 3). The decrease in the cell-associated matriptase-HAI-1 complex likely results from the shedding of the complex rather than as the result of any inhibition of matriptase zymogen activation, as demonstrated in Figure 3. Second, HAI-2 deletion caused a significant increase in the formation of activated prostasin-HAI-1 complexes (Fig. 4, lanes 2 and 3, bands b and c). This increase could result primarily from the substitution of HAI-2 by HAI-1 and the increase in prostasin zymogen activation, demonstrated in Figure 2. Third, HAI-2 deletion caused an increase in the level of prostasin-HAI-1 degradation complexes (Fig. 4, lanes 2 and 3, bands b and c), although the appearance of these shorter forms varied significantly from experiment to experiment. Finally, and most importantly, HAI-2 deletion caused a substantial decrease in the level of HAI-1 monomer (Fig. 4A, comparing lanes 2 and 3 with lane 1 for band d). This observation indicates that with the genetic ablation of HAI-2, HAI-1-mediated cellular anti-protease activity is also depleted in a non-genetic fashion. The loss of HAI-1 monomer is consistent with and supports the observation that there appears to be a significant increase in the level of enzymatically active prostasin (Fig. 2A, lanes 2 and 3). It is worth noting that there appears to be an inverse correlation between the level of HAI-1 monomer and the level of active prostasin. For example, HAI-2 KO2 cells express slightly higher HAI-1 monomer than HAI-2 KO1 cells (Fig. 4A, comparing lane 3 with lane 2 for band d). Higher level of active prostasin was seen in HAI-2 KO1 cells than HAI-2 KO2 cells (Fig. 2A, comparing lane 2 with lane 3 for active prostasin).
Figure 4 .

Targeted HAI-2 loss results in the depletion of HAI-1 monomer in Caco-2 cells. (A) Lysates were prepared from the parental (Parental, lane 1), HAI-2 KO1 (KO1, lane 2) and HAI-2 KO2 (KO2, lane 3) Caco-2 cells and equal amounts of protein were analyzed by western blot for HAI-1 species using the mAb M19. (B) and (C) HAI-2 Teton cells (C) and the control with empty virus (LVX-Control) (B) were treated with increasing concentrations of doxycycline, as indicated, to induce expression of the HAI-2 (C). Equal amounts of lysate proteins were analyzed by immunoblot for HAI-1 species using the HAI-1 mAb M19. The HAI-1 species, including 120 kDa matriptase-HAI-1 complex (band a’), 100 kDa prostasin-HAI-1 complex (band a), the two degraded fragments of prostasin-HAI-1 complex (bands b and c) and HAI-1 monomer, are indicated.
The doxycycline-inducible expression of HAI-2 was also used to investigate the loss of HAI-1-mediated anti-protease activity caused by HAI-2 loss (Fig. 4 B and C). In the control cells transduced with the empty virus, the addition of doxycycline did not alter the expression status of HAI-1, IE the very low level of HAI-1 monomer (Fig. 4B, band d) and the very high level of the prostasin-HAI-1 complexes (Fig. 4B, bands a, b and c). In the cells transduced with the HAI-2 virus, the addition of increasing amounts of doxycycline, caused a dose-dependent decrease in the level of prostasin-HAI-1 complexes (Fig. 4C). More importantly, the level of HAI-1 monomer steadily and then rapidly increased along with the increasing expression of HAI-2 and the rapid decrease in prostasin-HAI-1 complexes (Fig. 4C, band d). Thus, the loss of HAI-1-mediated anti-protease activity caused by targeted HAI-2 deletion can be restored by re-expression of HAI-2.
Targeted deletion of prostasin but not matriptase reverses the loss of HAI-1 monomer in HAI-2 KO1 Caco-2 cells
The depletion of HAI-1 monomer observed in HAI-2 knockout cells likely results from the binding of HAI-1 monomer with active matriptase or active prostasin or both. Thus, the roles of matriptase and prostasin in the non-genetic depletion of HAI-1 monomer were next determined by genetic ablation of either matriptase or prostasin in HAI-2 KO1 Caco-2 cells (Fig. 5). The impact of prostasin or matriptase deletion on HAI-1 status was analyzed by immunoblot of lysates prepared from the HAI-2/prostasin double knockout Caco-2 cells (Fig. 5, lanes 3, H2&P D. KO) and the HAI-2/matriptase double knockout Caco-2 cells (Fig. 5, lanes 4, H2&M D. KO). The parental cells (Fig. 5, lanes 1, Parental) and the HAI-2 single knockout cells (Fig. 5, lanes 2, HAI-2 S. KO), which were included in the analyses as a control. In the parental cells, HAI-1 monomer represented the major species, which is in stark contrast to the almost complete absence of HAI-1 monomer in the HAI-2 single knockout cells (Fig. 5A, HAI-1, comparing lane 2 with lane 1 for HAI-1 monomer). Almost identical data were also seen in Figure 4A (comparing lane 2 with lane 1). The genetic ablation of prostasin, which was demonstrated by the absence of prostasin protein in the double knockout cells (Fig. 5B, lane 3), dramatically affected the HAI-1 status, with HAI-1 monomer becoming the predominant species (Fig. 5A, lane 3). In marked contrast, the ablation of matriptase, which was demonstrated by the absence of matriptase protein in the double knockout cells (Fig. 5C, lane 4), appeared to have little effect on the HAI-1 status (Fig. 5A, lane 4), which was almost identical to that in the HAI-2 single knockout cells (Fig. 5A, lane 2). In both cells, the vast majority of the HAI-1 was detected in complexed forms and the almost complete absence of HAI-1 monomer (Fig. 5A, lanes 2 and 4). These data indicate that prostasin but not matriptase consumes HAI-1 monomer when HAI-2 is genetically deleted. Since equal amounts of lysate protein from the four cells were used for immunoblot analysis, the total cellular levels of HAI-1 were apparently higher in the parental cells compared with the three KO variants. The decrease in HAI-1 protein levels could be the result of reduced HAI-1 expression, but is more likely the result of an increase in HAI-1 shedding in the three KO variants compared with the parental cells. The depletion of HAI-1 monomer in HAI-2 KO Caco-2 cells is primarily the result of the presence of high levels of active prostasin, but not active matriptase. This is consistent with a functional linkage between prostasin and HAI-2 and the lack of such a linkage between matriptase and HAI-2. The increase in matriptase zymogen activation caused by targeted HAI-2 deletion does not contribute significantly to the depletion of HAI-1 monomer.
Figure 5 .

Prostasin but not matriptase depletes HAI-1 monomer. HAI-2 KO1 Caco-2 cells (HAI-2 S. KO, lanes 2) were used to generate HAI-2/prostasin double knockout cells (H2&P D. KO, lanes 3) and HAI-2/matriptase double knockout cells (H2&M D. KO, lanes 4) cells. Cell lysates were prepared from these cells and Caco-2 parental cells (Parental, lanes 1) and equal amount of proteins were analyzed by immunoblot for HAI-1 species using the mAb M19 (A), for prostasin species using the mAb YL11 and for matriptase species using the mAb M24 (C). The 120 kDa Matriptas-HAI-1 complex, the 100 kDa and degraded prostasin-HAI-1 complexes, prostasin complexes with HAI-1 or HA-2, active prostasin, zymogen prostasin, full-length matriptase and zymogen matriptase are indicated.
Targeted matriptase deletion appeared not to impact the status of prostasin protein. The prostasin protein band pattern in the HAI-2/matriptase double knockout cells (H2&M D. KO) was identical to that in the HAI-2 single knockout cells (HAI-2 S. KO; Fig. 5B comparing lane 4 with lane 2), although the total prostasin levels in the HAI-2 & matriptase double knockout cells might be slightly lower than in the HAI-2 single knockout cells. Both modified cell lines resembled one another with respect to the presence of enzymatically active prostasin and the absence of prostasin zymogen (Fig. 5B, lanes 2 and 4, free active prostasin), a situation which is in stark contrast to the abundant presence of prostasin zymogen and very low or undetectable levels of active prostasin in the parental cells (Fig. 5B, lane 1).
In contrast to the lack of impact of matriptase ablation on prostasin zymogen activation status, prostasin knockout appeared to strongly affect matriptase zymogen activation and subsequently shedding. In the parental cells (Fig. 5C, lane 1), cell-associated matriptase species included the 120 kDa matriptase-HAI-1 complex, 94 kDa full-length matriptase and 70 kDa matriptase zymogen. As described previously (Fig. 3A), HAI-2 loss enhanced matriptase zymogen activation and subsequently the shedding of matriptase, resulting a significant decrease in cell-associated matriptase level (Fig. 5C, lane 2). Prostasin ablation appeared to suppress the matriptase zymogen activation, as indicated by the presence of matriptase zymogen as the major species in the HAI-2/prostasin double knockout cells (Fig. 5C, lane 3), a situation identical to that in the parental cells (Fig. 5C, lane 1). As described above (Fig. 3) and in our previous publication (28), matriptase zymogen activation causes simultaneous matriptase shedding, resulting in reduced cell-associated matriptase. Prostasin deletion may, therefore, reverse the enhanced matriptase zymogen activation and shedding caused by targeted HAI-2 deletion. Thus, prostasin could promote matriptase zymogen activation in this context.
Prostasin enhances spontaneous but not acid-induced matriptase autoactivation
Since an autoactivation process has been identified as the primary mechanism by which matriptase zymogen is converted to the active protease (18,30), the mechanism underlying the apparent ability of prostasin to enhance matriptase zymogen activation was investigated next. As described above (Fig. 3) and in our previous study (28), a significant proportion of the matriptase is shed into the conditioned medium following matriptase zymogen activation. The ability of prostasin to enhance matriptase zymogen activation was verified using both cell lysates and conditioned medium prepared from three prostasin KO Caco-2 variants: one with prostasin KO alone and two with both HAI-2 and prostasin deleted (Fig. 6A). In contrast to the large proportion of matriptase species in the activated state in the Caco-2 parental cells (Fig. 6A, lanes 1 and 2, bands a + d + e/a + b + c + d + e + f; Fig. 3A, close to 40%), the level of activated matriptase in complexes with HAI-1 in the cell lysate and conditioned medium from prostasin KO Caco-2 cells was very low (Fig. 6A, lanes 3 and 4, bands a, d and e). The reduced ratio of activated to total matriptase was also observed in the two HAI-2 and prostasin double knockout Caco-2 variants (Fig. 6A, lanes 5–8, a + d + e/a + b + c + d + e + f). It is worth noting that as the result of reduced matriptase zymogen activation and subsequent shedding, matriptase zymogen levels accumulate in the prostasin knockout cells more than in the parental cells (Fig. 6A and B, comparing lanes 3 with lanes 1 for band c).
Figure 6 .

Prostasin promotes matriptase zymogen activation likely via the matriptase autoactivation machinery. (A) The parental (Parental, lanes 1 and 2), prostasin single knockout (Prostasin KO, lanes 3 and 4) and the two the HAI-2/prostasin double knockout variants (H2&P D. KO1, lane 5 and 6; H2&P D. KO2, lanes 7 and 8) Caco-2 cells were grown in serum-free medium for 24 h. Cell lysates were prepared in a consistent volume, and the conditioned medium were collected and concentrated to the same volume. Cell lysate protein concentration was determined, and samples containing the same amount of protein were prepared (Ly, lanes 1, 3, 5 and 7). Samples of the conditioned media (CM, lanes 2, 4, 6 and 8) of the same volume of the corresponding cell lysates were also prepared and all samples were analyzed by western blot for matriptase species using the mAb M24. (B) The parental and prostasin single knockout Caco-2 cells were exposed to a pH 6.0 buffer for 20 min to induce matriptase zymogen activation (Zymogen Activation +, lanes 2 and 4). The two samples and the non-activation controls (Zymogen Activation −, lanes 1 and 3) were analyzed by immunoblot for matriptase species using the mAb M24. The six different matriptase species are indicated. (C) Parental Caco-2 cells were engineered to express prostasin in a doxycycline-inducible manner (LVX-Prostasin, lanes 1–4) or with empty virus as the doxycycline treatment control (LVX-Control, lanes 5–8). These cells were treated with doxycycline (Dox +, lanes 3, 4, 7 and 8) or PBS as vehicle control (Dox −, lane 1, 2, 5 and 6). Following the induction of prostasin expression, these cells were treated with a pH 6.0 buffer to induce matriptase zymogen activation (Zymogen Activation +, lanes 2, 4, 6 and 8) or PBS for the control (Zymogen Activation −, lanes 1, 3, 5 and 7). Lysates were prepared and equal amount of proteins were analyzed by immunoblot for matriptase species (upper panels) and prostasin species (lower panels). (D) HAI-2/prostasin double KO1 cells were engineered to express prostasin in a doxycycline-inducible manner. In the absence of prostasin induction (Dox−, lanes 1 and 2), the cells were transiently exposed to PBS as non-activation control (Zymogen Activation −, lane 1) or a pH 6.0 buffer to induce matriptase zymogen activation (Zymogen Activation +, lane 2). The double KO cells were treated with doxycycline to induce prostasin expression in the absence of the induction of matriptase zymogen activation (Dox+, lane 3). Lysates were prepared and equal amount of proteins were analyzed by western blot for matriptase species using the M24 mAb (upper panel) or prostasin using the YL11 mAb (lower panel). The matriptase species and the prostasin species are indicated.
Consistent with the reduced matriptase zymogen activation in the cells with targeted prostasin deletion, the ability of prostasin to promote matriptase zymogen activation was also observed in the parental Caco-2 cells (Fig. 6C) and HAI-2/prostasin double KO Caco-2 variant (Fig. 6D), both of which were engineered to express prostasin in a doxycycline-inducible manner. Stimulation of the two cell lines with doxycycline induced very high levels of prostasin expression, much greater than the endogenous prostasin expression in the parental cells (Fig. 6C, lower panel, comparing lane 3 with lane 1) and in the HAI-2/prostasin double KO cells (Fig. 6D, lower panel, comparing lane 3 with lane 1). Along with the induction of prostasin expression, an increase in the levels of 120 kDa matriptase-HAI-1 complex and concomitant decrease in 70 kDa matriptase zymogen was observed (Fig. 6C and D, upper panels, comparing lanes 3 with lanes 1). In the control cells transduced with the empty virus control, the level of matriptase zymogen activation and the level of prostasin protein expression were not changed by doxycycline treatment (Fig. 6C, both panels, comparing lanes 7 with lanes 5). The suppression of matriptase zymogen activation caused by prostasin deletion and the increased matriptase zymogen activation caused by the induced prostasin over-expression provides strong evidence for the ability of prostasin to promote matriptase zymogen activation in Caco-2 cells.
The correlation between prostasin expression and matriptase zymogen activation suggests that prostasin may participate in matriptase zymogen activation by virtue of its enzymatic activity directly mediating the cleavage of matriptase at the activation cleavage site, as a direct upstream activator of matriptase. Alternatively, however, it could be that prostasin promotes matriptase zymogen activation in an indirect fashion, by which prostasin promotes matriptase zymogen activation via other cellular factors or events. These cellular factors or events could then subsequently induce matriptase zymogen activation via the autoactivation machinery, by which the intrinsic zymogen activity of matriptase executes the cleavage for activation. The impact of targeted prostasin deletion on the integrity of the matriptase zymogen activation machinery was, therefore, next investigated in the Caco-2 lines with different prostasin expression status. Transient exposure of cells to a mildly acidic buffer has been shown to be the most effective way to induce matriptase zymogen activation, which it does by accelerating the autoactivation machinery (31). Upon exposing to a pH 6.0 buffer for 20 min, matriptase zymogen can be rapidly converted to 120 kDa activated matriptase complex in Caco-2 cells expressing normal endogenous levels of prostasin, including the parental cells (Fig. 6B, comparing lane 2 with lane 1), Caco-2 cells transduced with the LVX-prostasin in the absence of doxycycline treatment (Fig. 6C, comparing lane 2 with lane 1) and Caco-2 cells transduced with the LVX-Control virus, regardless of the presence of doxycycline (Fig. 6C, comparing lane 6 with lane 5; lane 8 with lane 7). The appearance of 120 kDa matriptase-HAI-1 complex at the cost of 70 kDa matriptase zymogen was also seen in Caco-2 cells lacking prostasin expression, including the prostasin KO Caco-2 cells (Fig. 6B, comparing lane 4 with lane 3) and HAI-2/prostasin double KO Caco-2 cells (Fig. 6D, comparing lane 2 with lane 1). Furthermore, conversion of 70 kDa matriptase zymogen to 120 kDa matriptase-HAI-1 complex was also induced by exposure to pH 6.0 buffer in the presence of very high levels of exogenous prostasin induced by doxycycline treatment in LVX-prostasin Caco-2 cells (Fig. 6C, comparing lane 4 with lane 3). It is worth noting that the presence of high level prostasin promoted matriptase zymogen activation thereby leaving only very low levels of 70 kDa matriptase zymogen, the majority of which was the further converted to 120 kDa matriptase-HAI-1 complex by exposing the cells to a pH 6.0 buffer. Taken together, these data suggest that the integrity of matriptase autoactivation machinery remains intact regardless of the prostasin expression status in the cells. Prostasin, therefore, most likely regulates matriptase zymogen activation via an indirect mechanism that has yet to be elucidated, although it is true that a direct role via its proteolytic activity on matriptase zymogen activation cannot be completely excluded. It is also worth noting that prostasin zymogen activation was also induced along with the induction of matriptase zymogen activation by acid exposure of the cells, as indicated by the simultaneous decrease in the prostasin zymogen level and increase in the activated prostasin level (Fig. 6C, lower panel, comparing lane 2 with lane 1; lane 6 with lane 5 and lane 8 with lane 7). This increase in prostasin zymogen activation along with the induction of matriptase zymogen activation is consistent with the role of matriptase in prostasin zymogen activation, as an upstream activator (20,32).
Targeted deletion of HAI-2 does not cause a substantial reduction in the level of HAI-1 monomer in HaCT keratinocytes
A functional linkage between HAI-2 and prostasin has been seen in Caco-2 cells but not in HaCaT keratinocytes (24). It is of interest and potentially important for our understanding of the organ-selective damage caused by HAI-2 mutations in patients to study and compare the impact of targeted HAI-2 deletion on matriptase, prostasin and HAI-1 in skin epithelial cells. Four HaCaT variant cell lines with HAI-2 deletion were generated using the CRISPR methodology (Fig. 7). The lack of HAI-2 protein expression in the four knockout HaCaT variant lines (HAI-2 KO1-4) was verified by western blot analysis (Fig. 7A). HAI-2 was detected only in its monomer form (Fig. 7A, lane 1). The absence of prostasin-HAI-2 complex in the HaCaT cells is in stark contrast to the situation in Caco-2 cells (Fig. 1A, lane 1) and is consistent with the lack of functional linkage between HAI-2 and prostasin in skin and in human keratinocytes (17,24). The expression and zymogen activation status of matriptase, prostasin and HAI-1 in these cells were further analyzed by western blot. Like in Caco-2 cells, matriptase can be found in HaCaT cells in the 120 kDa HAI-1 complex form, the full-length form and the 70 kDa zymogen form (Fig. 7B). The appearance of 120 kDa matriptase-HAI-1 complex in the four HAI-2 KO variants but not in the parental cells demonstrates an increase in matriptase zymogen activation caused by HAI-2 loss, just as is seen in the Caco-2 cells. Similarly, an increase in zymogen activation and even in protein level was also observed for prostasin, as shown by the appearance of the prostasin-HAI-1 complex (Fig. 7C). The enhanced zymogen activation of both the serine proteases was also seen and verified by the appearance of 120 kDa matriptase-HAI-1 complex and prostasin-HAI-1 complexes, including the 100 kDa and the degraded forms in the immunoblot analysis for HAI-1 species (Fig. 7D). These data indicate that human keratinocytes respond to targeted HAI-2 deletion by increasing both matriptase and prostasin zymogen activation, in a similar way to Caco-2 colorectal adenocarcinoma cells. In spite of these similarities, however, the level of HAI-1 monomer remains very high in the four HAI-2 deleted HaCaT variants (Fig. 7D, lanes 2–5). Thus, although targeted HAI-2 deletion causes an increase in matriptase and prostasin zymogen activation in both Caco-2 and HaCaT cells, the level of HAI-1 monomer remains high in the HaCaT keratinocytes, a distinguishing difference associated with HAI-2 loss in the two epithelial cell lines, which possibly sheds light on the different severity of the phenotype observed in the tissues from which these cell lines originate in patients with SPINT 2 mutations.
Figure 7 .
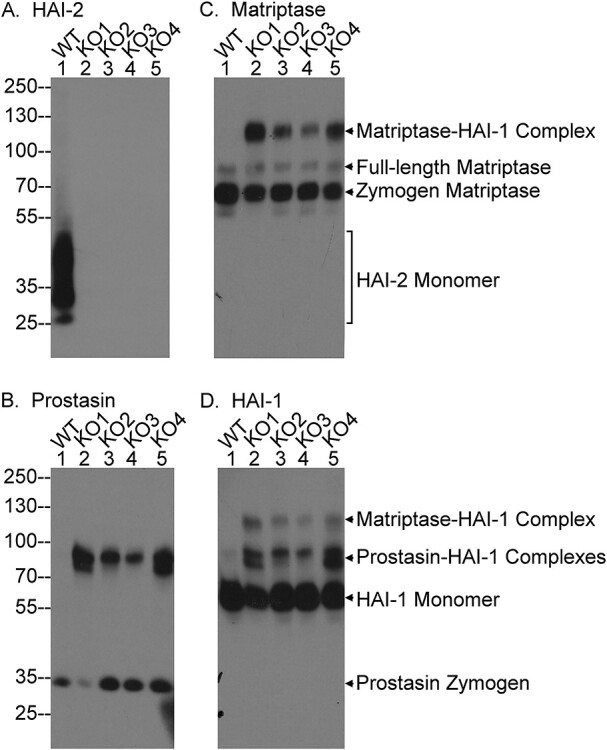
HaCaT keratinocytes with targeted deletion of HAI-2 retain HAI-1 largely in the monomer form. HAI-2 was deleted by CRISPR in HaCaT keratinocytes. Lysates were prepared from the parental (PAR., lanes 1) and four selected HAI-2 knockout HaCaT cells (KO1-KO4, lanes 2–5) and equal amount protein were analyzed by western blot for HAI-2 expression using the mAb DC16 (A; HAI-2), prostasin species using the YL11 mAb (B; Prostasin), HAI-1 species using the mAb M19 (C; HAI-1) and matriptase species using the mAb M24 (D; Matriptase). The three matriptase species, the two prostasin species, HAI-1 monomer and HAI-2 monomer are indicated.
Discussion
By systemically comparing the impact of targeted HAI-2 deletion on the proteolytic status of prostasin and matriptase, an imbalance favoring enhanced proteolysis caused by the loss of protease inhibitor was observed that is much more severe in Caco-2 cells than HaCaT cells. The severity is manifested by the prolonged presence of residual active prostasin and the depletion of HAI-1 monomer in the colorectal adenocarcinoma cells, but not in the keratinocytes. Caco-2 and HaCaT cells do resemble one another with respect to the increased matriptase and prostasin zymogen activation resulting from HAI-2 loss. The cell-type selective functional linkage between HAI-2 and prostasin in Caco-2 cells appears, however, to be the primary reason for the severe imbalance observed in Caco-2 cells but the HaCaT keratinocytes. This suggests that targeted deletion of HAI-2 will leave prostasin proteolytic activity unchecked in Caco-2 cells but not in HaCaT cells. The pre-existing constitutive, high levels of prostasin zymogen activation found in Caco-2 also likely contributes to the severe imbalance. It is important to note that constitutive, high-level prostasin zymogen activation has also been seen in human intestinal tissues as evidenced by the presence of high levels of activated prostasin complexes with the HAIs, which represent the majority of the total prostasin found in these tissues (24). This in vivo context for prostasin is likely what makes the GI tract so susceptible to HAI-2 loss, as a consequence of excessive prostasin proteolysis. Although prostasin is also activated at high levels in human skin, the tissue distribution and subcellular localization of the proteins does not support a functional relationship between prostasin and HAI-2 in human skin, where HAI-1 serves as the endogenous inhibitor of prostasin (17). This functional relationship is faithfully phenocopied in HaCaT keratinocyte cells. The lack of a functional linkage between prostasin and HAI-2 is likely what protects human skin and human keratinocytes from the potential hazards caused by excessive prostasin proteolysis when HAI-2 function is lost.
As a membrane-associated protease, prostasin is synthesized, undergoes maturation and likely performs physiological functions in specific sub-compartments of the secretory pathway and/or the extracellular milieu, including on the cell surface. The cell-type selective inhibition of active prostasin by HAI-2 has been attributed to whether HAI-2 is targeted into the close vicinity of prostasin. For example, both prostasin and HAI-2 have been detected near the terminal webs underneath the villi of human enterocytes (24). The physical proximity likely gives HAI-2 direct access to active prostasin for the establishment of functional linkage between the two proteins. In contrast, prostasin and HAI-2 are differentially expressed during the course of epidermal differentiation with HAI-2 primarily expressed in the basal and spinous keratinocytes and prostasin primarily in the granular keratinocytes (19). Furthermore, HAI-2 has been detected primarily in intracellular vesicles and prostasin is present in an un-identified organelle-like structure that forms and likely matures in concert with epidermal differentiation (19). The different subcellular localizations of these proteins mean that there is no functional linkage between prostasin and HAI-2 in keratinocytes. The important role that subcellular co-localization plays in functional linkages has, in fact, been also observed for the two forms of HAI-2. HAI-2 is expressed in two different forms which are present with a relatively similar ratio between them in different cell lines: one form with extensive terminal N-acetylglucosamine branching and the other form with oligomannose type glycosylation, without terminal N-acetylglucosamine branching (27). The two HAI-2 forms are targeted to different subcellular compartments with the form with extensive N-glycan branching detected in vesicle/granule structures and the form with light N-glycan detected in the perinuclear region. The HAI-2 form with extensive N-glycan branching but not the form with light N-glycan is co-localized with and, responsible for, prostasin inhibition in enterocytes. The lack of co-localization results in the HAI-2 form with light N-glycan not being an endogenous inhibitor of prostasin in the cells. Because the subcellular distribution and subcellular localization of HAI-1 also differs from that of HAI-2, the dual mechanism with both HAI-1 and HAI-2 participating in prostasin inhibition in Caco-2 suggests that HAI-1 and HAI-2 likely control prostasin in different subcellular compartments. Caco-2 cells, therefore, likely express prostasin in two different subcellular localizations, in which prostasin is differentially regulated with different physiological functions. With the targeted deletion of HAI-2, it is only that portion of prostasin that is under the control by HAI-2 is impacted. In response to HAI-2 deletion, Caco-2 cells might adjust the subcellular targeting for prostasin and HAI-1 so that some proportion of the HAI-2-regulated prostasin comes under the regulation of HAI-1, thereby leading to the increase in prostasin-HAI-1 complex detected in HAI-2 knockout Caco-2. If this is the case, the impact of HAI-2 deletion on cellular and subcellular events would become very complicated and cannot be explained simply by the enzyme-inhibitor relationship of HAI-2 and prostasin. Alternatively, the inhibition of this proportion of the HAI-2-regulated prostasin by HAI-1 in HAI-2 KO Caco-2 cells could occur after the lysis of cells for analysis, but this does not seem very likely due to measures taken to control of the temperature of the lysates. If this were the case, then the level of unchecked active prostasin caused by HAI-2 loss could be much higher in the living cells than that in the lysate.
Tissue-type selective phenomena associated with the functional relationship of prostasin and HAI-2 have been also observed in mice. Dysregulated prostasin proteolytic activity is responsible for placental defects and an early embryonic lethality in HAI-2 knockout mice (26,33). Prostasin proteolytic activity, however, appears to play no role in the early-onset intestinal failure caused by loss of HAI-2 (34). The differential role of prostasin enzymatic activity in the placental and intestinal defects caused by loss of HAI-2 indicates the enzyme-inhibitor relationship between HAI-2 and prostasin might be present in placenta but not in the intestinal tissues in mice. In spite of the resemblance in the tissue selectivity concerning the functional relationship between prostasin and HAI-2, human and rodents are surprisingly different with respect to the enzyme-inhibitor relationship in intestinal tissue. The relationship between HAI-2 and prostasin in human intestinal tissues has been supported by the presence of the enzyme-inhibitor complexes in the lysate of the intestinal tissues and the in vivo co-localization of the proteins near the brush borders of villus epithelial cells (24). The presence of enzyme-inhibitor complexes was initially observed in Caco-2 colon adenocarcinoma cells. This simple direct enzyme-inhibitor relationship is further supported by the robust increase in activated prostasin either in complex with HAI-1, or as the free active form, when HAI-2 is deleted in Caco-2 in the current study (Fig. 2). More recently, uncontrolled matriptase activity was reported to be responsible for the intestinal defects caused by loss of HAI-2 in mice (35). Although increased matriptase zymogen activation has been seen with targeted HAI-2 deletion in the current study, it is the active prostasin and not matriptase that depletes the HAI-1 monomer. Thus, it is likely that the unchecked active prostasin and not any increase in matriptase activity that contributes to the intestinal defects seen in the patients with SCSD. Although HAI-2 mutations have been identified and are associated with the pathogenesis of SCSD, all SCSD patients have at least one SPINT2 mutant allele in which the Kunitz domain 1 remains intact. It is the Kunitz domain 1 that is responsible for the inhibition of matriptase and prostasin. In fact, the vast majority of SCSD patients express HAI-2 mutants with missense point mutations of Kunitz domain 2. There are no SCSD patient lacking all expression of HAI-2, analogous to the loss of HAI-2 in HAI-2 knockout mouse or the Caco-2 cells with targeted deletion of HAI-2. It remains unclear and requires further investigated to establish whether and to what degree dysregulated prostasin or matriptase proteolytic activity is present in and contributes to the pathogenesis of SCSD.
In addition to the initial identification in SCSD patients (3), SPINT2 mutations have been reported in a proportion of patients with congenital tufting enteropathy [CTE; MIM#613217; (36–38)], majority of cases of which are caused by mutation of epithelial cell adhesion molecule—EPCAM (39). Distinct differences in subcellular ultrastructure and clinical manifestations have, however, been found between the two human genetic disorders. Although congenital diarrhea represents the primary syndrome for both CTE and SCSD, high sodium loss to the feces is much more common in SCSD than in CTE (38,40,41). Metabolic acidosis has been observed in patients with SCSD but is unusual in CTE (40,41). In CTE, the affected enterocytes exhibit a tear-drop configuration with rounding apical plasma membrane and likely with shortened microvilli and elongated desmosomes (42). The cytoplasmic organelles in these cells appear well preserved (43). In contrast, marked vacuolation has been observed in villous columnar epithelial cells in patients with SCSD with slightly mounded apical plasma membrane. Furthermore, distinct membranous whorls have been observed within vacuoles, lysosomal bodies and swollen mitochondria (40). Poorly developed desmosomes have been seen in duodenal biopsies of patient with SPINT2 mutation (38). Although enterocyte tufting has been found in some patients with SPINT2 mutations (36–38), the difference in subcellular ultrastructure and clinical manifestations clearly indicate a weak functional and pathophysiological linkage between HAI-2 and EpCAM. Recent studies, however, have indicated that increased matriptase rather than prostasin proteolytic activity caused by HAI-2 deficiency or mutation cleaves EpCAM and drives the onset and development of CTE (34,35,44,45). The conclusions of these studies are at odds with the conclusions of the current study in which targeted HAI-2 deletion causes an increased proteolysis primarily through uncontrolled prostasin rather than matriptase activity. The discrepancy most likely results from the different study models, the data quality and the interpretation of the results. As described previously, mouse models are of limited relevance for studying the biological role of matriptase in human skin due to the inverse expression pattern for matriptase through epidermal differentiation in human versus rodent skin (17–19,46,47). The mouse is also likely to be a model of limited relevance in studies of the biological role of HAI-2 in human intestinal tissues due to the fact that HAI-2 is an endogenous inhibitor of prostasin but not matriptase in human intestine (24), in contrast to the tight functional linkage between HAI-2 and matriptase and not prostasin in mouse intestines (34,35). The functional linkage among matriptase, HAI-2 and EpCAM in human intestinal epithelial cells was initially reported in a study by Wu et al. (44). The study described out a series of experiments, leading to the identification of EpCAM as a substrate of matriptase and the regulatory role of HAI-2 in matriptase-mediated EpCAM cleavage. In spite of the exciting conclusion that could shed light on the molecular events involved in the onset and development of CTE, the study is simply not consistent with the fact that HAI-2 is not targeted to the basolateral plasma membrane, which is where both matriptase and EpCAM are targeted in intestinal epithelial cells (24,25). The predominantly intracellular localization of HAI-2 has consistently been observed in different cells types including mammary epithelial cells (22), keratinocytes (19), enterocytes (24,48) and neoplastic B-cells (23). It is worth noting that in intestinal tissues HAI-2 has been independently detected by two different studies as present as intracytoplasmic granules beneath the apical plasma membrane (24,48). The lack of a functional linkage between the proteins is also supported by the lack of matriptase-HAI-2 complex in lysates prepared from Caco-2 cells or human intestinal tissues, in which activated matriptase was detected in complex with HAI-1 (24).
The experiments and data that support the involvement of matriptase in EpCAM cleavage are also problematic. Although matriptase is able to cleave EpCAM in solution and is co-localized with EpCAM on the cell surface, the correlation between matriptase expression and EpCAM degradation varied significantly from experiment to experiment. For example, in Figure 4 of the study by Wu et al. (44), the level of full-length EpCAM stably decreased as the level of matriptase increased, which was used as evidence for a role for matriptase in EpCAM cleavage. These data are, however, not supportive but, in fact, in opposition to the conclusion drawn. An inverse correlation of matriptase expression level with the combined level of full-length and cleaved EpCAM should have suggested that matriptase expression reduces the expression of EpCAM rather than that it promotes cleavage of EpCAM. The lack of correlation between matriptase expression and EpCAM cleavage is also supported by the inverse ratios of full-length versus cleaved EpCAM in Caco-2 cells between two independent experiments. In the experiment in Figure 4B of the study by Wu et al. (44), cleaved EpCAM is much more abundant than full-length EpCAM in the control Caco-2 cells. In another experiment presented in Figure 4C, full-length EpCAM is much more abundant than cleaved EpCAM in the control Caco-2 cells. Thus, it seems that the extent of EpCAM cleavage as assessed by the ratio of full-length to cleaved protein varies from experiment to experiment in Caco-2 cells.
In summary, the structural complementarity between serine protease domains and Kunitz domains is what renders matriptase and prostasin as the target proteases of the HAIs. Similarly, the trypsin-like activity of matriptase and prostasin confers on the two serine proteases the ability to activate one another. These biochemical relationships are important but are, however, not sufficient to explain the cellular and in vivo functional relationships among these four proteins. The membrane anchorage of these proteins renders their subcellular localization as an essential determinant of whether a biochemical functional relationship can be established at the cellular level and in vivo settings. HAI-1 has, therefore, been identified to be the default protease inhibitor for matriptase and prostasin, and HAI-2 to be a selective inhibitor only in some cells. The similarity and differences between Caco-2 colon adenocarcinoma cells versus HaCaT keratinocytes in the impact on these cells caused by targeted deletion of HAI-2 on prostasin and matriptase can be largely attributed to cell-type selective functional linkages. Along with the loss of HAI-2, the resultant free active prostasin overwhelms and depletes HAI-1 monomer resulting in a prolonged lifespan of the remaining free, active prostasin in Caco-2 cells, but not in HaCaT cells. The shared increase in matriptase zymogen activation caused by HAI-2 loss in both cell lines could result from a combination of the increased prostasin proteolysis and the increased ratio of the combined cellular proteases (prostasin and matriptase) relative to the combined protease inhibitors (the HAIs). It is worth noting that an indirect mechanism rather than the direct enzymatic activity of prostasin most likely contributes to matriptase zymogen activation when HAI-2 is deleted. Although it is the structural and biochemical aspects of the proteins that establish the possibility of functional linkages among prostasin, matriptase and the HAIs, their physiological relationships are also determined by the tissue distribution, subcellular localization and the cellular environments. The cell-type selective impact caused by HAI-2 loss sheds light on the organ-selective defects observed in SCSD patients. SCSD patients, however, express at least one HAI-2 mutant allele with a missense point mutation in the Kunitz domain 2, in which the structure and likely the biochemical function of the Kunitz domain 1 could remain intact. It remains unclear whether, in fact, excessive prostasin proteolytic activity is present in SCSD patients.
Materials and Methods
Cell lines
The colorectal adenocarcinoma cells line Caco-2 was obtained as a gift from Dr Toni Antalis of the University of Maryland Baltimore. The cells were cultured in RPMI-1640 medium (Lonza, Walkersville, MD), supplemented with 10% fetal bovine serum (FBS) at 37°C in a humidified atmosphere with 5% CO2. The level of free active prostasin in this lineage of Caco-2 cells is much lower than the Caco-2 cells used in our previous study (24). It is not clear what the reason is for the discrepancy between the two versions of Caco-2 cells, the identity of both lineages was verified by short tandem repeat based fingerprinting assay (not shown). The cells from Dr Antalis have been used in this study since they better mimic the negligible level of free active prostasin observed in human intestinal tissue lysates (24).
Western blot and antibodies
Protein samples, including cell lysates and conditioned media, were analyzed for the presence of the HAIs, matriptase and prostasin by immunoblot using the respective mAbs, as indicated. Lysates were prepared by lysing the cells with either 1% Triton 100 in phosphate buffered saline (PBS) or radioimmunoprecipitation assay (RIPA) buffer. There was no obvious difference in the results generated using lysates prepared with the two different lysis buffers. Ellman’s Reagent (5,5′-dithio-bis-[2-nitrobenzoic acid]) (DTNB) (Sigma-Aldrich, St. Louis MO, USA) at 1 mm was included in the lysis buffers to prevent cleavage of the disulfide bond which connects matriptase serine protease domain and the non-catalytic domain (49). After removal of insoluble debris by centrifugation, the protein concentration was determined by the Bio-Rad Protein Assay Dye Reagent (Bio-Rad Laboratories, Hercules, CA, USA) or the Pierce BCA Protein Assay (ThermoScientific, Rockford, IL USA) using an ELx808 microplate reader (BioTek, Winooski, VT USA). Equal amounts of lysate proteins were mixed with 5X sample loading buffer, to a final concentration of 1X, were run on 10% SDS-PAGE under non-reducing and non-boiled conditions, transferred to Amersham/Protran 0.45 um nitrocellulose (GE Healthcare Life Sciences, Marlborough, MA USA) and then blocked with 5% dry milk in PBS/0.1%Tween 20 (Sigma-Aldrich, St. Louis MO, USA).
In experiments where both lysates and conditioned media were analyzed, cell lysates were prepared in a given volume and the conditioned medium collected and concentrated to the same volume as the lysate using centrifugal filter units (Amicon Ultracel 10K, Millipore, Billerica, MA) following the manufacturer’s instructions. Lysate protein concentration was then measured as above and the volume of lysate required for the desired amount of protein calculated. Samples of conditioned medium of the same volume as the lysate were then prepared, and both samples subjected to western blot analysis as described. Blots were probed with primary antibody and goat anti-mouse antibody conjugated to horseradish peroxidase (HRP; Jackson ImmunoEesearch, West Grove, PA USA). The primary antibodies used were the prostasin mAb YL11, the matriptase mAb M24, the HAI-1 mAb M19 and the HAI-2 mAbs DC16. The generation and characterization of these mAbs can be found in our previous publications (7,22,27,50). Primary antibody binding was visualized using HRP-labeled secondary antibodies and Western Lightning enhanced chemiluminescence pro reagent (Thermo Fisher Scientific, Waltham, MA USA) either exposed to x-ray film for various times for optimal signal or using an Amersharm Imager 600.
Acid-induced matriptase activation
Matriptase zymogen activation was induced by transient exposure the cells to a pH 6.0 buffer as described previously (51–53). Briefly, cells are incubated with either control buffer (PBS), or phosphate buffer (pH 6.0) for 20 min, after which the cells and the conditioned buffer are separated and analyzed (54).
Targeted deletion of SPINT2, ST14 or prss8 in Caco-2 colon adenocarcinoma cells and HaCaT keratinocytes
CRISPR design
The first exon of SPINT2 (nucleotide 292 to 541 of NM_001166103) was queried in the CRISPR Target Finder algorithm (http://crispr.mit.edu/) to obtain two guide RNAs (gRNA) with unique nickase sites. In order to subclone each gRNA into pX335 (Addgene), the following oligonucleotides (IDT) were designed and annealed as previously described (54).
gRNA1 antisense 5′AAACGATCGCGAGACCCCAACGGC3′
gRNA1 sense 5′CACCGCCGTTGGGGTCTCGCGATC3′
gRNA2 antisense 5′AAACTGGCCAGCTCAGCCGAGACGC3′
gRNA2 sense 5′CACCGCGTCTCGGCTGAGCTGGCCA3′
The same procedure was conducted for SPINT1, ST14 and PRSS8 to obtain the following gRNAs for each gene:
SPINT1 (nucleotide 79 to 327 of NM_001032367.2)
gRNA1 antisense 5′AAACCGGTCAGCAACGGAGCTACCC3′
gRNA1 sense 5′CACCGGGTAGCTCCGTTGCTGACCG3′
gRNA2 antisense 5′AAACGCGCACGGTGGGGGACTCCAC3′
gRNA2 sense 5′CACCGTGGAGTCCCCCACCGTGCGC3′
ST14 (nucleotide 6 to 279 of NM_021978.4)
gRNA1 antisense 5′AAACCGAGCGGCCTCGGGGACCATC3′
gRNA1 sense 5′CACCGATGGTCCCCGAGGCCGCTCG3′
gRNA2 antisense 5′AAACCCCTTGCGGGCCCGATCGCTC3′
gRNA2 sense 5′CACCGAGCGATCGGGCCCGCAAGGG3′
PRSS8 (nucleotide 1589 to 1838 of U33446.1)
gRNA1 antisense 5′AAACGAAGGGGGTCCTGGGGCCTGC3′
gRNA1 sense 5′CACCGCAGGCCCCAGGACCCCCTTC3′
gRNA2 antisense 5′AAACCGACCGGAGTAATCCAAGATC3′
gRNA2 sense 5′CACCGATCTTGGATTACTCCGGTCG3′
Plasmid DNA for each gRNA construct was prepared using the HiSpeed Plasmid Midi kit following the manufacturer’s instructions (Qiagen).
Transfection and selection
Caco-2 cells and HaCaT cells were grown in DMEM (Lonza, Walkersville, MD USA) supplemented with 10% heat inactivated FBS (Sigma-Aldrich, St. Louis MO, USA). Cells were plated at 30–40% confluency on 10 cm dishes (Corning) 48 h prior to transfection. Cells were co-transfected with each gRNA construct and pEF6/V5 His (Invitrogen, Carlsbad, CA USA) according to the Lipofectamine 2000 instructions (LifeTechnologies, Frederick, MD USA) and as previously described (http://www.bio-protocol.org/e130). Briefly, 1.125 pmol of total plasmid DNA in a 0.5:0.5:0.2 ratio (gRNA1sense:gRNA1antisense:pEF6/V5 His) was added to 500 μl OptiMEM (Life Technologies, Frederick, MD USA). Separately, 40 μl of Lipofectamine 2000 was added to 500 μl OptiMEM. These two solutions were combined and incubated at room temperature for 30′. The media on the cells was replaced with 2.5 ml of serum-free DMEM, and the transfection mixture volume was raised to 2.5 ml with OptiMEM and then added to the cells dropwise. Six hours later 5 ml of DMEM supplemented with 20% FBS was added to the cells. Cells were grown another 48 h then split into 10 cm dishes at 1, 1:10, 1:100 dilutions in the presence of 10 ug/ml Blasticidin (LifeTechnologies) for 3–4 weeks. Individual clones were isolated by gently pushing the cells off the dish and immediate drawing them up with a pipette tip with a blunted and beveled bore. Each clone in 10 μl Dulbecco’s phosphate-buffered saline (DPBS; Lonza, Walkersville, MD USA) was then placed into a flat-bottomed 96-well plate (Corning, Kennebunk, ME USA) with 30 μl 0.05% Trypsin-0.53 mm ethylenediaminetetraacetic acid (EDTA; Sigma) for 5–10′ in a 37°C incubator. Cells clumps were disrupted by gentle pipetting and transferred to a new 96-well plate with 200 μl complete media without Blasticidin. Clones were expanded for the next 3–6 weeks and screened for HAI-2 expression by immunoblot.
Analysis of CRISPR-generated alleles
The sequences of several clones have been analyzed to confirm the presence of mutations that would correlate with a loss of gene expression. Genomic DNA was isolated from individual clones. In brief, approximately 5E5 trypsinized cells were washed in DPBS, then centrifuged at 5000 rpm for 5′ at room temperature in a microcentrifuge (Eppendorf). The supernatant was discarded, and cells were gently re-suspended by flicking the tube. 200 μl of proteinase K (PK) digestion buffer (50 mm Tris–HCl pH 8.0, 10 mm EDTA, 50 mm NaCl, 0.5% SDS, 0.5 μg/μl proteinase K (LifeTechnologies) was added and the tubes were incubated for 2 h at 37°C. An equal volume of digestion buffer without proteinase K was added, and then the lysates were extracted twice with an equal volume of 1:1 phenol:chloroform (Sigma) and once with chloroform. DNA was precipitated with addition of two volumes of 100% ethanol, washed with 70% ethanol, air-dried and re-suspended in water.
100 ng of genomic DNA was used as the PCR template with the following oligos (IDT):
5′ GCG AGT GAG GAG CAG ACC 3′
5′ GCT CCC AAA CCT CAT TTC AA 3′
OneTaq 2X Master Mix (NEB) was used according to the manufacturer’s instructions and with the following program: 95C 2′ 1 cycle; 95C 30s, 54C 30s, 72C 1′ for 35 cycles; 72C 5′; 4C 15′, on a MJ mini personal thermocycler (BioRad).
PCR products were isolated from 1.5% LMP/1X TBE agarose gel using the QIAquick gel extraction kit (Qiagen) and sequenced using the same PCR primers (Genewiz).
Exogenous protein expression with the Tet-On system
Plasmids
RC202044, a SPINT2/HAI-2 (NM_021102) Human Myc-DDK-Tagged ORF clone, was purchased from Origene (Rockville, MD). pLVX-Tet3G (TetON) and pLVX-TRE3G were purchased from Clontech/Takara. The generation of pCDNA3.1-SPINT1/HAI-1 and pCDNA3.1-ST14/Matriptase have been previously described (30). PRSS8/Prostasin cDNA was generated using the AMV-reverse transcription system (Promega, Madison, WI USA) and Caco-2 mRNA. Subsequent PCR amplification used Phusion High Fidelity PCR Master Mix (NEB) and the following oligos (IDT), forward 5′ATGGCCCAGAAGGGGGTC3′ and reverse 5′TCAGTGCTCGCTGAGCCATG3′. The PCR product was phosphorylated with T4 Polynucleotide Kinase (NEB) and then gel isolated prior to blunt-end ligation into pCDNA3.1 and pLVX-TRE3G using the Quick Ligation Kit (NEB). Both PRSS8 constructs were sequenced (Genewiz) prior to use.
Like PRSS8, SPINT2, SPINT1 and ST14 were subcloned into pLVX-TRE3G and used in co-transfections with pHRCMV8.2DR and pCMV-VSVG (both obtained from Dr Todd Waldman, Georgetown University) to produce lentiviruses in 293T as previously described (55).
Lentivirus production and infection
HEK 293T cells were grown in DMEM (Lonza) supplemented with10% FBS (Sigma) and plated at 60% confluency. pLVX-TRE3G-X or pLVX-Tet3G were mixed with pHRCMV8.2DR and pCMV-VSVG (a gift obtained from Dr Todd Waldman, Georgetown University) in a ratio of 1:1:0.1, respectively, and then co-transfected using Lipofectamine 2000 (Life technologies) according to manufacturer’s instructions. After 24 h media was replaced with DMEM supplemented with 15% FBS. Viral supernatants were collected 48 h after transfection, centrifuged, passed through a 0.45 μm syringe filter (Millipore) and then frozen at −80°C. HT1080 cells were used to determine the titer by serial dilution of the viral supernatants.
Caco-2 parental cells and variants were infected at an approximate MOI of 5 and 6 μg/ml polybrene in two rounds of selection. First, Tet3G (TetOn) virus carrying the neomycin resistance gene was used with subsequent selection using G418 (Life Technologies) at a concentration of 1000 μg/ml for at least two passages. Second, LVX-TRE3G-SPINT2, -SPINT1, -ST14 or -PRSS8 virus carrying the puromycin resistance gene was used to infect the G418-resistant population with subsequent selection using puromycin (Life Technologies) at a concentration of 2 μg/ml for at least two passages. LVX-TRE3G without insert was used as a negative control throughout this study. Doxycycline at variable concentrations (0–10 μg/ml) was added to the media for 48 h to induce expression of the gene of interest prior to harvesting of the cells and/or media. Conflict of Interest statement: There is no conflict of interest among all authors. The funders had no role in study design, data collection and analysis, the decision to publish, or the preparation of the manuscript.
Funding
This study was supported by National Cancer Institute (grant no. R01 CA 123223 to M.D.J. and C.Y.L.); Ministry of National Defense Medical Affairs Bureau, Taiwan (grant nos. MAB-108-079 and MAB-109-042 to J.-K.W.); Taipei City Hospital, Taipei, Taiwan (grant nos. 10801-62-068, 10901-62-056, TPCH-109-25, TPCH-110-28 to C.-H.L.).
Contributor Information
Robert B Barndt, Lombardi Comprehensive Cancer Center, Department of Oncology Georgetown University, Washington, DC 20057, USA.
Mon-Juan Lee, Lombardi Comprehensive Cancer Center, Department of Oncology Georgetown University, Washington, DC 20057, USA; Department of Bioscience Technology, Chang Jung Christian University, Tainan 71101, Taiwan; Department of Medical Science Industries, Chang Jung Christian University, Tainan 71101, Taiwan.
Nanxi Huang, Lombardi Comprehensive Cancer Center, Department of Oncology Georgetown University, Washington, DC 20057, USA.
Dajun D Lu, Lombardi Comprehensive Cancer Center, Department of Oncology Georgetown University, Washington, DC 20057, USA.
See-Chi Lee, Lombardi Comprehensive Cancer Center, Department of Oncology Georgetown University, Washington, DC 20057, USA.
Po-Wen Du, Lombardi Comprehensive Cancer Center, Department of Oncology Georgetown University, Washington, DC 20057, USA; Department of Biochemistry National Defense Medical Center, Taipei 114, Taiwan.
Chun-Chia Chang, School of Medicine, National Defense Medical Center, Taipei 114, Taiwan.
Ping-Feng B Tsai, School of Medicine, National Defense Medical Center, Taipei 114, Taiwan.
Yu-Siou K Huang, School of Medicine, National Defense Medical Center, Taipei 114, Taiwan.
Hao-Ming Chang, Department of Surgery, Tri-Service General Hospital, Taipei 114, Taiwan.
Jehng-Kang Wang, Department of Biochemistry National Defense Medical Center, Taipei 114, Taiwan.
Chih-Hsin Lai, Department of Dentistry Renai Branch, Taipei City Hospital, Taipei 106, Taiwan.
Michael D Johnson, Lombardi Comprehensive Cancer Center, Department of Oncology Georgetown University, Washington, DC 20057, USA.
Chen-Yong Lin, Lombardi Comprehensive Cancer Center, Department of Oncology Georgetown University, Washington, DC 20057, USA.
References
- 1.Kawaguchi, T., Qin, L., Shimomura, T., Kondo, J., Matsumoto, K., Denda, K. and Kitamura, N. (1997) Purification and cloning of hepatocyte growth factor activator inhibitor type 2, a Kunitz-type serine protease inhibitor. J. Biol. Chem., 272, 27558–27564. [DOI] [PubMed] [Google Scholar]
- 2.Chiu, Y.L., Wu, Y.Y., Barndt, R.B., Yeo, Y.H., Lin, Y.W., Sytwo, H.P., Liu, H.C., Xu, Y., Jia, B., Wang, J.K. et al. (2019) Aberrant regulation favours matriptase proteolysis in neoplastic B-cells that co-express HAI-2. J. Enzyme Inhib. Med. Chem., 34, 692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heinz-Erian, P., Müller, T., Krabichler, B., Schranz, M., Becker, C., Rüschendorf, F., Nürnberg, P., Rossier, B., Booth, I.W., Holmberg, C. et al. (2008) Mutations in SPINT2 cause a syndromic form of congenital sodium diarrhea. Am. J. Hum. Genet., 84, 188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bode, W. and Huber, R. (1992) Natural protein proteinase inhibitors and their interaction with proteinases. Eur. J. Biochem., 204, 433–451. [DOI] [PubMed] [Google Scholar]
- 5.Bode, W. and Huber, R. (2000) Structural basis of the endoproteinase-protein inhibitor interaction. Biochim. Biophys. Acta - Protein Struct. Mol. Enzymol., 1477, 241–252. [DOI] [PubMed] [Google Scholar]
- 6.Shimomura, T., Denda, K., Kitamura, A., Kawaguchi, T., Kito, M., Kondo, J., Kagaya, S., Qin, L., Takata, H., Miyazawa, K. et al. (1997) Hepatocyte growth factor activator inhibitor, a novel Kunitz-type serine protease inhibitor. J. Biol. Chem., 272, 6370–6376. [DOI] [PubMed] [Google Scholar]
- 7.Lin, C.Y., Anders, J., Johnson, M. and Dickson, R.B. (1999) Purification and characterization of a complex containing matriptase and a Kunitz-type serine protease inhibitor from human milk. J. Biol. Chem., 274, 18237–18242. [DOI] [PubMed] [Google Scholar]
- 8.Lai, C.H., Lai, Y.J.J., Chou, F.P., Chang, H.H.D., Tseng, C.C., Johnson, M.D., Wang, J.K. and Lin, C.Y. (2016) Matriptase complexes and prostasin complexes with HAI-1 and HAI-2 in human milk: significant proteolysis in lactation. PLoS One, 11, e0152904. doi: 10.1371/journal.pone.0152904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szabo, R., Kosa, P., List, K. and Bugge, T.H. (2009) Loss of matriptase suppression underlies Spint1 mutation-associated ichthyosis and postnatal lethality. Am. J. Pathol., 174, 2015–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szabo, R., Hobson, J.P., Christoph, K., Kosa, P., List, K. and Bugge, T.H. (2009) Regulation of cell surface protease matriptase by HAI2 is essential for placental development, neural tube closure and embryonic survival in mice. Development, 136, 2653–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carney, T.J., von der Hardt, S., Sonntag, C., Amsterdam, A., Topczewski, J., Hopkins, N. and Hammerschmidt, M. (2007) Inactivation of serine protease Matriptase1a by its inhibitor Hai1 is required for epithelial integrity of the zebrafish epidermis. Development, 134, 3461–3471. [DOI] [PubMed] [Google Scholar]
- 12.Mathias, J.R., Dodd, M.E., Walters, K.B., Rhodes, J., Kanki, J.P., Look, A.T. and Huttenlocher, A. (2007) Live imaging of chronic inflammation caused by mutation of zebrafish Hai1. J. Cell Sci., 120, 3372–3383. [DOI] [PubMed] [Google Scholar]
- 13.Friedrich, R., Fuentes-Prior, P., Ong, E., Coombs, G., Hunter, M., Oehler, R., Pierson, D., Gonzalez, R., Huber, R., Bode, W. et al. (2002) Catalytic domain structures of MT-SP1/matriptase, a matrix-degrading transmembrane serine proteinase. J. Biol. Chem., 277, 2160–2168. [DOI] [PubMed] [Google Scholar]
- 14.Liu, M., Yuan, C., Jensen, J.K., Zhao, B., Jiang, Y., Jiang, L. and Huang, M. (2017) The crystal structure of a multidomain protease inhibitor (HAI-1) reveals the mechanism of its auto-inhibition. J. Biol. Chem., 292, 8412–8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takeuchi, T., Harris, J.L., Huang, W., Yan, K.W., Coughlin, S.R. and Craik, C.S. (2000) Cellular localization of membrane-type serine protease 1 and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. J. Biol. Chem., 275, 26333–26342. [DOI] [PubMed] [Google Scholar]
- 16.Shipway, A., Danahay, H., Williams, J.A., Tully, D.C., Backes, B.J. and Harris, J.L. (2004) Biochemical characterization of prostasin, a channel activating protease. Biochem. Biophys. Res. Commun., 324, 953–963. [DOI] [PubMed] [Google Scholar]
- 17.Lee, S. P., Kao, C. Y., Chang, S. C., Chiu, Y. L., Chen, Y. J., Chen, M. H. G., Chang, C. C., Lin, Y. W., Chiang, C. P., Wang, J. K. et al. (2018) Tissue distribution and subcellular localizations determine in vivo functional relationship among prostasin, matriptase, HAI-1, and HAI-2 in human skin. PLoS One. doi: 10.1371/journal.pone.0192632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin, C.Y., Wang, J.K. and Johnson, M.D. (2020) The spatiotemporal control of human matriptase action on its physiological substrates: a case against a direct role for matriptase proteolytic activity in profilaggrin processing and desquamation. Hum. Cell, 33, 459–469. [DOI] [PubMed] [Google Scholar]
- 19.Chang, S.C., Chiang, C.P., Lai, C.H., Du, P.W.A., Hung, Y.S., Chen, Y.H., Yang, H.Y., Fang, H.Y., Lee, S.P., Tang, H.J. et al. (2020) Matriptase and prostasin proteolytic activities are differentially regulated in normal and wounded skin. Hum. Cell, 33, 990–1005. [DOI] [PubMed] [Google Scholar]
- 20.Chen, Y.W., Wang, J.K., Chou, F.P., Chen, C.Y., Rorke, E.A., Chen, L.M., Chai, K.X., Eckert, R.L., Johnson, M.D. and Lin, C.Y. (2010) Regulation of the matriptase-prostasin cell surface proteolytic cascade by hepatocyte growth factor activator inhibitor-1 during epidermal differentiation. J. Biol. Chem., 285, 31755–31762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su, H.C., Liang, Y.A., Lai, Y.J.J., Chiu, Y.L., Barndt, R.B., Shiao, F., Chang, H.H.D., Lu, D.D., Huang, N., Tseng, C.C. et al. (2016) Natural endogenous human matriptase and prostasin undergo zymogen activation via independent mechanisms in an uncoupled manner. PLoS One. doi: 10.1371/journal.pone.0167894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang, H.H.D., Xu, Y., Lai, H., Yang, X., Tseng, C.C., Lai, Y.J.J., Pan, Y., Zhou, E., Johnson, M.D., Wang, J.K. et al. (2015) Differential subcellular localization renders HAI-2 a matriptase inhibitor in breast cancer cells but not in mammary epithelial cells. PLoS One. doi: 10.1371/journal.pone.0120489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chiu, Y.L., Wu, Y.Y., Barndt, R.B., Lin, Y.W., Sytwo, H.P., Cheng, A., Yang, K., Chan, K.S., Wang, J.K., Johnson, M.D. et al. (2021) Differential subcellular distribution renders HAI-2 a less effective protease inhibitor than HAI-1 in the control of extracellular matriptase proteolytic activity. Genes Dis. doi: 10.1016/j.gendis.2020.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shiao, F., Liu, L.C.O., Huang, N., Lai, Y.J.J., Barndt, R.J., Tseng, C.C., Wang, J.K., Jia, B., Johnson, M.D. and Lin, C.Y. (2017) Selective inhibition of prostasin in human enterocytes by the integral membrane kunitz-type serine protease inhibitor HAI-2. PLoS One, 12, e0170944. doi: 10.1371/journal.pone.0170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friis, S., Sales, K.U., Godiksen, S., Peters, D.E., Lin, C.Y., Vogel, L.K. and Bugge, T.H. (2013) A matriptase-prostasin reciprocal zymogen activation complex with unique features: prostasin as a non-enzymatic co-factor for matriptase activation. J. Biol. Chem., 288, 19028–19039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szabo, R., Uzzun Sales, K., Kosa, P., Shylo, N.A., Godiksen, S., Hansen, K.K., Friis, S., Gutkind, J.S., Vogel, L.K., Hummler, E. et al. (2012) Reduced prostasin (CAP1/PRSS8) activity eliminates HAI-1 and HAI-2 deficiency-associated developmental defects by preventing matriptase activation. PLoS Genet. doi: 10.1371/journal.pgen.1002937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lai, Y.J.J., Chang, H.H.D., Lai, H., Xu, Y., Shiao, F., Huang, N., Li, L., Lee, M.S., Johnson, M.D., Wang, J.K. et al. (2015) N-glycan branching affects the subcellular distribution of and inhibition of matriptase by HAI-2/placental bikunin. PLoS One. doi: 10.1371/journal.pone.0132163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tseng, C.C., Jia, B., Barndt, R., Gu, Y., Chen, C.Y., Tseng, I.C., Su, S.F., Wang, J.K., Johnson, M.D. and Lin, C.Y. (2017) Matriptase shedding is closely coupled with matriptase zymogen activation and requires de novo proteolytic cleavage likely involving its own activity. PLoS One. doi: 10.1371/journal.pone.0183507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friis, S., Godiksen, S., Bornholdt, J., Selzer-Plon, J., Rasmussen, H.B., Bugge, T.H., Lin, C.Y. and Vogel, L.K. (2011) Transport via the transcytotic pathway makes prostasin available as a substrate for matriptase. J. Biol. Chem., 286, 5793–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oberst, M.D., Williams, C.A., Dickson, R.B., Johnson, M.D. and Lin, C.Y. (2003) The activation of matriptase requires its noncatalytic domains, serine protease domain, and its cognate inhibitor. J. Biol. Chem., 278, 26773–26779. [DOI] [PubMed] [Google Scholar]
- 31.Jia, B., Thompson, H.A., Barndt, R.B., Chiu, Y.L., Lee, M.J., Lee, S.C., Wang, J.K., Tang, H.J., Lin, C.Y. and Johnson, M.D. (2020) Mild acidity likely accelerates the physiological matriptase autoactivation process: a comparative study between spontaneous and acid-induced matriptase zymogen activation. Hum. Cell, 33, 1068–1080. [DOI] [PubMed] [Google Scholar]
- 32.Netzel-Arnett, S., Currie, B.M., Szabo, R., Lin, C.Y., Chen, L.M., Chai, K.X., Antalis, T.M., Bugge, T.H. and List, K. (2006) Evidence for a matriptase-prostasin proteolytic cascade regulating terminal epidermal differentiation. J. Biol. Chem., 281, 32941–32945. [DOI] [PubMed] [Google Scholar]
- 33.Mitchell, K.J., Pinson, K.I., Kelly, O.G., Brennan, J., Zupicich, J., Scherz, P., Leighton, P.A., Goodrich, L.V., Lu, X., Avery, B.J. et al. (2001) Functional analysis of secreted and transmembrane proteins critical to mouse development. Nat. Genet., 28, 241–249. [DOI] [PubMed] [Google Scholar]
- 34.Szabo, R. and Bugge, T.H. (2018) Loss of HAI-2 in mice with decreased prostasin activity leads to an early-onset intestinal failure resembling congenital tufting enteropathy. PLoS One, 13, e0194660. doi: 10.1371/journal.pone.0194660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szabo, R., Callies, L.L.K. and Bugge, T.H. (2019) Matriptase drives early-onset intestinal failure in a mouse model of congenital tufting enteropathy. Development. 146, doi: 10.1242/dev.183392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sivagnanam, M., Janecke, A.R., Müller, T., Heinz-Erian, P., Taylor, S. and Bird, L.M. (2010) Case of syndromic tufting enteropathy harbors SPINT2 mutation seen in congenital sodium diarrhea. Clin. Dysmorphol., 19, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salomon, J., Goulet, O., Canioni, D., Brousse, N., Lemale, J., Tounian, P., Coulomb, A., Marinier, E., Hugot, J.P., Ruemmele, F. et al. (2014) Genetic characterization of congenital tufting enteropathy: Epcam associated phenotype and involvement of SPINT2 in the syndromic form. Hum. Genet., 133, 299–310. [DOI] [PubMed] [Google Scholar]
- 38.Slae, M.A., Saginur, M., Persad, R., Yap, J., Lacson, A., Salomon, J., Canioni, D. and Huynh, H.Q. (2013) Syndromic congenital diarrhea because of the SPINT2 mutation showing enterocyte tufting and unique electron microscopy findings. Clin. Dysmorphol., 22, 118–120. [DOI] [PubMed] [Google Scholar]
- 39.Sivagnanam, M., Mueller, J.L., Lee, H., Chen, Z., Nelson, S.F., Turner, D., Zlotkin, S.H., Pencharz, P.B., Ngan, B.Y., Libiger, O. et al. (2008) Identification of EpCAM as the gene for congenital tufting enteropathy. Gastroenterology, 135, 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Müller, T., Wijmenga, C., Phillips, A.D., Janecke, A., Houwen, R.H.J., Fischer, H., Ellemunter, H., Frühwirth, M., Offner, F., Hofer, S. et al. (2000) Congenital sodium diarrhea is an autosomal recessive disorder of sodium/proton exchange but unrelated to known candidate genes. Gastroenterology, 119, 1506–1513. [DOI] [PubMed] [Google Scholar]
- 41.Goulet, O.J., Brousse, N., Canioni, D., Walker-Smith, J.A., Schmitz, J. and Phillips, A.D. (1998) Syndrome of intractable diarrhoea with persistent villous atrophy in early childhood: a clinicopathological survey of 47 cases. J. Pediatr. Gastroenterol. Nutr., 26, 151–161. [DOI] [PubMed] [Google Scholar]
- 42.Patey, N., Scoazec, J.Y., Cuenod-Jabri, B., Canioni, D., Kedinger, M., Goulet, O. and Brousse, N. (1997) Distribution of cell adhesion molecules in infants with intestinal epithelial dysplasia (tufting enteropathy). Gastroenterology, 113, 833–843. [DOI] [PubMed] [Google Scholar]
- 43.Sherman, P.M., Mitchell, D.J. and Cutz, E. (2004) Neonatal enteropathies: defining the causes of protracted diarrhea of infancy. J. Pediatr. Gastroenterol. Nutr., 38, 16–26. [DOI] [PubMed] [Google Scholar]
- 44.Wu, C.J., Feng, X., Lu, M., Morimura, S. and Udey, M.C. (2017) Matriptase-mediated cleavage of EpCAM destabilizes claudins and dysregulates intestinal epithelial homeostasis. J. Clin. Invest., 127, 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holt-Danborg, L., Vodopiutz, J., Nonboe, A.W., De Laffolie, J., Skovbjerg, S., Wolters, V.M., Müller, T., Hetzer, B., Querfurt, A., Zimmer, K.P. et al. (2019) SPINT2 (HAI-2) missense variants identified in congenital sodium diarrhea/tufting enteropathy affect the ability of HAI-2 to inhibit prostasin but not matriptase. Hum. Mol. Genet., 28, 828–841. [DOI] [PubMed] [Google Scholar]
- 46.Chen, Y.W., Wang, J.K., Chou, F.P., Wu, B.Y., Hsiao, H.C., Chiu, H., Xu, Z., Baksh, A.N.H., Shi, G., Kaul, M. et al. (2014) Matriptase regulates proliferation and early, but not terminal, differentiation of human keratinocytes. J. Invest. Dermatol., 134, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.List, K., Szabo, R., Molinolo, A., Nielsen, B.S. and Bugge, T.H. (2006) Delineation of matriptase protein expression by enzymatic gene trapping suggests diverging roles in barrier function, hair formation, and squamous cell carcinogenesis. Am. J. Pathol., 168, 1513–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kataoka, H., Itoh, H., Uchino, H., Hamasuna, R., Kitamura, N., Nabeshima, K. and Koono, M. (2000) Conserved expression of hepatocyte growth factor activator inhibitor type-2/placental bikunin in human colorectal carcinomas. Cancer Lett., 148, 127–134. [DOI] [PubMed] [Google Scholar]
- 49.Lee, M.S., Kiyomiya, K.I., Benaud, C., Dickson, R.B. and Lin, C.Y. (2005) Simultaneous activation and hepatocyte growth factor activator inhibitor 1-mediated inhibition of matriptase induced at activation foci in human mammary epithelial cells. Am. J. Physiol. Cell Physiol. 288, C932–C941. [DOI] [PubMed] [Google Scholar]
- 50.Benaud, C., Dickson, R.B. and Lin, C.Y. (2001) Regulation of the activity of matriptase on epithelial cell surfaces by a blood-derived factor. Eur. J. Biochem., 268, 1439–1447. [DOI] [PubMed] [Google Scholar]
- 51.Lee, M.S., Tseng, I.C., Wang, Y., Kiyomiya, K.I., Johnson, M.D., Dickson, R.B. and Lin, C.Y. (2007) Autoactivation of matriptase in vitro: requirement for biomembrane and LDL receptor domain. Am. J. Physiol. Cell Physiol. doi: 10.1152/ajpcell.00611.2006. [DOI] [PubMed] [Google Scholar]
- 52.Tseng, I.C., Xu, H., Chou, F.P., Li, G., Vazzano, A.P., Kao, J.P.Y., Johnson, M.D. and Lin, C.Y. (2010) Matriptase activation, an early cellular response to acidosis. J. Biol. Chem., 285, 3261–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang, J.K., Teng, I.J., Lo, T.J., Moore, S., Yeo, Y.H., Teng, Y.C., Kaul, M., Chen, C.C., Zuo, A.H., Chou, F.P. et al. (2014) Matriptase autoactivation is tightly regulated by the cellular chemical environments. PLoS One. doi: 10.1371/journal.pone.0093899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hsu, P.D., Scott, D.A., Weinstein, J.A., Ran, F.A., Konermann, S., Agarwala, V., Li, Y., Fine, E.J., Wu, X., Shalem, O., Cradick, T.J. et al. (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol., 31, 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Solomon, D.A., Kim, J.S., Cronin, J.C., Sibenaller, Z., Ryken, T., Rosenberg, S.A., Ressom, H., Jean, W., Bigner, D., Yan, H. et al. (2008) Mutational inactivation of PTPRD in glioblastoma multiforme and malignant melanoma. Cancer Res., 68, 10300–10306. [DOI] [PMC free article] [PubMed] [Google Scholar]