

Abstract
Cardiac TnC (cTnC) is highly conserved among mammals, and genetic variants can result in disease by perturbing Ca2+-regulation of myocardial contraction. Here, we report the molecular basis of a human mutation in cTnC's αD-helix (TNNC1-p.C84Y) that impacts conformational dynamics of the D/E central-linker and sampling of discrete states in the N-domain, favoring the “primed” state associated with Ca2+ binding. We demonstrate cTnC's αD-helix normally functions as a central hub that controls minimally frustrated interactions, maintaining evolutionarily conserved rigidity of the N-domain. αD-helix perturbation remotely alters conformational dynamics of the N-domain, compromising its structural rigidity. Transgenic mice carrying this cTnC mutation exhibit altered dynamics of sarcomere function and hypertrophic cardiomyopathy. Together, our data suggest that disruption of evolutionary conserved molecular frustration networks by a myofilament protein mutation may ultimately compromise contractile performance and trigger hypertrophic cardiomyopathy.
Cardiac TnC (cTnC) is highly conserved among mammals, and genetic variants can result in disease by perturbing Ca2+-regulation of myocardial contraction.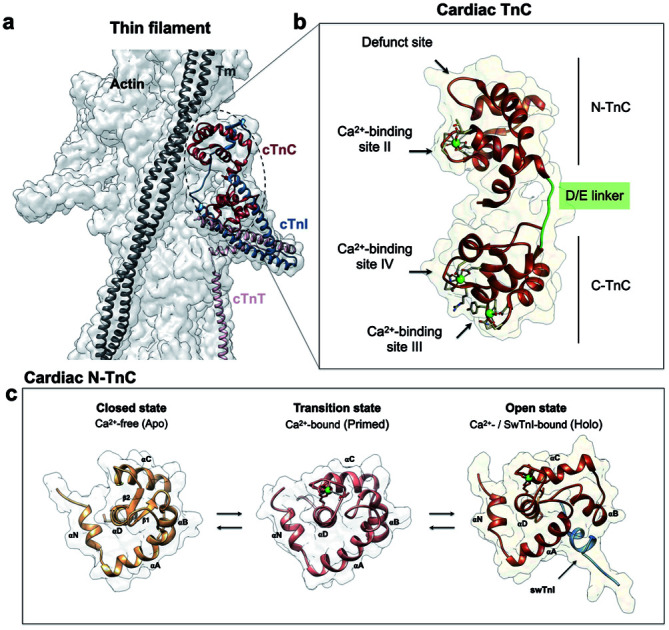
Introduction
Troponin (Tn) is a sarcomeric protein complex that plays a key role in Ca2+-mediated regulation of actomyosin force production in striated muscles. Downstream of an action potential, striated muscle contraction is activated upon an increase in intracellular free Ca2+ which promotes binding of Ca2+ to troponin C (TnC), triggering molecular rearrangements in thin filament proteins that favor formation of actomyosin cross-bridges.1 Regions of proteins or large protein complexes have coevolved to sustain essential communications between distant sites, a molecular process known as allostery.2 For instance, an extensive allosteric network maintains physical and dynamic signaling within the thin filament.3,4 In the heart, thin filament protein cardiac TnC (cTnC) plays a pivotal role in regulating both the systolic and diastolic phases of the cardiac cycle. Therefore, amino acid variations affecting Ca2+-binding affinity of cTnC can instigate cardiac dysfunction and pathological remodeling of the ventricular walls, typically in the form of dilated, hypertrophic, or restrictive cardiomyopathy (DCM, HCM, or RCM, respectively).5,6
The overall structure of cTnC consists of two globular domains, each containing EF-hand motifs comprised of helix–loop–helix structures that bind Ca2+ and/or Mg2+. The two domains of cTnC are connected by an intrinsically disordered structure, the D/E linker. The N and C domains are responsible for Ca2+ binding with lower and higher affinities, respectively. cTnC undergoes dynamic processes sampling a range of discrete intermediate states, i.e., the Ca2+-free state (apo) or closed conformation, the Ca2+-bound state or primed conformation, and the Ca2+- and swTnI-bound state or the open, active state. Of particular note, the open state in cTnC is induced when both Ca2+ and the switch TnI segment (swTnI, residues 147–163 in cTnI) bind to N-TnC.7,8 The sampling of the primed N-TnC to the open (or active) state involves the exposure of a hydrophobic patch to accommodate swTnI147–163 which facilitates release of the inhibitory TnI region (iTnI, residues 128–147 in cTnI) from F-actin. These dynamic events that are needed for Ca2+ to activate cross-bridge formation are essential for striated muscle contraction,1,9,10 and the underlying conformational transitions demonstrate the relevance of evolutionary selection of specific amino acids to function within the organized, native ensemble.
The principle of minimal frustration states that a biomolecule, driven by its polypeptide chain, populates low energy conformation that favors native folds and minimizes non-native contacts. Interestingly, hubs of minimally frustrated interactions are key elements that confer structural rigidity to protein domains.11 Whittaker et al. presented a link between conformational dynamics and frustration, indicating that non-native interactions often shift the kinetic equilibrium to transient trapping of intermediate species in the higher energy states, known as highly frustrated.12 Furthermore, highly frustrated residues are often clustered on protein surfaces and are involved in partner binding, with finely tuned compensation of the entropy and energy changes within the metastable ensemble. Moreover, local violations such as single point mutations, may disrupt the complexity of energy landscapes and interactions of the intermediate ensemble, and thus affect allosteric events and large-scale conformational changes that are particularly important for biomolecule function.13
Significant advances in the structural biology field enabled atomistic comprehension of cTn. Through NMR spectroscopy studies, valuable insights into the structure and dynamics of TnC and Tn complex partners have been elucidated.8,14,15 Later, Takeda et al. provided a high-resolution, X-ray crystal structure of the cTn core domain shedding light on the mechanism of Ca2+-regulation in cardiac muscle.7 Several MD simulations have focused on the crosstalk between allosteric communications among cTn subunits.4,16–19 Despite these achievements, the mechanism of activation of the thin filament remains elusive due to the lack of understanding how relatively small changes in TnC result in large-scale conformational changes within the thin filament upon Ca2+ activation. Cryo-EM studies revealed the structure of the whole thin filament at low and high Ca2+ levels, but the transitional pathway between the Ca2+-free and Ca2+-bound states is still inconclusive.9 Thus, a comprehensive mechanism of how the cTnC samples a range of discrete states – crucially for activation – is still lacking. Recent cryo-EM analysis of cardiac thin filaments at systolic Ca2+ levels10 confirmed presence of multiple structural states of the thin filament at physiological Ca2+ levels promising to be a valuable tool to bring high resolution NMR data into the context of the whole thin filament.
A study using paramagnetic relaxation enhancement showed that an HCM mutation in cTnC (A8V) induces a more open N-domain conformation in both apo and holo states.20 It is not clear what impact pathogenic variants have on this dynamic sampling of open states on muscle contraction and heart remodeling. In the present study, a previously reported, human HCM variant was the focus of a multi-scale investigation from the molecular level—using in vitro solution measurements and in silico molecular dynamics—to tissue and in vivo level studies using a knock-in mouse model. The altered residue (C84Y) is located at the end of the αD-helix and the beginning of the D/E linker in cTnC and increases myofilament Ca2+ sensitivity.21 The C84Y mutation illustrates how allosteric events drive cTnC sampling among open-closed states and ultimately impact myocardial contraction. Protein contacts exhibit a range of energy degree, or frustration, which controls the kinetics of switching between microstates. Evolution dictates an optimal frustration network to maintain protein function and manage the thermodynamic ensemble. Furthermore, the contact between nonnative interactions can destructively interfere with protein configurational landscapes.22 Our data suggest that a single residue change within the core of the minimally frustrated N-TnC hub directly loosens its structural rigidity, which impacts the conformational dynamics of other motifs within cTnC. This leads to altered Ca2+ sensitivity of isometric force and altered contractile kinetics, which trigger cardiomyopathy in a mouse model. Because the open state of cTnC precedes muscle contraction, understanding the dynamic processes of this seemingly inactive phase is an essential aspect of heart physiology and disease. We propose that the cTnC N-domain does not simply bind activating Ca2+, but responds allosterically to fine-tune myocardial contractility, perturbations in which can be deleterious.
Results
Sampling of the cardiac TnC open-like state
cTnC is essential within the thin filament (Fig. 1a). It responds through precise conformational changes triggered by Ca2+ and cTnI-binding (Fig. 1b and c). The discrete conformational states of N-cTnC relevant to this study are shown in (Fig. 1c and ESI Fig. 1a–c†). However, it remains unclear how this sensor tunes the ensemble of conformational states that exist during contraction, and how alterations and preference for specific states can adversely impact heart physiology, leading to disease. To address how cTnC alters its ensemble of conformational states from the closed to the active state, we first designed a Cys-to-Tyr substitution at position 84 (C84Y) of cTnC and characterized in solution whether this substitution can shift the equilibrium toward cTnC open state. To assess the degree of conformational change upon Ca2+ binding, the “openness” of the hydrophobic cleft was examined using fluorescence spectroscopy. Fluorescence generated by bis-ANS, a probe for nonpolar cavities of proteins, was measured at pCa 4 and 8, corresponding to the presence or absence of Ca2+, respectively. The Ca2+-dependent differences in bis-ANS binding indicate that Ca2+-induced conformational changes expose more hydrophobic regions in C84Y than unmodified cTnC (ESI Fig. 2a and b†).
Fig. 1. Troponin C is the Ca2+ sensor of cardiac muscle. (a) Cardiac thin filament organization. Density maps are from PDB structure 6KN8. (b) Enlargement of the cardiac TnC subunit from the cardiac troponin (Tn) complex (PDB code 1J1E), shown as a ribbon structure. Regulatory and structural domains are N-TnC and C-TnC, respectively. Green spheres represent the 3 Ca2+ ions of the Ca2+-saturated state. EF-hand residues participating in coordination of Ca2+ are shown as gray sticks. The D/E linker is colored green; (c) regulatory domain discrete states prior to contraction. Top view representations of N-TnC in the absence (PDB code 1SPY) or presence of Ca2+ (PDB code 1AP4). The open active state of cardiac TnC (PDB code 1MXL) is sampled in the presence of the switch peptide of the TnI subunit (swTnI).

To evaluate conformational differences associated with C84Y, we utilized nuclear magnetic resonance (NMR) and performed backbone assignment of C84Y in the Ca2+-bound state (Fig. 2a and ESI Fig. 2c†) followed by chemical shift perturbation analysis compared to WT cTnC (Fig. 2b). The most perturbed residues occurred within the αN-helix (αN, residues Y5 and V9), the αB-helix (αB, residues E40 and M45), Ca2+-binding site II (residues E66 and V72), and the D/E linker (residues D87 and D88). Superposition of N-cTnC structures in the primed and open active state revealed that conformational changes mostly occurred within the N-, B-, and C-helices, consistent with what was captured experimentally for C84Y by chemical shift analysis (Fig. 2 and ESI Fig. 1c†). These results suggest that C84Y shifts the native cTnC conformation to an average-weighted distribution of states that are more open compared to WT.
Fig. 2. Mutation-induced human cardiac TnC open-like state. (a) Superposition of 1H–15N HSQC spectra of WT (red) and C84Y (blue) in the primed state. Highlighted residues are those identified as undergoing significant structural changes; (b) chemical shift perturbation (CSP) analysis of C84Y against WT as a function of residue number. Residues deviating from the average values of CSP (Δδ) + 1 S.D. (Δδ + 1σ) and + 2 S.D. (Δδ + 2σ) are highlighted. Secondary content representation (αN–αH) are depicted as horizontal rectangles in the top. Ca2+-binding sites are shown as roman numerals; (c) regulatory (salmon) and structural (gray) cTnC domains show as sticks residues with significant structural changes due to the C84Y substitution. The N-helix (αN) and the D/E linker are colored blue.

In the absence of the swTnI peptide, which stabilizes the open state, we hypothesized that C84Y presents lower structural rigidity in solution. Thermostability measurements derived from circular dichroism (CD) and Kratky plots from chemical-induced unfolding of small-angle X-ray scattering (SAXS) illustrated that C84Y has lower structural stability compared to WT cTnC in solution (ESI Fig. 3a–e†). Melting temperature (Tm) values corresponding to the unfolding processes of the C- (1st) and then the N-domain (2nd) in the Ca2+-bound (primed) state, revealed decreased values for both domains upon C84Y substitution (ESI Fig. 3a–c†). In contrast to WT cTnC in the Ca2+-free (apo) state, C84Y exhibited one transition, indicative of cooperativity loss upon thermal unfolding (ESI Fig. 3a–c†).
Kratky plots derived from SAXS data are often used to infer subtle changes in the folding behavior of proteins against treatments. A Kratky plot with a bell-shaped distribution is indicative of a well-folded protein.23 By adding increasing amounts of denaturant to WT and C84Y in the Ca2+-free and -bound (primed) states, we noticed that, in contrast to WT, less denaturant was required to reduce the bell shaped Kratky plots for C84Y in either state (ESI Fig. 3d and e†). This shows that C84Y is less structurally stable than WT when subjected to chemical-induced unfolding. Because temperature is an important physical property impacting the range of conformational changes that can occur in proteins, we performed a local temperature variation analysis by 1H–15N heteronuclear single quantum correlation (HSQC) experiments (ESI Fig. 3f†). By measuring the temperature coefficients for the chemical shifts of the amide hydrogens (1H) as a function of residue position in the primary sequence (ESI Fig. 3g†), we uncovered several distinctions between WT and C84Y (ESI Fig. 3h–t†). Residues of C84Y located within the αN-helix (V9), the αB-helix (E40 and K43), the D/E central-linker (D87), and the αE-helix (L100 and N107) responded differently to temperature variations. This suggests that helices αN, αB, and the D/E linker of C84Y are prone to distinct conformational changes.
Because the C84Y substitution induces a less stable, open-like conformation in cTnC, we expected that it would affect its binding properties to cTnI. To test this hypothesis, we used 1H–15N HSQC spectroscopy to titrate two previously characterized consensus cTnI peptides referred to as the inhibitory peptide, iTnI128–147, and the switch peptide, swTnI147–163 (ESI Fig. 4a†). Surprisingly, regardless of whether the C84Y substitution was present, the peptides bound with equal affinity to cTnC (ESI Fig. 4b–g†) and with no apparent changes in their binding affinities for C84Y compared to WT (ESI Fig. 4h and i†). However, distinct regions in the D/E linker were perturbed when comparing WT vs. C84Y such as E96 and L97 for WT vs. E94, E95 and L97 for C84Y (ESI Fig. 4c†). This indicates that the lower structural rigidity and the open-like ensemble of states triggered by the C84Y substitution do not affect the binding properties between cTnC and cTnI consensus peptides, but rather are more likely explained by changes in conformational dynamics within cTnC (ESI Fig. 5†). Hence, we evaluated C84Y dynamics at different timescales.
Altogether, the C84Y substitution: (i) induces greater hydrophobic exposure of normally buried residues compared to WT; (ii) induces structural changes in several regions that are equally disturbed as the protein samples the active state; (iii) modulates temperature-induced conformational changes of some residues in these regions; (iv) reduces overall structural integrity of the protein; and (v) does not affect the binding of cTnI consensus peptides. Thus, our findings are suggestive that the C84Y substitution causes cTnC to sample an ensemble of states similar to the open state in solution.
Conformational dynamics of minimally frustrated residues in cTnC
To understand how the non-evolutionary, pathological introduction of tyrosine at position 84 in cTnC-C84Y impacts the funneled energy landscape driving protein folding, we examined how energetics were impacted within N-TnC structure. We performed a theoretical analysis of how extensively non-conservative substitutions shift the energetics of the protein and report a local mutational frustration index (see Methods). Then, we assessed the frustration levels of native cTnC contact pairs compared to the conformational dynamics of the mutation-induced ensemble of states. We performed this approach for the three previously solved, discrete conformational states of cardiac N-TnC (Fig. 3a–f). We discovered a net of minimally frustrated interactions governed by helix αD. This is an important finding because minimally frustrated nets confer structural rigidity to protein domains. Thus, it is expected that the pathological C84Y substitution impacts the conformational dynamics of other cTnC motifs. It is interesting to note that interactions within helix αC and those between helix αB and the B/C loop are highly frustrated, and these are the segments that participate in large scale motions toward the open state (Fig. 3a–c).
Fig. 3. Frustration analysis of discrete human cardiac TnC N-domain conformational states. (a–c) Tertiary frustration patterns of the closed, primed and open active states (PBD codes: 1SPY, 1AP4 and 1MXL), respectively, in two different views. Cα contact pairs within a distance of 5 Å are shown as red lines for highly frustrated interactions and green lines for minimally frustrated pairs; (d–f) contact maps for each studied case in (a–c), respectively. The color scale indicates the local mutational frustration index (see Methods).

In contrast to interactions within the defunct Ca2+ binding site I of cTnC, those within Ca2+-binding site II are highly frustrated (Fig. 3d–f). More interesting is the observation that in their open state, skeletal and cardiac TnC isoforms present segments with different frustration profiles (ESI Fig. 6†). For example, while sTnC presents highly frustrated interactions within both functional Ca2+-binding sites of the N-domain (ESI Fig. 6a and c†), the defunct site I of the cardiac isoform does not (ESI Fig. 6b and d†). Additionally, contact pairs within the cardiac D/E linker are minimally frustrated when compared to the corresponding skeletal segment (ESI Fig. 6c and d†). These findings may provide insights to explain how the evolutionary sequence diverged to acquire specialized functions in these two distinct muscle types.
We conducted additional simulations using a computational model that calculates the time-dependent behavior of fluctuations and conformational changes of proteins. To assess how C84Y altered sampling of the open state, we performed microsecond molecular dynamics (MD) simulations using the primed and open active discrete states of cardiac N-TnC and calculated the AB interhelical angles for each system (see Methods). The findings suggest that, independent of C84Y substitution, when cardiac N-TnC is bound to the swTnI peptide, the open conformation (AB interhelical angle < 110°) is significantly more favored (Fig. 4a and b), with major populations displaying interhelical angles ∼100°. In agreement with previous results (Fig. 2 and ESI Fig. 2a and b†), the MD simulations indicate that in the primed state, the C84Y substitution may increase the population of open structures relative to WT, although the difference was not statistically significant (Fig. 4a and b). The WT primed and open state trajectories were analyzed by principal component analysis (PCA). The first two principal components described 51% and 54% of the variance for the primed and open active systems, respectively, with the first principal component (PC1) accounting for 30% of the variance in both cases. Free energy landscapes based on the PC1 projections show a flatter distribution for the open active compared to the primed state, indicating that N-cTnC may be able to sample open conformations more readily when bound to the swTnI peptide (Fig. 4e and f). Strong correlations were observed between the PC1 projections and the AB interhelical angles (Fig. 4c, d and ESI Fig. 7a and b†). In an attempt to correlate our experimental evidence in isolated cTnC with theoretical models, we investigated whether the C84Y TnC impacts intra-TnC interactions for the intact troponin core domain (Tn, PDB code 1J1E) and thin filament–troponin (TF–Tn) structures (calcium-free state, PDB code 6KN7; calcium-bound state, PDB code 6KN8). We report the contact map analyses based on our simulation data (ESI Fig. 7c†). Data show significant contacts between helices N, A (residues M1-D25) and D (residues V72-M85), and between B (residues T38-M47) and C (residue Q50-T71) of the N-terminal domain. Comparable interactions are shown for residues L97-D113 and T129-E161 of the C-terminal domain. These interactions contribute to the packing of helical bundles in the TnC globular domains. Upon binding Ca2+ and swTnI peptide, interactions between G49-D65 with D73-S89 evident in the TF–Tn Ca2+-free state are disrupted in the intact Tn and TF–Tn Ca2+-bound states (ESI Fig. 7c†). These regions correspond to the two helices flanking the EF-hand Ca2+ binding site II in N-terminal domain of TnC; disruption of this interaction is commonplace in Ca2+-binding proteins and contributes to their high affinities for ions.24 Unique to the C84Y structures, we observe that the mutation enhances the intra-contacts between helix C (residues P52-T71) and helix D (residues D73-M85) in the intact Tn (ESI Fig. 7c†). This is consistent with our observations in the isolated TnC structure that C84Y alters interactions with helix D, which were shown to contribute to the protein's minimally frustrated interactions. Further, we observed similar AB-interhelical angles for the C84Y variant and WT structures, albeit those distributions may be slightly and insignificantly shifted toward higher angles relative to the WT equivalents (ESI Fig. 7d†). This is consistent with the root mean square fluctuations (RMSFs) that demonstrate comparable trends for WT and C84Y cTnC (ESI Fig. 7e†). In summary, the intact and TF–Tn simulations do not strongly evidence differences in the dynamics of the N-terminal domain. This is not surprising, in that our data for the isolated structure here and a previous study25 suggest AB opening dynamics occur at a microsecond timescale, which were not practicable for the intact and TF–Tn simulations.
Fig. 4. MD simulations and experimental dynamics. (a) AB interhelical angle distributions; (b) fractions under 110° (t-test: *, p = 0.015; **, p = 0.005); (c) comparison of the first principal component (PC1) projections with AB interhelical angles for WT primed; (d) comparison of the first principal component (PC1) projections with AB interhelical angles for WT open; (e) PC1-based free energy landscapes for WT primed; (f) PC1-based free energy landscapes for WT open; (g) R2/R1 relaxation ratio per residue for C84Y. Solid and dashed blue lines represent the R2/R1 avg. values among residues for C84Y and 1 S.D. from avg., respectively. Ca2+-binding sites and secondary structure elements are shown along the top.59-15N NOE data per residue. Residues with R2/R1 values > avg. + 1 S.D. are indicated by arrows. (h) Quantitative analysis of exchange dynamics in C84Y. (A) R2eff values for each residue obtained by CPMG relaxation dispersion in the holo state at 50 Hz (filled circles) and 1000 Hz (open circles). Upper panel, 25 °C; middle panel, 32 °C; lower panel, 37 °C. Residues experiencing intermediate exchange are indicated by red arrows.

To experimentally probe the conformational dynamics of C84Y and correlate the findings with the frustration levels of structures within cTnC obtained from MD simulations, we assessed the dynamics of C84Y cTnC in the Ca2+-bound (primed) state in different timescale regimens. We carried out nuclear spin relaxation experiments to measure the R1 (longitudinal) and R2 (transverse) rates derived from internuclear 15N–1H vectors of the protein backbone, that directly inform about protein diffusional properties within the fast, pico-to nano-second timescale. We observed that the N- and C-domains of C84Y cTnC present distinctly different diffusional properties in solution, with different correlation times and slightly lower R2/R1 values for the C-domain compared to the N-domain. This indicates that the tumbling of the two domains in solution occurs in a manner consistent with independent motion despite physical connection via the D/E linker. A group of residues located within the defunct site I (I26, G30, and C35) revealed R2/R1 ratios that were one standard deviation above the average values of the N-domain indicative of fast conformational exchange dynamics (Fig. 4g).
To assess the intermediate exchange dynamics of C84Y, we performed Carr-Purcell-Meiboom-Gill (CPMG) relaxation dispersion measurements at 25, 32, and 37 °C. These experiments are sensitive to millisecond exchange dynamics, which have been shown to be relevant for functional aspects of biomolecules.26 By observing the effective decay of magnetization R2eff, which reports on the effective transverse relaxation rate, with refocusing pulses of 50 and 1000 Hz, we pinpointed two classes of dynamic regimens: (i) fast-exchange motions in the microsecond timescale and (ii) intermediate exchange motions in the millisecond timescale. Residues undergoing fast-exchange motions are qualitatively observed as those presenting nearly identical values of R2eff for the 50 and 1000 Hz refocusing pulses one standard deviation above the average of residues. In contrast, intermediate exchange residues correspond to those in which R2eff of refocusing pulses are separated by a decay and values are one standard deviation above the average of all residues. At physiological temperature, we revealed that C84Y samples fast- and intermediate-exchange motions within the defunct site (residues L29, D33, and C35) and the D/E linker (residues D87, D88, S89, K90, S93, E94, and L97) (Fig. 4h and ESI Fig. 8†). The dynamic properties experimentally captured in these NMR measurements are of fundamental relevance to the mechanism by which this Ca2+ sensor is allosterically tuned during contraction. The Cys-to-Tyr substitution within the minimally frustrated hub of N-TnC loosens its structural rigidity and, because of that, the conformational dynamics of other TnC motifs including the D/E-linker and cTnC's defunct site I are allosterically altered (Fig. 4h and ESI Fig. 8†). Remarkably, these motifs have diverged through evolution to acquire specialized functions in skeletal and cardiac muscle.
Generation and in vivo characterization of a KI-Tnnc1-p.C84Y mouse model
To determine whether the C84Y allele associated with HCM in humans could be recapitulated in a small rodent model, we employed a gene-targeting strategy to generate KI-Tnnc1-p.C84Y mice (ESI Fig. 9,† see Methods). Interestingly, homozygous (Tnnc1C84Y/C84Y) mice were not obtained, indicating that the presence of two C84Y alleles is embryonically lethal. Therefore, all of our experiments were carried out using heterozygous (Tnnc1WT/C84Y) mice. Mice heterozygous for the C84Y allele (Tnnc1WT/C84Y) displayed in vivo functional and morphological features consistent with cardiomyopathy by 3 months of age (Fig. 5 and ESI Table 1†). Further evidence of disease in Tnnc1WT/C84Y mice was increased myocardial fibrosis by 8–9 months of age compared to WT (ESI Fig. 10†). Not only were indices of left ventricular hypertrophy increased in the 6 month-old Tnnc1WT/C84Y mice but overall, there was a significant increase in relative ventricle weight (RWT)% when considering multiple ventricular thickness measurements in these mice at this age. M-mode echocardiography revealed narrowing of the ventricular lumen by reduced LV diameter values in 3 month and 6 month-old Tnnc1WT/C84Y mouse hearts compared to age-matched WT controls. Doppler mitral flow measurements in Tnnc1WT/C84Y hearts provided evidence of diastolic dysfunction as early as 3 months of age, with significantly elevated isovolumetric contraction time (IVCT) (ms), an early indicator of asymptomatic diastolic dysfunction. The 3 month-old mice had increased index values for (IVRT + IVCT)/ET of 1.04 for WT hearts versus 0.786 for Tnnc1WT/C84Y hearts, whereas by 6 months of age the Tnnc1WT/C84Y mice had increased isovolumetric relaxation time (IVRT). Both ages of Tnnc1WT/C84Y mice had decreased mitral valve early peak flow velocity/atrial peak flow velocity (MV E/A) (Fig. 5). Additional informative echocardiographic parameters are reported in ESI Table 1.† Histopathological analysis of Tnnc1WT/C84Y mice at 14 months of age indicated advanced pathological remodeling with gross morphological features indicated by overt left ventricular hypertrophy of the myocardium compared to age-matched WT control mice (Fig. 5b). In concordance with human clinical data21 these cumulative results suggest that expression of a single C84Y allele at the Tnnc1 locus is sufficient to cause cardiomyopathy in mice.
Fig. 5. Knock-in mouse model bearing variant Tnnc1WT/C84Y recapitulates hypertrophic cardiomyopathy. (a) Echocardiography reveals differences in cardiac structure and function in 3 month-old knock-in Tnnc1WT/C84Y mice; (b) representative tissue slices from Tnnc1WT/WT and Tnnc1WT/C84Y mouse hearts stained with Masson's trichrome. Representative, longitudinally sectioned, stained hearts exhibit morphology of advanced cardiac disease in 14 month-old Tnnc1WT/C84Y but not Tnnc1WT/WT mouse hearts. Note the extensive hypertrophy of the left ventricle (LV) in Tnnc1WT/C84Y heart (circled area). (c–k) M-mode and pulsed wave Doppler measurements were analyzed in both the Tnnc1WT/WT and Tnnc1WT/C84Y mice at 3 and 6 months of age. (c)IVRT – isovolumetric relaxation time; (d) MV E/A – mitral valve early peak flow velocity – atrial peak flow velocity ratio; (e) CO – cardiac output; (f) SV – stroke volume; (g) LVPW – left ventricular posterior wall thickness during diastole (d) or; (h) systole (s); (i) diameter during diastole (d) or; (j) systole (s); (k) relative ventricular wall thickness; ANOVA with least significant difference post-hoc test was performed to determine significance. *p < 0.05; **p < 0.001 WT vs. C84Y within age time point. The heart rate was statistically significant lower in Tnnc1WT/C84Y compared to Tnnc1WT/WT (3 months, 504 vs. 450 bpm and 6 months 501 vs. 452 bpm, ESI Table 1†).

Tnnc1 WT/C84Y mice display HCM-related abnormalities in cardiac muscle mechanics
To determine whether myofilament dysfunction underlies the cardiac dysfunction observed in vivo, we carried out muscle mechanics experiments using permeabilized cardiac muscle preparations (CMPs) isolated from Tnnc1WT/C84Y or Tnnc1WT/WT mice at 6 months of age (ESI Fig. 11†). In line with the paradigm for most sarcomeric HCM variants, we observed significantly increased myofilament Ca2+ sensitivity of steady-state, isometric tension production for Tnnc1WT/C84Y CMPs (pCa50 = 5.72 ± 0.02) with a leftward shift of 0.16 pCa units compared to Tnnc1WT/WT control CMPs (pCa50 = 5.56 ± 0.02). There were no significant differences in maximum isometric tension (Fmax) or cooperativity of thin filament activation (nHill) (Fig. 6a and b, and ESI Table 2†). Measurements of maximal sinusoidal stiffness (SS max), which is used as an index of the overall number of cross-bridges, revealed no significant difference between from Tnnc1WT/C84Y (01.15 ± 0.21) and Tnnc1WT/WT (01.33 ± 0.25) (Fig. 6e and f, ESI Table 2†).
Fig. 6. Ca2+-dependence of steady-state isometric tension, kinetics of tension redevelopment (kTR) and sinusoidal stiffness (SS) in knock-in Tnnc1WT/C84Y CMPs. (a) Relative steady-state isometric force as a function of pCa. Force values were normalized to the maximum force generated by each individual CMP. (b) Normalized steady-state isometric force as a function of pCa. The C84Y force values (blue) were normalized to average maximum force produced by WT (red). (c) Ca2+-dependence of kTR. Dashed lines were drawn by eye. (d) kTR as a function of normalized steady-state isometric force. The force values were normalized to the maximum steady-state isometric force generated by WT. Both solid and dashed lines represent solutions obtained from a 3-state model describing force-kTR relationships: solid lines represent the best fit parameters (f, g, kOFF) and kON63 and dotted lines represent solutions when all four parameters were either increased by 1 or 3% (upper lines) or decreased by 1 or 3% (lower lines). (e) Ca2+-dependence of sinusoidal stiffness. (f) Sinusoidal stiffness as a function of normalized steady-state isometric force. Force values were normalized to the maximum steady-state isometric force generated by WT. Dashed lines were drawn by eye. Data are shown as mean ± S.E.M. (n = 5–6). Best fit parameter estimates from non-linear least square regression on the Hill equation (panels a, b and e) and the 3-state model for force-kTR relationships (panel d) are summarized in ESI Tables 2 and 3,† respectively.

Intriguingly, Ca2+ activation of the kinetics of isometric tension redevelopment (kTR) for Tnnc1WT/C84Y CMPs was more curvilinear than that obtained for WT (Fig. 6d). This was mainly due to slower kTR for Tnnc1WT/C84Y CMPs at submaximal Ca2+-activation because there was no significant difference in maximum kTR (Fig. 6c and d, and ESI Table 2†). Computational modeling based on a 3-state model of striated muscle contraction (see Methods) that accounts for the differences in Ca2+ sensitivity of isometric force (ESI Table 3†) suggests that Tnnc1WT/C84Y CMPs exhibit ∼2-fold faster rate of cross-bridge attachment (f) and ∼2-fold slower rate of cross-bridge detachment (g) (Fig. 6d, and ESI Table 3†). The more curvilinear relation between force and kTR for Tnnc1WT/C84Y CMPs (Fig. 6d) results from faster regulatory unit dynamics (specifically kOFF) (ESI Table 3†) combined with these changes in f and g. Taken together, we conclude that the Tnnc1-p.C84Y variant increases the magnitude while decreasing the kinetics of isometric tension production at submaximal Ca2+ activation in cardiac muscle, with no measurable impact on maximal values of the parameters measured or the overall number of cross-bridges.
Discussion
Despite our in-depth understanding of many aspects governing muscle physiology, a number of key topics remain vaguely delineated. Of particular significance for normal and pathophysiology of the heart, it is still not precisely known exactly how Ca2+ ions trigger contraction—especially not the atomistic details that dictate overall function—and how this process is modulated by single amino acid substitutions in cTnC such as C84Y that are associated with cardiomyopathy. Here, using a translational investigation, we (i) fashioned a system in which conformational dynamics reflect the intermolecular sampling of the open active state by cTnC in solution, (ii) investigated how the system behaves in terms of Ca2+ sensitivity and kinetics of force production within the context of the thin filament unit and the sarcomere, and (iii) determined the systemic effects in mouse models. We demonstrated how this cardiac sensor responds allosterically to Ca2+ binding when sampling conformations that favor the open state of the N-domain. To further define the consequences of the disturbance generated by the C84Y substitution, we also provide functional readouts of cardiac muscle function in a newly developed knock-in mouse model.
To experimentally investigate the cardiomyopathy variant, we introduced within the regulatory core (helix αD) of cTnC a Tyr side chain at position 84 in a bacterially expressed, recombinant protein for in vitro, solution experiments and also, via modification of the genome, in our mouse model. Because helix αD is the central hub connecting all other helices of the regulatory domain, and Tyr has a bulky side chain, we expected that C84Y would shift the conformational equilibrium toward a more open conformation. Our initial experiments revealed that C84Y cTnC exhibits Ca2+-induced conformational changes that lead to greater hydrophobic exposure in the N-domain, and structural changes in regions there are equally disturbed during open state sampling. It is worth mentioning that upon Ca2+ binding, the cTnC WT structure is known to be less affected than sTnC and binding of swTnI is required for the major structural displacement of BC helices toward the open conformation of cTnC. In this scenario, the NAD helices are mostly unaffected with a RMSD of 1.05 Å.8 Our MD simulations revealed an AB interhelical angle of ∼100° which was similar for both the WT and C84Y open states bound to swTnI. This agrees with previous measurements of 102 ± 5°,8 which supports the finding that the swTnI peptide stabilizes the open state regardless of whether C84Y is present when using NcTnC only. This is consistent with our swTnI peptide titrations as most of the structural changes observed were similar for WT and the C84Y (ESI Fig. 4†). However, chemical shift changes were observed for both N-terminal and D/E linker backbone atoms upon swTnI binding using full cTnC. In the primed state, C84Y revealed a dual distribution of trajectories with AB interhelical average angles of ∼110° and ∼150°, indicating that the substitution favors the ensemble of more open conformations (Fig. 4a). This is in line with greater hydrophobic exposure and the structural changes within helices αN and αB (Fig. 2 and ESI Fig. 2 a and b†). Because highly frustrated αB-αD tertiary interactions are progressively lost upon Ca2+-binding to the N-domain due to large motions toward the open state (Fig. 3a–f), this favoring of open conformations also explains why C84Y in either the Ca2+-free (apo) or Ca2+-bound (primed) state exhibits less structural rigidity than WT, as evidenced by temperature- and chemical-induced unfolding at lower temperatures or denaturant concentrations, respectively (ESI Fig. 3†).
In sTnC, αN stabilizes the protein and participates in the Ca2+-switching mechanism.27 Further, the structural C-terminal domain is also impacted,28 an observation that was subsequently demonstrated to occur in cTnC.29 In fact, cTnC can assume bent conformations in solution, which is partially due to the flexible nature of its central helix, the D/E linker.30,31 The flexibility of this linker is of pivotal importance to the association of both domains, and the dynamic repertoire that TnC adopts during activation of cardiac muscle contraction. Studies aimed toward elucidating the functionality of this linker utilized truncations of the skTnC linker, which impaired its flexibility and regulation of the actomyosin ATPase.32–34 In calmodulin (CaM), transient interdomain association leads to compact states and is similarly modulated by the length and rigidity of the central linker.35 In the context of TnC's binding partner TnI, when cTnI1–80 is bound to cardiac C-TnC, the D/E linker binds the inhibitory cTnI peptide, iTnI129–149.36 This is aligned with our titrations using iTnI128–147 that affect a cluster of residues located at the beginning of αE (E94, E95, E96, and L97) (ESI Fig. 4c†). Further, a large decrease in cTnC linker flexibility has been noted upon binding of TnI1–211, indicating that conformational dynamics is a fundamental aspect of TnC–TnI interactions during activation of contraction.37
The crystal structure of the core domain of the Ca2+-saturated cardiac Tn complex revealed a chopstick-like TnI configuration and also some of the TnC–TnI interactions of the open TnC active state.7 Some highly flexible Tn segments were not present or were not resolved in the crystal structure, thus indicating that Tn conformational dynamics remains an unexplored avenue for tuning the activation of muscle contraction. The concept of population shifts or re-distribution of conformational states introduces the dependence of protein function on conformational flexibility.38 In fact, there are examples of proteins (other than Tn) in which increased flexibility of local segments favors conformational transitions of ensembles.39 Our results point out the dynamic behavior of the EF-hand loop in the defunct site I and the D/E linker (Fig. 4i and j). By introducing C84Y and shifting the conformational ensemble toward a more open state, the open active state of WT is mimicked in solution. Therefore, we assessed how cTnC behaves allosterically within the context of the BC helices moving apart from the NAD unit, a sine qua non condition for activation of muscle contraction. Frustration analysis of the cTnC hinge mechanism accommodating the swTnI peptide revealed that most of the interactions within the BC unit are highly frustrated (Fig. 3). Large-scale structural rearrangements such as those occurring in functional hinges have been shown to involve highly frustrated segments in the protein adenylate kinase.40 Our data support a model in which cTnC behaves allosterically in the open state prior to muscle activation. This allostery involves a large-amplitude, rigid-body motion of the highly frustrated BC helices driven away from the NAD unit accompanied by local rearrangements of minimally frustrated dynamic segments such as defunct site I and the D/E linker to alleviate the high-stress transition toward the open state.
Our solution experiments and computational simulations allowed us to explore cTnC's sampling of discrete dynamic states and demonstrated that the C84Y variant, due to its location at the end of αD-helix, impairs allosteric communication within cTnC and its conformational dynamics. The concept of minimal frustration, referring to a hypothesis that was developed to quantify the dominance of interactions that stabilize specific native structures over other interactions that favor topologically distinct, non-native structures that serve as energy traps,41 is readily applicable to these observations. It helps us to understand how a bulky amino acid within a minimally frustrated region of cTnC could negatively alter the balance of microstates between closed and open conformations by introducing nonnative interactions. Nevertheless, studies of the whole thin filament using cryo-EM are necessary to understand how transitions between the discrete dynamic states in the TnC that we revealed are related to the large-scale conformational changes in within the whole thin filament upon activation by Ca2+.
Conflicting molecular contacts could explain why some mutations lead to adverse outcomes and help to clarify some higher-level characteristics of cardiomyopathy observed in our Tnnc1WT/C84Y mouse model. In particular, the observation of faster dynamics in regions of cTnC-C84Y in solution, combined with frustration analysis (Fig. 4 and ESI Fig. 6 and 7†), provide significant molecular insights into the altered (faster) dynamics at higher levels of structural organization—specifically, thin filament regulatory units that contain cTnC—inferred from both altered Ca2+ sensitivity of isometric force combined with analysis of the force-kTR data (Fig. 6d and ESI Table 3†). The force-kTR relation (Fig. 6d) informs about both cross-bridge cycling kinetics (ESI Table 3†) and dynamics of individual regulatory units.42 Comparing force-kTR relations between our Tnnc1WT/C84Y (Fig. 6d) with Tnnc1A8V/A8V mouse models,43 we note that HCM variants in Tnnc1 do not necessarily cause exactly the same alterations in Ca2+-activation kinetics. The low-force end (left end) of the force-kTR relation, in particular, is indicative of dynamics of individual regulatory units,42 with slower dynamics leading to an elevation of the force-kTR relation at low force, and faster dynamics leading to lowering of the force-kTR relation at low force42,44 as observed here for Tnnc1WT/C84Y compared with Tnnc1WT/WT despite no significant change in Fmax (Fig. 6d). This in turn is responsible for faster dynamics of the whole heart observed in vivo in Tnnc1WT/C84Y mice (ESI Table 1†).
Investigation of cTnC-C84Y therefore provides a platform to visualize how cTnC responds allosterically to Ca2+ binding and its molecular partners, and how this leads to alterations at the tissue and organ systems levels. Our physiological measurements confirm early pathological changes in the hearts of Tnnc1WT/C84Y mice, which showed early indications of diastolic dysfunction and pathological ventricular hypertrophy. In aged Tnnc1WT/C84Y mice, left ventricular hypertrophy and myocardial fibrosis were visibly evident. Our previous studies have shown that cTnC variant residues located in the C-terminus also impact D/E linker dynamics and Ca2+ sensitivity of contraction.3 Furthermore, the C-terminus of cTnT allosterically modulates contraction through binding to cTnC.45 Mutations influencing frustration networks can affect allosteric events either directly or indirectly and may be a unifying mechanism dictating changes in Ca2+ affinity of site II that cause HCM.3,43 In summary, our findings demonstrate the importance of studying cardiomyopathic mutations at multiple scales of biological complexity and provide a framework for identifying novel therapeutic targets aimed at directly modulating the sarcomere in disease.
Methods
Recombinant protein preparation
The pET-3d vectors containing the human sequence of WT or C84Y cardiac TnC were heat shocked into BL21 DE3 strain and prepared as previously described.3 Briefly, recombinant, overexpressed protein was obtained by lysis of E. coli in 50 mM Tris–Cl (pH 7.8), 6 M urea, 1 mM EDTA, and 1 mM of fresh dithiothreitol (DTT). Clarified supernatants were loaded onto a Q-sepharose column and eluted by a linear gradient from 0 to 600 mM KCl. Fractions were dialyzed overnight against 50 mM Tris–Cl (pH 7.5), 50 mM NaCl, 1 mM MgCl2, 1 mM CaCl2, and 1 mM DTT, and loaded onto a Superdex 75 preparation grade column to achieve purity higher than 95%. 15N- and 15N–13C-labeled C84Y was produced as described.3
Fluorescence spectroscopy
Fluorescence emission spectra of 2 μM bis-ANS upon excitation at 360 nm were recorded at 440–580 nm and at 15 °C. Purified cTnC WT and C84Y proteins were mixed in 200 mM MOPS (pH 7), 150 mM KCl, 2 mM EGTA, and 1 mM DTT to reach 2 μM final concentration. The pCa 8 and 4 values were calculated using a pCa calculator program46 considering 2 mM free Mg2+.
NMR spectroscopy and backbone assignment
We acquired a set of 3D-experiments composed of HNCaCb, CbCa(CO)NH, HN(Ca)CO, and HNCO using 15N–13C-labeled C84Y cTnC at 25 °C in a Bruker 700 MHz spectrometer equipped with a triple resonance cryogenic probe. The composition of the buffer used was 20 mM MOPS (pH 7.0), 6 mM CaCl2, 1 mM MgCl2, 100 mM KCl, and 10 mM DTT containing 10% D2O. Peak assignments in the cTnC C84Y 1H–15N HSQC spectrum was guided by superposition of previous WT peak assignments. 3D-experiments were used to unambiguously confirm peak correlations not clearly distinguished by WT vs. C84Y spectra superposition. The CCPN suite was used for backbone peak assignments.
Circular dichroism (CD)
Thermostability assessment of 0.1 mg ml−1 WT or C84Y cTnC was carried out in a Chirascan spectropolarimeter. Thermograms were acquired at 222 nm from 20 to 90 °C at a rate of 1 °C min−1 every 0.2 °C in a 1 mm quartz cell. Buffer composition was 20 mM MOPS (pH 7.0), 100 mM KCl, and 1 mM EGTA for the Ca2+-free (apo) state, and addition of 2 mM MgCl2 and 1 mM CaCl2 to yield free Ca2+ and Mg2+ concentration of 0.1 mM and 2 mM, respectively, for the Ca2+-bound (primed) state. Mean residue ellipticity values in deg. cm2 dmol−1 res−1 were calculated from [θ]MRE = θ × 0.1(MW/n)/l × c where θ is ellipticity in millidegrees, MW is the molecular weight in Daltons, n is the number of peptide bonds, l is the path length in cm, and c is the concentration in mg ml−1.
Small angle X-ray scattering (SAXs) acquisition and analysis
SAXS data were collected on the SAXS1 beam line of the National Synchrotron Light Laboratory (Campinas, Brazil) as previously described.3 Parasitic scattering and buffer contribution were properly subtracted for each condition. Chemical-induced unfolding of 2 to 4 mg ml−1 cardiac TnC was performed with urea concentrations of 1, 3, 5, and 7 M. The buffer composition was 200 mM MOPS (pH 7.0), 100 mM KCl, and 1 mM EGTA for the Ca2+-free (apo) state and in addition of 2 mM MgCl2 and 1 mM CaCl2 to yield free Ca2+ and Mg2+ concentrations of 0.1 mM and 2 mM, respectively, for the Ca2+-bound (primed) state. Kratky plots were obtained by multiplying the scattering intensity I(s) by the scattering vector, s squared and the results were plotted as a function of s.47
Temperature analysis by NMR spectroscopy
We acquired 1H–15N heteronuclear single quantum correlation (HSQC) spectra of WT and C84Y cTnC at increasing temperatures ranging from 15 to 50 °C in a Bruker 700 MHz spectrometer. 1H–15N HSQC spectroscopy generates 2D spectra with one axis for protons (1H) in amide hydrogens and the other for a heteronucleus, 15N in this case, and the spectra contain one peak for each unique proton attached to each heteronucleus. This provides a unique fingerprint for each protein and allows detection of residues perturbed by temperature that vary between the C84Y variant compared to WT. The temperature dependence of 1H chemical shift (δ) for each 1H–15N peak correlation was fitted using linear regression, and temperature coefficients were obtained from the slope in ppb °C−1. Because of the linear dependence of δ to temperature increments, NMR assignment of 1H–15N HSQC spectra at increasing temperatures was done by tracking δ at 25 °C as a reference.
TnI peptide titrations by NMR spectroscopy
Switch (sw) and inhibitory (i) TnI peptides were synthesized by Genscript (Piscataway, Inc.) with purity higher than 95% and contained acetylation and amidation at the N- and C-terminal ends, respectively. A shorter switch TnI peptide (swTnI147–163) precipitated at concentrations above ∼50 μM and thus data from higher concentrations were excluded from analysis. To circumvent this, we performed most experiments with a longer and more soluble switch TnI peptide (swTnI144–165).
We acquired 1H–15N HSQC spectra at 37 °C of WT and C84Y cTnC (primed state) in the absence of peptides, and in the presence of increasing concentrations of each peptide (15, 40, 70, 120, 150, and 200 μM). Chemical shift analysis (CSA) comparing WT against C84Y or WT/C84Y peptide-bound against peptide-free cTnC was performed by the following equation: CSP = [(ΔδH)2 + 0.1(ΔδN)2]0.5 where ΔδH and ΔδN are the chemical shift variations of 1H and 15N, respectively, for the two conditions studied.
Dynamic properties
Longitudinal (R1) and transverse (R2) relaxation rates of C84Y were acquired from 1H–15N HSQC spectra with relaxation delays of 20, 50, 100, 200, 250, 500, 750, 1000, and 1500 ms for R1 and 16, 48, 80, 112, 144, 176, 208, 240, 272, and 304 ms for R2. Carr-Purcell-Meiboom-Gill (CPMG) relaxation dispersion experiments were acquired in a Bruker 700 MHz spectrometer at 25, 32, and 37 °C. Effective R2 relaxation rates (R2eff) as a function of residues were obtained from 1H–15N HSQC spectra acquired at CPMG frequencies of 50 and 1000 MHz to assess intermediate exchange dynamics of C84Y. R2eff was calculated from the following equation R2eff = −1/T ln(I0/ICPMG) where T is the relaxation time and I0 and ICPMG are the intensities of resonances obtained from experiments in the absence or presence of the CPMG pulse block, respectively.
Molecular dynamics (MD) simulations
TnC–TnI peptide
MD simulations were performed with GROMACS 2018.48 Starting structures were the best representative conformer from the 1AP4 (ref. 49) (WT TnC) and 1MXL8 (WT TnC bound to the swTnI147–162 peptide) ensembles, models 14 and 18, respectively. Position 84 was changed to Tyr for the C84Y trajectories using Chimera,50 and selecting the most probable rotamer conformation. The CHARMM36m force field was used with the CHARMM TIP3P water model.51 Na+ and Cl− ions were added to achieve charge neutrality and a physiological salt concentration of 0.15 M. System energy was minimized by steepest descent and propagated by leapfrog integration with a 2 fs time step. Temperature and pressure were regulated at 310 K and 1 bar using v-rescale and Parrinello-Rahman algorithms, respectively.52 Six replicates of each system were simulated with different initial atom velocity distributions for 3 μs apiece, giving 18 μs total sampling per system, and an aggregate simulation time of 72 μs. Analysis was performed on the concatenated trajectories (6 × 3 μs) for each system. AB interhelical angles were calculated using VGM53,54 using residues 14–25 and 38–47 for helices A and B, respectively. Open states were defined by an AB interhelical angle < 110°.16 Principal component analysis (PCA) was performed as described previously55 using the Cα atoms of the cTnC core region (residues 4–85).
Troponin complex and thin filament
MD simulations of the troponin (Tn) complex were performed based on the crystal structure resolved at 3.3 Å resolution by Takeda et al.7 and deposited in the Protein Databank (PDB ID 1J1E). We followed the procedure outlined in ref. 56 to add residues in TnI (residues 1–34, 137–146 and 192–210) and TnT (residues 280–288) that were not resolved in the Takeda et al. structure. The modeled structure, hereafter referred to as ‘intact Tn’, thus consisted of TnC 1–161, TnI 1–172, and TnT 236–285. Additionally, we simulated the Tn complex bound to tropomyosin and actin from the thin filament structure resolved via electron microscopy. We refer to these structures as the thin filament–troponin (TF–Tn) configuration. We used two structures corresponding to the Ca2+-free and Ca2+- bound states (PDB 6KN7 and 6KN8 9), which were resolved at 6.6 and 4.8 Å, respectively. Thus we have the TF–Tn Ca2+-bound model and TF–Tn Ca2+-free model. To maintain a computationally tractable system, we limited the structures to Tn as well as the two adjacent actins and tropomyosin helices. This led to a truncated system with 1330 residues. Given the low resolution of the 6KN7 and 6KN8 structures, the positions of bound Ca2+ ions were not resolved. We therefore placed two Ca2+ ions in 6KN7 sites III and IV (apo, C-terminal domain) and in 6KN8 sites II, III and IV (holo, N- and C-terminal domains) based on their positions in the Takeda et al.7 Tn structure. Lastly, we utilized the tyrosine rotamer at position C84Y, based on our simulations of the isolated N-terminal domains (1MXL and 1AP4).
These Tn structures were solvated using TIP3P waters57 and assuming a 14 Å margin around the protein for the periodic box. The ionic strength was set to 0.15 M NaCl after adding counter ions to neutralize the charge using the CHARMM36m force field.51 The MD protocol was otherwise identical to that used in 1MXL/1AP4 simulations, with exception that triplicate, minimum 100 ns simulations were performed. We performed the RMSF calculations and contact map analyses using the CPPTRAJ program.58 A contact was defined with an atom-atom distance cutoff of 7 Å for residue pairs separated by at least 6 residues. In the RMSF calculations, all frames were first aligned to the first frame using the backbone atoms of TnC residues M1- L100. All code written in support of this publication is publicly available at https://github.com/huskeypm/pkh-lab-analyses/(2021-TnSims).
Spatial localization of frustration nets
We used the Frustratrometer (http://www.frustratometer.tk/)59 to localize the level of frustration for each residue interaction. The theoretical calculation generates a frustration index, that measures “how favorable a particular contact is relative to sets of possible interactions, normalized using the variance of the energy distribution”.60 The color scale indicates the local mutational frustration where the minimally, neutral and highly frustrated sites are shown in green, gray and red, respectively. PDB models (Fig. 3: 1SPY, 1AP4 and 1MXL); (ESI Fig. 7†: 1TCF, 1J1E).
Animal procedures
The experimental protocol involving mice was reviewed and approved by the Florida State University (FSU) Animal Care and Use Committee (ACUC). Animal handling and experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals outlined by the National Institutes of Health (NIH). Animals were housed in a temperature-controlled vivarium on a 12:12 h light/dark cycle at FSU with ad libitum access to water and normal chow. For terminal procedures, mice were humanely sacrificed by rapid cervical dislocation under anesthesia with isoflurane, USP (Patterson Veterinary). Ages of the mice ranged from 3–14 months and mixed sexes were used as indicated in each respective figure legend.
Genetically engineered Tnnc1-p.C84Y mouse model
A gene-targeting approach was utilized to alter one Tnnc1 gene in the mouse genome to express Tnnc1-p.C84Y by homologous recombination in murine 129/SvEV-derived embryonic stem (ES) cells. The sequential overlapping polymerase chain reaction (PCR)-based method was used to generate the genetic variant in exon 4 of a genomic clone encompassing the entire murine Tnnc1 gene. The construct was subcloned into the “Targeting Vector” containing an MC1 neo cassette flanked by two loxP sites and thymidine kinase (TK) under the control of 3′ phosphoglycerate kinase (PGK).
Ella-Cre mice were on an FVB/N strain; Tnnc1WT/C84Y and Tnnc1WT/WT maintained on a C57BL/6 background.
Mouse genotyping was performed using tail DNA by PCR analysis with the following primers:
Tnnc1_WT_Forward: AGTCTGAGGAGGAGCTGTCG.
Tnnc1_WT_Reverse: GTCTCACCTCTCATTGGATGC (460 bp fragment).
Tnnc1_Neo_Forward: AGGACATAGCGTTGGCTACC (253 bp fragment).
Tnnc1_C84Y: CGTGGATTGCTGGAATGC.
Echocardiography
A Vevo 2100 high-resolution in vivo imaging system (Fujifilm Sonosite Inc., Toronto ON, Canada) was used to perform echocardiography (ECHO) analysis as previously described.61 Briefly, experimental mice were subjected to light anesthesia with 2% isoflurane (Patterson Veterinary). M-mode imaging of the parasternal short axis view permitted evaluation of wall thickness and left ventricular systolic/diastolic function. Mitral valve flow parameters were collected using pulsed-wave spectral Doppler mode via four-chamber viewing of the heart.
Histopathology
Whole mouse hearts were surgically excised and perfused in 10% buffered formaldehyde solution as previously described.61 Fixed hearts were sliced and stained by IDEXX Incorporated (Boston MA, USA). Images were acquired on a Nikon AZ 100M microscope and analysis was performed in a blinded manner using NIH ImageJ software. For Masson's trichrome imaging hearts were stained using Sigma-Aldrich Trichome stain (Masson) kit HT15-1KT according to the manufacturer's directions.
Cardiac muscle mechanics
Preparation and compositions of the buffered Ca2+ solutions and methods for assessment of cardiac muscle mechanical properties were previously described in detail.43 Briefly, left ventricle papillary muscle bundles were dissected from freshly excised Tnnc1WT/C84Y or Tnnc1WT/WT mouse hearts at 6 months of age and permeabilized with 1% Triton X-100 (v/v) in relaxing (pCa 8.0) solution for 4 h at 4 °C. These permeabilized cardiac muscle preparations (CMPs) were stored at −20 °C in 51% glycerol (v/v) in pCa 8.0 and used for experiments within 1 week. Force was measured using a force transducer (Aurora Scientific Inc. model 403A) and length was controlled using a high-speed servomotor (Aurora Scientific Inc. model 322C). Ca2+-activated steady-state, isometric tension generation, kinetics of isometric tension redevelopment (kTR), sinusoidal stiffness (SS) measurements (0.2% L0 peak-to-peak at 100 Hz) and mathematical modeling of kTR were carried out as previously described.43 Steady-state data were fitted with either 2-, 3- or 4-parameter Hill equations in SigmaPlot (v.12.0) as indicated in each respective figure legend, whereas estimations of the 3-state model parameters were computed in MatLab as previously described.43,45
Statistics and reproducibility
Ca2+ titrations by fluorescence spectroscopy and thermostability measurements by CD are expressed as mean ± S.E.M. Sample size was set to n = 3 of at least two protein batches.
SAXS analysis was performed by taking six frames of 5 s each and 2 s delay per frame. Frames were superimposed to inspect for radiation damage. Because successive frames revealed no radiation effects, the six replicates were averaged to enhance the signal-to-noise ratio. Scattering in each urea condition was performed twice (sample size, n = 2) from two independent protein batches. Data shown are from one of the samples.
Residues selected in chemical shift perturbation analysis (CSP) from NMR data refer to those that deviate from the average values of CSP (Δδ) among residues + 1 S.D. (Δδ + 1σ) and + 2 S.D. (Δδ + 2σ).
Histopathology images, echocardiography data and contractile parameters were obtained from both female and male Tnnc1WT/WT and Tnnc1WT/C84Y mice. Data are presented as mean ± S.E.M. Statistical significance was tested using Student's t-test in which alpha p < 0.05 (ECHO) and p < 0.02 (contractile parameters) were considered to be significant.
Data availability statement
All data and biological materials generated in this study are available from the corresponding authors upon request. The source data underlying Fig. 2b, 4g–h, 5, 6 and Supplementary Fig. 2a–b, 3a–s, 4b–c, 4h–i, 11b–i are provided in an excel file as a supplementary material.
Author contributions
M. A. M., G. A. P. d. O., and J. R. P. conceptualization; M. A. M., M. L.-V., A. H. M., B. S., K. M. D. J., E. C., I. C. V., P. M. K.-H., and G. A. P. d. O. data curation; M. A. M., M. L.-V., A. H. M., B. S., K. M. D. J., E. C., M. S. P., I. C. V., P. B. C., P. M. K.-H., and G. A. P. d. O. formal analysis; M. A. M., G. A. P. d. O., and J. R. P. supervision; M. A. M., M. L.-V., A. H. M., B. S., K. M. D. J., E. C., M. S. P., I. C. V., V. E. G., P. B. C., P. M. K.-H., G. A. P. d. O., and J. R. P. investigation; M. A. M., M. L.-V., A. H. M., B. S., J. R. J., K. M. D. J., E. C., M. S. P., I. C. V., P. B. C., P. M. K.-H., G. A. P. d. O., and J. R. P. methodology; M. A. M., and G. A. P. d. O. writing-original draft; M. A. M., G. A. P. d. O., and J. R. P. project administration; M. A. M., M. L.-V., A. H. M., B. S., J. R. J., K. M. D. J., E. C., M. S. P., J. L. S., V. E. G., P. B. C., P. M. K.-H., G. A. P. d. O., and J. R. P. writing-review and editing; E. A. C., J. L. S., P. M. K.-H., G. A. P. d. O., and J. R. P. resources; J. L. S., P. M. K.-H., G. A. P. d. O., and J. R. P. funding acquisition.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We gratefully acknowledge Dr Diego Ferreiro for critically reading our manuscript and providing helpful feedback. We thank Michael Osei Assibey for the technical and animal husbandry assistance. This work was supported by grants from the American Heart Association 19PRE34380628 awarded to JRJ and National Institutes of Health, National Heart, Lung and Blood Institute F31HL137408 awarded to KMDJ, HL128683 awarded to JRP and Maximizing Investigators' Research Award (MIRA) (R35) from the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) under grant number R35GM124977 awarded to PK-H. This work used the Extreme Science and Engineering Discovery Environment (XSEDE),62 which is supported by National Science Foundation grant number ACI-1548562. This research was also supported by the Pew Charitable Trusts Foundation to GAPdO as well as by the Carlos Chagas Filho Foundation for Research Support in the State of Rio de Janeiro (FAPERJ), grants 210.008/2018, 202840/2018, the National Council for Scientific and Technological Development (CNPq), and the National Institute of Science and Technology for Structural Biology and Bioimaging (INCT), grants 465395/2014-7 and 402321/2016-2 to JLS. This work was aided by the Brazilian Synchrotron Light Laboratory (LNLS) under proposals SAXS1-14202 and RMN-15571.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01886h
References
- Risi C. et al., Ca2+-induced movement of tropomyosin on native cardiac thin filaments revealed by cryoelectron microscopy. Proc. Natl. Acad. Sci. U. S. A. 2017;114:6782–6787. doi: 10.1073/pnas.1700868114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. A. et al., Allosteric switch regulates protein-protein binding through collective motion. Proc. Natl. Acad. Sci. U. S. A. 2016;113:3269–3274. doi: 10.1073/pnas.1519609113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques M. A. et al., Allosteric Transmission along a Loosely Structured Backbone Allows a Cardiac Troponin C Mutant to Function with Only One Ca2+ Ion. J. Biol. Chem. 2017;292:2379–2394. doi: 10.1074/jbc.M116.765362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams M. R. Lehman S. J. Tardiff J. C. Schwartz S. D. Atomic resolution probe for allostery in the regulatory thin filament. Proc. Natl. Acad. Sci. U. S. A. 2016;113:3257–3262. doi: 10.1073/pnas.1519541113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willott R. H. et al., Mutations in Troponin that cause HCM, DCM AND RCM: what can we learn about thin filament function? J. Mol. Cell. Cardiol. 2010;48:882–892. doi: 10.1016/j.yjmcc.2009.10.031. [DOI] [PubMed] [Google Scholar]
- Tadros H. J. et al., Meta-analysis of cardiomyopathy-associated variants in troponin genes identifies loci and intragenic hotspots that are associated with worse clinical outcomes. J. Mol. Cell. Cardiol. 2020;142:118–125. doi: 10.1016/j.yjmcc.2020.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S. Yamashita A. Maeda K. Maeda Y. Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- Li M. X. Spyracopoulos L. Sykes B. D. Binding of cardiac troponin-I147-163 induces a structural opening in human cardiac troponin-C. Biochemistry. 1999;38:8289–8298. doi: 10.1021/bi9901679. [DOI] [PubMed] [Google Scholar]
- Yamada Y. Namba K. Fujii T. Cardiac muscle thin filament structures reveal calcium regulatory mechanism. Nat. Commun. 2020;11:153. doi: 10.1038/s41467-019-14008-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risi C. M. et al., The structure of the native cardiac thin filament at systolic Ca2+ levels. Proc. Natl. Acad. Sci. U. S. A. 2021;118(13):e2024288118. doi: 10.1073/pnas.2024288118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreiro D. U. Komives E. A. Wolynes P. G. Frustration, function and folding. Curr. Opin. Struct. Biol. 2018;48:68–73. doi: 10.1016/j.sbi.2017.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker S. B. Clayden N. J. Moore G. R. NMR characterisation of the relationship between frustration and the excited state of Im7. J. Mol. Biol. 2011;414:511–529. doi: 10.1016/j.jmb.2011.09.038. [DOI] [PubMed] [Google Scholar]
- Ferreiro D. U. Komives E. A. Wolynes P. G. Frustration in biomolecules. Q. Rev. Biophys. 2014;47:285–363. doi: 10.1017/S0033583514000092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. X. et al., NMR studies of Ca2+ binding to the regulatory domains of cardiac and E41A skeletal muscle troponin C reveal the importance of site I to energetics of the induced structural changes. Biochemistry. 1997;36:12519–12525. doi: 10.1021/bi971222l. [DOI] [PubMed] [Google Scholar]
- Hwang P. M. Cai F. Pineda-Sanabria S. E. Corson D. C. Sykes B. D. The cardiac-specific N-terminal region of troponin I positions the regulatory domain of troponin C. Proc. Natl. Acad. Sci. U. S. A. 2014;111:14412–14417. doi: 10.1073/pnas.1410775111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens C. M. et al., Changes in the dynamics of the cardiac troponin C molecule explain the effects of Ca2+-sensitizing mutations. J. Biol. Chem. 2017;292:11915–11926. doi: 10.1074/jbc.M116.770776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamora J. E. Papadaki M. Messer A. E. Marston S. B. Gould I. R. Troponin structure: its modulation by Ca2+ and phosphorylation studied by molecular dynamics simulations. Phys. Chem. Chem. Phys. 2016;18:20691–20707. doi: 10.1039/c6cp02610a. [DOI] [PubMed] [Google Scholar]
- Lindert S. Cheng Y. Kekenes-Huskey P. Regnier M. McCammon J. A. Effects of HCM cTnI mutation R145G on troponin structure and modulation by PKA phosphorylation elucidated by molecular dynamics simulations. Biophys. J. 2015;108:395–407. doi: 10.1016/j.bpj.2014.11.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewan S. McCabe K. J. Regnier M. McCulloch A. D. Lindert S. Molecular Effects of cTnC DCM Mutations on Calcium Sensitivity and Myofilament Activation-An Integrated Multiscale Modeling Study. J. Phys. Chem. B. 2016;120:8264–8275. doi: 10.1021/acs.jpcb.6b01950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordina N. M. et al., Effects of calcium binding and the hypertrophic cardiomyopathy A8V mutation on the dynamic equilibrium between closed and open conformations of the regulatory N-domain of isolated cardiac troponin C. Biochemistry. 2013;52:1950–1962. doi: 10.1021/bi4000172. [DOI] [PubMed] [Google Scholar]
- Landstrom A. P. et al., Molecular and functional characterization of novel hypertrophic cardiomyopathy susceptibility mutations in TNNC1-encoded troponin C. J. Mol. Cell. Cardiol. 2008;45:281–288. doi: 10.1016/j.yjmcc.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluber A. Burt T. A. Clementi C. Size and topology modulate the effects of frustration in protein folding. Proc. Natl. Acad. Sci. U. S. A. 2018;115:9234–9239. doi: 10.1073/pnas.1801406115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doniach S. Changes in biomolecular conformation seen by small angle X-ray scattering. Chem. Rev. 2001;101:1763–1778. doi: 10.1021/cr990071k. [DOI] [PubMed] [Google Scholar]
- Grabarek Z. Structural Basis for Diversity of the EF-hand Calcium-binding Proteins. J. Mol. Biol. 2006;359:509–525. doi: 10.1016/j.jmb.2006.03.066. [DOI] [PubMed] [Google Scholar]
- Lindert S. Kekenes-Huskey P. M. McCammon J. A. Long-timescale molecular dynamics simulations elucidate the dynamics and kinetics of exposure of the hydrophobic patch in troponin C. Biophys. J. 2012;103:1784–1789. doi: 10.1016/j.bpj.2012.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loria J. P. Berlow R. B. Watt E. D. Characterization of enzyme motions by solution NMR relaxation dispersion. Acc. Chem. Res. 2008;41:214–221. doi: 10.1021/ar700132n. [DOI] [PubMed] [Google Scholar]
- Smith L. Greenfield N. J. Hitchcock-DeGregori S. E. The effects of deletion of the amino-terminal helix on troponin C function and stability. J. Biol. Chem. 1994;269:9857–9863. [PubMed] [Google Scholar]
- Smith L. Greenfield N. J. Hitchcock-DeGregori S. E. Mutations in the N- and D-helices of the N-domain of troponin C affect the C-domain and regulatory function. Biophys. J. 1999;76:400–408. doi: 10.1016/S0006-3495(99)77206-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordina N. M. et al., Interdomain orientation of cardiac troponin C characterized by paramagnetic relaxation enhancement NMR reveals a compact state. Protein Sci. 2012;21:1376–1387. doi: 10.1002/pro.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badr M. A. Pinto J. R. Davidson M. W. Chase P. B. Fluorescent Protein-Based Ca2+ Sensor Reveals Global, Divalent Cation-Dependent Conformational Changes in Cardiac Troponin C. PloS One. 2016;11:e0164222. doi: 10.1371/journal.pone.0164222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell A. P. Sykes B. D. Interaction of troponin I and troponin C. Use of the two-dimensional nuclear magnetic resonance transferred nuclear Overhauser effect to determine the structure of the inhibitory troponin I peptide when bound to skeletal troponin C. J. Mol. Biol. 1991;222:405–421. doi: 10.1016/0022-2836(91)90219-v. [DOI] [PubMed] [Google Scholar]
- Babu A. Rao V. G. Su H. Gulati J. Critical minimum length of the central helix in troponin C for the Ca2+ switch in muscular contraction. J. Biol. Chem. 1993;268:19232–19238. [PubMed] [Google Scholar]
- Dobrowolski Z. Xu G. Q. Hitchcock-DeGregori S. E. Modified calcium-dependent regulatory function of troponin C central helix mutants. J. Biol. Chem. 1991;266:5703–5710. [PubMed] [Google Scholar]
- Ramakrishnan S. Hitchcock-DeGregori S. E. Investigation of the structural requirements of the troponin C central helix for function. Biochemistry. 1995;34:16789–16796. doi: 10.1021/bi00051a029. [DOI] [PubMed] [Google Scholar]
- Anthis N. J. Clore G. M. The length of the calmodulin linker determines the extent of transient interdomain association and target affinity. J. Am. Chem. Soc. 2013;135:9648–9651. doi: 10.1021/ja4051422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott M. B. Dvoretsky A. Gaponenko V. Rosevear P. R. Cardiac troponin I inhibitory peptide: location of interaction sites on troponin C. FEBS Lett. 2000;469:168–172. doi: 10.1016/s0014-5793(00)01271-0. [DOI] [PubMed] [Google Scholar]
- Abbott M. B. et al., Regulatory domain conformational exchange and linker region flexibility in cardiac troponin C bound to cardiac troponin I. J. Biol. Chem. 2000;275:20610–20617. doi: 10.1074/jbc.M909252199. [DOI] [PubMed] [Google Scholar]
- Gunasekaran K. Ma B. Nussinov R. Is allostery an intrinsic property of all dynamic proteins? Proteins. 2004;57:433–443. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- Endrizzi J. A. Beernink P. T. Alber T. Schachman H. K. Binding of bisubstrate analog promotes large structural changes in the unregulated catalytic trimer of aspartate transcarbamoylase: implications for allosteric regulation. Proc. Natl. Acad. Sci. U. S. A. 2000;97:5077–5082. doi: 10.1073/pnas.090087197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit A. Verkhivker G. M. The energy landscape analysis of cancer mutations in protein kinases. PLoS One. 2011;6:e26071. doi: 10.1371/journal.pone.0026071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzul F. O. Vasilchuk D. Makhatadze G. I. Evidence for the principle of minimal frustration in the evolution of protein folding landscapes. Proc. Natl. Acad. Sci. U. S. A. 2017;114:E1627–E1632. doi: 10.1073/pnas.1613892114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase P. B. Martyn D. A. Hannon J. D. Isometric force redevelopment of skinned muscle fibers from rabbit activated with and without Ca2+ Biophys. J. 1994;67:1994–2001. doi: 10.1016/S0006-3495(94)80682-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Martinez D. et al., Structural and functional impact of troponin C-mediated Ca2+ sensitization on myofilament lattice spacing and cross-bridge mechanics in mouse cardiac muscle. J. Mol. Cell. Cardiol. 2018;123:26–37. doi: 10.1016/j.yjmcc.2018.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnier M. Rivera A. J. Chase P. B. Smillie L. B. Sorenson M. M. Regulation of skeletal muscle tension redevelopment by troponin C constructs with different Ca2+ affinities. Biophys. J. 1999;76:2664–2672. doi: 10.1016/S0006-3495(99)77418-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J. R. et al., The intrinsically disordered C terminus of troponin T binds to troponin C to modulate myocardial force generation. J. Biol. Chem. 2019;294:20054–20069. doi: 10.1074/jbc.RA119.011177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dweck D. Reyes-Alfonso Jr A. Potter J. D. Expanding the range of free calcium regulation in biological solutions. Anal. Biochem. 2005;347:303–315. doi: 10.1016/j.ab.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Mertens H. D. Svergun D. I. Structural characterization of proteins and complexes using small-angle X-ray solution scattering. J. Struct. Biol. 2010;172:128–141. doi: 10.1016/j.jsb.2010.06.012. [DOI] [PubMed] [Google Scholar]
- Abraham M. J. et al., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. [Google Scholar]
- Spyracopoulos L. et al., Calcium-induced structural transition in the regulatory domain of human cardiac troponin C. Biochemistry. 1997;36:12138–12146. doi: 10.1021/bi971223d. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F. et al., UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Huang J. et al., CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods. 2017;14:71–73. doi: 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussi G. Donadio D. Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Yap K. L. Ames J. B. Swindells M. B. Ikura M. Diversity of conformational states and changes within the EF-hand protein superfamily. Proteins. 1999;37:499–507. doi: 10.1002/(sici)1097-0134(19991115)37:3<499::aid-prot17>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Yap K. L. Ames J. B. Swindells M. B. Ikura M. Vector geometry mapping. A method to characterize the conformation of helix-loop-helix calcium-binding proteins. Methods Mol. Biol. 2002;173:317–324. doi: 10.1385/1-59259-184-1:317. [DOI] [PubMed] [Google Scholar]
- Cino E. A. Choy W. Y. Karttunen M. Characterization of the Free State Ensemble of the CoRNR Box Motif by Molecular Dynamics Simulations. J. Phys. Chem. B. 2016;120:1060–1068. doi: 10.1021/acs.jpcb.5b11565. [DOI] [PubMed] [Google Scholar]
- Cheng Y. et al., Computational studies of the effect of the S23D/S24D troponin I mutation on cardiac troponin structural dynamics. Biophys. J. 2014;107:1675–1685. doi: 10.1016/j.bpj.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L. Chandrasekhar J. Madura J. D. Impey R. W. Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- Roe D. R. Cheatham III T. E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Parra R. G. et al., Protein Frustratometer 2: a tool to localize energetic frustration in protein molecules, now with electrostatics. Nucleic Acids Res. 2016;44:W356–W360. doi: 10.1093/nar/gkw304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreiro D. U. Hegler J. A. Komives E. A. Wolynes P. G. Localizing frustration in native proteins and protein assemblies. Proc. Natl. Acad. Sci. U. S. A. 2007;104:19819–19824. doi: 10.1073/pnas.0709915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins A. S. et al., In Vivo Analysis of Troponin C Knock-In (A8V) Mice: Evidence that TNNC1 Is a Hypertrophic Cardiomyopathy Susceptibility Gene. Circ.: Cardiovasc. Genet. 2015;8:653–664. doi: 10.1161/CIRCGENETICS.114.000957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towns J. et al., XSEDE: Accelerating Scientific Discovery. Comput. Sci. Eng. 2014;16:62–74. [Google Scholar]
- Pinto J. R. et al., Strong cross-bridges potentiate the Ca2+ affinity changes produced by hypertrophic cardiomyopathy cardiac troponin C mutants in myofilaments: a fast kinetic approach. J. Biol. Chem. 2011;286:1005–1013. doi: 10.1074/jbc.M110.168583. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and biological materials generated in this study are available from the corresponding authors upon request. The source data underlying Fig. 2b, 4g–h, 5, 6 and Supplementary Fig. 2a–b, 3a–s, 4b–c, 4h–i, 11b–i are provided in an excel file as a supplementary material.