

Abstract
Viral infection of the central nervous system (CNS) can cause lasting neurological decline in surviving patients and can present with symptoms resembling Parkinson’s disease (PD). The mechanisms underlying postencephalitic parkinsonism remain unclear but are thought to involve increased innate inflammatory signaling in glial cells, resulting in persistent neuroinflammation. We therefore studied the role of glial cells in regulating neuropathology in postencephalitic parkinsonism by studying the involvement of astrocytes in loss of dopaminergic neurons and aggregation of α–synuclein protein following infection with western equine encephalitis virus (WEEV). Infections were conducted in both wildtype mice and in transgenic mice lacking NFκB inflammatory signaling in astrocytes. For two months following WEEV infection, we analyzed glial activation, neuronal loss and protein aggregation across multiple brain regions, including the substantia nigra pars compacta (SNpc). These data revealed that WEEV induces loss of SNpc dopaminergic neurons, persistent activation of microglia and astrocytes that precipitates widespread aggregation of α-synuclein in the brain of C57BL/6 mice. Microgliosis and macrophage infiltration occurred prior to activation of astrocytes and was followed by opsonization of α-synuclein protein aggregates in the cortex, hippocampus and midbrain by the complement protein, C3. Astrocyte-specific NFκB knockout mice had reduced gliosis, α-synuclein aggregate formation and neuronal loss. These data suggest that astrocytes play a critical role in initiating PD-like pathology following encephalitic infection with WEEV through innate immune inflammatory pathways that damage dopaminergic neurons, possibly by hindering clearance of α-synuclein aggregates. Inhibiting glial inflammatory responses could therefore represent a potential therapy strategy for viral parkinsonism.
Keywords: Alphaviruses, Western equine encephalitis virus, Viral encephalitis, Neurodegeneration, Glia, Neuroinflammation, Alpha-synuclein, Parkinson’s Disease
Introduction
Parkinson’s disease (PD) affects both the central and peripheral nervous systems but is primarily characterized by the loss of voluntary motor function due to the degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc). Epidemiological and experimental evidence suggests that genetic susceptibility and exposure to environmental agents such as pesticides and viruses are possible risk factors for PD and related neurodegenerative diseases. There have been multiple reports of parkinsonism observed among human survivors of encephalitic viral infection (Jang et al., 2009b). Following the 1918 Spanish Flu pandemic, nearly every patient who suffered from acute encephalitis following infection with H1N1 went on to develop postencephalitic parkinsonism (Dourmashkin, 1997; Reid et al., 2001). Additionally, many neurotropic viruses, such as West Nile virus (WNV), Japanese encephalitis virus (JEV), H5N1 influenza A and St. Louis encephalitis virus can cause Parkinson’s-like pathology in humans, including aggregation of α-synuclein, neuronal loss and inflammatory activation of microglia and astrocytes (Clarke et al., 2014; Elizan et al., 1978; Jang et al., 2009a; Jang et al., 2009b). Earlier studies reported that Western equine encephalitis virus (WEEV) can cause parkinsonism in humans following encephalitic infection (Mulder et al., 1951; Palmer and Finley, 1956; Schultz et al., 1977). This was highlighted by an outbreak of WEEV in Colorado where multiple patients presented with PD-like symptoms following encephalitic infection, including tremor, cognitive deterioration and cogwheel rigidity (Schultz et al., 1977). Antibodies against mosquito-borne viruses have been reported in patients with von Economo’s postencephalitic parkinsonism and in idiopathic PD, suggesting a viral etiology in some instances (Mulder et al., 1951; Palmer and Finley, 1956). Both virally-induced parkinsonism and PD present with inflammatory activation of microglia and astrocytes, suggesting common neuroinflammatory responses between the encephalitic and idiopathic forms of the diease (Barcia, 2013). Although the cause of post-encephalitic parkinsonism is not fully understood, these studies and others suggest that neuroinflammatory activation of glial cells following viral infection could be a critical initiating factor (Lesteberg and Beckham, 2019).
Studies of the pathophysiology and progression of PD increasingly focus on the role played by reactive glial cells. Neuroinflammation could be a key factor connecting viral encephalitis and the development of parkinsonian neurological symptoms (Caggiu et al., 2019). Clinical evidence suggests a significant role for microglial-derived inflammatory mediators in PD, including the presence of inflammatory cytokines in cerebral spinal fluid (CSF), plasma and in PET imaging studies showing sustained neuroinflammatory activation of microglia in the midbrain of PD patients (Duffy et al., 2018; Gerhard et al., 2006; Lindqvist et al., 2013; Mogi et al., 1996). Likewise, inflammatory activation of astrocytes is associated with initiation and progression of neurodegeneration through increased release of inflammatory mediators that damage associated neurons (Martinez et al., 2017). Reactive neurotoxic astrocytes (those described as having an A1 phenotype) release multiple factors that modulate neuronal morphology and synaptic function, as well as the function of microglia. Among the factors released by reactive astrocytes, complement C3 protein modulates microglial activity through the C3/C3aR axis, thereby regulating phagocytic activity and protein aggregation dynamics in the brain (Clarke et al., 2018; Lian et al., 2016; Liddelow and Barres, 2017; Liddelow et al., 2017; Zhang et al., 2010; Zhang et al., 2014). C3 is transcriptionally regulated in astrocytes by NFκB and has differing effects on microglial activity depending on whether C3 is expressed acutely or chronically. Acute expression induces microglia phagocytosis and clearance of protein aggregates, whereas chronic expression of C3 suppresses microglial clearance of aggregated proteins (Lian et al., 2016). Thus, chronic phenotypic activation of key A1 genes such as C3 during encephalitic viral infection could directly alter both the inflammatory and phagocytic activity of microglia and promote the formation of neurotoxic of α-synuclein aggregates.
We recently demonstrated that intranasal infection with WEEV induced selective loss of dopaminergic neurons in the SNpc, associated with persistent microgliosis, astrogliosis, and the development of proteinase-K resistant α-synuclein aggregates that spread into the cortex, hippocampus, and midbrain by 8 weeks post-intranasal infection (8 weeks post-infection (WPI) in wild-type CD-1 mice (Bantle et al., 2019). The majority of α-synuclein aggregates were located amongst dying neurons surrounded by phagocytotic microglia. However, whether viral-mediated loss of dopaminergic neurons following acute infection with WEEV is due in part to an increased neuroinflammatory response of microglia and astrocytes remains to be determined.
In the current study, we examined whether neuroinflammatory activation of glia is an initiating event in selective loss of dopaminergic neurons and α-synuclein protein aggregation following encephalitic WEEV infection. Astrocyte-specific NFκB KO mice generated in our laboratory were infected with WEEV to interrogate the contribution of NFκB-regulated gene expression in astrocytes as a causative event in neuronal injury from WEEV infection. Astrocyte-specific KO of NFκB drastically reduced loss of dopaminergic neurons and α-synuclein aggregation following infection, suggesting that innate immune inflammatory signaling in astrocytes regulates both neuroinflammation and protein aggregation that precedes neuronal injury. These data suggest that glial inflammation from viral infection contributes to neurodegeneration associated with post-encephalitic parkinsonism.
Materials and Methods
DsRed and Firefly Luciferase Expressing WEEV Viral Constructs.
Construction of recombinant WEEV (MacMillan strain) reporter viruses was previously described (Logue et al., 2009; Phillips et al., 2013; Phillips et al., 2016). Briefly, a duplication of the subgenomic promoter (SGP) sequence (nucleotides 7341–7500 of viral genome) of the WEEV McMillan strain was used to express firefly luciferase or DsRed. Plasmids were purified by QIAprep Spin MiniPrep Kit (Qiagen, Valencia, CA USA) and RNA was transcribed in vitro using a T7 RNA polymerase (MAXIscript™ kit, Life Technologies, Grand Island, NY USA). BHK-21 cells (2×107 in 400 μL) were transfected with 20 μL of total RNA using an ECM 630 electroporator (BTX Harvard Apparatus, Holliston, MA USA). The rescued virus was stored at −80°C before plaquing. All plaque titration assays were run in duplicate in Vero cells before experimental use as previously described (Liu et al., 1970).
Viral infections in wildtype and astrocyte Specific NFκB knock-out mice.
All animals were housed on a 12 hr light/dark cycle in a temperature-controlled room (maintained at 22-24°C) and had access to standard chow/water ad libitum. We generated astrocyte-specific NFκB knockout mice as previously described by crossing mice expressing the human Gfap-Cre+/− mice with Ikk2-loxP+/+ mice (Kirkley et al., 2019). Progeny were backcrossed four generations to produce Gfap-Cre+/−/Ikk2F/F (KO) or Gfap-Cre−/−/Ikk2F/F (WT) animals for the study. Infections with recombinant WEEV were performed as previously described (Bantle et al., 2019). Six-week-old male and female C57BL/6 mice (Charles River Labs, Wilmington, MA USA) or astrocyte-specific NFκB knockout mice were housed in a biosafety level 3 (BSL-3) facility at the Infectious Disease Research Center at Colorado State University. Mice were anesthetized with isoflurane (Minrad Inc, Bethlehem, PA USA). We intranasally administered 20 μL of either DsRed-expressing or luciferase-expressing WEEV at a concentration of 1×104 PFU/ml to anesthetized mice. All infected mice were euthanized at four days-post-infection and used to characterize the viral spread of WEEV. Mice infected with luciferase-expressing WEEV received a subcutaneous 150 mg/kg dose of luciferin and were analyzed using an IVIS 200 bioluminescence imaging system (Xenogen, Alameda, CA) 10–15 minutes later. Uninfected mice were used as an imaging control to adjust for background signal. The exposure time was 2 min under standard settings for the camera, and the Living Image 3.0 software (Caliper Life Sciences, Waltham, MA USA) was used to analyze and process images taken using the IVIS 200 camera. A threshold for significant BLM was established using negative imaging controls at 5×103 p/s/cm2/sr. Total light emission from each mouse was determined by creating a region of interest (ROI) of standard size for each mouse and collecting light emission data. All mice were handled in compliance with the PHS Policy and Guide for the Care and Use of Laboratory Animals, and all animal protocols used in the study were reviewed and approved by the Animal Care and Use Committee at Colorado State University (Permit #11-2605A).
Generating anti-Alphavirus E1 Treatment Serum.
We vaccinated rabbits with recombinant WEEV McMillan strain E1-ectodomain antigen (10 μg antigen/dose), which was diluted in an immunological adjuvant comprised of polyI:C (dsRNA analog), ODN 1826 (unmethylated CpG DNA) (InVivoGen), and TiterMax Gold to a final concentration of 0.1 mg/mL. A total of four doses were administered every two weeks before rabbits were terminally bled for serum collection. After collection, serum was heat inactivated at 56°C for 30 minutes and stored at −80°C. Naïve serum was also collected from control animals. All anti-E1 serum was tested against E1 antigen on coated titration plates before its use in the study and was found to have reciprocal antibody titer measurement greater than 26,000 reciprocal value, as previously reported (Bantle et al., 2019).
Tissue Processing and Sectioning.
One, two, four, and eight weeks after infection with luciferase-expressing WEEV, animals were terminally anesthetized with isoflurane and transcardially perfused. The brains were then extracted, fixed in 4% paraformaldehyde at 4°C and later processed for paraffin embedding and sectioned at 10 microns on a microtome through the anatomic midbrain and mounted on polyionic slides (Superfrost-plus, Fisher Scientific) (Miller et al., 2011; Smeyne et al., 2016).
Immunofluorescence Staining and Imaging.
Coronal and sagittal sections were deparaffinized and immunostained using anti-tyrosine hydroxylase (TH; 1:500; Millipore AB152) to identify dopaminergic neurons, anti-glial fibrillary acidic protein (GFAP; 1:500; DAKO Z0334) to identify astrocytes and anti-calcium adaptor binding protein 1 (Iba-1; 1:250; WAKO 016-20001) to label microglia, per our previously published methods (Hammond et al., 2017; Miller et al., 2011). Sections were visualized by automated montage imaging of individual 10X frames of each immunostained section using a Hammatsu Flash4.0 digital CMOS camera, ProScan III stage controller (Prior, Rockland, MA USA) and CellSens Dimension software (version 1.12, Olympus, Center Valley, PA, USA). Regions of interest representing distinct anatomical nuclei were analyzed on each composite montage image.
Stereological Assessment of neurons and glial cells.
Methodologies for imaging and counting dopaminergic and total neurons in the substantia nigra, as well as microglia and astrocytes, were adapted from our previously reported studies (Baquet et al., 2009; Tapias et al., 2013). To determine the number of dopaminergic neurons and microglia in the SNpc, deparaffinized sections were double-immunostained for TH and Iba-1 (mouse monoclonal anti-tyrosine hydroxylase and rabbit polyclonal Iba-1 (Wako Chemicals; 1:500)), using secondary antibodies including biotinylated mouse IgG (for TH, 1:1000) or biotinylated rabbit IgG (for Iba-1, 1:1000). Diaminobenzindine (DAB) or a VIP kit (Vector labs) reaction was used to yield a brown (TH) or a purple (IBA-1) chromogen, respectively. All tissue sections were counterstained with Nissl substance and Neutral Red to identify anatomical landmarks. Stereological analysis of dopaminergic neurons and microglia in the SNpc was performed as previously described (Baquet et al., 2009). To determine the number of resting and activated microglia, Iba-1-positive microglia in the SNpc were analyzed using our previously published morphological criteria to distinguish resting and activated microglia from infiltrating macrophages (Graeber and Streit, 2010; Sadasivan et al., 2015; Smeyne et al., 2016; Tansey, 2010). The number of microglia in the right and left SNpc were summed to provide an estimate of the total number of resting and active microglia in the SNpc (Bantle et al., 2019). The investigator was blinded to the individual treatment groups when counting cells.
Analysis of catecholamines and monoamines.
Brain samples were prepared and analyzed using high performance liquid chromatography with electrochemical detection, as previously described (De Miranda et al., 2015). Sample analysis for DA, 3, 4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), serotonin (5-HT) and 5-hydroxindoleacetic acid (5-HIAA) was performed at the VBI Neurochemistry Core, Vanderbilt University School of Medicine. All tissue samples from each experimental group were coded and blinded for unbiased analysis.
Behavioral Analysis.
All mice were acclimated to handling two weeks prior to infection and habituated to the video trackway with repeated runs prior to baseline measurements the day before infections with WEEV (Gouveia and Hurst, 2013; Stuart and Robinson, 2015). Multiple neurobehavioral parameters, including stride length, run duration, step cycle, cadence, duty cycle and stop time were measured using a custom-made real-time video gait analysis system, as previously described (Hammond et al., 2018). All behavioral testing was performed on uninfected and infected mice on days 0, 14, 28 and 56. All parameter values were subtracted from the baseline measurements obtained on day 0 to normalize across all time points and treatment groups.
Immunohistochemical Staining and Pathological Scoring of Phospho-serine 129+ Alpha-Synuclein Protein Aggregates.
Sections were processed for immunohistochemistry using a Leica Bond RXm automated staining system according to the manufacturer’s protocols. Antigen retrieval was performed in Bond Epitope Retrieval Solution 2 for 20 minutes at 37 °C. Reactive synuclein cell/cell aggregates were stained using a mouse monoclonal antibody against phosphorylated α-synuclein at Ser129, (pSyn#64, FUJIFILM Wako Chemicals) (Jang et al., 2009a). Antibodies for phospho-Eif2α (Cell Signaling Technology, 119A11, Rabbit mAb #3597) and phospho-PERK (Cell Signaling Technology, 16F8, Rabbit mAb #3179) were used at 1:500 dilution. Immunostaining and neuropathological scoring of P129+ protein aggregates were conducted in the cortex, hippocampus and midbrain on infected and uninfected brain sections by a veterinary pathologist blinded to the treatment groups using scoring methodology was adapted from previous reports (Rey et al., 2016; Rey et al., 2018). We assessed the presence of P129+ inclusions on two coronal sections per animal that were 20 μm in thickness, spaced at 100 μm intervals within the SNpc, with an N=6-8 mice at each timepoint for each treatment group (N=6-8 mice per group). Each section was analyzed at a 20× magnification using an Olympus IX71 microscope (Center Valley, PA) with Retiga 2000R (Qimaging, Surrey, BC, Canada) and Qcolor3 (Olympus) camera and slidebook software (v6.0, Intelligent Imaging Innovations, Inc., Denver, CO) for image acquisition and analysis. A score of 1 to 5 was assigned to each brain region from a single coronal brain section and scored as follows: 1 = no aggregates, 2 = very sparse or one to two P129+ cells, 3 = mild with less than ten P129+ cells, 4 = dense with more than ten P129+ cells, 5 = very dense with more than 20 P129+ cells involved. The two scores obtained for each individual section were averaged for each mouse.
Quantification of C3 and P129+ Alpha-Synuclein aggregates.
Formalin-fixed, paraffin embedded mouse brain sections from the SNpc of infected mice were mounted on glass slides and immunofluorescently labeled on a Leica Bond RXM automated robotic staining system using Bond Epitope Retrieval Solution 2 for 20 minutes. Sections were then incubated with primary antibodies for mouse alpha synuclein phosphorylated Ser129 (P129) antibody (1:1000, clone pSyn#64, FUJIFILM Wako Chemicals) and rat anti-C3/C3b (1:500; Abcam 11871). Secondary antibodies included anti-mouse IgG AlexaFluor-488 and anti-rat IgG AlexaFluor-555. Whole brain fluorescent montage images of labeled tissue sections were visualized using a 20x objective and an automated Olympus BX51 fluorescence microscope equipped with a Hamamatsu ORCA-flash 4.0 LT CCD camera and collected using Olympus Cellsens software (v 1.15). Quantitative analysis was performed on dual- or triple-labeled fluorescence images generated by montage imaging of an entire coronal brain section, with individual images acquired using an Olympus Plan Apochromat 10X air objective (0.40 N.A.). The cortex, hippocampus and midbrain were delineated by anatomical landmarks and referenced to the Allen brain atlas. A region of interest (ROI) was generated around all P129+ cell aggregates in each neuroanatomical region of interest and the mean fluorescence intensity of C3/C3b was then obtained within each P129+ ROI. Duplicate tissue sections were stained and averaged for each mouse (N=6-8 mice at each time point for each treatment group). The investigator was blinded from all experimental groups for data acquisition. All slides were scanned under the same conditions for magnification, exposure time, excitation intensity and camera gain.
Image analysis of reactive astrocytes.
Formalin-fixed, paraffin embedded 8 μm brain sections were labeled by immunofluorescence staining as described above using a Leica Bond RXMm automated robotic staining system. Sections were then incubated with primary antibodies for anti-S100β (rabbit polyclonal, 1:500; Abcam 212816) and anti-C3 (mouse monoclonal, 1:50; Abcam 11871). Secondary antibodies included anti-rabbit IgG Alexafluor-647 and anti-rat IgG Alexafluor-555. To detect C3+ cells co-localizing with S100β+ astrocytes, individual ROIs were created around all S100β+ astrocytes within the SNpc and the mean C3 intensity was measured within each S100β+ astrocyte. Each brain was analyzed in duplicate, with a total of 6-8 mice per group at each time point for each treatment group. All slides were scanned under the same conditions for magnification, exposure time, excitation intensity, and camera gain.
Generation of Representative Normalized Pathological Overlays.
To generate the representative overlays at each time point, the total number of dopaminergic neurons, microglia, macrophages, astrocytes cellular counts and P129 pathological scores from infected mice were normalized to controls according to the following equation: [((1-0.2)*((x-y)/(y-z)))+0.2]*100=normalized percentage value (Gopal Krishna Patro and Kumar Sahu, 2015). The control values obtained for each pathological parameter were averaged and then subtracted from the individual experimentally infected animal values yielding a pathological representative of activation (x). The minimum (y) and maximum (z) values were determined for each activation parameter dataset. Normalization was then performed by determining the difference between the control subtracted experimental infected values and the minimum overall value. The total obtained from this calculation serves as the numerator. The range of the data set was then determined by obtaining the difference from the minimum (y) and maximum (z) values, this would serve as the denominator. To account for phenotype associated with normal aging, a baseline value of twenty percent was factored into the equation for glial activation, protein aggregation and neurodegeneration. The value obtained from this overall calculation was then multiplied by 100 to represent total percentage activation of each parameter. The respective percentages at each time point (1 week, 2 weeks and 8 weeks) were averaged and plotted using spline curve fitting within GraphPad software (version 9.1.0; Graph Pad Software, San Diego, CA).
Statistical Analysis.
All data was presented as mean +/− SEM, unless otherwise noted. Experimental values from each mean were analyzed with a Grubb’s (α=0.05) test for exclusion of significant outliers. Differences between each experimental group were analyzed using an unpaired t-test or a one-way ANOVA with Tukey’s post hoc test for multiple comparisons. Significance was identified as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. All statistical analysis was conducted using Prism (version 6.0; Graph Pad Software, San Diego, CA).
Results
WEEV selectively infects neurons throughout the brain and induces persistent microgliosis and dopaminergic neuronal loss in the substantia nigra pars compacta.
To determine the viral dissemination of WEEV following intranasal infection, we infected six-week-old C57Bl/6 mice with either WEEV-DsRed (Fig. 1A) or WEEV-Luc (Fig. 1H). Following infection, 100% of mice developed severe locomotor abnormalities and became moribund by 4 DPI. Histopathological analysis of brain tissue from infected mice revealed that WEEV showed selective tropism for neuronal cell populations in the entorhinal cortex and hippocampus, as well as dopaminergic neurons in the nigrostriatal pathway (Fig. 1B–G). WEEV infection was associated with widespread astrogliosis (Fig. 1B,E) and microgliosis (Fig. 1C,F), with WEEV co-localizing with TH+ dopaminergic neurons (Fig. 1D,G). All microglia and astrocytes were devoid of WEEV-DsRed. In C57Bl/6 mice infected with WEEV, there was viral dissemination throughout multiple brain regions, similar to the pattern of viral spread throughout the neuroaxis that we recently reported in outbred CD-1 mice (Bantle et al., 2019). Using luciferase-expressing WEEV (Fig. 1H), passive immunotherapy was conducted to limit the extent of infection so that mice would survive beyond 3 DPI and the long-term effects of post-encephalitic inflammation could be observed (Bantle et al., 2019). In mice given passive immunotherapy using anti-E1 polyclonal rabbit immune serum at 12hr and 48hr post-infection, virus was completely cleared by 8 WPI (Fig. 1I–L). Mice that did not maintain a sufficient encephalitic infection were removed from the study (such as mouse No. 2, 8 WPI, Fig. 1L). The anti-E1 passive immunotherapy regimen was optimized to establish a consistent level of viral encephalitis within the previously identified range of acceptable luciferase-activity (Fig. 1M). More than 80% of mice receiving anti-E1 passive immunotherapy treatment at 12hr and 48hr survived following intranasal infection with WEEV-Luc (Fig. 1N), whereas mice intranasally infected without immunotherapy treatment died at 4 DPI. This treatment regimen was thereafter used for every animal throughout the study.
Figure 1. Intranasal inoculation with recombinant WEEV coupled with immunotherapy facilitates viral propagation and persistent infection throughout the CNS without incapacitating the mice.

(A) Schematic illustration of recombinant viral sequence expressing dsRed used and the associated treatment scheme. SPG - subgenomic promoter internal initiation site, UTR - untranslated region, DsRed – destabilized red fluorescent protein. Intranasal inoculation results in a persistent infection, with WEEV replicating in neurons and not in glia in the CNS. (B - D) Entire sagittal sections of mouse brain were imaged by digital montaging following intranasal inoculation with DsRed-expressing WEEV (1×104 PFU/ml) and co-immunostained for the astrocyte marker, glial fibrillary acidic protein GFAP (B), the microglia marker, ionized calcium binding adaptor molecule IBA1 (C) or the dopaminergic neuronal marker, tyrosine hydroxylase TH (D). High resolution insets depict cellular staining in the substantia nigra, hippocampus and olfactory bulb. Coronal sections (E - G) were imaged separately, with high resolution insets depicting region-specific cellular staining with dsRed and GFAP, IBA1 and TH (1-3). (H) Schematic illustration of recombinant viral sequence expressing firefly luciferase used and the associated treatment scheme. Pseudo-colored images of luciferase-activity following infection with WEEV-Luc were collected at Ohr (I), 12hr PI (J), 48hr PI (K) and 8WPI (L). (M) Following intranasal infection with WEEV-Luc, mice were treated with anti-E1 passive immunotherapy at 12hr and 48hr post-infection to maintain a consistent infection within established level of total lumen flux (red lines). (N) Survival curves for control mice (blue symbols), mice receiving I.N. WEEV + immunotherapy (green symbols) and mice receiving I.N. WEEV without immunotherapy (red symbols).
We next interrogated the neuroinflammatory and neurodegenerative consequences of encephalitic infection with WEEV over 8 weeks following intranasal inoculation (Fig. 2). In a recent study we reported that WEEV induces selective loss of dopaminergic neurons in the SNpc by 8 WPI with widespread gliosis and α-synuclein aggregation in outbred CD-1 mice (Bantle et al., 2019), but the temporal sequence of these events remained to be determined. To elucidate the order of these pathological events following infection with WEEV, we characterized neuropathological changes in the SNpc at 1, 2, 4 and 8WPI. Stereological assessment of dopaminergic neurons, microglia and peripheral monocyte infiltration in the SNpc indicated a significant loss of dopaminergic neurons at 1, 2, 4 and 8 WPI (Fig. 2A–I). Dual immunostaining for IBA1+ microglia indicated robust activation of this cell type proximal to degenerating neurons at all timepoints (Fig. 2B,D,F,H). These ameboid and activated microglial cells emerged by 1 WPI and persisted to 8 WPI, after the inflammatory stimulus had subsided and subsequent virus was completely cleared from the brain (Fig. 1L). In addition, infiltration of peripheral monocytes was noted within one week of infection with WEEV. Microglia were discriminated from infiltrating peripheral macrophages by a previously established protocol that differentiates based on size and morphological differences, with slides read blinded by a pathologist (Baquet et al., 2009; Sadasivan et al., 2012; Sadasivan et al., 2015; Sadasivan et al., 2017; Smeyne et al., 2016). These data demonstrate that microgliosis and macrophage infiltration followed the same time course, peaking at 1WPI and decreasing from 4 to 8WPI, when actively replicating virus was no longer detectable in the brain (Fig. 2J–M). To examine the temporal relationship between these pathological findings, stereological counts were conducted for each cell type over the time course of infection (Fig. 2N–P) and then normalized to create a representative overlay of these findings. (Fig. 2Q). These data demonstrated that the initiation of neuronal loss coincided with the peak of microglia activation and macrophage infiltration at 1 WPI and continued to progress up to 8 WPI.
Figure 2. Intranasal infection with WEEV causes dopaminergic neuronal loss, microgliosis and invastion of peripheral macrophages in the substantia nigra pars compacta.

(A-H) Representative IHC images of the substantia nigra pars compacta (SNpc) from mice infected or mock-infected with saline at 1,2,4, and 8WPI. (I) Stereological assessment of dopaminergic neurons in the SNpc. (J) Stereological assessment of microglia in the SN. (K-M) Quantification of resting and active microglia and infiltrating peripheral macrophages. (N-P) Linear dot-plot representation of cell counts following encephalitic infection with WEEV at 1,2,4, and 8WPI. (Q) Normalized cell count overlay of dopaminergic neurons (green), microgliosis (grey), and macrophage infiltration (blue) at 1, 2, and 8WPI. (*p<0.05, ** p<0.005, ***p<0.0005, n=6-8 per group)
Encephalitic WEEV infection is associated with PD-like neurochemical and neurobehavioral abnormalities.
Given the extensive inflammatory activation of microglia and loss of dopaminergic neurons, we assessed whether these pathological findings correlated with alterations in neurotransmitter content in the nigrostriatal pathway, as well as with locomotor dysfunction consistent with a parkinsonian behavioral phenotype (Fig. 3). By 8 WPI, we observed a significant decrease of dopamine (DA, Fig. 3A) in the SNpc, as well as its metabolites, 3,4-dihydroxyphenylacetic acid (DOPAC Fig. 3B), homovanillic acid (HVA, Fig. 3C) and 3-methoxytyramine (3-MT, Fig. 3D), as well as an increase in the ratio of DOPAC/DA (Fig. 3D). No change was detected in levels of serotonin (5-HT) in the SNpc (Fig. 3F). In the striatum (ST), there was not a significant loss in DA levels following infection with WEEV (Fig. 3G), although levels were trending downward at both 4 and 8 WPI. The DA metabolites, DOPAC and HVA, were decreased in the ST by 8 WPI (Fig. 3H,I) but there was no change the DOPAC:DA ratio or in levels of 5-HT (Fig. 3K,L).
Figure 3. Encephalitic infection with WEEV alters catecholamine homeostasis in the brain with associated neurobehavioral abnormalities in C57Bl/6 mice.
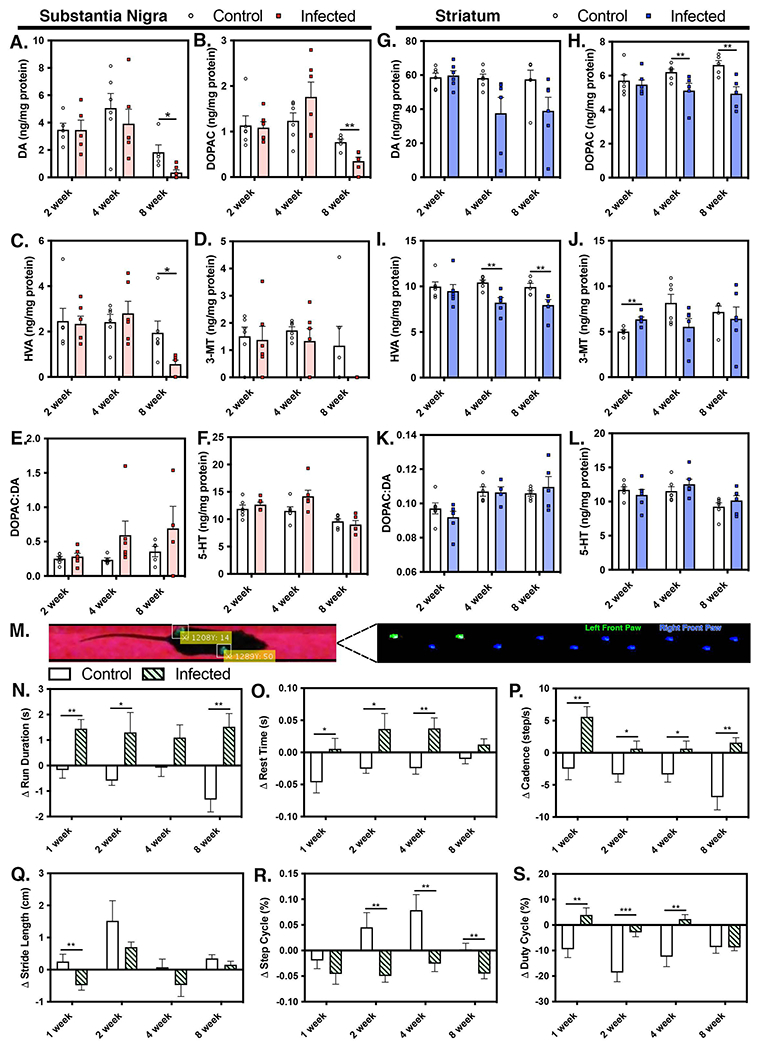
Levels of catecholamines and associated metabolites were measured in substantia nigra and striatum by high performance liquid chromatography (HPLC) analysis and electrochemical detection. (A, G) Dopamine (DA) levels in the substantia nigra (SN) and striatum (ST), respectively, along with DA metabolites 3,4-dihydroxyphenyl-acetic acid (DOPAC) (B, H), homovanillic acid (HVA) (C, I), and 3-methoxytyramine (3MT) (D, J) were measured at 2, 4 and 8WPI, as well as the DOPAC:DA ratio (E, K). Levels of serotonin (5-HT) (F, L) were also determined at 2, 4, and 8WPI in each brain region. (M) The gait and locomotor function of freely moving animals was analyzed at 1, 2, 4 and 8WPI. Parameters evaluated included run duration (N), rest time (O), cadence (P), stride length (Q), step cycle (R) and duty cycle (S) and were used to identify neurological changes associated with WEEV infection. (*p<0.05, ** p<0.005, ***p<0.0005, n=4-8 per group)
To determine how these neurochemical changes in the basal ganglia impacted locomotor function in WEEV-infected mice, we examined multiple parameters associated with gait and locomotion in freely moving animals on a fixed trackway using a video-based system under BSL-3 conditions. Pawprints were detected by total internal reflection and captured for analysis in software written in Matlab (R2021a, Mathworks, Natick, MA)(Fig. 3M). Persistent locomotor and gait abnormalities were observed following encephalitic infection with WEEV. Run Duration (Fig. 3N) increased throughout the course of infection and Rest Time correspondingly increased (Fig. 3O). There was an overall increase in Cadence (Fig. 3P) and an early decrease in Stride Length (Fig. 3Q), that trended down throughout the remainder of the observation period through 8 WPI. Step Cycle (Fig. 3R) and Duty Cycle (Fig. 3S) are inversely correlated and were negatively impacted throughout the course of infection compared to mock-infected control animals.
Encephalitic WEEV infection increases the number of neurotoxic A1 astrocytes expressing complement C3.
Astrogliosis is a well described pathological feature of PD and related synucleinopathies (Elizan and Casals, 1991; Silva da Costa et al., 2012). To characterize the inflammatory state of astrocytes following encephalitic infection with WEEV, we quantified the total number of astrocytes in the SNpc, as well as the number of astrocytes co-expressing complement C3, a marker for neurotoxic A1 astrocytes. Because Sl00β is ubiquitously expressed in astrocytes, we used Sl00β and C3 co-expression to delineate reactive (A1) and non-reactive (A2) astrocytes in the SNpc (Clarke et al., 2018; Liddelow and Barres, 2017; Liddelow et al., 2017). The number of S100β+ astrocytes in the SNpc increased by 1 WPI, compared to mock-infected controls, and stayed elevated throughout the course of infection (Fig. 4A–H,I). C3 expression in S100β+ astrocytes increased at 2 WPI, peaking at approximately 4 WPI (Fig. 4J). Multiple comparisons between treatment groups by one-way ANOVA (Fig. 4K) revealed significant differences in the number of Sl00β+ astrocytes expressing C3 between time points (p=0.0039), with C3 expression remaining elevated at 4 and 8 WPI above the levels observed at both 2 and 4 WPI.
Figure 4. Encephalitic infection with WEEV induces neuroinflammatory activation of astrocytes in the SNpc.

(A-H) Representative 10X montage immunofluorescence images of control and infectd mice at 1, 2, 4 and 8 WPI. Sections were co-immunostained with S100β (cyan), anti-C3 (red), and DAPI (blue) with 40X high magnification insets of the SNpc. (I) Quantification of S100β + astrocytes in the SNpc at 1,2,4, and 8 WPI. (J) Quantitative analysis of C3 intensity within astrocyte-specific regions of interest (ROIs) localized to the SNpc in control and infected animals at all time points. (K) Time-dependent determination of C3 levels in S100β + astrocytes spanning all time-points in infected animals. (*p<0.05, ** p<0.005, ***p<0.0005, n=6-8 per group)
WEEV induces α-synuclein protein aggregation in the cortex, hippocampus and midbrain of infected C57Bl/6 mice.
Development of α-synuclein protein aggregates is an important pathological feature of PD (Jellinger, 2008). We previously reported the formation of proteinase-K resistant α-synuclein aggregates in the entorhinal cortex, hippocampus and midbrain of outbred CD-1 mice following intranasal WEEV infection by 8 WPI (Bantle et al., 2019). To better characterize the temporal development of protein aggregates in brain tissue of C57Bl/6 mice, we examined levels of phosphorylated α-synuclein (phosphoserine 129/P129) at distinct timepoints following infection with WEEV using a pathological scoring system to quantify the severity of protein aggregation (Anderson et al., 2006; Rey et al., 2016) (Fig. 5). Immunohistochemical staining and neuropathological scoring of P129+ protein aggregates was conducted in the cortex, hippocampus and midbrain of infected and uninfected brain sections by a veterinary pathologist blinded to the treatment groups, and pathological scoring methodology was adapted from previous reports (Rey et al., 2016; Rey et al., 2018). A score of 1 to 5 was assigned to each brain region and scored as follows: 1 = no aggregates, 2 = very sparse and one to two P129+ cells, 3 = mild with less than ten P129+ cells, 4 = dense with more than ten P129+ cells, 5 = very dense with more than 20 P129+ cells involved. At 1 WPI there were no differences in the extent of P129 synuclein staining between control and infected groups (Fig. 5A–E). Strikingly, we saw P129+ α-synuclein protein aggregates as early as 2 WPI in WEEV-infected wildtype mice in the cortex, hippocampus and midbrain (Fig. 5F–J). P129+ synuclein aggregates reached a maximal extent between 2 - 4 WPI, with the largest aggregates observed in the entorhinal cortex at 4 WPI, followed by hippocampus and midbrain (Fig. 5F–O). The extent and severity of P129+ aggregates began to decrease by 8 WPI (Fig. 5P–T), although P129+ synuclein inclusions were still evident in neuronal soma in each brain region at 8 WPI.
Figure 5. WEEV induces rapid formation of α-synuclein protein plaques in the cortex, hippocampus and midbrain of surviving wild-type mice.

Representative images and pathological scoring of P129+ immunohistochemical staining from mice infected for 1WPI (A-E), 2WPI (F-J), 4WPI (K-O) and 8WPI (P-T) with representative high magnification 40X inset images. The average pathological score in the cortex, hippocampus and midbrain at each timepoint is shown is black (A,F,K,P) as well as in each image panel. (*p<0.05, ** p<0.005, ***p<0.0005, n=6-8 per group). Brain sections were immunolabeled for expression of phospho-Eif2 (U,V) and phospho-PERK (W,X) and imaged by fluorescence micrscopy.
To determine whether the observed increase in P129+ synuclein aggregates was associated with activation of the unfolded protein response (UPR) in dopaminergic neurons, we looked for the presence of the phosphorylated (active) forms of eukaryotic initiation factor 2α (eIF2α) and double-stranded RNA-activated protein kinase-like ER kinase (PERK) within TH+ neurons in the SNpc. As shown by the data in Fig. 5U, no phospho-eIF2α was detected in dopaminergic neurons in the SNpc in control animals at 4 WPI, the timepoint at which maximum deposition of P129+ aggregates was seen in infected mice. In contrast, animals infected with WEEV showed the presence of phospho-eIF2α in TH+ neurons in the SNpc, seen primarily as intracellular puncta present in perinuclear areas, as well as in some surrounding non-neuronal cells (Fig. 5V). Similarly, phospho-PERK was not detected in dopaminergic neurons in control mice but was strongly induced in WEEV-infected mice (Fig. 5W,X).
Opsonization of α-synuclein protein plaques with complement C3
Based on the observation that P129+ protein aggregates began to clear from the brains of infected mice by 8 WPI, we next investigated whether C3 produced by reactive astrocytes could bind to and opsonize P129+ α-synuclein protein aggregates for clearance in the cortex, hippocampus and midbrain following WEEV infection (Fig. 6). Using co-immunofluorescence labeling, we observed that P129+ α-synuclein protein aggregates stained positively with C3b in the cortex (Fig. 6A–C), hippocampus (Fig. 6D–F) and midbrain (Fig. 6G–I), with the greatest increase relative to baseline noted in the midbrain at 8 WPI. There were no P129+ α-synuclein protein aggregates detected in control mice at any timepoint and minimal background expression of C3b was observed. Quantitative analysis of C3b intensity within P129+ α-synuclein protein aggregates (Fig. 6J) indicated a difference in the extent of C3b opsonized P129+ inclusions between brain region (Two-way ANOVA, p<0.0001) and timepoint (p<0.0382), with the greatest magnitude increase seen in the substantia nigra within the basal midbrain. Whether C3b binding to α-synuclein protein plaques is a protective mechanism to facilitate clearance or a pathological consequence of the A1 astrocyte phenotype remains to be determined.
Figure 6. Encephalitic infection with WEEV induces opsonization of α-synuclein protein aggregates in the cortex, hippocampus and midbrain with astrocyte-derived complement C3 protein.

Immunofluorescence images of complement C3b (red) co-localization with P129+ protein aggregates (green) were analyzed in the cortex (A-C), hippocampus (D-F) and midbrain (G-I) at various times following infection with WEEV. Nuclei were counterstained with DAPI (blue). High magnification 40X inset images are depicted from each region. Representative images panels are presented from 2 WPI, the time of maximal P129 aggregation. (J) Quantification of C3b deposition co-localizing with protein aggregates within each brain region was determined at 2, 4 and 8WPI by analyzing the amount of C3b co-localizing with regions of interest detecting P129+ aggregates. (*p<0.05, **p<0.005, ***p<0.0005, n=6-8 per group)
Genetic knockout of NFκB in astrocytes reduces virus-induced α-synuclein aggregation and decreases neuronal loss and gliosis.
Previous reports have shown that reactive A1 astrocytes are the source of C3 in the brain and that NFκB-dependent expression of C3 in astrocytes regulates amyloid-beta expression, microgliosis and neuronal loss in models of Alzheimer’s disease (Lian et al., 2016; Loeffler et al., 2006; Shi et al., 2017; Vasek et al., 2016). We therefore determined the effects of selective genetic ablation of NFκB in astrocytes on glial activation, neuronal loss and P129+ α-synuclein immunoreactivity in hGfap-cre+/−/Ikk2F/F (NFκB KO) transgenic mice developed in our laboratory (Kirkley et al., 2019) following infection with WEEV at 8 WPI (Fig. 7). In WT Ikk2F/F mice, infection with WEEV induced the same extensive P129+ α-synuclein protein aggregates by 8 WPI in the cortex, hippocampus and midbrain that were observed in the C57Bl/6 background strain (Fig. 7B–D). There was a significant reduction of P129+ α-synuclein plaque load following WEEV infection in hGfap-cre+/−/Ikk2F/F KO mice in the cortex, hippocampus and midbrain compared to WT Ikk2F/F controls (Fig. 7E–J). Histopathological examination of KO mice infected with WEEV revealed marginally detectable levels of P129+ α-synuclein protein aggregates by 8 WPI in the entorhinal cortex and hippocampus (Fig. 7H,I), with nearly undetectable staining for P129+ aggregates in the midbrain (Fig. 7J). Pathological scoring of P129+ α-synuclein protein aggregates revealed differences between genotype and treatment across brain regions, with the greatest decrease the extent of P129+ staining occurring in the midbrain of infected KO mice, relative to infected WT mice (Fig. 7K).
Figure 7. Genetic knockout of NFκB in astrocytes reduces gliosis and α-synuclein aggregation throughout the brain.

(A) Schematic illustration of the Cre-recombinase under control of the human glial fibrillary acidic protein promoter (hGFAP). Hemizygous GFAP-Cre mice were crossed with I kappa B kinase 2 (Ikk2)-loxP mice, which facilitated selective deletion of IKK2 in astrocytes and provided a cell-specific knockout of NF-κB in astrocytes. hGFAP-cre+/−/IKK2fl/fl (KO) or hGFAP-cre−/−/IKK2fl/fl (WT) animals were intranasally infected with WEEV or mock-infected (control) with saline and treated with anti-E1immunotherapy. (B-D) IHC images with high magnification 40X inset images of P129+ staining from infected hGFAP-cre−/−/IKK2fl/fl WT mice, (E-G) uninfected control hGFAP-cre+/−/IKK2fl/fl KO mice and (H-J) infected hGFAP-cre+/−/IKK2fl/fl KO mice. (K) Pathological scoring of multiple brain regions was performed at 8WPI to quantify the extent of P129+ aggregation in each treatment group. (*p<0.05, **p<0.005, n=6-8 per group).
Concomitant to the decrease in P129+ α-synuclein protein aggregates observed in KO mice infected with WEEV, hGfap-cre+/−/Ikk2F/F KO mice showed no loss of dopaminergic neurons in the substantia nigra (Fig. S1 A). There was also dramatically reduced microgliosis in the cortex and hippocampus of KO mice infected with WEEV (Fig. S1 B–C), as well and an overall reduction in the number of activated astroglia in the midbrain of infected KO mice when compared to infected WT mice (Fig. S1 E–G). Additionally, infected hGfap-cre+/−/Ikk2F/F KO mice appeared to have drastically reduced motor deficits and an improved clinical symptoms when compared to infected hGfap-cre+/−/Ikk2F/F WT mice. Considering that virus is cleared from the CNS at 8 WPI, these data provide additional support for the importance of cell-cell signaling between microglia and astrocytes in modulating the chronic neuroinflammatory responses to encephalitic infection with WEEV.
To further assess how microgliosis, macrophage infiltration, dopaminergic neuronal loss, astrogliosis and α-synuclein protein aggregates were temporally related in the SNpc following encephalitic infection with WEEV, we generated normalized overlay graphs for these pathological findings by modeling the kinetics of each histopathological response (Fig. 8). From these overlays, we were able to determine that microgliosis and macrophage infiltration (Fig. 8A) preceded the maximal extent of astrocyte activation (Fig. 8B), which in turn preceded maximum α-synuclein protein aggregation in the SNpc (Fig. 8C). Loss of dopamine neurons (Fig. 8D), initiated during the initial stages of viral infection, progressed subsequent to neuroinflammatory activation of glial cells and formation of α-synuclein protein aggregates, even after clearance of virus. This can be seen from the composite overlay image in Fig. 8E, which clearly demonstrates the kinetics of glial activation and α-synuclein protein aggregation precedes the maximum extent of dopamine neuron loss. From this, we were able to construct timeline of cellular responses to WEEV (Fig. 8F), depicting how infection leads to rapid activation of resident microglia and recruitment of monocytes from blood, followed by NFκB-dependent activation of astrocytes and α-synuclein aggregation that ultimately promote the loss of dopaminergic neurons in the SNpc.
Figure 8. Microgliosis and peripheral macrophage infiltration precede astrogliosis and formation of phospho-Ser(129)-α-synuclein protein aggregates following intranasal infection with WEEV.

The kinetics of cellular responses to WEEV infection in the substantia nigra were modeled by curve fitting normalized data sets to generate representative plots of (A) monocyte infiltration and microglial activation, (B) astrocyte activation, (C) accumulation of P129+ protein aggregates and (D) dopaminergic neurodegeneration. (E) Combined overlays of each response during the 8-week course of infection. (F) Schematic representation of time-dependent cellular responses to WEEV infection indicated that glial activation and α-synuclein aggregation precede degeneration of dopaminergic neurons. (n=6-8 per group)
Discussion
Environmental agents, including viruses, can produce parkinsonian neurological symptoms in people (Hayase and Tobita, 1997; Jang et al., 2009a; Jang et al., 2009b; McCall et al., 2001). Viral infections are also implicated in the onset and progression of multiple neurodegenerative diseases (Bantle et al., 2019; Mattson, 2004; Sadasivan et al., 2017). Indeed, a recent multiscale network analysis of the late-onset Alzheimer’s-associated virome in postmortem tissues indicated a strong correlation between histopathological and molecular indices of disease and the presence of herpes virus (Readhead et al., 2018). An association between viral encephalitis and parkinsonism has been noted for nearly a century (Lewy, 1932) but the underlying mechanisms have surprisingly remained obscure. Multiple reports posit that pathogenic microbes could be etiological factors in neurodegenerative disease and increasing evidence suggests that proteins involved in neurodegeneration, such as α-synuclein, ß-amyloid, Tau and the prion protein PrPc act as antimicrobial peptides that modulate the neuroinflammatory response to reduce the microbial load in the brain (Beatman et al., 2015; Chida et al., 2018; Eimer et al., 2018). In the present study, we found that encephalitic WEEV infection in C57Bl/6 mice by intranasal inoculation resulted in rapid neuronal tract-based dissemination from the olfactory bulb to the cortex, hippocampus and basal ganglia by four days post-infection, whereas other regions of the nigrostriatal pathway, such as the striatum, were largely free of virus (Fig. 1). Why only select neurons are initially infected is unclear but a similar pattern has been observed by stereotactic administration of α-synuclein preformed fibrils into the olfactory bulb of mice (Rey et al., 2016). Likewise, striatal injection of preformed synuclein fibrils shows similar regional selectivity, with little impact on neuronal physiology in other brain regions like the hippocampus, despite widespread protein aggregation (Nouraei et al., 2018). WEEV is a non-promoter driven RNA virus and shows a regional pattern of infectivity likely based on expression of cell surface receptors as well as the intracellular environment in neurons in the basal midbrain. The highly oxidative metabolic state of neurons in this brain region may also favor viral replication (Pacelli et al., 2015), potentially through enhanced activation of NFκB (Yeh et al., 2019). However, it has also been suggested that specific neuroanatomical areas of the brain exhibit an innate ability to restrict viral infection, which could influence the selective pattern of propagation of WEEV between the cortex, hippocampus and basal midbrain (Gullberg et al., 2015; Kostuk et al., 2019; Phillips et al., 2016).
Viruses potently stimulate a neuroinflammatory phenotype in glia through activation of innate immune signaling emanating from pattern recognition receptors, including Toll-like receptors, which likely initiates a lingering inflammatory state in infected brain regions even after WEEV is completely cleared from the brain (Bantle et al., 2019). The persistence of glial activation 8 WPI after the virus was cleared from brain (Fig. 2) suggests that neuroinflammatory activation of glia could represent a form of innate immunological memory in the brain to prevent recurrent microbial infection. Previous data support this assertion, in which altered patterns of histone acetylation in astrocytes and microglia correlated with the severity of the glial inflammatory response in the SNpc after infection with H1N1 (Bantle et al., 2021). Following inoculation with WEEV, we observed progressive loss of dopaminergic neurons, altered dopamine metabolism and gait abnormalities consistent with a parkinsonian phenotype (Fig. 2, Fig. 3). Interestingly, intranasal infection with WEEV results in more severe viral replication in the substantia nigra than the striatum (Phillips et al., 2016), which may explain the more severe alteration in dopamine and serotonin metabolism in this brain region relative to the striatum (Fig. 2A–L). The overall loss of neurons in the SNpc, as well as the persistent neurobehavioral deficits observed at 8 WPI, support a progressive neurodegenerative lesion in the basal midbrain, rather than merely decreased expression of tyrosine hydroxylase without overt neuronal loss (Alam et al., 2017). Notably, activation of microglia preceded both aggregation of α-synuclein and maximal loss of dopamine neurons, with the peak of microgliosis coinciding with the maximum extent of macrophage infiltration (Fig. 2Q). These data strongly suggest that neuroinflammatory activation of microglia was the initiating event in promoting protein aggregation of α-synuclein following infection with WEEV.
Phenotypic staining for reactive A1 astrocytes supports this conclusion. We characterized the inflammatory response of astrocytes in the brain following infection with WEEV by colocalizing the expression of C3 in S100β+ astrocytes to identify neurotoxic A1 astrocytes (Fig. 4). Expression of complement C3 in astrocytes began at 2 WPI and persisted through 8 WPI, with the highest levels of expression in the midbrain. The number of reactive astrocytes peaked in the midbrain within 2 WPI, soon after the peak of microgliosis, and remained elevated through 8 WPI (Fig. 4). Encephalitic viral infection can cause a phenotypic switch in astrocytes and induce a long-lasting neurotoxic and proinflammatory phenotype, termed A1 (astrogliosis), in contrast to a neurotrophic A2 phenotype (Liddelow et al., 2017). Neurotoxic A1 astrocytes may help to combat recurrent encephalitic infections but can also magnify neuronal injury (Hirsch and Hunot, 2009). Notably, A1 astrocytes uniquely express a number of neurotoxic NFκB-regulated proteins, including complement C3 (Lian et al., 2015; Lian et al., 2016). Complement C3 has been shown to produce neuronal ER-stress and intracellular neuronal calcium perturbation, as well as opsonization of P129+ α-synuclein protein aggregates in the substantia nigra (Loeffler et al., 2006). However, the total number of S100β+ astrocytes increased prior to the maximum expression of C3, suggesting a phase of initial activation followed by a phenotypic switch to a reactive C3-expressing A1 astrocyte.
We previously reported that WEEV causes selective loss of dopaminergic neurons despite widespread dissemination of the virus (Bantle et al., 2019). This was also observed in the current study, where extensive intra- and extracellular α-synuclein aggregates were present within 2 – 4 WPI in the cortex and hippocampus, with primarily intracellular α-synuclein aggregates in the substantia nigra (Fig. 5). The size and extent of these aggregates correlated directly with the degree of viral replication at four days post-infection (Fig. 1B,E) within each neuroanatomical region. To determine whether infection with WEEV directly activated the unfolded protein response (UPR) in dopaminergic neurons, we also measured levels of phosphorylated Eif2α and PERK in TH+ neurons in the SNpc, both critical mediators of the ER stress response to unfolded proteins associated with neurodegeneration. PERK is constitutively bound to GRP78 in an inactive state until sensing misfolded proteins, upon which it dissociates and phosphorylates the translational inhibitor, Eif2α, in order to reduce the cellular burden of misfolded proteins (Bond et al., 2020). Levels of phosphorylated PERK and Eif2α were increased in TH+ neurons in the SNpc at 4 WPI (Fig. 5U–X), suggesting that WEEV-induced cellular stress directly induces the UPR in these neurons. Indeed, it was reported that infection with closely related Japanese encephalitis virus caused activation of PERK and subsequent ATF4/CHOP-dependent apoptosis in neurons (Wang et al., 2019), supporting that WEEV-induced activation of the UPR in dopaminergic neurons is an important pathological mechanism in this brain region.
The mechanisms driving selective loss of dopaminergic neurons in the midbrain are not entirely known but the lack of neuronal degeneration in the cortex and hippocampus, despite extensive aggregation of α-synuclein (Fig. 5), raises the possibility that α-synuclein could be neuroprotective or anti-viral in these brain regions. Support for this hypothesis comes from related studies demonstrating that α-synuclein KO mice had more severe encephalitic infection and increased neuroinflammation from West Nile Virus, suggesting that α-synuclein may have a neuroprotective effect (Beatman et al., 2015; Clarke et al., 2014; Lesteberg and Beckham, 2019). Other studies demonstrate that α-synuclein expression increases following infection with RNA viruses, which can restrict viral replication by directly binding to virus or by inducing an inflammatory response in glia (Beatman et al., 2015; Clarke et al., 2014; Lesteberg and Beckham, 2019). Interestingly, α-synuclein expression in enteric neurons increases with viral infection and stimulates innate immune activation of dendritic cells (Stolzenberg et al., 2017). This parallels our findings in mice infected with WEEV and lends further support for the immune function of α-synuclein in the brain. Selective vulnerability of neurons in the SNpc could also be influenced by expression of the D2 dopamine receptor (D2R). Neurons expressing D2R are more vulnerable to infection with alphaviruses such as West Nile and Japanese equine encephalitis (Malik et al., 2014; Simanjuntak et al., 2017), thought to involve D2R-dependent activation of phospholipase C (PLC) and excessive release of intracellular Ca2+ that activates ER stress. This D2R/PLC mechanism appears to be important for pathogenicity in brain of other RNA viruses, such as HIV (Gaskill et al., 2014) and H1N1 influenza (Zhu et al., 2014). Thus, replication of WEEV in the basal midbrain could enhance ER stress and thereby promote aggregation of α-synuclein.
It was noted that P129+ α-synuclein aggregates in the cortex, hippocampus and midbrain were coated with C3 (Fig. 6), suggesting that NFκB-dependent overproduction of C3 by astrocytes promotes targeting of P129+ aggregates for microglial phagocytosis (Bodea et al., 2014; Linnartz and Neumann, 2013; Schafer et al., 2012). The highest levels of C3-expressing astrocytes were noted in the substantia nigra, which could be a selective response to the vulnerability of this brain region to infection with alphaviruses (Malik et al., 2014; Simanjuntak et al., 2017). Moreoever, C3 exacerbates ER stress and contributes to inflammatory activation of macrophage lineage cells (Chaumonnot et al., 2021), which could also contribute to neuronal injury in this brain region. To directly test the role of astrocyte-derived C3 in neuronal injury following WEEV infection, we used astrocyte-specific NFκB KO mice generated in our laboratory, in which the human Gfap promoter is used to drive expression of Cre recombinase in astrocytes (hGfap-cre+/−/Ikk2F/F) to inhibit inflammatory signaling through IKK2/NFκB (Kirkley et al., 2019). These mice showed dramatically decreased aggregation of α-synuclein in all brain regions examined, with similarly reduced levels of microgliosis and dopaminergic neuronal loss (Fig. 7). The precise mechanism by which NFκB KO in astrocytes protects dopaminergic neurons from the neuroinflammatory effects of WEEV is not yet entirely clear but likely favors a neurotrophic A2 phenotype while suppressing an A1 phenotype, thereby mitigating the damaging effects of inflammatory cytokine and chemokine release and facilitating trophic support to surrounding neurons (Kostuk et al., 2019; Liddelow et al., 2017). This strongly suggests that glial inflammatory processes modulate the neurotoxicity of WEEV and subsequently regulate aggregation of α-synuclein. This also indicates that astrocyte-derived signaling molecules are critical to the development of microgliosis following WEEV infection, as we previously reported for the astrocyte-derived chemokine, CCL2, in a model of environmentally induced neuroinflammation (Popichak et al., 2018) and as suggested in studies of astrocyte-microglia interactions in other inflammatory models of neurodegeneration (Lian et al., 2016; Liddelow and Barres, 2017).
Collectively, these data indicate that following infection with WEEV, the majority of α-synuclein aggregation occurs subsequent to activation of microglia and astrocytes. This contrasts studies reporting that α-synuclein activates microglia and astroglia prior to loss of dopaminergic neurons (Bruck et al., 2016). It is likely that both mechanisms occur simultaneously during viral infection with WEEV, where inflammation from activated glial cells promotes aggregation of α-synuclein, thereby further stimulating innate immune inflammatory signaling in glial cells. To examine this, we modeled temporal changes in the various cellular responses to WEEV over the course of infection by normalizing each response to its respective minimum and maximum within the substantia nigra (Fig. 8A–D). This enabled us to construct a single overlay plot and timeline for each parameter (Fig. 8E,F). These data clearly demonstrate that microglia and invading macrophages are the first to activate following infection with WEEV, followed by astrocytes and then by the peak of α-synuclein aggregation. Maximal loss of dopaminergic neurons occurs subsequent to these events and was prevented by NFκB KO in astrocytes. As such, these data suggest that neuroinflammatory activation of glia play a causal role in the onset and progression of virally induced parkinsonism. Moreover, considering that the peak of microgliosis preceded α-synuclein aggregate formation in wildtype mice and that NFκB KO in astrocytes mitigated both microgliosis and α-synuclein aggregation, it is possible that inflammatory activation of microglia represents a critical threshold for initiating protein aggregation and neurodegeneration. This is consistent with recent studies reporting that Lewy Body-like pathology in the pre-formed fibril model of Parkinson’s disease is associated with inflammation and immune cell infiltration prior to degeneration of dopaminergic neurons (Earls et al., 2019).
These data support the conclusion that reactive neurotoxic astrocytes increase the selective vulnerability of dopaminergic neurons in the substantia nigra to injury from viral infection, as suggested in models of Parkinson’s disease examining regional differences in glial-mediated neuronal injury (Kostuk et al., 2019). The model of WEEV-induced neurodegeneration has the potential to elucidate molecular mechanisms relevant to neuroinflammatory injury in Parkinson’s and related diseases, particularly with respect to the role of glial activation as an initiating factor in neuronal injury.
Supplementary Material
Supplemental Figure 1. Genetic knockout of NF-κB in astrocytes reduces gliosis across multiple brain regions and prevents dopaminergic neuronal loss in the SNpc. hGFAP-cre+/−/IKK2fl/fl (KO) or hGFAP-cre−/−/IKK2fl/fl (WT) animals were intranasally infected with WEEV or mock-infected with saline (KO control) and aged to 8 WPI. (A) Dopaminergic neurons were quantified in the SNpc and microgliosis and astrogliosis were quantified in the cortex (B, E), hippocampus (C, F), and midbrain (D, G). (*p<0.05, ** p<0.005, ***p<0.0005, n=6-8 per group)
Highlights.
Infection with Western equine encephalitis virus causes dopaminergic neurodegeneration and α-synuclein aggregation in C57Bl/6 mice associated with activation of the unfolded protein response in neurons.
Activation of microglia and astrocytes occurs prior to α-synuclein aggregation and loss of dopamine neurons.
Phenotypic activation of astrocytes to A1 phenotype results in production of complement C3 that opsonizes α-synuclein aggregates.
Cell-specific knockout of NFκB in astrocytes prevents neuroinflammatory activation of microglia and protects against both α-synuclein aggregation and loss of dopamine neurons.
Acknowledgements
We thank the veterinary staff of Laboratory Animal Resources at Colorado State University for outstanding care of the animals used in these studies. In addition, the authors wish to recognize the invaluable assistance and advice of Susi Bennett of the Regional Biocontainment Laboratory at Colorado State University for help with WEEV constructs.
Funding information
This work was supported by NIH grants R01ES021656 (RBT, RJS) and R21ES024183 (RBT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interest
The authors declare no competing interest
Competing interests
The authors declare that there exist no competing interests or conflicts, disclosed or otherwise. All research and data will be made freely accessible, per the guidelines of the National Institutes of Health.
Collin M. Bantle
Savannah M. Rocha
C. Tenley French
Aaron T. Phillips
Kevin Tran
Kenneth E. Olson
Todd A. Bass
Tawfik Aboellail
Richard J. Smeyne
Ronald B. Tjalkens
References
- Alam G, et al. 2017. Single low doses of MPTP decrease tyrosine hydroxylase expression in the absence of overt neuron loss. Neurotoxicology. 60, 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JP, et al. 2006. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 281, 29739–52. [DOI] [PubMed] [Google Scholar]
- Bantle CM, et al. 2019. Infection with mosquito-borne alphavirus induces selective loss of dopaminergic neurons, neuroinflammation and widespread protein aggregation. NPJ Parkinsons Dis. 5, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantle CM, et al. 2021. Manganese exposure in juvenile C57BL/6 mice increases glial inflammatory responses in the substantia nigra following infection with H1N1 influenza virus. PLoS One. 16, e0245171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquet ZC, et al. 2009. A comparison of model-based (2D) and design-based (3D) stereological methods for estimating cell number in the substantia nigra pars compacta (SNpc) of the C57BL/6J mouse. Neuroscience. 161, 1082–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcia C, 2013. Glial-mediated inflammation underlying parkinsonism. Scientifica (Cairo). 2013, 357805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatman EL, et al. 2015. Alpha-Synuclein Expression Restricts RNA Viral Infections in the Brain. J Virol. 90, 2767–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodea LG, et al. 2014. Neurodegeneration by activation of the microglial complement-phagosome pathway. J Neurosci. 34, 8546–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond S, et al. 2020. The Integrated Stress Response and Phosphorylated Eukaryotic Initiation Factor 2alpha in Neurodegeneration. J Neuropathol Exp Neurol. 79, 123–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruck D, et al. 2016. Glia and alpha-synuclein in neurodegeneration: A complex interaction. Neurobiol Dis. 85, 262–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caggiu E, et al. 2019. Inflammation, Infectious Triggers, and Parkinson’s Disease. Front Neurol. 10, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaumonnot K, et al. 2021. The HSP GRP94 interacts with macrophage intracellular complement C3 and impacts M2 profile during ER stress. Cell Death Dis. 12, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chida J, et al. 2018. Prion protein protects mice from lethal infection with influenza A viruses. PLoS Pathog. 14, e1007049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LE, et al. 2018. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci U S A. 115, E1896–E1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke P, et al. 2014. Death receptor-mediated apoptotic signaling is activated in the brain following infection with West Nile virus in the absence of a peripheral immune response. J Virol. 88, 1080–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Miranda BR, et al. 2015. Novel para-phenyl substituted diindolylmethanes protect against MPTP neurotoxicity and suppress glial activation in a mouse model of Parkinson’s disease. Toxicol Sci. 143, 360–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dourmashkin RR, 1997. What caused the 1918-30 epidemic of encephalitis lethargica? J R Soc Med. 90, 515–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy MF, et al. 2018. Lewy body-like alpha-synuclein inclusions trigger reactive microgliosis prior to nigral degeneration. J Neuroinflammation. 15, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earls RH, et al. 2019. Intrastriatal injection of preformed alpha-synuclein fibrils alters central and peripheral immune cell profiles in non-transgenic mice. J Neuroinflammation. 16, 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eimer WA, et al. 2018. Alzheimer’s Disease-Associated beta-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron. 99, 56–63 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elizan TS, et al. 1978. Antibodies against arboviruses in postencephalitic and idiopathic Parkinson’s disease. Arch Neurol. 35, 257–60. [DOI] [PubMed] [Google Scholar]
- Elizan TS, Casals J, 1991. Astrogliosis in von Economo’s and postencephalitic Parkinson’s diseases supports probable viral etiology. J Neurol Sci. 105, 131–4. [DOI] [PubMed] [Google Scholar]
- Gaskill PJ, et al. 2014. Dopamine receptor activation increases HIV entry into primary human macrophages. PLoS One. 9, e108232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard A, et al. 2006. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis. 21, 404–12. [DOI] [PubMed] [Google Scholar]
- Gopal Krishna Patro S, Kumar Sahu K, 2015. Noramlization: A Preporcessing Stage [Google Scholar]
- Gouveia K, Hurst JL, 2013. Reducing mouse anxiety during handling: effect of experience with handling tunnels. PLoS One. 8, e66401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeber MB, Streit WJ, 2010. Microglia: biology and pathology. Acta Neuropathol. 119, 89–105. [DOI] [PubMed] [Google Scholar]
- Gullberg RC, et al. 2015. Oxidative stress influences positive strand RNA virus genome synthesis and capping. Virology. 475, 219–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond SL, et al. 2017. Cellular selectivity of AAV serotypes for gene delivery in neurons and astrocytes by neonatal intracerebroventricular injection. PLoS One. 12, e0188830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond SL, et al. 2018. The Nurr1 Ligand,1,1-bis(3′-Indolyl)-1-(p-Chlorophenyl)Methane, Modulates Glial Reactivity and Is Neuroprotective in MPTP-Induced Parkinsonism. J Pharmacol Exp Ther. 365, 636–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayase Y, Tobita K, 1997. Influenza virus and neurological diseases. Psychiatry Clin Neurosci. 51, 181–4. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Hunot S, 2009. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 8, 382–97. [DOI] [PubMed] [Google Scholar]
- Jang H, et al. 2009a. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci U S A. 106, 14063–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang H, et al. 2009b. Viral parkinsonism. Biochim Biophys Acta. 7, 714–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA, 2008. Neuropathological aspects of Alzheimer disease, Parkinson disease and frontotemporal dementia. Neurodegener Dis. 5, 118–21. [DOI] [PubMed] [Google Scholar]
- Kirkley KS, et al. 2019. Genetic suppression of IKK2/NF-kappaB in astrocytes inhibits neuroinflammation and reduces neuronal loss in the MPTP-Probenecid model of Parkinson’s disease. Neurobiol Dis. 127, 193–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostuk EW, Cai J, Iacovitti L, 2019. Subregional differences in astrocytes underlie selective neurodegeneration or protection in Parkinson’s disease models in culture. Glia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesteberg KE, Beckham JD, 2019. Immunology of West Nile Virus Infection and the Role of Alpha-Synuclein as a Viral Restriction Factor. Viral Immunol. 32, 38–47. [DOI] [PubMed] [Google Scholar]
- Lewy FH, 1932. Die Entstehung der Einschlußkörper und ihre Bedeutung für die systematische Einordnung der sogenannten Viruskrankheiten. Deutsche Zeitschrift für Nervenheilkunde. 124, 93–100. [Google Scholar]
- Lian H, et al. 2015. NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron. 85, 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian H, et al. 2016. Astrocyte-Microglia Cross Talk through Complement Activation Modulates Amyloid Pathology in Mouse Models of Alzheimer’s Disease. J Neurosci. 36, 577–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Barres BA, 2017. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity. 46, 957–967. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, et al. 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindqvist D, et al. 2013. Cerebrospinal fluid inflammatory markers in Parkinson’s disease--associations with depression, fatigue, and cognitive impairment. Brain Behav Immun. 33, 183–9. [DOI] [PubMed] [Google Scholar]
- Linnartz B, Neumann H, 2013. Microglial activatory (immunoreceptor tyrosine-based activation motif)- and inhibitory (immunoreceptor tyrosine-based inhibition motif)-signaling receptors for recognition of the neuronal glycocalyx. Glia. 61, 37–46. [DOI] [PubMed] [Google Scholar]
- Liu C, et al. 1970. A comparative study of the pathogenesis of western equine and eastern equine encephalomyelitis viral infections in mice by intracerebral and subcutaneous inoculations. J Infect Dis. 122, 53–63. [DOI] [PubMed] [Google Scholar]
- Loeffler DA, Camp DM, Conant SB, 2006. Complement activation in the Parkinson’s disease substantia nigra: an immunocytochemical study. J Neuroinflammation. 3, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue CH, et al. 2009. Virulence variation among isolates of western equine encephalitis virus in an outbred mouse model. J Gen Virol. 90, 1848–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik R, et al. 2014. Dengue encephalopathy - still an enigma? J Infect Dev Ctries. 8, 1076–8. [DOI] [PubMed] [Google Scholar]
- Martinez EM, et al. 2017. Editor’s Highlight: Nlrp3 Is Required for Inflammatory Changes and Nigral Cell Loss Resulting From Chronic Intragastric Rotenone Exposure in Mice. Toxicol Sci. 159, 64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, 2004. Infectious agents and age-related neurodegenerative disorders. Ageing Res Rev. 3, 105–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall S, et al. 2001. Influenza RNA not detected in archival brain tissues from acute encephalitis lethargica cases or in postencephalitic Parkinson cases. J Neuropathol Exp Neurol. 60, 696–704. [DOI] [PubMed] [Google Scholar]
- Miller JA, et al. 2011. 1,3-Dinitrobenzene-induced metabolic impairment through selective inactivation of the pyruvate dehydrogenase complex. Toxicol Sci. 122, 502–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogi M, et al. 1996. Interleukin (IL)-1 beta, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci Lett. 211, 13–6. [DOI] [PubMed] [Google Scholar]
- Mulder DW, Parrott M, Thaler M, 1951. Sequelae of western equine encephalitis. Neurology. 1, 318–27. [DOI] [PubMed] [Google Scholar]
- Nouraei N, et al. 2018. Critical appraisal of pathology transmission in the alpha-synuclein fibril model of Lewy body disorders. Exp Neurol. 299, 172–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacelli C, et al. 2015. Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr Biol. 25, 2349–60. [DOI] [PubMed] [Google Scholar]
- Palmer RJ, Finley KH, 1956. Sequelae of encephalitis; report of a study after the California epidemic. Calif Med. 84, 98–100. [PMC free article] [PubMed] [Google Scholar]
- Phillips AT, et al. 2013. Bioluminescent imaging and histopathologic characterization of WEEV neuroinvasion in outbred CD-1 mice. PLoS One. 8, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips AT, et al. 2016. Entry Sites of Venezuelan and Western Equine Encephalitis Viruses in the Mouse Central Nervous System following Peripheral Infection. J Virol. 90, 5785–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popichak KA, et al. 2018. Glial-neuronal signaling mechanisms underlying the neuroinflammatory effects of manganese. J Neuroinflammation. 15, 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Readhead B, et al. 2018. Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron. 99, 64–82 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid AH, et al. 2001. Experimenting on the past: the enigma of von Economo’s encephalitis lethargica. J Neuropathol Exp Neurol. 60, 663–70. [DOI] [PubMed] [Google Scholar]
- Rey NL, et al. 2016. Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J Exp Med. 213, 1759–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey NL, et al. 2018. Spread of aggregates after olfactory bulb injection of alpha-synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropathol. 135, 65–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadasivan S, et al. 2012. Methylphenidate exposure induces dopamine neuron loss and activation of microglia in the basal ganglia of mice. PLoS One. 7, e33693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadasivan S, et al. 2015. Induction of microglia activation after infection with the non-neurotropic A/CA/04/2009 H1N1 influenza virus. PLoS One. 10, e0124047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadasivan S, et al. 2017. Synergistic effects of influenza and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) can be eliminated by the use of influenza therapeutics: experimental evidence for the multi-hit hypothesis. NPJ Parkinsons Dis. 3, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, et al. 2012. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 74, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz DR, Barthal JS, Garrett C, 1977. WESTERN EQUINE ENCEPHALITIS WITH RAPID ONSET OF PARKINSONISM. Neurology. 27, 1095–1096. [DOI] [PubMed] [Google Scholar]
- Shi Q, et al. 2017. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci Transl Med. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva da Costa L, et al. 2012. Mitochondrial bioenergetic alterations in mouse neuroblastoma cells infected with Sindbis virus: implications to viral replication and neuronal death. PLoS One. 7, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simanjuntak Y, et al. 2017. Japanese Encephalitis Virus Exploits Dopamine D2 Receptor-phospholipase C to Target Dopaminergic Human Neuronal Cells. Front Microbiol. 8, 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeyne RJ, et al. 2016. Assessment of the Effects of MPTP and Paraquat on Dopaminergic Neurons and Microglia in the Substantia Nigra Pars Compacta of C57BL/6 Mice. PLoS One. 11, e0164094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolzenberg E, et al. 2017. A Role for Neuronal Alpha-Synuclein in Gastrointestinal Immunity. J Innate Immun. 9, 456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart SA, Robinson ES, 2015. Reducing the stress of drug administration: implications for the 3Rs. Sci Rep. 5, 14288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey MG, 2010. Inflammation in neuropsychiatric disease. Neurobiol Dis. 37, 491–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapias V, Greenamyre JT, Watkins SC, 2013. Automated imaging system for fast quantitation of neurons, cell morphology and neurite morphometry in vivo and in vitro. Neurobiol Dis. 54, 158–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasek MJ, et al. 2016. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature. 534, 538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, et al. 2019. Japanese Encephalitis Virus Induces Apoptosis and Encephalitis by Activating the PERK Pathway. J Virol. 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh JX, et al. 2019. NF-kappaB Activation Promotes Alphavirus Replication in Mature Neurons. J Virol. 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Hoppe AD, Swanson JA, 2010. Coordination of Fc receptor signaling regulates cellular commitment to phagocytosis. Proc Natl Acad Sci U S A. 107, 19332–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, et al. 2014. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 34, 11929–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Ly H, Liang Y, 2014. PLC-gamma1 signaling plays a subtype-specific role in postbinding cell entry of influenza A virus. J Virol. 88, 417–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Genetic knockout of NF-κB in astrocytes reduces gliosis across multiple brain regions and prevents dopaminergic neuronal loss in the SNpc. hGFAP-cre+/−/IKK2fl/fl (KO) or hGFAP-cre−/−/IKK2fl/fl (WT) animals were intranasally infected with WEEV or mock-infected with saline (KO control) and aged to 8 WPI. (A) Dopaminergic neurons were quantified in the SNpc and microgliosis and astrogliosis were quantified in the cortex (B, E), hippocampus (C, F), and midbrain (D, G). (*p<0.05, ** p<0.005, ***p<0.0005, n=6-8 per group)