

In this review, Beatty et al. discuss recent advances in our understanding of the biological underpinnings of pancreatic ductal adenocarcinoma (PDAC) and dissect therapeutic targets that are intrinsic to PDAC and those that are defined by noncancer cells, including stromal cells, immune cells, and microbes.
Keywords: genetics, metabolism, microbiome, PDAC, pancreatic cancer, pancreatic tumor microenvironment, targeted therapy, therapeutic resistance
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a leading cause of cancer-related mortality in the United States and has only recently achieved a 5-yr survival rate of 10%. This dismal prognosis reflects the remarkable capacity of PDAC to effectively adapt to and resist therapeutic intervention. In this review, we discuss recent advances in our understanding of the biological underpinnings of PDAC and their implications as targetable vulnerabilities in this highly lethal disease.
Pancreatic ductal adenocarcinoma (PDAC) is a devastating disease with a 5-yr overall survival of 10% (Siegel et al. 2021). A hallmark of PDAC is its remarkable therapeutic resistance. This biology has been attributed to several key features including genetic instability, metabolic aberrations (Perera and Bardeesy 2015; Halbrook and Lyssiotis 2017), immune suppression (Balachandran et al. 2019), and a heterogeneous and hostile microenvironment that is marked by dense fibrosis and low vascularity (Ligorio et al. 2019). Improved understanding of the mechanisms underlying therapeutic resistance has revealed novel targets with potential promise.
Although chemotherapy remains the mainstay of treatment for PDAC, new efforts are now focused on intervening on aberrant genetic programs, disrupting DNA repair mechanisms (Golan et al. 2019; O'Reilly et al. 2020), exploiting metabolic vulnerabilities (Daemen et al. 2015; Perera and Bardeesy 2015; Ying et al. 2016), unraveling the harmful contributions of the microenvironment (Beatty et al. 2017), and leveraging the connection between cancer and the microbiome (Riquelme et al. 2019). In this regard, PDAC has historically been considered as a single entity, but more recently, genetic variants such as those harboring microsatellite instability or mutations in BRCA genes have confirmed that PDAC is a conglomerate of multiple subtypes (Collisson et al. 2019). To this end, targetable vulnerabilities have emerged in recent years that show promise in taming PDAC even in the absence of cytotoxic therapies (Golan et al. 2019; Pishvaian et al. 2020). This review focuses on efforts to dissect therapeutic targets that are intrinsic to PDAC and those that are defined by noncancer cells, including stromal cells, immune cells, and microbes.
Cancer cell-intrinsic targets
The mutational landscape of pancreatic adenocarcinoma
Over a decade ago, pioneering work identified 12 core signaling pathways that are genetically altered in PDAC (Jones et al. 2008). Since then, numerous genome sequencing studies have been performed to broadly define the genomic landscape of PDAC. Each study, powered by progress in sequencing technology, has contributed an incremental advancement in our knowledge of PDAC etiology and progression, clarified the relationship between primary and metastatic lesions, and provided the rationale for a more personalized approach to treatment. PDAC is driven largely by mutations in four genes; namely, the oncogene KRAS and the tumor suppressor genes TP53, CDKN2A, and SMAD4. Mutations in these four genes were initially identified prior to the next-generation sequencing (NGS) era by candidate gene approaches and have been confirmed in all subsequent genomic studies. Oncogenic mutations in KRAS are almost ubiquitous in PDAC and are present in 92%–95% of cases (Bailey et al. 2016; Chan-Seng-Yue et al. 2020), depending on the study. Activating mutations in KRAS are considered the initiating event in PDAC carcinogenesis and are evident in early premalignant lesions (pancreatic intraepithelial neoplasia [PanIN]). In addition, multiple clonal and subclonal KRAS mutations are concurrently detected in a small percentage of cases, suggesting convergent evolution of multiple tumor clones (The Cancer Genome Atlas Research Network 2017). Other mutations in genes regulating the RAS-MAPK pathway are observed in 60% of KRAS wild-type PDACs, confirming the critical role of this pathway in PDAC etiology. Mutations in the tumor suppressor genes TP53, CDKN2A, or SMAD4 occur in ∼50%–75% of patients. Alterations in these genes accumulate in the primary lesion according to a stepwise progression model during a period of several years, although a single chromothripsis event may accelerate PDAC progression by promoting the simultaneous copy loss of several tumor suppressor genes in a percentage of cases (Notta et al. 2016). In addition to mutations in the four genes listed above, a large number of additional recurrent mutations are present at a low frequency (<5%–10%). Notably, the combinations of such mutations are largely different among samples, making PDAC a remarkably heterogeneous disease. As a result, it is not surprising that therapeutic strategies using an unselected approach may have limited the opportunity to identify effective therapies. Here, we highlight some of the genetic alterations and accompanying aberrant signaling pathways that identify therapeutic targets that are beginning to be exploited clinically.
DNA damage repair (DDR) pathways
DNA damage occurs due to endogenous errors in replication or as a result of exogenous factors such as ionizing radiation or chemotherapeutic drugs. Since accurate repair of such lesions is crucial to maintain genome integrity, multiple and redundant pathways exist to repair DNA breaks (Sancar et al. 2004). In PDAC cells, the inability to repair DNA lesions promotes genomic instability and enhances the mutational rate, which eventually drives tumor evolution and progression (Feldmann et al. 2011; Drosos et al. 2017). On the other hand, loss of function in one or more DDR genes sensitizes cells to certain types of DNA-damaging chemotherapy. Indeed, the presence of BRCA1 and BRCA2 mutations was first noted to increase sensitivity to platinum-based chemotherapy in breast (Tutt et al. 2018) and ovarian cancer (Cass et al. 2003; Bolton et al. 2012) and, more recently, in PDAC (Golan et al. 2014; Waddell et al. 2015).
The therapeutic implications of the DDR pathway have led to the development of DDR targeting drugs based on the concept of synthetic lethality. In particular, the effectiveness of a specific DDR targeting drug to cause cell death is dependent on the genetic background of the cells with respect to competency in DNA damage response pathways. A prototypical example of therapeutic exploitation of synthetic lethality has been the use of poly-ADP ribose polymerase (PARP) inhibitors to treat tumors defective in HR repair (Sancar et al. 2004). PARP inhibitors (PARPis) “trap” PARP to the DNA and lead to stalling of replication forks due to the accumulation of unrepaired single-strand breaks (Helleday 2011). Stalled replication forks degrade into cytotoxic double-strand breaks (DSBs) if not corrected by appropriate repair mechanisms, which are essentially absent in BRCA1/2 mutated cells. Scientific studies have determined that a synthetic lethal relationship exists in certain HR-incompetent cells that render them PARPi-responsive (Bryant et al. 2005; Farmer et al. 2005).
Molecular analyses of PDAC have identified a significant PDAC subtype, constituting up to 20%–25% of cases, that is characterized by recurrent mutations in genes involved in DNA damage repair, such as BRCA1, BRCA2, PALB2, and ATM. This subtype of PDAC has emerged as a defined and targetable biological entity (Waddell et al. 2015; Bailey et al. 2016) and is characterized by an “unstable genome,” as well as increased chemosensitivity (Waddell et al. 2015). Notably, genes associated with DNA damage repair are frequently mutated in the germline in patients with PDAC and are mutated at higher frequency in familial PDAC (Bartsch et al. 2012).
Small retrospective clinical studies initially demonstrated that patients with HR-deficient PDAC due to germline mutations in BRCA1 and BRCA2 have a survival benefit when treated with platinum-based chemotherapy regimens (Golan et al. 2019; O'Reilly et al. 2020; Wattenberg et al. 2020). In parallel, clinical trials conducted in patients with HR-deficient breast and ovarian cancers have shown treatment efficacy with PARP inhibitors (Coleman et al. 2019; Tung et al. 2020). These two independent observations led to the examination of the potential therapeutic benefit of combining chemotherapy and PARP inhibitors in PDAC. Specifically, a randomized multicenter phase 2 trial of gemcitabine and cisplatin with or without the PARP inhibitor veliparib was conducted in patients with PDAC who harbor germline mutations in BRCA1/2 or PALB2 (O'Reilly et al. 2020). The addition of veliparib to cisplatin and gemcitabine was found to not be superior to chemotherapy alone, and the triple combination was notable for increased hematologic toxicity. As an alternative approach, a novel trial design of maintenance therapy was also tested in patients with BRCA1/2-mutant PDAC. Here, the Pancreas Cancer Olaparib Ongoing (POLO) study was designed to assess the efficacy of maintenance therapy with the PARPi olaparib in patients with PDAC harboring a germline BRCA1 or BRCA2 mutation following disease stabilization with platinum-based chemotherapy (Golan et al. 2019). The advantage of the POLO trial over the previously failed combination trial was a design built to leverage the therapeutic benefit of two types of promising therapy for germline BRCA1/2 patients but instead using them sequentially rather than concomitantly to mitigate the toxicity profile of the combination. Progression-free survival was the primary endpoint and determined to be significantly prolonged in the olaparib group (7.4 mo vs. 3.8 mo, P = 0.004), paving the way for regulatory approval by the FDA for olaparib in this setting. Notably, this reflects the first successful phase 3 trial of a biomarker-driven strategy for the treatment of PDAC. Beyond germline BRCA1 and BRCA2 mutations, it is possible that other biomarkers, such as somatic BRCA1, BRCA2, or PALB2 mutations, may identify patients responsive to PARPi maintenance therapy and ongoing clinical studies (NCT02511223, NCT02677038) in small cohorts of patients are in progress. Specific efforts to target ATM mutations in PDAC are also underway (NCT02511223) (Gout et al. 2021).
Not all patients with germline BRCA mutations respond to platinum-containing regimens or PARPi. In this regard, emerging data suggest that biallelic inactivation of DDR genes is associated with a favorable therapeutic response and that genomic hallmarks such as an unstable genome (Waddell et al. 2015), single-base signature 3 (Polak et al. 2017), or the HRDetect composite model score (Davies et al. 2017) may help to better predict patients who will respond to these agents. Development of improved predictive biomarkers may also help define those patients with DDR mutations, beyond germline BRCA1 and BRCA2, who are likely to respond to other DDR inhibitors, including those targeting ATM, ATR, WEE1, and CHK1 that are currently being evaluated in the clinical trial setting (Cleary et al. 2020).
Cell-intrinsic features are one determinant of treatment response in patients with DDR mutant PDAC. However, recent data have generated an increasing appreciation for the contribution of the tumor microenvironment in defining the therapeutic response to DNA damage. For instance, double-strand DNA breaks can stimulate up-regulation of PD-L1 expression by cancer cells (Sato et al. 2017), and similarly, PARP inhibition can increase PD-L1 expression in cancer cells (Jiao et al. 2017). DNA damage has also been shown to activate immune signaling through the introduction of genomic double-stranded DNA (dsDNA) into the cytosol and engagement of the cGAS/STING dsDNA-sensing pathway (Kwon and Bakhoum 2020). In response, cGAS generates cyclic dinucleotides that are sensed by STING, resulting in subsequent activation of interferon regulatory factor 3 (IRF3), nuclear factor κB (NF-κB), and the STAT6 signaling pathways to induce a robust type I interferon-driven proinflammatory cytokine response (Kwon and Bakhoum 2020). In a BRCA1-deficient breast cancer model, treatment with a PARP inhibitor in vivo augmented DNA damage and activated the cGAS/STING pathway, resulting in increased CD8+ T-cell infiltration into tumors (Pantelidou et al. 2019). Consequently, the potential value of combining DDR inhibitors with immunotherapy is beginning to be tested in multiple clinical trials (Seeber et al. 2019; Vinayak et al. 2019).
DNA alterations in cancer can also arise via defects in the DNA mismatch repair pathway (MMR), which functions to identify and repair mismatched DNA base pairs. A small but notable subgroup of patients with PDAC (1%–2%) harbor a mutation in a distinct set of DNA mismatch repair genes (MLH1, MSH2, MSH6, and PMS2), either due to the presence of a pathogenic germline mutation in one of the MMR genes (Lynch syndrome) or via a somatic MMR gene mutational event. Loss of MMR function leads to microsatellite instability (Connor et al. 2017) and elevated tumor mutational burden, resulting in a high level of neoantigen expression. Consequently, these tumors are more visible to immune surveillance and show significantly improved responses to PD‐1 blockade, which was approved by the FDA for use in this specific patient population (Connor et al. 2017). The presence of a MMR-deficient PDAC can be discerned by immunohistochemical assays for MSH1, MLH1, MSH6, and PMS2 expression or by use of next-generation genomic sequencing (Wimmer et al. 2014). It is important for all PDAC tumors to be tested for MMR deficiency, as these patients may have up to a 40% response rate to immune checkpoint inhibition and may derive survival benefit (Le et al. 2015).
Oncogenic KRAS mutations
Given the frequency of KRAS mutations in PDAC, there has been intense interest in mutant KRAS as a therapeutic target. To date, direct blockade of oncogenic KRAS has been challenging, due to a lack of ability to identify an adequate binding pocket for small molecule inhibitors (Kessler et al. 2019). Recently, identification of a small pocket within the KRASG12C mutant along with the possibility of creating a stable covalent bond with a mutant cysteine residue led to the development of the first selective inhibitors of KRASG12C (Hong et al. 2020a). This mutation is most frequent in lung adenocarcinomas (14%), and early phase I/II clinical trials using KRASG12C inhibitors demonstrated significant responses in lung cancer patients (Hong et al. 2020a). However, only 1%–2% of PDAC cases harbor KRASG12C mutations. In the examination of 12 patients with KRASG12C mutated PDAC treated with a selective inhibitor, disease stability, albeit transient, was seen in several patients, with one patient achieving a response lasting >10 mo (Hong et al. 2020b). Currently, the rest of the oncogenic KRAS isoforms remain undruggable, although concerted research efforts to develop KRASG12D inhibitors are underway, particularly given that 55% of PDAC harbor this specific KRAS mutant.
Attempts to indirectly target KRAS mutant tumors through inhibition of downstream effectors of KRAS, such as the RAF-MEK-ERK signaling cascade, have been largely ineffective due to activation of compensatory feedback loops resulting in adaptive resistance (Drosten and Barbacid 2020). Nonetheless, newer strategies show promise, such as targeting of the SHP2 protein-tyrosine phosphatase, an important mediator of cellular signaling through the RAS/MAPK pathway that is thought to act via activation of SOS1-regulated RAS-GTP loading (Hofmann et al. 2020). Preclinical studies using SHP2 inhibitors and SHP2/MEK inhibitor combinations prevented adaptive resistance in multiple cancer models expressing mutant KRAS (Fedele et al. 2018), and SHP2 inhibitors are being explored in phase 1 clinical trials. Recently, a small molecule SOS1 inhibitor (BI-3406) has been developed that prevents SOS1:KRAS binding, reducing the formation of GTP-loaded RAS (Hofmann et al. 2020). In preclinical xenograft studies, the combination of BI-3406 with the MEK inhibitor trametinib resulted in tumor regression in multiple KRAS-driven cancer models, leading to this combinatorial regimen now being tested in a phase 1 clinical trial in patients with advanced KRAS mutant tumors (NCT04111458). A third promising approach involves combining MEK inhibitors with autophagy inhibitors (NCT04132505) based on preclinical activity seen in PDAC models (Bryant et al. 2019; Kinsey et al. 2019) and is discussed in the metabolism section of this review.
The minority of PDAC patients who harbor wild-type KRAS tumors, constituting 6% of cases, is also a subset of interest. In the absence of an oncogenic KRAS mutation, PDACs have been found to exhibit alterations in other RAS pathway genes or oncogenic drivers, such as BRAF mutations, ERBB2 amplification, and the presence of NTRK gene fusions (Aguirre et al. 2018). While rare, some of these alternative drivers are potentially targetable with existing therapies.
Transcriptional subtypes of pancreatic adenocarcinoma
Recent studies using bulk RNA sequencing have proposed multiple gene expression classifications of PDAC (Collisson et al. 2011; Moffitt et al. 2015; Bailey et al. 2016; The Cancer Genome Atlas Research Network 2017; Puleo et al. 2018; Chan-Seng-Yue et al. 2020). When including the full spectrum of neoplastic cellularity seen in PDAC samples along with standardization of computational methodology, two tumor-specific PDAC subtypes have emerged, basal-like/squamous and classical, that have been validated across multiple studies in both primary and metastatic tumor samples (The Cancer Genome Atlas Research Network 2017). Basal-like/squamous tumors are associated with a significantly worse prognosis compared with classical tumors and exhibit a higher pathological grade (Puleo et al. 2018) and a poorer response to standard chemotherapy (Chan-Seng-Yue et al. 2020). To simplify subtype analysis in the clinical setting, IHC classifiers have been developed as surrogates to transcriptional subtyping and show that high GATA6 expression is associated with the classical subtype (O'Kane et al. 2020), and basal-like/squamous tumors have elevated expression of nuclear GLI1 (Puleo et al. 2018) and the basal marker KRT17 (Roa-Peña et al. 2019). Further analysis of individual tumors has led to the recognition that in fact, a continuum exists between basal-like/squamous and classical tumors, with single-cell sequencing demonstrating that most tumors harbor both basal-like/squamous and classical tumor cells. The varying proportions of these cells create a transcriptional continuum at the bulk RNA-sequencing level. Where a particular patient tumor may sit on this continuum is an outcome of the ratio of these subpopulations of cells (Chan-Seng-Yue et al. 2020). More recent work has built upon the initial identification of the subtypes to define putative master regulators of the basal-like/squamous subtype, including ΔNp63 (Somerville et al. 2018), GLI2 (Adams et al. 2019), and EZH2 (Patil et al. 2020), as well as the classical subtype (a GATA6-mediated gene regulatory network involving HNF1A and HNF4A) (Kloesch et al. 2021). In addition, subtype plasticity has been documented in response to drug treatments (Porter et al. 2019; Gabitova-Cornell et al. 2020), highlighting an increasing appreciation that these states are likely dynamic and interchangeable. The relevance of transcriptomic subtyping, both at the initiation of and in response to treatment, is an active area of investigation and will help determine whether this level of molecular detailing will provide benefit to patients.
Precision medicine for pancreatic cancer
The identification of pancreatic tumor subtypes and the possibility of performing comprehensive molecular profiling of tumors in a period of weeks have now opened the way for large studies (COMPASS, Know Your Tumor, PancSEQ, IMPaCT, and others) aimed to evaluate the feasibility of real-time molecular profiling and its impact on the clinical management of PDAC (Fig. 1; Chantrill et al. 2015; Aguirre et al. 2018; Chan-Seng-Yue et al. 2020; Pishvaian et al. 2020). In these studies, results were returned to the treating clinician within several weeks, and feasibility was in most cases >90%. These results indicate that time-sensitive prospective molecular profiling of PDAC is feasible within the clinical setting. Of note, this requires proper coordination of multiple disciplines and that dedicated procedures for tissue procurement and processing are in place.
Figure 1.

Precision medicine in pancreatic cancer. (Top) PDAC patients exhibit a breadth of tumor biology with generally poor responses to standard chemotherapy. Precision medicine, rooted in a multiomic approach to tumor/patient sequencing, can uncover potential avenues for targeted therapies. (Bottom) Adoption of a precision medicine approach coupled with matched therapies can substantially improve survival of patients with pancreatic cancer. (Bottom figure adapted from Pishvaian et al. 2020, © 2020, with permission from Elsevier.)
Actionable targets have been identified in many patients with PDAC (ranging from 28.5% to 49%), although the number of actionable targets and the criteria used to define them differ across studies. As an example, actionable targets defined as somatic alterations with a FDA-approved biomarker in another cancer indication limit the percentage of PDAC patients with actionable targets to <10%. Broadening the definition to include any somatic mutation for which there is clinical or preclinical evidence that suggests response to a drug increases the percentage of patients with an actionable target to 30% (Lowery et al. 2017). In the Know Your Tumor study (Pishvaian et al. 2020) with an analysis cohort of 677 patients with PDAC who received at least one line of therapy and had adequate longitudinal follow-up, 189 patients were identified with actionable mutations, and of these, 46 (24%) received a matched therapy while 143 (76%) did not. Although the number of patients who were able to obtain a matched therapy were small, these patients derived substantial therapeutic benefit, with a significant increase in mean overall survival (2.6 yr vs. 1.5 yr; P = 0.0004). Limitations in receiving a matched therapy included aggressiveness of disease, lack of a biomarker-directed clinical trial, or other logistical or economic issues. To advance our understanding of genomic alterations and classifications to predict therapeutic responses, the deployment of platform trials, such as Precision Promise (NCT04229004), that capture molecular data before and on treatment in well-annotated cohorts will assist in the development of molecular information as therapeutic biomarkers to help match therapies to patients most likely to respond to a specific treatment.
Cell-intrinsic metabolic mechanisms of therapeutic resistance
Pancreatic cancer cells use several cell-intrinsic metabolic processes to combat the challenges imposed by aberrant growth and proliferation. These include adaptations that facilitate nutrient acquisition, the rewiring of central carbon metabolism to support bioenergetics and biosynthesis, activation of pathways that inhibit oxidative stress and cell death, and evasion of the immune system. These mechanisms similarly act to afford pancreatic cancer cells with protection against therapy. Below, we discuss these mechanisms and highlight instances where they have been described to have a direct role in therapeutic resistance.
Metabolism
Metabolism is rewired in pancreatic cancer cells to facilitate the demands of cell growth and proliferation (Perera and Bardeesy 2015; Halbrook and Lyssiotis 2017). Deregulated signaling downstream from mutant KRAS and tumor suppressor loss are key contributors to alterations in metabolic pathways. For example, P53 mutations reprogram mitochondrial metabolism to promote malignant gene expression (Morris et al. 2019). Similarly, mutant KRAS signaling drives nutrient uptake and diversion into alternate biosynthetic and bioenergetic pathways (Ying et al. 2012; Kamphorst et al. 2013; Son et al. 2013; Viale et al. 2014). Pancreatic cancer cells also demonstrate increased glucose metabolism through the nonoxidative pentose phosphate pathway, which facilitates the production of nucleic acids (Ying et al. 2012; Shukla et al. 2017). This biology has implications in directing treatment resistance in PDAC. For example, pancreatic cancer cells respond to gemcitabine chemotherapy by further enhancing the oxidative pentose phosphate pathway to produce high levels of pyrimidine derivatives, including cytosine monophosphate. Thus, pancreatic cancer cells are flooded with cytidylates, which directly compete with the phosphorylation and activation of gemcitabine. Gemcitabine resistance can also emerge due to the expression and activity of complex I of the mitochondrial electron transport chain (Masoud et al. 2020). In this regard, inhibition of complex I with phenformin potentiates the activity of gemcitabine in several preclinical models of PDAC. Taken together, rewiring of metabolism is a common feature that has been shown to evolve in gemcitabine-treated PDAC tumors and to afford therapeutic resistance (Fig. 2A).
Figure 2.

Metabolic mechanisms of therapeutic resistance in PDA. (A) Cell-autonomous mechanisms of therapeutic resistance. PDAC cells enhance the production of nucleic acids from glucose through the pentose phosphate pathway (PPP) to promote resistance to gemcitabine. Macropinocytosis and autophagy provide nutrients (e.g. Fe2+, amino acids) to support biosynthesis and survival. Autophagy also removes MHC-I from the cell surface to impair recognition by the antitumor immune system. Malic enzyme 1 (ME1)-derived NADPH and the nuclear factor erythroid 2-related factor 2 (NRF2) pathway promote resistance to reactive oxygen species (ROS). NRF2 is transcriptionally activated by mutant KRAS and post-translationally stabilized by the ataxia-telangiectasia group D-associated protein (ATDC)-mediated binding and inhibition of Kelch-like ECH-associated protein 1 (KEAP1). (B) Tumor microenvironment-mediated mechanisms of therapeutic resistance. Deoxycytidine (dC) derived from cancer-associated fibroblasts (CAFs) and tumor-associated macrophages (TAMs) promotes resistance to gemcitabine. Pyruvate derived from CAFs and circulating asparagine (Asn) promote resistance to mitochondrial inhibitors.
Nutrient acquisition
Pancreatic tumors are marked by a robust fibroinflammatory response, and the associated stromal cells deposit considerable extracellular matrix (Whittle and Hingorani 2019). This matrix avidly retains water, leading to high interstitial fluid pressure and vascular collapse (DuFort et al. 2016). Consequently, cancer cells and their surrounding microenvironment have limited access to blood-derived nutrients and oxygen. This observation raised an early question concerning pancreatic tumor metabolism and, specifically, how metabolism and growth pathways are fueled. Over the past 10 yr, the consensus from many studies is that cancer cells combat nutrient austerity by activating scavenging and recycling pathways.
Macropinocytosis is the regulated uptake of bulk extracellular fluid, more colloquially known as cell drinking (Canton 2018). This is a critical mechanism by which pancreatic cancer cells obtain the nutrients that support their growth and survival (Commisso et al. 2013). Several subsequent studies expounded upon this observation (Kamphorst et al. 2015; Davidson et al. 2017) and revealed how macropinocytosis is integrated with progrowth signaling pathways (Palm et al. 2015; King et al. 2020). Indeed, numerous reports have now illustrated specific micronutrients that can be scavenged from the tumor microenvironment (Sousa et al. 2016; Olivares et al. 2017; Hollinshead et al. 2020; Kim et al. 2020; Parker et al. 2020) to enhance cancer cell survival, and these are discussed in detail in the following section.
Autophagy
Among the nutrient recycling pathways, autophagy and its role in regulating therapeutic resistance in PDAC are the most well characterized. Autophagy is a regulated process in which internal proteins and organelles are selectively degraded to regulate protein and organelle homeostasis and provide nutrient building blocks to support bioenergetics (Rabinowitz and White 2010). Intracellular cargo is captured in autophagosomes, which fuse with lysosomes to break large molecules into their constituent parts (e.g. sugars, lipids, and amino acids). When autophagy is activated following nutrient deprivation, this can produce metabolites to fuel energy production (Fig. 2A). Similarly, autophagy is activated in states of cell stress, used to target damaged organelles for destruction, and used to regulate signaling programs. Classic examples of this include mitochondrial or DNA damage, such as those induced by chemotherapy and radiotherapy, and more recent nuanced examples illustrate how specific programs can be regulated by autophagy (e.g. iron homeostasis) (Mancias et al. 2014; Kremer et al. 2020).
Both murine and human pancreatic cancer cells depend on constitutive autophagy, where its inhibition is growth inhibitory and can potentiate the activity of gemcitabine chemotherapy (Yang et al. 2011). In a recent study using a genetically engineered mouse model of PDAC in which autophagy can be conditionally inactivated, autophagy was shown to be required for PDAC tumor initiation and maintenance (Yang et al. 2018). This and related studies illustrate that autophagy, a process that is normally activated in response to nutrient limitation or stress, is constitutively active in PDAC, even in cell lines grown in vitro in nutrient-rich conditions and without stress. This paradoxical finding is because PDAC transcriptionally activates a lysosome and autophagy response, which is mediated by the MiT/TFE family of transcription factors (Perera et al. 2015). Through this mechanism, amino acid levels in pancreatic cancer cells can be maintained to support cellular homeostasis and metabolic reprogramming.
Elevation in basal autophagy is observed even in metabolically quiescent PDAC (Viale et al. 2014; Alcalá et al. 2020). Furthermore, genetic inhibition of KRAS was unexpectedly found to enhance autophagy (Viale et al. 2014). Extinguishing mutant KRAS activity was shown to trigger metabolic stress and, as a result, activated autophagy to address the metabolic needs of pancreatic cancer cells even in the absence of deregulated nutrients downstream from mutant KRAS signaling.
More recently, studies of the interplay of oncogenic signaling and autophagy have extended to include the MAPK pathway that is downstream from KRAS signaling. Inhibition of MAPK signaling, like the genetic extinction of mutant KRAS, potently increases autophagy (Bryant et al. 2019; Kinsey et al. 2019). This finding aligns with our understanding of MAPK in PDAC and classical autophagy models. Specifically, downstream from mutant KRAS signaling, MAPK drives nutrient uptake and utilization in PDAC (Ying et al. 2012; Son et al. 2013). Thus, its extinction with MAPK inhibitors engages a catabolic nutrient program, consistent with autophagy induction. Furthermore, in the setting of MAPK inhibition, autophagy becomes a critical process for maintaining cellular homeostasis, thereby identifying a therapeutic context that might be exploited. Indeed, pharmacologic and genetic methods of autophagy inhibition have been shown to potently synergize with MAPK pathway inhibition in preclinical PDAC models.
The function of constitutively elevated autophagy serves several protective roles in PDAC. As described above, and consistent with the classical role of autophagy, pancreatic cancer cells use autophagy to manage the myriad of stressors in the tumor microenvironment, including nutrient and oxygen limitation as well as replicative stress. However, this explanation is unsatisfactory, particularly since pancreatic cancer cells grown in vitro under nutrient-replete conditions continue to maintain high basal autophagy. This implies that autophagy may also be a hard-wired program with additional biological significance. To this end, basal autophagy in PDAC has been found to remove major histocompatibility complex (MHC) class I molecules from the cell surface and target them for degradation in the lysosome (Fig. 2A; Yamamoto et al. 2020). As such, pancreatic cancer cells are less capable of presenting antigen and thus being recognized by the immune system. Accordingly, the combination of autophagy inhibition with immune checkpoint inhibitors was found to synergize in impairing tumor growth in immune-competent preclinical PDAC models in a CD8+ T-cell-dependent fashion. Similarly, another recent study using a CRISPR/Cas9 screen in a murine PDAC model found that autophagy can protect cancer cells from TNFα-dependent killing by CD8+ T cells (Zhu et al. 2021). In summary, pancreatic cancer cells may use metabolic programs as a means to evade immune elimination and, in doing so, reduce the efficacy of immunotherapy.
Based on these important functions, several clinical trials testing the utility of autophagy inhibitors in PDAC are ongoing. Results from phase I/II trials indicate that autophagy inhibition is well tolerated, and modest responses have been observed in some studies (Piffoux et al. 2020). Chloroquine and hydroxychloroquine are among the most widely used autophagy inhibitors in preclinical studies and are the only drugs targeting autophagy that are approved clinically (Mauthe et al. 2018). It is important to note, however, that the pharmacodynamic properties of hydroxychloroquine limit achievement of intratumoral therapeutic doses of the drug and thus activity (Kimmelman and White 2017). The development of new autophagy inhibitors is an active area. Nevertheless, new trials with hydroxychloroquine are ongoing and include combination treatment with chemotherapy (e.g. NCT01506973 and NCT04524702) or MAPK pathway inhibitors (NCT04386057, NCT04145297, NCT03825289, and NCT04132505). In addition, it will be important to establish how hydroxychloroquine performs in combination with immunotherapies, as well as to determine the efficacy of novel autophagy inducers in combination studies with chemotherapy, MAPK pathway inhibitors, and immunotherapies.
Antioxidant programs
The hypoxic nature of PDAC tumors imposes challenges on the maintenance of redox balance. To counteract this, mutant Kras activity drives pathways that protect cells from reactive oxygen species (ROS), a byproduct of metabolism. Excess ROS have the potential to be toxic to cells if their levels are not tightly regulated (Purohit et al. 2019). To this end, pancreatic cancer cells activate transcriptional and metabolic antioxidant pathways to tolerate the high rate of ROS generation, leading to elevated ROS flux. Ultimately, this leads to both overutilization and dependence on antioxidant pathways to maintain redox balance.
There are two well-characterized mechanisms by which ROS is managed in PDAC (Fig. 2A). First, rewired metabolism downstream from mutant KRAS leads to enhanced generation of the antioxidant NADPH from malic enzyme 1 (ME1) (Son et al. 2013). Inhibition of this pathway promotes the tumoricidal activity of radiotherapy (Nelson et al. 2020). Second, mutant KRAS signaling drives expression of the NRF2 transcription factor program (DeNicola et al. 2011). NRF2 is a master regulator of antioxidant defense, whose activity results in both activation of antioxidant enzymes and regulation of metabolic fluxes to promote a reduced cellular environment (Hayes and Dinkova-Kostova 2014). In addition, NRF2 regulation protects pancreatic cancer cells from oxidative stress and the cytotoxic activity of chemotherapy (Purohit et al. 2021). Specifically, ATDC, an oncogene highly expressed in human PDAC, binds to KEAP1, the negative regulator of NRF2, leading to NRF2 stabilization and enhanced activity.
Pancreatic cancer cells also must maintain reduced glutathione (GSH) pools to tolerate the high rate of ROS generation. GSH is a principle cellular antioxidant tripeptide composed of glutamate, glycine, and cysteine, where cysteine serves as the rate-limiting amino acid in GSH biosynthesis. Thus, access to cysteine is required to maintain antioxidant capacity. For example, in a genetically engineered mouse model of PDAC, inhibition of cysteine uptake was shown to promote ferroptosis, an oxidative form of cell death (Badgley et al. 2020). In this study, inhibition of cysteine uptake was induced by systemic deletion Slc7a11, which encodes the xCT subunite of the cystine antiporter, system xC−. Notably, ferroptotic cell death could be reversed by systemic administration of cell-permeable cysteine (N-acetyl cysteine [NAC]). This finding highlights the unique metabolic demands on redox balance in PDAC and illuminates new therapeutic opportunities to target cysteine import and metabolism and/or to perpetuate inherent oxidative stress.
Cancer cell-extrinsic targets
Metabolic resistance to therapy by the tumor microenvironment
The deregulated metabolic programs in PDAC are strongly influenced by the tumor microenvironment (TME). Hallmarks of metabolic dysfunction in the TME are nutrient deficiency, accumulation of metabolic waste products, and disruption in pH, oxygenation, and the redox state (Lyssiotis and Kimmelman 2017; Sullivan et al. 2019). Historically, studies on therapeutic resistance imparted by the TME have focused on insufficiencies in drug delivery (DuFort et al. 2016), and resistance to immune therapies have centered on the function of signaling factors and signal transduction receptors in cell–cell communication (Balachandran et al. 2019). However, several recent studies now indicate that metabolites also contribute significantly to therapeutic resistance.
Chemoresistance
Tumor-associated macrophages (TAMs) can constitute upward of 40% of the cellular content in PDAC tumors (Steele et al. 2020) and are well-described mediators of immune suppression and therapeutic resistance. Macrophages can be endowed with either protumor or antitumor functions (Wattenberg and Beatty 2020) and are often classified by metabolic programs, for example, how cells metabolize the amino acid arginine (Murray et al. 2014). In PDAC, metabolites released by cancer cells promote polarization of naïve macrophages into TAMs. For example, metabolites released from pancreatic cancer cells participate in programming glucose and glutamine metabolism in TAMs, which then facilitates release of pyrimidine species at micromolar levels. Among these pyrimidines, TAM-released deoxycytidine can impair the cytotoxic activity of gemcitabine against pancreatic cancer cells. Mechanistically, this occurs through metabolic competition for the rate-limiting step in gemcitabine activation; namely, phosphorylation by deoxycytidine kinase. Genetic or pharmacologic depletion of TAMs dramatically sensitizes tumors to gemcitabine. Consistent with this, patients with a low macrophage burden in their tumors respond significantly better to gemcitabine than those with a high macrophage burden (Halbrook et al. 2019). Similarly, pancreatic cancer-associated fibroblasts (CAFs) have been shown to release deoxycytidine, which promotes gemcitabine resistance by pancreatic cancer cells (Dalin et al. 2019). Thus, metabolites produced by cells within the tumor microenvironment can influence the sensitivity of PDAC to cytotoxic stress (Fig. 2B).
Recently, efforts to intervene on metabolism have turned to the development of drugs that target the mitochondria of pancreatic as well as other cancers (Vasan et al. 2020). Several of these have shown varying degree of promise in preclinical models (Viale et al. 2014; Daemen et al. 2015; Alistar et al. 2017; Rajeshkumar et al. 2017; Ashton et al. 2018; Molina et al. 2018; Masoud et al. 2020). Inhibitors of complex I of the electron transport chain (ETC) are arguably the most well established. A primary mechanism by which ETC inhibitors mediate their growth inhibitory effects is through increasing NADH/NAD+ ratio accumulation, which blocks metabolism. However, while these inhibitors exhibit potent cytostatic and even cytotoxic activity in vitro, their activity in vivo is limited and context dependent. One explanation for this discordant biology seen with ETC inhibitors comes from the recent observation that pyruvate can provide partial rescue of ETC inhibition (Gui et al. 2016). In this context, pyruvate is used to oxidize NADH to NAD+ and thereby relieve this “brake” on metabolism. While pyruvate is present in serum at a low concentration, additional studies have illustrated that pyruvate is abundantly released by pancreatic CAFs and that this is a mechanism of therapeutic resistance imparted by the TME (Fig. 2B; Datta et al. 2020; Kerk et al. 2020). Thus, future preclinical and clinical studies using mitochondrial inhibitors will need to consider both circulating pyruvate and the composition of the TME as well as associated TME-derived pyruvate.
Like pyruvate, asparagine has similarly been shown to promote therapeutic resistance and support tumor growth upon inhibition of complex I (Fig. 2B; Halbrook et al. 2020; Krall et al. 2020). Asparagine promotes therapeutic resistance and permits tumor growth by supporting biosynthesis and aspartate availability, in both mTOR-dependent and -independent manners. The excitement for these findings stems from the translatability of targeting asparagine in patients with cancer. Asparagine can be depleted systemically, and thus intratumorally, by diet (Krall et al. 2021) or through treatment with L-asparaginase. Notably, L-asparaginase has been used therapeutically for decades in hematologic cancers, and recent on-going clinical trials are testing its efficacy in PDAC with chemotherapy (NCT03665441). Furthermore, these studies suggest the potential for combining asparaginase and complex I targeted therapies (Halbrook et al. 2020; Krall et al. 2021).
Immune resistance
Antitumor immune cells require nutrients to support proliferation and execute effector functions. To facilitate immune evasion, and thus tumor survival, neoplastic and nonneoplastic cells in the TME compete with immune cells for nutrients. Given that immune cells tend to be less adapted for nutrient competition, this may be an important mechanism by which the antitumor immune response is impaired (Lyssiotis and Kimmelman 2017). For example, the availability of several amino acids and amino acid catabolites influence Treg and cytotoxic T-cell activity (Kelly and Pearce 2020). In particular, arginine and tryptophan are required for CD8+ T-cell expansion. However, cancer cells and other cell types in the TME promote the catabolism of these amino acids. Arginine catabolism into proline positively influences collagen production, and thus extracellular matrix deposition, and tryptophan catabolism produces kynurenine, an aryl hydrocarbon receptor agonist that drives Treg differentiation and immunosuppression (Lyssiotis and Kimmelman 2017). Kynurenine can be produced from tryptophan by the indolamine dioxygenases (IDOs). However, the targeting of this amino acid catabolism axis has yet to demonstrate reproducible benefit in patients (Van den Eynde et al. 2020). Ongoing studies aim to identify the appropriate context and combinations to deploy IDO inhibitors clinically for PDAC (NCT03006302, NCT02077881). For example, a recent study in PDA has shown that GM-CSF-secreting, allogeneic pancreatic tumor whole-cell vaccine (GVAX) can induce IDO1 expression in PDAC tumors, which then promotes immune suppression (Blair et al. 2019). To this end, combining IDO1 inhibition with GVAX potentiates therapeutic efficacy in a preclinical model. These results reveal a potential approach to harness IDO inhibitors for PDAC. Collectively, these studies illustrate the significance of metabolites derived from the pancreatic TME, as well as the importance and ubiquity of metabolic cooperation and competition processes in promoting resistance to various therapies in PDAC.
Neuronal support for PDAC
Over recent years, data have emerged that implicate the sensory and sympathetic nervous systems as promoters of PDAC development and progression. Pancreatic cancer cells may actively promote axonal ingrowth into tumors via secretion of nerve growth factor (NGF) to leverage neurons for the delivery of key nutrients to the poorly perfused TME. For example, it has been demonstrated that axons from the dorsal root ganglion can release the amino acid serine into the pancreatic TME, which is essential for proper messenger RNA translation and thus the survival of cancer cells (Banh et al. 2020). Interestingly, in a subset of human PDAC that lacks the capability of de novo serine synthesis, Banh et al. (2020) observed increased NGF signaling and, consequently, a denser tumor innervation.
NGF has also been implicated as part of the tumor-supportive role of the sympathetic nervous system in PDAC, offering a possible link between chronic stress and PDAC development and growth. Elevated norepinephrine levels as part of the stress response can activate the adrenergic β-2 receptor (ADRB2) on PDAC cells and mediate epithelial proliferation (Renz et al. 2018a) as well as NGF release, further promoting ingrowth of sympathetic nerve fibers and effectively creating a positive feedback loop. Conversely, the parasympathetic nervous system appears to have a constraining effect on PDAC by, at least in part, cholinergic signaling through the muscarinic type 1 receptor CHRM1 and downstream inhibition of the MAPK/EGFR pathway in cancer cells (Renz et al. 2018b).
Targeting of these newly uncovered pathways that regulate cross-talk between pancreatic tumors and the surrounding nerves has shown some therapeutic efficacy in murine PDAC models (Renz et al. 2018a; Banh et al. 2020). Either antagonism of ADRB2 or the inhibition of TRK1, the receptor for NGF, was able to block neuronal ingrowth and concomitantly stunt PDAC growth. These approaches have the potential for clinical translation, since adrenergic signaling can be mitigated with the well-studied drug class of β-blockers, and TRK1 can be targeted with its inhibitor larotrecinib, an FDA-approved drug in clinical use for solid tumors with NTRK fusions.
The tumor microenvironment in pancreatic cancer
The stromal microenvironment that surrounds pancreatic cancer is a fundamental determinant of its biology and treatment resistance. Formation of this microenvironment is triggered at the earliest stages of cancer conception and evolves during cancer development under the instruction of cancer cell-intrinsic mechanisms as well as in response to cues communicated by infiltrating cell populations, such as leukocytes and fibroblasts (Stone and Beatty 2019). This coevolutionary process that occurs between cancer cells and the stroma ultimately supports immune evasion, metastasis, and remarkable resilience to cytotoxic stress imposed by therapeutics (Balachandran et al. 2019). In recent years, our understanding of the signaling pathways that direct this biology has spawned new treatment opportunities but also emphasized that simply disrupting stromal elements can signal compensatory and nonredundant mechanisms that maintain and foster pancreatic cancer progression. Here, we discuss determinants of the stromal microenvironment, their role in directing cancer cell fate and immune evasion, and their potential to serve as therapeutic targets.
Determinants of the stromal response
Mouse modeling has provided key insights into the evolution of the stromal microenvironment in pancreatic cancer. For instance, the combination of experimentally induced pancreatic inflammation and KRAS activation in pancreatic epithelial cells is sufficient to trigger PanIN formation, development of a desmoplastic reaction, and progression to invasive PDAC (Guerra et al. 2007; Gidekel Friedlander et al. 2009). Interestingly, inactivation of KRAS at an early stage of cancer conception causes not only lesion regression but also resolution of the desmoplastic stroma (Collins et al. 2012; Ying et al. 2012). Other genetic aberrations, such as TP53 inactivation and MYC activation in cancer cells, are also key switches that trigger formation of a stromal microenvironment (Guerra et al. 2007; Sodir et al. 2020). Notably, inactivation of MYC has been shown to cause disassembly of the stroma with concomitant cancer cell death (Sodir et al. 2020). Together, these findings illustrate the importance of oncogenes and tumor suppressor genes as determinants of the tumor microenvironment in pancreatic cancer.
Cellular contexture of the tumor microenvironment
The microenvironment that surrounds pancreatic cancer is composed of multiple cell populations and is dominated by fibroblasts and leukocytes. Fibroblast cell populations arise, at least in part, from resident cells found in the pancreas that expand and transition into cancer-associated fibroblasts (Sahai et al. 2020). Several functionally distinct subclasses of CAFs have now been identified in mouse and human pancreatic cancer and classified as myofibroblast-CAF (myCAF), inflammatory-CAF (iCAF), and antigen-presenting-CAF (apCAF) (Collins et al. 2012; Öhlund et al. 2017; Helms et al. 2020). These CAF populations are separated spatially within the microenvironment and contribute to pancreatic cancer cellular heterogeneity, metastasis, immune evasion, and chemotherapy resistance (Olive et al. 2009; Feig et al. 2013; Waghray et al. 2016; Öhlund et al. 2017; Biffi et al. 2019; Lee et al. 2019). For example, iCAFs are recognized as a significant source of IL-6 in the microenvironment and have the ability (1) to stimulate STAT3 activation in cancer cells, which then drives pancreatic cancer progression (Öhlund et al. 2017), and (2) to instruct the formation of a niche environment in the liver that ultimately supports metastatic spread (Lee et al. 2019). CAF-derived factors (e.g. TGF-β) also coordinate a phenotypical shift in cancer cells to invasive epithelial-to-mesenchymal transition (EMT) and proliferative phenotypes that are linked to MAPK and STAT3 signaling and associated with increased metastatic potential (Ligorio et al. 2019). Taken together, CAFs are a heterogeneous population of cells with important functions in shaping pancreatic cancer biology and its surrounding microenvironment.
Like CAFs, resident myeloid cells are also present in the normal pancreas. These embryonically derived macrophages expand during cancer progression and exhibit a profibrotic phenotype suggestive of their role in remodeling the extracellular matrix that is instructed by cancer cells and CAFs (Zhu et al. 2017). Bone marrow-derived monocytes and neutrophils are also recruited from the circulation into the stroma by CCR2 and CXCR2 ligands, respectively (Sanford et al. 2013; Steele et al. 2016; Nywening et al. 2018). These cells contribute to the myeloid response and are recognized for their role in treatment resistance to immune and cytotoxic therapies (Mitchem et al. 2013; Nywening et al. 2016; Kalbasi et al. 2017).
In contrast to myeloid cells and fibroblasts, which more diffusely infiltrate PDAC, T and B cells are found focally within the stroma that surrounds pancreatic cancer cells and within tertiary lymphoid structures (TLSs), which are detected sporadically throughout tumors (Stromnes et al. 2017). Notably, TLSs, when found in surgically resected PDAC, associate with a favorable prognosis (Hiraoka et al. 2015; Beatty et al. 2017). However, T and B cells are usually excluded from direct interaction with cancer cells. In contrast, mouse modeling has shown that a more pronounced T-cell infiltrate is present within early PanIN lesions and is comprised of CD4+ T-cell subsets, which contribute to an immunosuppressive microenvironment by repressing the activity of CD8+ T cells (Zhang et al. 2014). CD4+ T cells that infiltrate PanIN are comprised of both regulatory T cells (Tregs) and helper T cells that produce IL-17 (Th17 cells). Selective elimination of Tregs during PanIN development accelerates progression due to a compensatory CCR1-dependent inflammatory response driven by increased myeloid cell infiltration and a decrease in CAFs (Zhang et al. 2020b). In contrast, neutralization of IL-17, which is released by Th17 cells and γδ T cells, prevents PanIN progression (McAllister et al. 2014). Consistent with this, IL-17 facilitates recruitment of neutrophils, which, by releasing neutrophil extracellular traps (NETs), can exclude CD8+ T-cell infiltration into tumors (Zhang et al. 2020a). Together, these findings identify an intricate balance between CD4+ T-cell subsets that contribute influentially to the stromal inflammatory response that surrounds PanIN lesions and their propensity to progress to invasive PDAC.
Remarkable stromal heterogeneity is a hallmark of PDAC. The underlying mechanisms that direct the cellular contexture of the stroma are numerous and complex. In this regard, mouse models have shown a critical role for cancer cell-intrinsic pathways that instruct the recruitment of cells into the stromal microenvironment. For instance, KRAS activation up-regulates GM-CSF in pancreatic epithelial cells to recruit myeloid cells with immunosuppressive properties (Bayne et al. 2012; Pylayeva-Gupta et al. 2012). MYC activation also instructs the recruitment of myeloid cell populations (Sodir et al. 2020). In addition, PTEN loss in cancer cells promotes NF-kB activation and a cytokine response that coordinates protumorigenic inflammation (Ying et al. 2011). Cancer cell production of G-CSF may also restrict productive immunosurveillance by impairing bone marrow development of dendritic cells, which are essential to the generation of tumor-reactive T cells (Meyer et al. 2018). In general, the molecular wiring of a cancer cell can shape the contexture of the stromal microenvironment, and many cancer cell-intrinsic determinants, including USP22, EPHA2, and CXCL1 among others, have been identified that favor a myeloid-rich and T-cell-poor community that thwarts the efficacy of cancer therapies (Li et al. 2018, 2020; Markosyan et al. 2019). Notably, common to each of these determinants is their role in regulating tumor inflammation. For this reason, strategies to disrupt the inflammatory response to PDAC by inhibiting myeloid cell recruitment have garnered significant interest. However, disrupting one chemoattractant pathway can trigger a compensatory one with equally immunosuppressive properties. For example, blocking recruitment of CCR2+ inflammatory monocytes into tumors signals for increased infiltration by CXCR2+ granulocytes and vice versa (Nywening et al. 2018). This finding illustrates the pliability of the tumor microenvironment but also its loyalty to supporting tumor progression.
Stromal-directed therapies
Strategies that disrupt the stromal response to pancreatic cancer can impact treatment efficacy. For example, depletion of matrix components, such as hyaluronan and type I collagen, improves the activity of gemcitabine chemotherapy in mouse models (Olive et al. 2009; Provenzano et al. 2012; Jacobetz et al. 2013). These strategies though have not shown clinical activity in combination with more intensive chemotherapy regimens in patients (Hingorani et al. 2018; Ramanathan et al. 2019; Van Cutsem et al. 2020). This observation may reflect the reciprocal relationship between fibrosis and vascularity. For instance, sustained depletion of sonic hedgehog, a soluble ligand produced by cancer cells and that drives formation of a fibroblast-rich stroma, incites the development of tumors with enhanced VEGF-dependent angiogenesis that then support increased cancer cell proliferation (Lee et al. 2014; Rhim et al. 2014; Hingorani et al. 2018). This finding shows that some elements of the tumor microenvironment can act to restrain pancreatic cancer progression. However, this is balanced by other stromal components that aim to foster tumor growth. For example, blockade of leukemia inhibitory factor (LIF), a paracrine molecule released by pancreatic stellate cells and that acts on cancer cells, slows pancreatic cancer growth in mouse models and combines with chemotherapy to improve outcomes (Shi et al. 2019). Similarly, disrupting signaling pathways such a focal adhesion kinase (FAK) and IL1 receptor-associated kinase 4 (IRAK4) can shift the tumor microenvironment in pancreatic cancer mouse models from treatment resistant to sensitive (Zhang et al. 2018). FAK is activated in pancreatic cancer cells as well as the surrounding stroma and engages a chemokine network that recruits myeloid cells and fibroblasts (Jiang et al. 2016). Interestingly, FAK activity has been correlated with IRAK4 activation, such that overexpression of IRAK4 in tumor cells drives FAK signaling (Dodhiawala et al. 2020). IRAK4 expression in CAFs also triggers NF-kB activity and tumor fibrosis, which culminate in increased cancer cell proliferation, survival, and resistance to chemotherapy (Zhang et al. 2018). Consistent with these data, pancreatic cancer cells have been found to respond to microbial-dependent activation of TLR4 by releasing IL-1β, which then promotes the activation and secretory phenotype of quiescent pancreatic stellate cells (Das et al. 2020). Together, this signaling cascade orchestrates an immunosuppressive microenvironment.
An alternative approach to inhibiting elements of the tumor microenvironment in PDAC is to redirect the biology of tumor-infiltrating cells. For example, CD40 and CD11b agonists can shift the biology of tumor-infiltrating myeloid cells toward an antitumor and immunostimulatory phenotype that then unveils the activity of cytotoxic and T-cell immune therapies (Beatty et al. 2011, 2015; Long et al. 2016; Panni et al. 2019). This finding supports pliability as a central theme of the tumor microenvironment in PDAC. However, remarkable resilience has been shown to prevail, suggesting that merely initiating a shift in the character of the tumor microenvironment is insufficient and that strategies to maintain this biology will be needed. This concept of induction followed by maintenance therapy is supported by studies in other solid cancers where immunotherapy produces a benefit when administered after induction chemotherapy (Grivas et al. 2019; Powles et al. 2020).
Harnessing the immune response
Whereas immunotherapy has been successful in other solid cancers, it has not yet translated to PDAC except for microsatellite instability (MSI)-high tumors, which represent 1% of all PDAC (Le et al. 2017; Balachandran et al. 2019). Defining strategies capable of overcoming T-cell exclusion has been a priority for leveraging the potential of immunotherapy in PDAC. In this regard, multiple therapeutic challenges have been identified that impact the success of immune surveillance and the disposition of the microenvironment that surrounds pancreatic cancer cells (Fig. 3). For example, poor T-cell surveillance may be driven by a lack of priming for tumor-reactive T cells, poor recruitment of T cells and their limited expansion within tumors, or both (Beatty and O'Hara 2016). Consistent with this, mouse models show that pancreatic cancer impairs dendritic cell (DC) biology, which is associated with poor T-cell priming (Hegde et al. 2020; Lin et al. 2020). Deficiencies in DCs and T cells are seen in patients, indicating that the health of the immune system in PDAC is compromised (Meyer et al. 2018; Xu et al. 2019; Lin et al. 2020). This poor immune health can be improved by systemic CD40 activation with enhanced T-cell-priming activity seen in some transplantable models of murine PDAC (Beatty et al. 2011; Lin et al. 2020). However, a CD40 agonist alone, and even in combination with chemotherapy, fails to trigger productive T-cell immunity in models of spontaneous PDAC (Beatty et al. 2011). This exclusion of T cells can be overcome by depleting phagocytic cells residing outside of tumors (Beatty et al. 2015). Additionally, recent work has shown that deficiencies in DCs that limit T-cell activation can be resolved using a FLT3 ligand that stimulates DC development in the bone marrow. Combing a FLT3 ligand with a CD40 agonist to “license” DCs with T-cell stimulatory properties can subsequently trigger productive T-cell immunity in mouse models of PDAC. Together, this combination of FLT3 ligand and a CD40 agonist suggests that defects in both DC function and abundance are limiting for generating tumor-reactive T cells (Hegde et al. 2020; Lin et al. 2020).
Figure 3.

Stromal determinants and therapeutic challenges to intervening on PDAC oathogenesis. The stromal compartment in PDAC is shaped by tumor-infiltrating leukocytes recruited from the peripheral blood. These leukocytes coordinate therapeutic resistance and aid in PDAC progression and metastasis. Shown are therapeutic challenges established by this dynamic interaction between host and tumor. (1) Within the peripheral blood, deficiencies in immune health are observed that may limit the efficacy of immunotherapy. (2) Tumors recruit immune-suppressive cells and exclude effector T cells. (3) Within tumors, genetic aberrations in the cancer cells instruct the formation of a microenvironment marked by immunosuppression, nutrient deprivation, and a desmoplastic reaction. (4) The tumor microenvironment supports the metastatic cascade. (5) Tumors produce factors that alter host physiology and condition distant organs for increased metastatic susceptibility.
However, T cells that infiltrate PDAC tumors ultimately encounter a hostile microenvironment and acquire a hypofunctional state associated with up-regulation of immunoregulatory molecules (Moon et al. 2014; Stromnes et al. 2015). Consistent with this, adoptive cell therapy studies using chimeric antigen receptor (CAR)-modified T cells indicate that T-cell trafficking and expansion within tumors are also impaired in patients with PDAC (Beatty et al. 2018; Haas et al. 2019). To this end, efforts to improve the activity of T cells in PDAC will likely need to surmount multiple barriers associated with immune health, T-cell priming, T-cell infiltration, and T-cell hypofunction.
The microbiome in pancreatic cancer
Cancer can affect the integrity of the epithelial barrier and, in doing so, expose tissues to commensal organisms. In this regard, the gut microbiome has been shown to influence cancer and immune biology with implications on therapeutic outcomes. The gut microbiome is a complex ecosystem in constant communication with its host and is vitally important for intestinal epithelial, energy, hormone, and immune cell homeostasis (Zitvogel et al. 2018). Notably, the microbiome has been implicated as a key determinant of epithelial cancer biology where it can influence the efficacy of immunotherapy and shape cancer-associated inflammation (Gopalakrishnan et al. 2018; Routy et al. 2018).
The microbiome and its relationship with carcinogenesis and antitumor immunity have sparked interest in understanding its role in pancreatic cancer. The pancreas and microbiome interface through local, intestinal, and systemic factors (Sun et al. 2015; Stenwall et al. 2019). Consistent with this, distinct changes in the composition and diversity of the gut microbiome (Ren et al. 2017), and even the oral flora (Michaud et al. 2013; Fan et al. 2018), have been seen in patients with PDAC. Pancreas-intrinsic microbiota have also recently been identified in the context of general health, pancreatitis, and PDAC. This observation challenges a long-held belief that the pancreas is a sterile organ (Geller et al. 2017; Thomas et al. 2018; Riquelme et al. 2019). Notably, in PDAC, vastly expanded numbers of bacteria suggest that the pancreatic TME is uniquely suited for microbial colonization and expansion. However, it remains unclear whether bacteria actively shape, or simply coevolve with, the overall state of the TME. Nonetheless, recent observational data show that specific microbes within the tumors associate with long-term survival (Riquelme et al. 2019) after pancreaticoduodenectomy and may even confer resistance to chemotherapy (Geller et al. 2017).
The human pancreas harbors a microbiome
The pancreas stands in direct anatomic communication with the duodenum via the major and minor papillae. However, antimicrobial properties of pancreatic digestive juices, unidirectional flow, and an intact sphincter of Oddi have been thought to maintain the pancreas as a sterile site. Recently, several groups have demonstrated bacteria in the normal pancreas, in the setting of pancreatitis, and in PDAC (Geller et al. 2017; Thomas et al. 2018; Riquelme et al. 2019). While the pancreatic microbiome is similar in its composition across various disease states (Thomas et al. 2018), the PDAC TME shows a roughly 1000-fold expansion (Geller et al. 2017) of bacteria. The most abundant class of bacteria present is Gammaproteobacteria, which are highly prevalent in the duodenal flora, suggesting direct bacterial translocation via the ampulla of Vater as a predominant source of colonization. In support of this hypothesis, higher numbers of bacteria are observed in the pancreases of patients with preoperative biliary tract instrumentation. Other potential routes of bacterial colonization are controversial but include hematogenous spread via the portal venous circulation and trafficking through mesenteric lymph nodes (Diehl et al. 2013).
The relationship between the gut microbiome and biology in the pancreatic TME is an active area of investigation. Efforts to alter the microbiome in tumors have focused on the use of fecal microbial transfer. In this regard, matched fecal and tumor samples from three patients undergoing resection were shown to have a 25% overlap between the two bacterial communities. Remarkably, the transfer of human stool into antibiotic-treated mice resulted in a 40% engraftment of human-derived bacteria in the murine gut. Subsequent orthotopic tumor implantation into these mice revealed ∼5% of the PDAC microbiome to be derived from the original human donor (Riquelme et al. 2019). In addition, homing of the relatively small fraction of donor bacteria to the pancreas shifted the intratumoral microbiome to a distinct taxonomic profile. Collectively, these data suggest that the pancreas is endowed with a unique microbiome, derived at least in part from the intestine, and that it can be manipulated using fecal microbial transfer.
Gut and pancreatic microbiome act as biomarkers
The microbiome has recently been shown to associate with outcomes in patients with PDAC. Specifically, a retrospective analysis showed that bacterial ribosomal 16S subunit sequencing of archived tissue specimens can be used to distinguish short-term survivors (STSs; <5 yr) and long-term survivors (LTSs; >5 yr) after surgical resection (Riquelme et al. 2019). Higher intratumoral α diversity, defined as the number of bacterial species within a sample, correlates with greater CD8+ T-cell infiltration, higher granzyme B expression, and long-term survival. In this study, three genera (Pseudoxanthomonas, Saccharopolyspora, and Streptomyces) and the species Bacillus clausii were enriched in LTS, and the combination of these four taxa produced a prediction model of survival outcome. Intriguingly, fecal microbial transfer of stool from LTS into mice challenged with orthotopic tumor implantation recapitulated the inflammatory immune infiltrate observed in human samples and conferred tumor protection. Taken together, the data imply that the intratumoral microbiome in PDAC may be a useful prognostic biomarker and a correlate of the immunogenicity of PDAC. In addition to the intratumoral microbiome, the gut microbiome is also altered in patients with PDAC. For example, one study profiled stool samples from 85 stage I/II PDAC patients and 57 matched healthy controls in a Chinese population and observed decreased α diversity in the context of cancer, independent of anatomic location of the tumor (head versus tail) or presence of malignant biliary obstruction. On the phylum level, the PDAC gut microbiome was significantly enriched for Bacteroidetes, with a reduction in Firmicutes and Proteobacteria. This dysbiotic change can result in higher lipopolysaccharide and reduced short chain fatty acid production (Ren et al. 2017). The investigators of this study further compiled 40 individual genera enriched in PDAC into a prediction model that achieved 85% accuracy in predicting the presence of pancreatic cancer, supporting the microbiome as a diagnostic biomarker. To this end, unique microbial signatures may offer an early, noninvasive detection method for identifying individuals at high-risk for PDAC development. However, some challenges will need to be considered, including a myriad of confounding variables of PDAC such as obstructive jaundice and the resulting lack of bile and pancreatic juices in the intestine, endocrine and exocrine dysfunction, biliary stenting/instrumentation, antibiotic administration, and dietary and lifestyle factors.
Implications of correlative and preclinical data
Currently, the overwhelming proportion of human microbiome studies is correlative in nature and offers a limited understanding of causal relationships of the complex interplay of host–cancer–microbiome. For example, no conclusive evidence exists to delineate phenotypical contributions of the gut versus the pancreatic tumor microbiome, simply because selective ablation of one bacterial community cannot be achieved experimentally. In this regard, one study deployed subcutaneous xenotransplants of human PDAC cell lines, which are devoid of an intrinsic microbiome, into immune-deficient mice (Thomas et al. 2018). Microbial ablation with antibiotics in these mice resulted in lower rates of successful engraftment, reduced tumor growth, cancer cell-intrinsic transcriptomic changes, and increased infiltration by CD45+ leukocytes. A second study found in immunocompetent mice injected with pancreatic cancer cell lines that antibiotic treatment reduced tumor size and enhanced tumor infiltration by IFNγ+ T helper cells and cytotoxic T cells (Sethi et al. 2018). Finally, using the KRASG12D/PTENlox/+ genetic mouse model of PDAC, microbial ablation with oral antibiotics or germ-free husbandry was shown to delay progression of PanIN lesions to PDAC. Together, these data suggest a mechanistic role for the gut microbiome in defining pancreatic tumor pathogenesis.
Proposed mechanisms of host–microbiome–cancer interactions
Mechanisms by which the microbiome exerts its potent influence over the pancreatic TME are likely to be multifactorial (Fig. 4). One mechanism involves the ligation of innate toll-like receptors (TLRs) by bacterial-derived, pathogen-associated molecular patterns (PAMPs), which then leads to modulation of local and systemic inflammation. For example, the prototypical bacterial antigen lipopolysaccharide can ligate its cognate receptor TLR4 on tumor cells and induce IL-1β production. IL-1β can subsequently activate pancreatic stellate cells and orchestrate an immunosuppressive TME. Conversely, IL-1β blockade has been found to increase IFNγ and granzyme B-expressing CTLs and, by doing so, enhance the antitumor activity of anti-PD-1 therapy (Das et al. 2020). Other TLRs have also been implicated in regulating the biology of cancer cells, cancer-associated fibroblasts, and immune cells (Grimmig et al. 2016; Dajon et al. 2017). Thus, TLR signaling triggered by bacteria may promote inflammation-driven oncogenesis and restrict antitumor immunity in established cancers.
Figure 4.

Overview over the pleotropic effects of the microbiome on the pancreatic tumor microenvironment. (1) TLR ligation by pancreas-intrinsic bacteria-derived peptides skews the immune system toward a tolerogenic phenotype. (2) Bacterial metabolites and peptides from the gut microbiome further promote a tolerogenic immune infiltrate. (3) Bacteria produce cytidine deaminase, which metabolizes gemcitabine to its inactive form difluorodeoxyuridine (dFdU). (4) Digestive juices and antimicrobioal peptides (AMPs) shape the intestinal flora, which then influence the pancreatic TME. (5) Bacterial peptides with similarity to human proteins may trigger cognate T-cell priming and activation of antitumor immunity.
Bacterial metabolites can directly shape host and tumor metabolism (Cani et al. 2019), induce direct cellular damage (Rooks and Garrett 2016), and regulate tolerogenic immune cell recruitment. Short chain fatty acids, produced by bacterial fermentation of carbohydrates in the colon, positively regulate antimicrobial peptide production in the pancreas, which in turn recruits TGF-β-producing macrophages, regulatory DCs, and Tregs (Sun et al. 2015). Similarly, microbial-derived secondary bile acids can induce direct cellular damage but also repress natural killer T-cell (NKT) infiltration and activation in primary and secondary liver tumors (Ma et al. 2018), implicating a potential role in metastatic PDAC. Bacteria can also contribute to drug metabolism and confer resistance to chemotherapy. For instance, Gammaproteobacteria (e.g. E. coli and P. aeruginosa), the most abundant class of microbes in the pancreas, express the enzyme cytidine deaminase, which can metabolize gemcitabine to its inactive form difluorodeoxyuridine. In an experimental model of subcutaneous colon cancer implantation, E. coli inoculation into tumor-bearing mice conferred gemcitabine resistance, whereas antibiotic ablation of the tumor microbiome restored gemcitabine efficacy (Geller et al. 2017).
Bacteria may also influence the immunogenicity of PDAC and, in doing so, impact T-cell infiltration into tumors. For instance, the quantity and quality of neoantigens in human PDAC along with the degree of CD8+ T-cell infiltration into tumors have been shown to correlate with survival (Balachandran et al. 2017). Notably, identified neoantigens in human PDAC tumors from long-term survivors show homology with bacterial-derived epitopes, suggesting that T-cell entrainment or activation may occur through molecular mimicry of the microbiome.
Cumulatively, these data underscore the multifaceted interaction of the microbiome with PDAC. TLRs, metabolites, chemotherapy inactivation, and molecular mimicry are distinct mechanisms (Fig. 4) that may be exploited by the microbiome for regulating the pathogenesis of PDAC. However, as we deepen our understanding of the role of bacteria in PDAC, the significance of the virome and mycobiome remains largely unexplored. In addition, future studies will need to disentangle the complexity of interactions that the microbiome may have on PDAC biology to realize potential therapeutic opportunities.
Support from other epithelial cancers
The field of microbiome research in PDAC has only begun to unravel the complex fabric of host–microbiota–cancer interactions. To this end, the therapeutic potential of targeting the microbiome in PDAC remains ill-defined. However, translational work from other human cancers highlights potential avenues for leveraging the microbiome as a treatment strategy. For instance, stools from non-small cell lung cancer (NSCLC) and renal cell carcinoma patients responding to immune checkpoint blockade (ICB) show a higher bacterial αdiversity as well as enrichment for Akkermansia muciniphila (Routy et al. 2018). Intriguingly, fecal microbial transplant of stool from an ICB “responder” patient or selective administration of A. mucinophila into tumor-bearing mice can improve outcomes to ICB. Similar observations have been made in melanoma patients (Gopalakrishnan et al. 2018; Matson et al. 2018), suggesting that the microbiome may be a strategy for potentiating the activity of ICB. However, there are limitations to extrapolating these data to PDAC. Notably, pancreatic cancer has so far been largely resistant to ICB (Royal et al. 2010; Brahmer et al. 2012; Weiss et al. 2017), and thus, identification of a “responder” microbiome has proven challenging. Additionally, the administration of antibiotics in nongastrointestinal malignancies is generally associated with worse outcomes (Routy et al. 2018) after ICB, whereas preclinical data in PDAC support an opposite effect. Nonetheless, the gut and tumor microbiota have emerged as clear pivotal determinants in cancer, and their therapeutic implications warrant continued investigation in PDAC.
Conclusions
Pancreatic ductal adenocarcinoma is characterized by remarkable therapeutic resistance driven by cancer cell-intrinsic and -extrinsic pathways. The tumor microenvironment that surrounds PDAC is pliable and resilient. It represents a bidirectional evolution arising from cross-talk between cancer cells and the host and is influenced by cancer and host genetics, the immune system, the microbiome, fibrosis, and the metabolic state of cells within and outside of tumors (Fig. 5). These key biological determinants of PDAC form the basis for ongoing therapeutic interventions. Here, we highlighted recent advances in deciphering key pathways in PDAC and their targetable components. Notably, it has become increasingly evident that no one single target will emerge as the Achilles’ heel of PDAC. To this end, our review underscores the need for a “precision medicine” approach. We propose that therapies will need to be patient-centric and tailored based on high-throughput analysis of genetic alterations, as well as transcriptomic, metabolomic, immunologic, and microbial profiling. To achieve this mission, a coordinated effort involving clinicians, basic scientists, patients, advocacy agencies, and industry partners will be necessary. Together, this strategy rooted in sound science and rigorous clinical trial design holds promise for improving outcomes for patients with PDAC.
Figure 5.
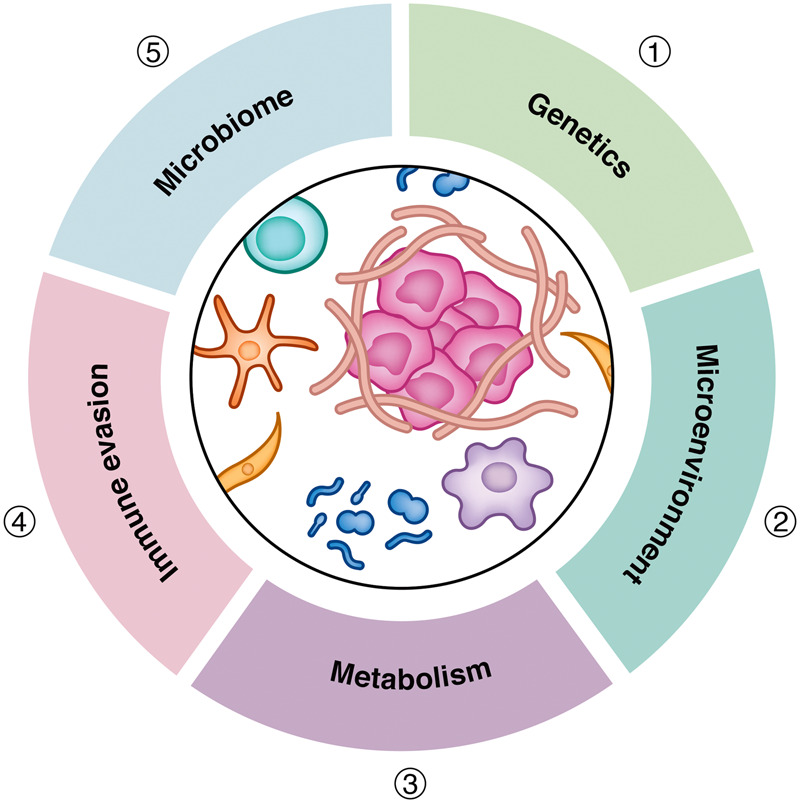
Key determinants of therapeutic resistance in pancreatic cancer. Therapeutic resistance in pancreatic ductal adenocarcinoma is influenced by a myriad of biological pathways directed by (1) genetics, including alterations in oncogenes and tumor suppressor genes; (2) the microenvironment, including fibrosis and poor vascularity, which are hallmarks of pancreatic cancer and contribute to limit drug delivery and impact the contexture of the host immune response; (3) metabolism, including metabolites that shape tumor and host biology as well as cancer cell sensitivity to cytotoxic agents; (4) immune evasion, the capacity to avoid detection and elimination by T cells and other effector immune cell populations; and (5) the microbiome, including gut and intratumoral microbes as well as their byproducts.
Acknowledgments
This work was supported by grants from the National Cancer Institute (R01CA245005, R01CA131045, and R01CA206105 to D.M.S.; R37CA237421, R01CA248160, and R01CA244931 to C.A.L; and R01CA245323, R01CA197916, and U01CA224193 to G.L.B.).
Footnotes
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.348523.121.
Competing interest statement
G.L.B. reports prior or active roles as consultant/advisory board member for Seattle Genetics (now Seagen), Aduro Biotech, AstraZeneca, Bristol-Myers Squibb, Cour Pharmaceuticals, Boehringer Ingelheim, Janssen, Monopteros, Genmab, Merck, Nano Ghosts, Pancreatic Cancer Action Network, and BiolineRx; reports receiving commercial research grants from Incyte, Bristol-Myers Squibb, Verastem, Halozyme, Biothera, Hibercell, Newlink, Novartis, Arcus Biosciences, and Janssen; and is an inventor of intellectual property and recipient of royalties related to CAR T cells that are licensed by the University of Pennsylvania to Novartis and Tmunity Therapeutics. C.A.L. reports prior or active roles as a consultant/advisory board member, as well as receiving research grant funding from Astellas. He is an inventor on patents pertaining to Kras-regulated metabolic pathways, redox control pathways in pancreatic cancer, and targeting GOT1 as a therapeutic approach. D.M.S. reports prior or active roles as a consultant/advisory board member for Celgene, Merck, Tyme, Cyteir, Immunovia, and Interpace and reports receiving research grant funding from Tyme, Repare Therapeutics, Angiodynamics, Sanofi, Cyteir Therapeutics, and Tempus.
References
- Adams CR, Htwe HH, Marsh T, Wang AL, Montoya ML, Subbaraj L, Tward AD, Bardeesy N, Perera RM. 2019. Transcriptional control of subtype switching ensures adaptation and growth of pancreatic cancer. Elife 8: e45313. 10.7554/eLife.45313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre AJ, Nowak JA, Camarda ND, Moffitt RA, Ghazani AA, Hazar-Rethinam M, Raghavan S, Kim J, Brais LK, Ragon D, et al. 2018. Real-time genomic characterization of advanced pancreatic cancer to enable precision medicine. Cancer Discov 8: 1096–1111. 10.1158/2159-8290.CD-18-0275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcalá S, Sancho P, Martinelli P, Navarro D, Pedrero C, Martín-Hijano L, Valle S, Earl J, Rodríguez-Serrano M, Ruiz-Cañas L, et al. 2020. ISG15 and ISGylation is required for pancreatic cancer stem cell mitophagy and metabolic plasticity. Nat Commun 11: 2682. 10.1038/s41467-020-16395-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alistar A, Morris BB, Desnoyer R, Klepin HD, Hosseinzadeh K, Clark C, Cameron A, Leyendecker J, D'Agostino R, Topaloglu U, et al. 2017. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: a single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol 18: 770–778. 10.1016/S1470-2045(17)30314-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS. 2018. Oxidative phosphorylation as an emerging target in cancer therapy. Clin Cancer Res 24: 2482–2490. 10.1158/1078-0432.CCR-17-3070 [DOI] [PubMed] [Google Scholar]
- Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, Sagalovskiy IR, Ma A, Kapilian J, Firl CEM, et al. 2020. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368: 85–89. 10.1126/science.aaw9872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, et al. 2016. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531: 47–52. 10.1038/nature16965 [DOI] [PubMed] [Google Scholar]
- Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, Remark R, Herbst B, Askan G, Bhanot U, Senbabaoglu Y, et al. 2017. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 551: 512–516. 10.1038/nature24462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balachandran VP, Beatty GL, Dougan SK. 2019. Broadening the impact of immunotherapy to pancreatic cancer: challenges and opportunities. Gastroenterology 156: 2056–2072. 10.1053/j.gastro.2018.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banh RS, Biancur DE, Yamamoto K, Sohn ASW, Walters B, Kuljanin M, Gikandi A, Wang H, Mancias JD, Schneider RJ, et al. 2020. Neurons release serine to support mRNA translation in pancreatic cancer. Cell 183: 1202–1218.e25. 10.1016/j.cell.2020.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch DK, Gress TM, Langer P. 2012. Familial pancreatic cancer—current knowledge. Nat Rev Gastroenterol Hepatol 9: 445–453. 10.1038/nrgastro.2012.111 [DOI] [PubMed] [Google Scholar]
- Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, Vonderheide RH. 2012. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 21: 822–835. 10.1016/j.ccr.2012.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, O'Hara M. 2016. Chimeric antigen receptor-modified T cells for the treatment of solid tumors: defining the challenges and next steps. Pharmacol Ther 166: 30–39. 10.1016/j.pharmthera.2016.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al. 2011. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 331: 1612–1616. 10.1126/science.1198443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, Winograd R, Evans RA, Long KB, Luque SL, Lee JW, Clendenin C, Gladney WL, Knoblock DM, Guirnalda PD, et al. 2015. Exclusion of T cells from pancreatic carcinomas in mice is regulated by Ly6Clow F4/80+ extratumoral macrophages. Gastroenterology 149: 201–210. 10.1053/j.gastro.2015.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, Eghbali S, Kim R. 2017. Deploying immunotherapy in pancreatic cancer: defining mechanisms of response and resistance. Am Soc Clin Oncol Educ Book 37: 267–278. 10.1200/EDBK_175232 [DOI] [PubMed] [Google Scholar]
- Beatty GL, O'Hara MH, Lacey SF, Torigian DA, Nazimuddin F, Chen F, Kulikovskaya IM, Soulen MC, McGarvey M, Nelson AM, et al. 2018. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology 155: 29–32. 10.1053/j.gastro.2018.03.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, Preall J, Tuveson DA. 2019. IL1-Induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov 9: 282–301. 10.1158/2159-8290.CD-18-0710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair AB, Kleponis J, Thomas DL 2nd, Muth ST, Murphy AG, Kim V, Zheng L. 2019. IDO1 inhibition potentiates vaccine-induced immunity against pancreatic adenocarcinoma. J Clin Invest 129: 1742–1755. 10.1172/JCI124077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton KL, Chenevix-Trench G, Goh C, Sadetzki S, Ramus SJ, Karlan BY, Lambrechts D, Despierre E, Barrowdale D, McGuffog L, et al. 2012. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA 307: 382–390. 10.1001/jama.2012.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. 2012. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366: 2455–2465. 10.1056/NEJMoa1200694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. 2005. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434: 913–917. 10.1038/nature03443 [DOI] [PubMed] [Google Scholar]
- Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, Gunda V, Pierobon M, Waters AM, George SD, et al. 2019. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med 25: 628–640. 10.1038/s41591-019-0368-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2017. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 32: 185–203.e13. 10.1016/j.ccell.2017.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cani PD, Van Hul M, Lefort C, Depommier C, Rastelli M, Everard A. 2019. Microbial regulation of organismal energy homeostasis. Nat Metab 1: 34–46. 10.1038/s42255-018-0017-4 [DOI] [PubMed] [Google Scholar]
- Canton J. 2018. Macropinocytosis: new insights into Its underappreciated role in innate immune cell surveillance. Front Immunol 9: 2286. 10.3389/fimmu.2018.02286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cass I, Baldwin RL, Varkey T, Moslehi R, Narod SA, Karlan BY. 2003. Improved survival in women with BRCA-associated ovarian carcinoma. Cancer 97: 2187–2195. 10.1002/cncr.11310 [DOI] [PubMed] [Google Scholar]
- Chan-Seng-Yue M, Kim JC, Wilson GW, Ng K, Figueroa EF, O'Kane GM, Connor AA, Denroche RE, Grant RC, McLeod J, et al. 2020. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat Genet 52: 231–240. 10.1038/s41588-019-0566-9 [DOI] [PubMed] [Google Scholar]
- Chantrill LA, Nagrial AM, Watson C, Johns AL, Martyn-Smith M, Simpson S, Mead S, Jones MD, Samra JS, Gill AJ, et al. 2015. Precision medicine for advanced pancreas cancer: the individualized molecular pancreatic cancer therapy (IMPaCT) trial. Clin Cancer Res 21: 2029–2037. 10.1158/1078-0432.CCR-15-0426 [DOI] [PubMed] [Google Scholar]
- Cleary JM, Aguirre AJ, Shapiro GI, D'Andrea AD. 2020. Biomarker-guided development of DNA repair inhibitors. Mol Cell 78: 1070–1085. 10.1016/j.molcel.2020.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RL, Fleming GF, Brady MF, Swisher EM, Steffensen KD, Friedlander M, Okamoto A, Moore KN, Efrat Ben-Baruch N, Werner TL, et al. 2019. Veliparib with first-line chemotherapy and as maintenance therapy in ovarian cancer. N Engl J Med 381: 2403–2415. 10.1056/NEJMoa1909707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MA, Bednar F, Zhang Y, Brisset JC, Galbán S, Galbán CJ, Rakshit S, Flannagan KS, Adsay NV, Pasca di Magliano M. 2012. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 122: 639–653. 10.1172/JCI59227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L, et al. 2011. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 17: 500–503. 10.1038/nm.2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collisson EA, Bailey P, Chang DK, Biankin AV. 2019. Molecular subtypes of pancreatic cancer. Nat Rev Gastroenterol Hepatol 16: 207–220. 10.1038/s41575-019-0109-y [DOI] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. 2013. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497: 633–637. 10.1038/nature12138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor AA, Denroche RE, Jang GH, Timms L, Kalimuthu SN, Selander I, McPherson T, Wilson GW, Chan-Seng-Yue MA, Borozan I, et al. 2017. Association of distinct mutational signatures with correlates of increased immune activity in pancreatic ductal adenocarcinoma. JAMA Oncol 3: 774–783. 10.1001/jamaoncol.2016.3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daemen A, Peterson D, Sahu N, McCord R, Du X, Liu B, Kowanetz K, Hong R, Moffat J, Gao M, et al. 2015. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc Natl Acad Sci 112: E4410–E4417. 10.1073/pnas.1501605112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dajon M, Iribarren K, Cremer I. 2017. Toll-like receptor stimulation in cancer: a pro- and anti-tumor double-edged sword. Immunobiology 222: 89–100. 10.1016/j.imbio.2016.06.009 [DOI] [PubMed] [Google Scholar]
- Dalin S, Sullivan MR, Lau AN, Grauman-Boss B, Mueller HS, Kreidl E, Fenoglio S, Luengo A, Lees JA, Vander Heiden MG, et al. 2019. Deoxycytidine release from pancreatic stellate cells promotes gemcitabine resistance. Cancer Res 79: 5723–5733. 10.1158/0008-5472.CAN-19-0960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Shapiro B, Vucic EA, Vogt S, Bar-Sagi D. 2020. Tumor cell-derived IL1β promotes desmoplasia and immune suppression in pancreatic cancer. Cancer Res 80: 1088–1101. 10.1158/0008-5472.CAN-19-2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta R, Lau AN, Sivanand S, Florek L, Wyckoff J, Skala MC, Vander Heiden MG. 2020. Interactions with stromal cells promote a more oxidized cancer cell redox state in pancreatic tumors. bioRxiv 10.1101/2020.10.20.347658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Jonas O, Keibler MA, Hou HW, Luengo A, Mayers JR, Wyckoff J, Del Rosario AM, Whitman M, Chin CR, et al. 2017. Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat Med 23: 235–241. 10.1038/nm.4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, Ramakrishna M, Martin S, Boyault S, Sieuwerts AM, et al. 2017. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med 23: 517–525. 10.1038/nm.4292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al. 2011. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475: 106–109. 10.1038/nature10189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl GE, Longman RS, Zhang JX, Breart B, Galan C, Cuesta A, Schwab SR, Littman DR. 2013. Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX3CR1hi cells. Nature 494: 116–120. 10.1038/nature11809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodhiawala PB, Khurana N, Zhang D, Cheng Y, Li L, Wei Q, Seehra K, Jiang H, Grierson PM, Wang-Gillam A, et al. 2020. TPL2 enforces RAS-induced inflammatory signaling and is activated by point mutations. J Clin Invest 130: 4771–4790. 10.1172/JCI137660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosos Y, Escobar D, Chiang MY, Roys K, Valentine V, Valentine MB, Rehg JE, Sahai V, Begley LA, Ye J, et al. 2017. ATM-deficiency increases genomic instability and metastatic potential in a mouse model of pancreatic cancer. Sci Rep 7: 11144. 10.1038/s41598-017-11661-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten M, Barbacid M. 2020. Targeting the MAPK pathway in KRAS-driven tumors. Cancer Cell 37: 543–550. 10.1016/j.ccell.2020.03.013 [DOI] [PubMed] [Google Scholar]
- DuFort CC, DelGiorno KE, Hingorani SR. 2016. Mounting pressure in the microenvironment: fluids, solids, and cells in pancreatic ductal adenocarcinoma. Gastroenterology 150: 1545–1557.e2. 10.1053/j.gastro.2016.03.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Alekseyenko AV, Wu J, Peters BA, Jacobs EJ, Gapstur SM, Purdue MP, Abnet CC, Stolzenberg-Solomon R, Miller G, et al. 2018. Human oral microbiome and prospective risk for pancreatic cancer: a population-based nested case-control study. Gut 67: 120–127. 10.1136/gutjnl-2016-312580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434: 917–921. 10.1038/nature03445 [DOI] [PubMed] [Google Scholar]
- Fedele C, Ran H, Diskin B, Wei W, Jen J, Geer MJ, Araki K, Ozerdem U, Simeone DM, Miller G, et al. 2018. SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discov 8: 1237–1249. 10.1158/2159-8290.CD-18-0444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, et al. 2013. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci 110: 20212–20217. 10.1073/pnas.1320318110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann G, Karikari C, dal Molin M, Duringer S, Volkmann P, Bartsch DK, Bisht S, Koorstra JB, Brossart P, Maitra A, et al. 2011. Inactivation of Brca2 cooperates with Trp53R172H to induce invasive pancreatic ductal adenocarcinomas in mice: a mouse model of familial pancreatic cancer. Cancer Biol Ther 11: 959–968. 10.4161/cbt.11.11.15534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabitova-Cornell L, Surumbayeva A, Peri S, Franco-Barraza J, Restifo D, Weitz N, Ogier C, Goldman AR, Hartman TR, Francescone R, et al. 2020. Cholesterol pathway inhibition induces TGF-β signaling to promote basal differentiation in pancreatic cancer. Cancer Cell 38: 567–583.e11. 10.1016/j.ccell.2020.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, Gavert N, Zwang Y, Cooper ZA, Shee K, et al. 2017. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 357: 1156–1160. 10.1126/science.aah5043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidekel Friedlander SY, Chu GC, Snyder EL, Girnius N, Dibelius G, Crowley D, Vasile E, DePinho RA, Jacks T. 2009. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell 16: 379–389. 10.1016/j.ccr.2009.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan T, Kanji ZS, Epelbaum R, Devaud N, Dagan E, Holter S, Aderka D, Paluch-Shimon S, Kaufman B, Gershoni-Baruch R, et al. 2014. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer 111: 1132–1138. 10.1038/bjc.2014.418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, Park JO, Hochhauser D, Arnold D, Oh DY, et al. 2019. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med 381: 317–327. 10.1056/NEJMoa1903387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC, et al. 2018. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359: 97–103. 10.1126/science.aan4236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gout J, Perkhofer L, Morawe M, Arnold F, Ihle M, Biber S, Lange S, Roger E, Kraus JM, Stifter K, et al. 2021. Synergistic targeting and resistance to PARP inhibition in DNA damage repair-deficient pancreatic cancer. Gut 70: 743–760. 10.1136/gutjnl-2019-319970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimmig T, Moench R, Kreckel J, Haack S, Rueckert F, Rehder R, Tripathi S, Ribas C, Chandraker A, Germer CT, et al. 2016. Toll like receptor 2, 4, and 9 signaling promotes autoregulative tumor cell growth and VEGF/PDGF expression in human pancreatic cancer. Int J Mol Sci 17: 2060. 10.3390/ijms17122060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivas P, Monk BJ, Petrylak D, Reck M, Foley G, Guenther S, Hennessy D, Makris C, Moehler M. 2019. Immune checkpoint inhibitors as switch or continuation maintenance therapy in solid tumors: rationale and current state. Target Oncol 14: 505–525. 10.1007/s11523-019-00665-1 [DOI] [PubMed] [Google Scholar]
- Guerra C, Schuhmacher AJ, Cañamero M, Grippo PJ, Verdaguer L, Pérez-Gallego L, Dubus P, Sandgren EP, Barbacid M. 2007. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 11: 291–302. 10.1016/j.ccr.2007.01.012 [DOI] [PubMed] [Google Scholar]
- Gui DY, Sullivan LB, Luengo A, Hosios AM, Bush LN, Gitego N, Davidson SM, Freinkman E, Thomas CJ, Vander Heiden MG. 2016. Environment dictates dependence on mitochondrial complex I for NAD+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab 24: 716–727. 10.1016/j.cmet.2016.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas AR, Tanyi JL, O'Hara MH, Gladney WL, Lacey SF, Torigian DA, Soulen MC, Tian L, McGarvey M, Nelson AM, et al. 2019. Phase I study of lentiviral-transduced chimeric antigen receptor-modified T cells recognizing mesothelin in advanced solid cancers. Mol Ther 27: 1919–1929. 10.1016/j.ymthe.2019.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbrook CJ, Lyssiotis CA. 2017. Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell 31: 5–19. 10.1016/j.ccell.2016.12.006 [DOI] [PubMed] [Google Scholar]
- Halbrook CJ, Pontious C, Kovalenko I, Lapienyte L, Dreyer S, Lee HJ, Thurston G, Zhang Y, Lazarus J, Sajjakulnukit P, et al. 2019. Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell Metab 29: 1390–1399.e6. 10.1016/j.cmet.2019.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbrook CJ, Thurston G, McCarthy A, Nelson BS, Sajjakulnukit P, Krall AS, Mullen PJ, Zhang L, Batra S, Viale A, et al. 2020. Clonal heterogeneity supports mitochondrial metabolism in pancreatic cancer. bioRxiv 10.1101/2020.05.15.098368 [DOI] [Google Scholar]
- Hayes JD, Dinkova-Kostova AT. 2014. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 39: 199–218. 10.1016/j.tibs.2014.02.002 [DOI] [PubMed] [Google Scholar]
- Hegde S, Krisnawan VE, Herzog BH, Zuo C, Breden MA, Knolhoff BL, Hogg GD, Tang JP, Baer JM, Mpoy C, et al. 2020. Dendritic cell paucity leads to dysfunctional immune surveillance in pancreatic cancer. Cancer Cell 37: 289–307.e9. 10.1016/j.ccell.2020.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleday T. 2011. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol 5: 387–393. 10.1016/j.molonc.2011.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms E, Onate MK, Sherman MH. 2020. Fibroblast heterogeneity in the pancreatic tumor microenvironment. Cancer Discov 10: 648–656. 10.1158/2159-8290.CD-19-1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Zheng L, Bullock AJ, Seery TE, Harris WP, Sigal DS, Braiteh F, Ritch PS, Zalupski MM, Bahary N, et al. 2018. HALO 202: randomized phase II study of PEGPH20 plus Nab-paclitaxel/gemcitabine versus Nab-paclitaxel/gemcitabine in patients with untreated, metastatic pancreatic ductal adenocarcinoma. J Clin Oncol 36: 359–366. 10.1200/JCO.2017.74.9564 [DOI] [PubMed] [Google Scholar]
- Hiraoka N, Ino Y, Yamazaki-Itoh R, Kanai Y, Kosuge T, Shimada K. 2015. Intratumoral tertiary lymphoid organ is a favourable prognosticator in patients with pancreatic cancer. Br J Cancer 112: 1782–1790. 10.1038/bjc.2015.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann MH, Gmachl M, Ramharter J, Savarese F, Gerlach D, Marszalek JR, Sanderson MP, Kessler D, Trapani F, Arnhof H, et al. 2020. BI-3406, a potent and selective SOS1-KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK inhibition. Cancer Discov 11: 142-157. 10.1158/2159-8290.CD-20-0142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollinshead KER, Parker SJ, Eapen VV, Encarnacion-Rosado J, Sohn A, Oncu T, Cammer M, Mancias JD, Kimmelman AC. 2020. Respiratory supercomplexes promote mitochondrial efficiency and growth in severely hypoxic pancreatic cancer. Cell Rep 33: 108231. 10.1016/j.celrep.2020.108231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, Falchook GS, Price TJ, Sacher A, Denlinger CS, et al. 2020a. KRASg12c inhibition with sotorasib in advanced solid tumors. N Engl J Med 383: 1207–1217. 10.1056/NEJMoa1917239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong DS, Kuo J, Sacher AG, Barlesi F, Besse B, Kuboki Y, Dy GK, Dembla V, Krauss JC, Burns TF, et al. 2020b. Codebreak 100: phase I study of AMG 510, a novel KRASG12C inhibitor, in patients (pts) with advanced solid tumors other than non-small cell lung cancer (NSCLC) and colorectal cancer (CRC). J Clin Oncol 38: 3511–3511. 10.1200/JCO.2020.38.15_suppl.3511 [DOI] [Google Scholar]
- Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, Feig C, Nakagawa T, Caldwell ME, Zecchini HI, et al. 2013. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 62: 112–120. 10.1136/gutjnl-2012-302529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, et al. 2016. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med 22: 851–860. 10.1038/nm.4123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, Hsu JL, Yu WH, Du Y, Lee HH, et al. 2017. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res 23: 3711–3720. 10.1158/1078-0432.CCR-16-3215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. 2008. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321: 1801–1806. 10.1126/science.1164368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalbasi A, Komar C, Tooker GM, Liu M, Lee JW, Gladney WL, Ben-Josef E, Beatty GL. 2017. Tumor-derived CCL2 mediates resistance to radiotherapy in pancreatic ductal adenocarcinoma. Clin Cancer Res 23: 137–148. 10.1158/1078-0432.CCR-16-0870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, Thompson CB, Rabinowitz JD. 2013. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci 110: 8882–8887. 10.1073/pnas.1307237110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA, Bar-Sagi D, et al. 2015. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 75: 544–553. 10.1158/0008-5472.CAN-14-2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly B, Pearce EL. 2020. Amino assets: how amino acids support immunity. Cell Metab 32: 154–175. 10.1016/j.cmet.2020.06.010 [DOI] [PubMed] [Google Scholar]
- Kerk SA, Lin L, Myers AL, Chen B, Sajjakulnukit P, Robinson A, Thurston G, Nelson BS, Kemp SB, Steele NG, et al. 2020. The pancreatic tumor microenvironment buffers redox imbalance imposed by disrupted mitochondrial metabolism. bioRxiv 10.1101/2020.08.07.238766 [DOI] [Google Scholar]
- Kessler D, Gmachl M, Mantoulidis A, Martin LJ, Zoephel A, Mayer M, Gollner A, Covini D, Fischer S, Gerstberger T, et al. 2019. Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci 116: 15823–15829. 10.1073/pnas.1904529116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim PK, Halbrook CJ, Kerk SA, Wisner S, Kremer DM, Sajjakulnukit P, Hou SW, Thurston G, Anand A, Yan L, et al. 2020. Hyaluronic acid fuels pancreatic cancer growth. bioRxiv 10.1101/2020.09.14.293803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmelman AC, White E. 2017. Autophagy and tumor metabolism. Cell Metab 25: 1037–1043. 10.1016/j.cmet.2017.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King B, Araki J, Palm W, Thompson CB. 2020. Yap/Taz promote the scavenging of extracellular nutrients through macropinocytosis. Genes Dev 34: 1345–1358. 10.1101/gad.340661.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, Schuman SS, Shea JE, Seipp MT, Yap JT, et al. 2019. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med 25: 620–627. 10.1038/s41591-019-0367-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloesch B, Ionasz V, Paliwal S, Hruschka N, Martinez de Villarreal J, Ollinger R, Mueller S, Dienes HP, Schindl M, Gruber ES et al. 2021. A GATA6-centred gene regulatory network involving HNFs and ΔNp63 controls plasticity and immune escape in pancreatic cancer. Gut gutjnl-2020-321397. 10.1136/gutjnl-2020-321397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall AS, Mullen PJ, Surjono F, Momcilovic M, Schmid EW, Halbrook CJ, Thambundit A, Mittelman SD, Lyssiotis CA, Shackelford DB, et al. 2020. Asparagine signals mitochondrial respiration and can be targeted to impair tumour growth. bioRxiv 10.1101/2020.03.17.995670 [DOI] [Google Scholar]
- Krall AS, Mullen PJ, Surjono F, Momcilovic M, Schmid EW, Halbrook CJ, Thambundit A, Mittelman SD, Lyssiotis CA, Shackelford DB, et al. 2021. Asparagine couples mitochondrial respiration to ATF4 activity and tumor growth. Cell Metab 33: 1013–1026.E6. 10.1016/j.cmet.2021.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer DM, Nelson BS, Lin L, Yarosz EL, Halbrook CJ, Kerk SA, Sajjakulnukit P, Myers A, Thurston G, Hou SW, et al. 2020. GOT1 inhibition primes pancreatic cancer for ferroptosis through the autophagic release of labile iron. bioRxiv 10.1101/2020.02.28.970228 [DOI] [Google Scholar]
- Kwon J, Bakhoum SF. 2020. The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer Discov 10: 26–39. 10.1158/2159-8290.CD-19-0761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. 2015. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 372: 2509–2520. 10.1056/NEJMoa1500596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et al. 2017. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357: 409–413. 10.1126/science.aan6733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JJ, Perera RM, Wang H, Wu DC, Liu XS, Han S, Fitamant J, Jones PD, Ghanta KS, Kawano S, et al. 2014. Stromal response to hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci 111: E3091–E3100. 10.1073/pnas.1411679111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Stone ML, Porrett PM, Thomas SK, Komar CA, Li JH, Delman D, Graham K, Gladney WL, Hua X, et al. 2019. Hepatocytes direct the formation of a pro-metastatic niche in the liver. Nature 567: 249–252. 10.1038/s41586-019-1004-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, Richman LP, Lin JH, Sun YH, Rech AJ, et al. 2018. Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity 49: 178–193.e7. 10.1016/j.immuni.2018.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yuan S, Norgard RJ, Yan F, Yamazoe T, Blanco A, Stanger BZ. 2020. Tumor cell-intrinsic USP22 suppresses antitumor immunity in pancreatic cancer. Cancer Immunol Res 8: 282–291. 10.1158/2326-6066.CIR-19-0661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligorio M, Sil S, Malagon-Lopez J, Nieman LT, Misale S, Di Pilato M, Ebright RY, Karabacak MN, Kulkarni AS, Liu A, et al. 2019. Stromal microenvironment shapes the intratumoral architecture of pancreatic cancer. Cell 178: 160–175.e27. 10.1016/j.cell.2019.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Huffman AP, Wattenberg MM, Walter DM, Carpenter EL, Feldser DM, Beatty GL, Furth EE, Vonderheide RH. 2020. Type 1 conventional dendritic cells are systemically dysregulated early in pancreatic carcinogenesis. J Exp Med 217: e20190673. 10.1084/jem.20190673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long KB, Gladney WL, Tooker GM, Graham K, Fraietta JA, Beatty GL. 2016. IFNγ and CCL2 cooperate to redirect tumor-infiltrating monocytes to degrade fibrosis and enhance chemotherapy efficacy in pancreatic carcinoma. Cancer Discov 6: 400–413. 10.1158/2159-8290.CD-15-1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery MA, Jordan EJ, Basturk O, Ptashkin RN, Zehir A, Berger MF, Leach T, Herbst B, Askan G, Maynard H, et al. 2017. Real-time genomic profiling of pancreatic ductal adenocarcinoma: potential actionability and correlation with clinical phenotype. Clin Cancer Res 23: 6094–6100. 10.1158/1078-0432.CCR-17-0899 [DOI] [PubMed] [Google Scholar]
- Lyssiotis CA, Kimmelman AC. 2017. Metabolic interactions in the tumor microenvironment. Trends Cell Biol 27: 863–875. 10.1016/j.tcb.2017.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, Agdashian D, Terabe M, Berzofsky JA, Fako V, et al. 2018. Gut microbiome–mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360: eaan5931. 10.1126/science.aan5931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. 2014. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509: 105–109. 10.1038/nature13148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markosyan N, Li J, Sun YH, Richman LP, Lin JH, Yan F, Quinones L, Sela Y, Yamazoe T, Gordon N, et al. 2019. Tumor cell-intrinsic EPHA2 suppresses anti-tumor immunity by regulating PTGS2 (COX-2). J Clin Invest 129: 3594–3609. 10.1172/JCI127755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masoud R, Reyes-Castellanos G, Lac S, Garcia J, Dou S, Shintu L, Abdel Hadi N, Gicquel T, El Kaoutari A, Diémé B, et al. 2020. Targeting mitochondrial complex I overcomes chemoresistance in high OXPHOS pancreatic cancer. Cell Rep Med 1: 100143. 10.1016/j.xcrm.2020.100143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, Luke JJ, Gajewski TF. 2018. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359: 104–108. 10.1126/science.aao3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ, Coppes RP, Engedal N, Mari M, Reggiori F. 2018. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 14: 1435–1455. 10.1080/15548627.2018.1474314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister F, Bailey JM, Alsina J, Nirschl CJ, Sharma R, Fan H, Rattigan Y, Roeser JC, Lankapalli RH, Zhang H, et al. 2014. Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell 25: 621–637. 10.1016/j.ccr.2014.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MA, Baer JM, Knolhoff BL, Nywening TM, Panni RZ, Su X, Weilbaecher KN, Hawkins WG, Ma C, Fields RC, et al. 2018. Breast and pancreatic cancer interrupt IRF8-dependent dendritic cell development to overcome immune surveillance. Nat Commun 9: 1250. 10.1038/s41467-018-03600-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud DS, Izard J, Wilhelm-Benartzi CS, You DH, Grote VA, Tjønneland A, Dahm CC, Overvad K, Jenab M, Fedirko V, et al. 2013. Plasma antibodies to oral bacteria and risk of pancreatic cancer in a large European prospective cohort study. Gut 62: 1764–1770. 10.1136/gutjnl-2012-303006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, et al. 2013. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 73: 1128–1141. 10.1158/0008-5472.CAN-12-2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, Rashid NU, Williams LA, Eaton SC, Chung AH, et al. 2015. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 47: 1168–1178. 10.1038/ng.3398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, McAfoos T, Morlacchi P, Ackroyd J, Agip AA, et al. 2018. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med 24: 1036–1046. 10.1038/s41591-018-0052-4 [DOI] [PubMed] [Google Scholar]
- Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, Kapoor V, Scholler J, Puré E, Milone MC, et al. 2014. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res 20: 4262–4273. 10.1158/1078-0432.CCR-13-2627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris J, Yashinskie JJ, Koche R, Chandwani R, Tian S, Chen CC, Baslan T, Marinkovic ZS, Sánchez-Rivera FJ, Leach SD, et al. 2019. α-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 573: 595–599. 10.1038/s41586-019-1577-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, et al. 2014. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41: 14–20. 10.1016/j.immuni.2014.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson BS, Lin L, Kremer DM, Sousa CM, Cotta-Ramusino C, Myers A, Ramos J, Gao T, Kovalenko I, Wilder-Romans K, et al. 2020. Tissue of origin dictates GOT1 dependence and confers synthetic lethality to radiotherapy. Cancer Metab 8: 1. 10.1186/s40170-019-0202-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notta F, Chan-Seng-Yue M, Lemire M, Li Y, Wilson GW, Connor AA, Denroche RE, Liang SB, Brown AM, Kim JC, et al. 2016. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 538: 378–382. 10.1038/nature19823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, Toriola AT, Nieman RK, Worley LA, Yano M, et al. 2016. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol 17: 651–662. 10.1016/S1470-2045(16)00078-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, Jacobs RC, Ye J, Patel AA, Gillanders WE, et al. 2018. Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 67: 1112–1123. 10.1136/gutjnl-2017-313738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, Corbo V, Oni TE, Hearn SA, Lee EJ, et al. 2017. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 214: 579–596. 10.1084/jem.20162024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Kane GM, Grünwald BT, Jang GH, Masoomian M, Picardo S, Grant RC, Denroche RE, Zhang A, Wang Y, Lam B, et al. 2020. GATA6 expression distinguishes classical and basal-like subtypes in advanced pancreatic cancer. Clin Cancer Res 26: 4901–4910. 10.1158/1078-0432.CCR-19-3724 [DOI] [PubMed] [Google Scholar]
- Olivares O, Mayers JR, Gouirand V, Torrence ME, Gicquel T, Borge L, Lac S, Roques J, Lavaut MN, Berthezène P, et al. 2017. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat Commun 8: 16031. 10.1038/ncomms16031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, et al. 2009. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324: 1457–1461. 10.1126/science.1171362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly EM, Lee JW, Zalupski M, Capanu M, Park J, Golan T, Tahover E, Lowery MA, Chou JF, Sahai V, et al. 2020. Randomized, multicenter, phase II trial of gemcitabine and cisplatin with or without veliparib in patients with pancreas adenocarcinoma and a germline BRCA/PALB2 mutation. J Clin Oncol 38: 1378–1388. 10.1200/JCO.19.02931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, Thompson CB. 2015. The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell 162: 259–270. 10.1016/j.cell.2015.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panni RZ, Herndon JM, Zuo C, Hegde S, Hogg GD, Knolhoff BL, Breden MA, Li X, Krisnawan VE, Khan SQ, et al. 2019. Agonism of CD11b reprograms innate immunity to sensitize pancreatic cancer to immunotherapies. Sci Transl Med 11: eaau9240. 10.1126/scitranslmed.aau9240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, Visal T, Li MK, Pinto J, Castrillon JA, et al. 2019. PARP inhibitor efficacy depends on CD8+ T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov 9: 722–737. 10.1158/2159-8290.CD-18-1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker SJ, Amendola CR, Hollinshead KER, Yu Q, Yamamoto K, Encarnación-Rosado J, Rose RE, LaRue MM, Sohn ASW, Biancur DE, et al. 2020. Selective alanine transporter utilization creates a targetable metabolic niche in pancreatic cancer. Cancer Discov 10: 1018–1037. 10.1158/2159-8290.CD-19-0959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil S, Steuber B, Kopp W, Kari V, Urbach L, Wang X, Küffer S, Bohnenberger H, Spyropoulou D, Zhang Z, et al. 2020. EZH2 regulates pancreatic cancer subtype identity and tumor progression via transcriptional repression of GATA6. Cancer Res 80: 4620–4632. 10.1158/0008-5472.CAN-20-0672 [DOI] [PubMed] [Google Scholar]
- Perera RM, Bardeesy N. 2015. pancreatic cancer metabolism: breaking it down to build it back up. Cancer Discov 5: 1247–1261. 10.1158/2159-8290.CD-15-0671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, et al. 2015. Transcriptional control of autophagy–lysosome function drives pancreatic cancer metabolism. Nature 524: 361–365. 10.1038/nature14587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piffoux M, Eriau E, Cassier PA. 2020. Autophagy as a therapeutic target in pancreatic cancer. Br J Cancer 124: 333–344. 10.1038/s41416-020-01039-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pishvaian MJ, Blais EM, Brody JR, Lyons E, DeArbeloa P, Hendifar A, Mikhail S, Chung V, Sahai V, Sohal DPS, et al. 2020. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the know your tumor registry trial. Lancet Oncol 21: 508–518. 10.1016/S1470-2045(20)30074-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak P, Kim J, Braunstein LZ, Karlic R, Haradhavala NJ, Tiao G, Rosebrock D, Livitz D, Kübler K, Mouw KW, et al. 2017. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat Genet 49: 1476–1486. 10.1038/ng.3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter RL, Magnus NKC, Thapar V, Morris R, Szabolcs A, Neyaz A, Kulkarni AS, Tai E, Chougule A, Hillis A, et al. 2019. Epithelial to mesenchymal plasticity and differential response to therapies in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci 116: 26835–26845. 10.1073/pnas.1914915116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powles T, Park SH, Voog E, Caserta C, Valderrama BP, Gurney H, Kalofonos H, Radulović S, Demey W, Ullén A, et al. 2020. Avelumab maintenance therapy for advanced or metastatic urothelial carcinoma. N Engl J Med 383: 1218–1230. 10.1056/NEJMoa2002788 [DOI] [PubMed] [Google Scholar]
- Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. 2012. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 21: 418–429. 10.1016/j.ccr.2012.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puleo F, Nicolle R, Blum Y, Cros J, Marisa L, Demetter P, Quertinmont E, Svrcek M, Elarouci N, Iovanna J, et al. 2018. Stratification of pancreatic ductal adenocarcinomas based on tumor and microenvironment features. Gastroenterology 155: 1999–2013.e3. 10.1053/j.gastro.2018.08.033 [DOI] [PubMed] [Google Scholar]
- Purohit V, Simeone DM, Lyssiotis CA. 2019. Metabolic regulation of redox balance in cancer. Cancers (Basel) 11: 955. 10.3390/cancers11070955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit V, Wang L, Yang H, Li J, Ney GM, Gumkowski ER, Vaidya AJ, Wang A, Bhardwaj A, Zhao E, et al. 2021. ATDC binds to KEAP1 to drive NRF2-mediated tumorigenesis and chemoresistance in pancreatic cancer. Genes Dev 35: 218–233. 10.1101/gad.344184.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. 2012. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 21: 836–847. 10.1016/j.ccr.2012.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JD, White E. 2010. Autophagy and metabolism. Science 330: 1344–1348. 10.1126/science.1193497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajeshkumar NV, Yabuuchi S, Pai SG, De Oliveira E, Kamphorst JJ, Rabinowitz JD, Tejero H, Al-Shahrour F, Hidalgo M, Maitra A, et al. 2017. Treatment of pancreatic cancer patient-derived xenograft panel with metabolic inhibitors reveals efficacy of phenformin. Clin Cancer Res 23: 5639–5647. 10.1158/1078-0432.CCR-17-1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan RK, McDonough SL, Philip PA, Hingorani SR, Lacy J, Kortmansky JS, Thumar J, Chiorean EG, Shields AF, Behl D, et al. 2019. Phase IB/II randomized study of FOLFIRINOX plus pegylated recombinant human hyaluronidase versus FOLFIRINOX alone in patients with metastatic pancreatic adenocarcinoma: SWOG S1313. J Clin Oncol 37: 1062–1069. 10.1200/JCO.18.01295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Jiang J, Xie H, Li A, Lu H, Xu S, Zhou L, Zhang H, Cui G, Chen X, et al. 2017. Gut microbial profile analysis by miSeq sequencing of pancreatic carcinoma patients in China. Oncotarget 8: 95176–95191. 10.18632/oncotarget.18820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renz BW, Takahashi R, Tanaka T, Macchini M, Hayakawa Y, Dantes Z, Maurer HC, Chen X, Jiang Z, Westphalen CB, et al. 2018a. β2 adrenergic-neurotrophin feedforward loop promotes pancreatic cancer. Cancer Cell 33: 75–90.e7. 10.1016/j.ccell.2017.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renz BW, Tanaka T, Sunagawa M, Takahashi R, Jiang Z, Macchini M, Dantes Z, Valenti G, White RA, Middelhoff MA, et al. 2018b. Cholinergic signaling via muscarinic receptors directly and indirectly suppresses pancreatic tumorigenesis and cancer stemness. Cancer Discov 8: 1458–1473. 10.1158/2159-8290.CD-18-0046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, et al. 2014. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25: 735–747. 10.1016/j.ccr.2014.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, Quesada P, Sahin I, Chandra V, San Lucas A, et al. 2019. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell 178: 795–806.e12. 10.1016/j.cell.2019.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roa-Peña L, Leiton CV, Babu S, Pan CH, Vanner EA, Akalin A, Bandovic J, Moffitt RA, Shroyer KR, Escobar-Hoyos LF. 2019. Keratin 17 identifies the most lethal molecular subtype of pancreatic cancer. Sci Rep 9: 11239. 10.1038/s41598-019-47519-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooks MG, Garrett WS. 2016. Gut microbiota, metabolites and host immunity. Nat Rev Immunol 16: 341–352. 10.1038/nri.2016.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, et al. 2018. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359: 91–97. 10.1126/science.aan3706 [DOI] [PubMed] [Google Scholar]
- Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, Sherry RM, Topalian SL, Yang JC, Lowy I, et al. 2010. Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother 33: 828–833. 10.1097/CJI.0b013e3181eec14c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T, et al. 2020. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer 20: 174–186. 10.1038/s41568-019-0238-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar A, Lindsey-Boltz LA, Ünsal-Kaçmaz K, Linn S. 2004. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 73: 39–85. 10.1146/annurev.biochem.73.011303.073723 [DOI] [PubMed] [Google Scholar]
- Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, Mitchem JB, Plambeck-Suess SM, Worley LA, Goetz BD, et al. 2013. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res 19: 3404–3415. 10.1158/1078-0432.CCR-13-0525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, Nuryadi E, Sekine R, Oike T, Kakoti S, et al. 2017. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun 8: 1751. 10.1038/s41467-017-01883-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeber A, Puccini A, Xiu J, Goldberg RM, Grothey A, Shields AF, Salem ME, Battaglin F, El-Deiry WS, Tokunaga R, et al. 2019. Association of BRCA-mutant pancreatic cancer with high tumor mutational burden (TMB) and higher PD-L1 expression. J Clin Oncol 37: 4133–4133. 10.1200/JCO.2019.37.15_suppl.4133 [DOI] [Google Scholar]
- Sethi V, Kurtom S, Tarique M, Lavania S, Malchiodi Z, Hellmund L, Zhang L, Sharma U, Giri B, Garg B, et al. 2018. Gut microbiota promotes tumor growth in mice by modulating immune response. Gastroenterology 155: 33–37.e6. 10.1053/j.gastro.2018.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Gao W, Lytle NK, Huang P, Yuan X, Dann AM, Ridinger-Saison M, DelGiorno KE, Antal CE, Liang G, et al. 2019. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 569: 131–135. 10.1038/s41586-019-1130-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla SK, Purohit V, Mehla K, Gunda V, Chaika NV, Vernucci E, King RJ, Abrego J, Goode GD, Dasgupta A, et al. 2017. MUC1 and HIF-1α signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell 32: 392. 10.1016/j.ccell.2017.08.008 [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Fuchs HE, Jemal A. 2021. Cancer statistics, 2021. CA Cancer J Clin 71: 7–33. 10.3322/caac.21654 [DOI] [PubMed] [Google Scholar]
- Sodir NM, Kortlever RM, Barthet VJA, Campos T, Pellegrinet L, Kupczak S, Anastasiou P, Swigart LB, Soucek L, Arends MJ, et al. 2020. MYC instructs and maintains pancreatic adenocarcinoma phenotype. Cancer Discov 10: 588–607. 10.1158/2159-8290.CD-19-0435 [DOI] [PubMed] [Google Scholar]
- Somerville TDD, Xu Y, Miyabayashi K, Tiriac H, Cleary CR, Maia-Silva D, Milazzo JP, Tuveson DA, Vakoc CR. 2018. TP63-mediated enhancer reprogramming drives the squamous subtype of pancreatic ductal adenocarcinoma. Cell Rep 25: 1741–1755.e7. 10.1016/j.celrep.2018.10.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. 2013. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496: 101–105. 10.1038/nature12040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, et al. 2016. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536: 479–483. 10.1038/nature19084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, Foth M, Bryson S, McDaid K, Wilson Z, et al. 2016. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell 29: 832–845. 10.1016/j.ccell.2016.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele NG, Carpenter ES, Kemp SB, Sirihorachai VR, The S, Delrosario L, Lazarus J, Amir E-a, Gunchick V, Espinoza C, et al. 2020. Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer. Nature Cancer 1: 1097–1112. 10.1038/s43018-020-00121-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenwall A, Ingvast S, Skog O, Korsgren O. 2019. Characterization of host defense molecules in the human pancreas. Islets 11: 89–101. 10.1080/19382014.2019.1585165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone ML, Beatty GL. 2019. Cellular determinants and therapeutic implications of inflammation in pancreatic cancer. Pharmacol Ther 201: 202–213. 10.1016/j.pharmthera.2019.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromnes IM, Schmitt TM, Hulbert A, Brockenbrough JS, Nguyen H, Cuevas C, Dotson AM, Tan X, Hotes JL, Greenberg PD, et al. 2015. T cells engineered against a native antigen can surmount immunologic and physical barriers to treat pancreatic ductal adenocarcinoma. Cancer Cell 28: 638–652. 10.1016/j.ccell.2015.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromnes IM, Hulbert A, Pierce RH, Greenberg PD, Hingorani SR. 2017. T-cell localization, activation, and clonal expansion in human pancreatic ductal adenocarcinoma. Cancer Immunol Res 5: 978–991. 10.1158/2326-6066.CIR-16-0322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, Dennstedt EA, Vander Heiden MG, Muir A. 2019. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 8: e44235. 10.7554/eLife.44235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Furio L, Mecheri R, van der Does AM, Lundeberg E, Saveanu L, Chen Y, van Endert P, Agerberth B, Diana J. 2015. Pancreatic β-cells limit autoimmune diabetes via an immunoregulatory antimicrobial peptide expressed under the influence of the gut microbiota. Immunity 43: 304–317. 10.1016/j.immuni.2015.07.013 [DOI] [PubMed] [Google Scholar]
- Thomas RM, Gharaibeh RZ, Gauthier J, Beveridge M, Pope JL, Guijarro MV, Yu Q, He Z, Ohland C, Newsome R, et al. 2018. Intestinal microbiota enhances pancreatic carcinogenesis in preclinical models. Carcinogenesis 39: 1068–1078. 10.1093/carcin/bgy073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung NM, Robson ME, Ventz S, Santa-Maria CA, Nanda R, Marcom PK, Shah PD, Ballinger TJ, Yang ES, Vinayak S, et al. 2020. TBCRC 048: phase II study of olaparib for metastatic breast cancer and mutations in homologous recombination-related genes. J Clin Oncol 38: 4274–4282. 10.1200/JCO.20.02151 [DOI] [PubMed] [Google Scholar]
- Tutt A, Tovey H, Cheang MCU, Kernaghan S, Kilburn L, Gazinska P, Owen J, Abraham J, Barrett S, Barrett-Lee P, et al. 2018. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: the TNT Trial. Nat Med 24: 628–637. 10.1038/s41591-018-0009-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Cutsem E, Tempero MA, Sigal D, Oh DY, Fazio N, Macarulla T, Hitre E, Hammel P, Hendifar AE, Bates SE, et al. 2020. Randomized phase III trial of pegvorhyaluronidase alfa with Nab-paclitaxel plus gemcitabine for patients with hyaluronan-high metastatic pancreatic adenocarcinoma. J Clin Oncol 38: 3185–3194. 10.1200/JCO.20.00590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Eynde B, van Baren N, Baurain J-F. 2020. Is there a clinical future for IDO1 inhibitors after the failure of epacadostat in melanoma? Ann Rev Cancer Biol. 4: 241–256. 10.1146/annurev-cancerbio-030419-033635 [DOI] [Google Scholar]
- Vasan K, Werner M, Chandel NS. 2020. Mitochondrial metabolism as a target for cancer therapy. Cell Metab 32: 341–352. 10.1016/j.cmet.2020.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sánchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, et al. 2014. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514: 628–632. 10.1038/nature13611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinayak S, Tolaney SM, Schwartzberg L, Mita M, McCann G, Tan AR, Wahner-Hendrickson AE, Forero A, Anders C, Wulf GM, et al. 2019. Open-label clinical trial of niraparib combined with pembrolizumab for treatment of advanced or metastatic triple-negative breast cancer. JAMA Oncol 5: 1132–1140. 10.1001/jamaoncol.2019.1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al. 2015. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518: 495–501. 10.1038/nature14169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waghray M, Yalamanchili M, Dziubinski M, Zeinali M, Erkkinen M, Yang H, Schradle KA, Urs S, Pasca Di Magliano M, Welling TH, et al. 2016. GM-CSF mediates mesenchymal–epithelial cross-talk in pancreatic cancer. Cancer Discov 6: 886–899. 10.1158/2159-8290.CD-15-0947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattenberg MM, Beatty GL. 2020. Overcoming immunotherapeutic resistance by targeting the cancer inflammation cycle. Semin Cancer Biol 65: 38–50. 10.1016/j.semcancer.2020.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattenberg MM, Asch D, Yu S, O'Dwyer PJ, Domchek SM, Nathanson KL, Rosen MA, Beatty GL, Siegelman ES, Reiss KA. 2020. Platinum response characteristics of patients with pancreatic ductal adenocarcinoma and a germline BRCA1, BRCA2 or PALB2 mutation. Br J Cancer 122: 333–339. 10.1038/s41416-019-0582-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss GJ, Waypa J, Blaydorn L, Coats J, McGahey K, Sangal A, Niu J, Lynch CA, Farley JH, Khemka V. 2017. A phase Ib study of pembrolizumab plus chemotherapy in patients with advanced cancer (PembroPlus). Br J Cancer 117: 33–40. 10.1038/bjc.2017.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle MC, Hingorani SR. 2019. Fibroblasts in pancreatic ductal adenocarcinoma: biological mechanisms and therapeutic targets. Gastroenterology 156: 2085–2096. 10.1053/j.gastro.2018.12.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer K, Kratz CP, Vasen HF, Caron O, Colas C, Entz-Werle N, Gerdes AM, Goldberg Y, Ilencikova D, Muleris M, et al. 2014. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the european consortium ‘care for CMMRD’ (C4CMMRD). J Med Genet 51: 355–365. 10.1136/jmedgenet-2014-102284 [DOI] [PubMed] [Google Scholar]
- Xu J, Sai H, Li Y, Jordan AC, McGettigan SE, Chen JH, Bedoya F, Fraietta JA, Gladney WL, Melenhorst JJ, et al. 2019. Peripheral blood T-cell fitness is diminished in patients with pancreatic carcinoma but can be improved with homeostatic cytokines. Cell Mol Gastroenterol Hepatol 8: 656–658.e6. 10.1016/j.jcmgh.2019.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, Sohn ASW, Mukhopadhyay S, Lin EY, Parker SJ, et al. 2020. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 581: 100–105. 10.1038/s41586-020-2229-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, et al. 2011. Pancreatic cancers require autophagy for tumor growth. Genes Dev 25: 717–729. 10.1101/gad.2016111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, Wang X, Deng J, Hai J, Yang S, Wong KK, et al. 2018. Autophagy sustains pancreatic cancer growth through both cell-autonomous and nonautonomous mechanisms. Cancer Discov 8: 276–287. 10.1158/2159-8290.CD-17-0952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Elpek KG, Vinjamoori A, Zimmerman SM, Chu GC, Yan H, Fletcher-Sananikone E, Zhang H, Liu Y, Wang W, et al. 2011. PTEN is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-κB-cytokine network. Cancer Discov 1: 158–169. 10.1158/2159-8290.CD-11-0031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. 2012. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149: 656–670. 10.1016/j.cell.2012.01.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Dey P, Yao W, Kimmelman AC, Draetta GF, Maitra A, DePinho RA. 2016. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 30: 355–385. 10.1101/gad.275776.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yan W, Mathew E, Bednar F, Wan S, Collins MA, Evans RA, Welling TH, Vonderheide RH, di Magliano MP. 2014. CD4+ T lymphocyte ablation prevents pancreatic carcinogenesis in mice. Cancer Immunol Res 2: 423–435. 10.1158/2326-6066.CIR-14-0016-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Li L, Jiang H, Li Q, Wang-Gillam A, Yu J, Head R, Liu J, Ruzinova MB, Lim KH. 2018. Tumor–stroma IL1β-IRAK4 feedforward circuitry drives tumor fibrosis, chemoresistance, and poor prognosis in pancreatic cancer. Cancer Res 78: 1700–1712. 10.1158/0008-5472.CAN-17-1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chandra V, Riquelme Sanchez E, Dutta P, Quesada PR, Rakoski A, Zoltan M, Arora N, Baydogan S, Horne W, et al. 2020a. Interleukin-17-induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J Exp Med 217: e20190354. 10.1084/jem.20190354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lazarus J, Steele NG, Yan W, Lee HJ, Nwosu ZC, Halbrook CJ, Menjivar RE, Kemp SB, Sirihorachai VR, et al. 2020b. Regulatory T-cell depletion alters the tumor microenvironment and accelerates pancreatic carcinogenesis. Cancer Discov 10: 422–439. 10.1158/2159-8290.CD-19-0958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, Cullinan DR, Luo J, Bearden AR, Lavine KJ, et al. 2017. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity 47: 597. 10.1016/j.immuni.2017.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XG, Chudnovskiy A, Baudrier L, Prizer B, Liu Y, Ostendorf BN, Yamaguchi N, Arab A, Tavora B, Timson R, et al. 2021. Functional genomics In vivo reveal metabolic dependencies of pancreatic cancer cells. Cell Metab 33: 211–221.e6. 10.1016/j.cmet.2020.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, Ma Y, Raoult D, Kroemer G, Gajewski TF. 2018. The microbiome in cancer immunotherapy: diagnostic tools and therapeutic strategies. Science 359: 1366–1370. 10.1126/science.aar6918 [DOI] [PubMed] [Google Scholar]